

Abstract
A failure of iron to appropriately regulate liver hepcidin production is central to the pathogenesis of hereditary hemochromatosis. SMAD1/5 transcription factors, activated by bone morphogenetic protein (BMP) signaling, are major regulators of hepcidin production in response to iron; however, the role of SMAD8 and the contribution of SMADs to hepcidin production by other systemic cues remains uncertain. Here, we generated hepatocyte Smad8 single (Smad8;Alb-Cre+), Smad1/5/8 triple (Smad158;Alb-Cre+), and littermate Smad1/5 double (Smad15;Alb-Cre+) knockout mice to investigate the role of SMAD8 in hepcidin and iron homeostasis regulation and liver injury. We found that Smad8;Alb-Cre+ mice exhibited no iron phenotype, whereas Smad158;Alb-Cre+ mice had greater iron overload than Smad15;Alb-Cre+ mice. In contrast to the sexual dimorphism reported for wildtype mice and other hemochromatosis models, hepcidin deficiency and extrahepatic iron loading were similarly severe in Smad15;Alb-Cre+ and Smad158;Alb-Cre+ females compared with males. Moreover, epidermal growth factor (EGF) failed to suppress hepcidin in Smad15;Alb-Cre+ hepatocytes. Conversely, hepcidin was still increased by lipopolysaccharide in Smad158;Alb-Cre+ mice, although lower basal hepcidin resulted in lower maximal hepcidin. Finally, unlike most mouse hemochromatosis models, Smad158;Alb-Cre+ developed liver injury and fibrosis at 8 weeks. Liver injury and fibrosis were prevented in Smad158;Alb-Cre+ mice by a low iron diet and were minimal in iron-loaded Cre- mice.
Conclusion:
Hepatocyte Smad1/5/8 knockout mice are a new model of hemochromatosis that encompass liver injury and fibrosis seen in human disease. These mice reveal the redundant, but critical, role of SMAD8 in hepcidin and iron homeostasis regulation; establish a requirement for SMAD1/5/8 in hepcidin regulation by testosterone and EGF, but not inflammation; and suggest a pathogenic role for both iron loading and SMAD1/5/8 deficiency in liver injury and fibrosis.
Keywords: Hemochromatosis, hepcidin, liver fibrosis, testosterone, epidermal growth factor
INTRODUCTION
Iron is tightly regulated for its functional necessity to most living organisms, but toxicity when in excess. The iron hormone hepcidin controls systemic iron homeostasis by triggering degradation of the iron exporter ferroportin, thereby limiting iron entry into the circulation (1). The importance of hepcidin was recognized when mutations were linked to juvenile hemochromatosis, characterized by early onset tissue iron loading that leads to organ dysfunction including cirrhosis, hypogonadism, cardiomyopathy, and diabetes mellitus (2). Subsequently, the inability of iron to appropriately induce hepcidin was recognized as the common pathogenic mechanism for other genetic causes of hemochromatosis, including HFE, transferrin receptor 2 (TFR2), and hemojuvelin (HJV) mutations (3).
BMP-SMAD signaling is a central pathway for hepcidin regulation by iron (4). Tissue iron stores induce BMP6 and BMP2 ligand expression in liver endothelial cells (5–7). Secreted BMPs bind type I and type II BMP receptors and the co-receptor HJV on hepatocytes to phosphorylate receptor-activated SMAD transcription factors (R-SMADs), which complex with common mediator SMAD4, bind the hepcidin promoter and induce transcription (4). HFE and TFR2, which are implicated in sensing serum iron levels (4), are also thought to regulate hepcidin by interacting with the SMAD signaling pathway (8–11).
In addition to iron, numerous other systemic cues are integrated by the liver to regulate hepcidin production (12). Inflammation induces hepcidin to limit iron availability to infectious microorganisms (12). Erythropoietic demand suppresses hepcidin to increase iron availability for erythropoiesis (12). Both testosterone and estrogen suppress hepcidin expression, but through different mechanisms, which may help explain sex differences in iron loading and liver disease progression in hemochromatosis patients (13–16). Several growth factors, including EGF and hepatocyte growth factor (HGF) also suppress liver hepcidin production (17). Interestingly, many of these systemic signals are reported to intersect with the BMP-SMAD pathway to regulate hepcidin, although the molecular mechanisms are not fully understood (4).
Inflammation increases hepcidin production primarily by inducing interleukin-6 (IL-6), which activates STAT3 phosphorylation to stimulate hepcidin transcription (12). BMP R-SMAD phosphorylation is also activated by inflammation (18, 19), and inhibiting BMP-SMAD signaling lowers hepcidin in the context of inflammation (20–23), suggesting crosstalk between the IL-6-STAT3 and BMP-SMAD pathways. One mechanism proposed for this crosstalk is inflammatory-mediated induction of the BMP/TGF-β superfamily ligand Activin B, which can utilize BMP type I receptors to phosphorylate BMP R-SMADs and induce hepcidin in hepatocytes (18, 19). However, inflammation still induces hepcidin in Activin B knockout mice (24), raising questions about the contribution of this pathway in vivo. STAT3 and SMADs were also proposed to interact at the hepcidin promoter (25). However, the functional contribution of BMP R-SMADs to hepcidin induction by inflammation in vivo has not been definitively demonstrated.
Compared with females, male mice exhibit lower basal hepcidin and more severe hepcidin deficiency and consequent extrahepatic iron loading in many hemochromatosis models (5, 6, 15, 26). Testosterone was demonstrated to account for these observed sex differences by suppressing hepcidin expression (15). However, there are conflicting reports regarding how testosterone reduces hepcidin (15, 16). One model proposed that testosterone decreases hepcidin production by enhancing hepatic EGF receptor (EGFR) signaling and reducing R-SMAD phosphorylation (15). In a second study, EGF was also implicated in hepcidin suppression; however, EGF did not impact R-SMAD phosphorylation, but rather reduced R-SMAD nuclear localization (17). Finally, a third group proposed that testosterone promotes the association of R-SMADs with the androgen receptor, thereby interfering with R-SMAD binding to the hepcidin promoter (16). A common thread for all of these studies is crosstalk with the BMP-SMAD pathway; however, a functional role for BMP R-SMADs in hepcidin regulation by testosterone and EGF has not been demonstrated in vivo.
Three R-SMADs, SMAD1, SMAD5, and SMAD8, are activated by BMP signals. Recently, we showed that mice with an ablation of both Smad1 and Smad5 in hepatocytes (Smad15;Alb-Cre+) exhibited hepcidin deficiency and iron overload, demonstrating the essential role of SMAD1/5 signaling in hepcidin regulation and systemic iron homeostasis (27) . SMAD8 was not initially studied because SMAD8 knockdown did not impact hepcidin expression in cell culture screening assays (27). However, SMAD1 and SMAD5 were differently active in vitro and in vivo, likely due to copy number variations in the different model systems (27). Moreover, hepcidin was further suppressed by neutralizing BMP antibodies in Smad15;Alb-Cre+ mice (27), suggesting the presence of residual BMP-SMAD signaling. Notably, SMAD8 has weaker transcriptional activity than SMAD1/5 and was not only reported to stimulate BMP signaling, but also to suppress BMP signaling in some contexts by forming a heterodimer with SMAD1 (28).
Here, we generated hepatocyte-specific Smad8 single- and Smad1/5/8 triple-knockout mice to investigate whether SMAD8 functions to accelerate or antagonize BMP signaling and hepcidin expression in vivo. We also used these mouse models to gain further mechanistic insight into how inflammation, testosterone, and EGF regulate hepcidin production as well as the pathogenesis of liver injury in hemochromatosis.
METHODS
Animals
Animal protocols were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital (MGH). Hepatocyte conditional Smad8 single-knockout (Smad8fl/fl;Alb-Cre+) mice; Smad1, Smad5, and Smad8 triple-knockout (Smad158;Alb-Cre+) mice; Smad1 and Smad5 double-knockout (Smad15;Alb-Cre+) mice; and littermate Cre- controls were generated by crossing mice harboring a LoxP-flanked allele of Smad8 (Smad8fl/wt) on a mixed C57BL/6J x CD1 background (29) (Medical Research Council) with mice expressing a Cre transgene driven by a hepatocyte-specific albumin promoter (5) on a C57BL/6J background (Jackson Laboratory) or previously generated hepatocyte Smad15;Alb-Cre+ mice (27) as described in Fig. S1. After weaning, mice were maintained on a Prolab 5P75 Isopro RMH 3000 house diet (380 ppm iron). Where indicated, mice received a low (2–6 ppm; #TD.80396), sufficient (48 ppm; #TD.80394) or high iron (2% carbonyl iron; #TD.08496) diet (Envigo) for 3–5 weeks. Alternatively, mice received 1 intraperitoneal dose of 1 mg/kg lipopolysaccharides (LPS, serotype 055:B5; Sigma) in PBS, 5 daily oral doses of 200 mg/kg EGFR inhibitor Gefitinib (ZD1839, Selleckchem) in DMSO, or an equivalent volume of vehicle.
RNA isolation, reverse transcription, and qRT-PCR
RNA was isolated using QIAshredder and RNeasy Mini Kit (Qiagen). First-strand cDNA was synthesized from 1 μg RNA using the High capacity RNA-to-cDNA Kit (Applied Biosystems). qPCR was performed using the PowerUp SYBR Green Master Mix on the QuantStudio3 Real-Time PCR system (Applied Biosystems) using primers in Table S1. Relative quantities were determined by the standard curve method. Transcript levels were normalized to Rpl19 as an internal control.
Iron analysis, biochemical assays and hepcidin ELISA
Serum iron and unsaturated iron binding capacity were measured by colorimetric assays (Pointe Scientific) to calculate transferrin saturation according to manufacturer’s instructions. Tissue nonheme iron concentrations (in μg/g wet weight) were determined as described previously (27). Serum alanine aminotransferase (ALT) and total bilirubin were measured by the MGH Veterinary Clinical Pathology Laboratory. Liver hydroxyproline (Sigma MAK008) and malondialdehyde (Abcam ab118970) were determined by colorimetric assays and serum hepcidin by ELISA (Intrinsic LifeSciences HMC-001) according to manufacturers’ instructions.
Primary hepatocyte culture
Primary hepatocytes were isolated and cultured as previously described (27). Where indicated, cells were treated with 20 ng/ml mouse EGF (R&D Systems 2028-EG) in serum-free growth medium for 24 hours, 100 nM LDN-193189 (Sigma SML0559) in 2% 2-hydroxypropyl-β-cyclodextrin (Sigma H107) in 10% FBS growth medium for 24 hours, or 1% FBS serum starvation overnight followed by 2 ng/ml mouse IL-6 (R&D Systems 406-ML) in 1% FBS growth medium for 6 hours.
Histology
Tissues were fixed in 10% formalin for 24 hours, embedded in paraffin, sectioned at 6 μm, and stained for ferric iron (blue) using an iron stain kit with nuclear fast red counterstain according to manufacturer’s instructions (American MasterTech #KTIRO). Collagen was stained for 1 hour using picrosirius red solution (0.1% Direct Red 80 (Sigma #365548) in 1.3% saturated picric acid (Sigma #P6744)) after 8 min haematoxylin nuclei stain. Slides were washed with 0.5% acetic acid twice before dehydration and mounting. Images were acquired using a Zeiss LSM 800 Airyscan confocal microscope. Sirius red positivity was quantitated using ImageJ (NIH) by dividing the total area calculated under polarized light (after subtracting major veins) by total area calculated under brightfield.
Immunoblot
Liver lysates were prepared and immunoblots performed as described previously (27) using rabbit anti-phosphorylated EGFR (1:500; 2234S; Cell Signaling), rabbit anti-EGFR (1:1000; 4267S; Cell Signaling), rabbit anti-phosphorylated SMAD5 (1:500; ab92698; Abcam), rabbit anti-SMAD5 (1:1000; ab40771; Abcam), or mouse anti-actin (1:20,000 MAB1501; Millipore) antibodies. Chemiluminescence quantification of scanned films was performed using ImageJ 1.46v.
Statistics
Data are presented as scatter plots or box plots. Means were compared by Student’s paired or unpaired t test, one-way or two-way ANOVA with Tukey’s post-hoc test using Prism 7 (GraphPad). Data with unequal variances were log transformed prior to statistical analysis. P < 0.05 was considered significant.
RESULTS
Liver Smad8 mRNA expression is regulated by dietary iron in wildtype mice
Since SMAD8 is positively regulated by BMP signaling in vitro (27) and liver BMP signaling is regulated by dietary iron in mice (30), we examined the effects of a low (2–6 ppm), sufficient (48 ppm) or high iron (2% carbonyl) diet on Smad8 mRNA expression in C57BL/6 wildtype male mice. Mice fed a high iron diet had a 5-fold increase, whereas mice fed a low iron diet had a 45% reduction, in Smad8 mRNA levels compared to mice on an iron sufficient diet (Control, Fig. 1A).
Fig. 1. Iron regulates liver Smad8 expression, but hepatocyte Smad8 conditional knockout mice (Smad8fl/fl;Alb-Cre+) exhibit minimal to no iron loading.

(A) Relative Smad8 expression was measured by qRT-PCR in livers of 7-week-old C57BL/6 male mice after receiving a low (2–6 ppm), sufficient (48 ppm; Control) or high iron (2% carbonyl) diet for 3 weeks (n= 8 per group). (B, top) Schematic depictions of the lox-P-flanked (floxed) Smad8 allele and the allele after Cre recombinase-mediated excision. F and R indicate forward and reverse primers used for PCR genotyping. (B, bottom) PCR analysis of genomic DNA extracted from total liver of Smad8fl/fl;Alb-Cre+ mice and littermate Cre- controls at 8 weeks of age. (C) Relative Smad8 mRNA levels in the total livers of Smad8fl/fl;Alb-Cre+ mice and littermate Cre- controls was measured by qRT-PCR to confirm hepatocyte Smad8 ablation in conditional knockout mice (n= 5–8 per group). (D) Serum iron (left), transferrin saturation (Tf sat, right), (E) hepatic nonheme iron concentrations (left) and liver hepcidin (Hamp) expression (right) were quantified in 8-week-old male and female Smad8fl/fl;Alb-Cre+ mice compared with their respective littermate Cre- controls (n= 5–8 per group). (F) Liver Smad8 and Hamp mRNA were measured by qRT-PCR in male and female Smad8fl/fl;Alb-Cre+ mice and littermate Cre- controls at 10 days of age (n=4–7 per group). All transcript levels were normalized to Rpl19, and the average of the respective littermate Cre- control mice was set to 1. Data are presented as scatter plots with mean ± SEM or box plots with min to max whiskers. *P < 0.05, **P < 0.01, ***P < 0.001 relative to mice fed an iron sufficient diet by one-way ANOVA with Tukey’s post hoc test or to the respective Cre- controls by Student’s t test.
Hepcidin expression and iron levels are not changed in hepatocyte Smad8fl/fl;Alb-Cre+ single-knockout mice
Next, we generated mice with a conditional knockout of hepatocyte Smad8 to determine its functional role in hepcidin regulation and iron homeostasis in vivo. Excision of LoxP-flanked Smad8 was confirmed by PCR of total liver genomic DNA (Fig. 1B). qRT-PCR demonstrated >95% lower total liver Smad8 mRNA levels in Smad8fl/fl;Alb-Cre+ compared with Cre- mice (Fig. 1C). Serum iron, serum transferrin saturation, liver iron, and liver hepcidin (Hamp) mRNA levels were unchanged in 8-week-old Smad8fl/fl;Alb-Cre+ compared with Cre- mice, with the exception of a subtle increase in liver iron in Smad8fl/fl;Alb-Cre+ males (Fig. 1D–E).
We previously reported a similar phenotype of minimal to no iron overload with unchanged Hamp expression in 8-week-old hepatocyte single Smad1 (Smad1fl/fl;Alb-Cre+) and Smad5 (Smad5fl/fl;Alb-Cre+) knockout mice (27); however, hepcidin levels were significantly lower in both Cre+ mouse strains compared with Cre- controls before weaning and in primary hepatocyte cultures, when not exposed to the hepcidin stimulatory effect of the standard rodent diet (27). In contrast, Hamp mRNA did not differ in primary hepatocytes (Fig. S2) or 10-day-old Smad8fl/fl;Alb-Cre+ mouse livers (Fig. 1F) compared with Cre- mice. Moreover, hepcidin expression and serum and tissue iron loading were similarly increased in Smad8fl/fl;Alb-Cre+ and Cre- mice fed a high iron diet (Fig. S3). Together, these data suggest that whereas SMAD1 and SMAD5 contribute to basal hepcidin expression, SMAD8 is dispensable for both basal and iron-induced hepcidin expression when SMAD1 and SMAD5 are present. Notably, basal Smad8 expression was 30- and 3-fold lower than Smad1 and Smad5 by quantitative copy number analysis in mouse hepatocytes (Fig. S4).
Hepatocyte Smad158;Alb-Cre+ triple-knockout mice have more severe iron overload than Smad15;Alb-Cre+ double-knockout mice
To investigate how SMAD8 functions in the absence of SMAD1 and SMAD5, we generated littermate hepatocyte Smad158;Alb-Cre+ triple-knockout and Smad15;Alb-Cre+ double-knockout mice on the same genetic background. qRT-PCR demonstrated that total liver Smad1 and Smad5 mRNA levels were equally reduced (>70%) in both Smad15;Alb-Cre+ and Smad158;Alb-Cre+ compared with Cre- controls (Fig. S5A–B). As previously reported (27), liver Smad8 mRNA was modestly reduced (~50%) in Smad15;Alb-Cre+ mice, most likely due to the regulation of SMAD8 by SMAD1/5 signaling, but was greatly reduced (~95%) in Smad158;Alb-Cre+ mice (Fig. S5C). All Cre+ mice exhibited high serum iron, high transferrin saturation, liver iron loading, and low Hamp mRNA levels relative to liver iron content compared with Cre- controls (Fig. 2A–E). However, serum and liver iron were significantly higher in sex-matched Smad158;Alb-Cre+ compared with Smad15;Alb-Cre+ mice (Fig. 2A,C). Moreover, basal liver Hamp expression and Hamp mRNA relative to liver iron content were both significantly lower in Smad158;Alb-Cre+ compared with Smad15;Alb-Cre+ males, with a similar trend in females (Fig. 2D–E). Additionally, Smad158;Alb-Cre+ mice exhibited significant extrahepatic iron loading in pancreas and heart that began as early as 5 weeks (Fig. S6) and progressed at 8 weeks of age (Fig. 3A–C). In contrast, no significant extrahepatic iron loading was seen in 8-week-old Smad15;Alb-Cre+ mice on a house diet (Fig. 3A–C), although extrahepatic iron loading could be induced by high iron diet (Fig. S7). Finally, spleen iron was more dramatically reduced in Smad158;Alb-Cre+ mice compared with Smad15;Alb-Cre+ mice (Fig. 3D). Thus, SMAD8 has an important functional role in hepcidin and iron homeostasis regulation in the absence of SMAD1 and SMAD5.
Fig. 2. Smad1/5/8 triple-knockout mice (Smad158;Alb-Cre+) exhibit more severe hepcidin deficiency and iron overload compared with Smad1/5 double-knockout mice (Smad15;Alb-Cre+).

(A) Serum iron, (B) Tf sat, (C) liver iron concentration, (D) liver Hamp mRNA and (E) liver Hamp mRNA levels relative to liver iron concentration were measured in 8-week-old male and female Smad15;Alb-Cre+ and Smad158;Alb-Cre+ mice and their respective littermate Cre- controls (n=5–8 per group). Transcript levels were normalized to Rpl19, and the average of male Smad15;Alb-Cre- control mice was set to 1. Data are presented as box plots with min to max whiskers. Results were compared across genotype and sex by two-way ANOVA with Tukey’s post-hoc test. Means without a common superscript differ significantly (P < 0.05).
Fig. 3. Eight-week-old Smad158;Alb-Cre+, but not Smad15;Alb-Cre+, mice exhibit extrahepatic iron loading in pancreas, heart and kidney with similar severity in females and males.
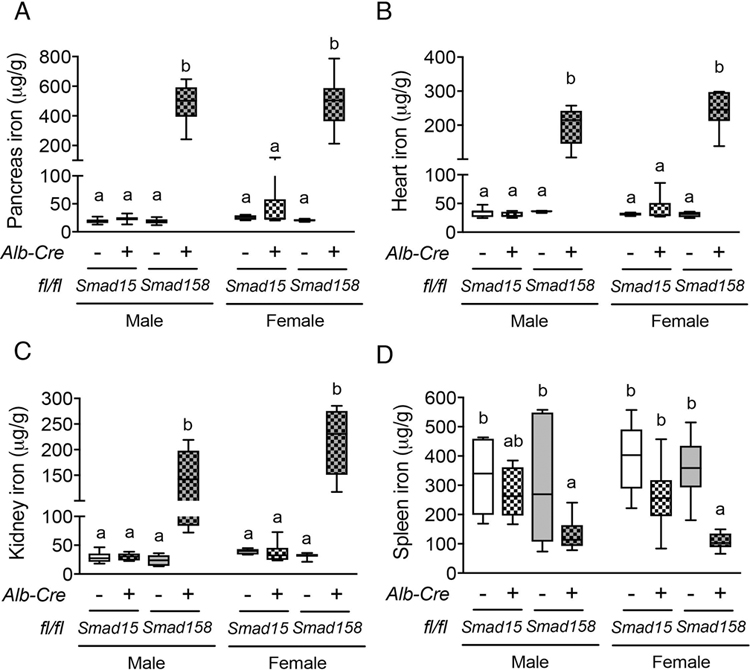
Eight-week-old male and female Smad15;Alb-Cre+ and Smad158;Alb-Cre+ mice and their respective littermate Cre- controls were analyzed for tissue iron in (A) pancreas, (B) heart, (C) kidney and (D) spleen (n=5–8 per group). Data are presented in box plots with min to max whiskers. Results were compared across genotype and sex by two-way ANOVA with Tukey’s post-hoc test. Means without a common superscript differ significantly (P < 0.05).
Hepcidin deficiency and extrahepatic iron loading are not greater in Smad15;Alb-Cre+ and Smad158;Alb-Cre+ males compared to females
Prior studies reported that wildtype males exhibited lower hepcidin expression than females, and lower hepcidin engendered greater extrahepatic iron loading in male compared with female Bmp6, Bmp2, and Hjv global or conditional knockout mice (5, 6, 15, 26). We therefore compared hepcidin and iron homeostasis parameters in male versus female Smad15;Alb-Cre+ and Smad158;Alb-Cre+ mice. As previously reported for wildtype mice (15), basal Hamp mRNA was lower in male compared with female Cre- mice (Fig. S8). However, when normalized to liver iron, Hamp mRNA was not lower in Smad15;Alb-Cre+ or Smad158;Alb-Cre+ males compared with females of the same genotype and in fact was lower in Smad15;Alb-Cre+ females than males (Fig. 2E). Moreover, liver iron levels were higher in both Cre+ females compared with genotype-matched males and serum iron was higher in Smad158;Alb-Cre+ females (Fig. 2A,C). Finally, although extrahepatic iron loading was not seen in 8-week-old Smad15;Alb-Cre+ mice, Smad158;Alb-Cre+ females exhibited similar or greater extrahepatic iron loading as males (Fig. 3A–C, Fig. S6B–C).
EGFR inhibition does not increase hepcidin in male mice
Testosterone-mediated hepcidin suppression was demonstrated to account for the sexual dimorphism in wildtype and Bmp6 knockout mice (15, 16). In one study, activation of EGFR signaling and suppression of SMAD5 phosphorylation (15) were proposed as mechanisms. Consistent with the prior study (15), male Smad15;Alb-Cre+ and Smad158;Alb-Cre+ mice had increased Egfr mRNA compared to genotype-matched females (Fig. 4A). However, in contrast to the prior report (15), although administration of the EGFR inhibitor gefitinib reduced EGFR phosphorylation (Fig. 4B), gefitinib did not increase Hamp mRNA or SMAD5 phosphorylation in wildtype male mice (Fig. 4C–D). Thus, although the absence of more severe hepcidin deficiency and extrahepatic iron loading in Smad15;Alb-Cre+ and Smad158;Alb-Cre+ males compared with females provides in vivo evidence for a critical functional role for BMP R-SMADs in hepcidin suppression by testosterone, we could not confirm a functional role for EGFR signaling in testosterone-mediated hepcidin suppression.
Fig. 4. The EGFR inhibitor gefitinib does not reverse hepcidin suppression in male mice, but hepatocyte ablation of Smad1 and Smad5 abolishes hepcidin suppression by EGF.

(A) Eight-week-old male and female Smad15;Alb-Cre+ and Smad158;Alb-Cre+ mice and their respective littermate Cre- controls were analyzed for hepatic Egfr mRNA (n=5–8 per group). (B-D) C57BL/6 wildtype male mice at 7 weeks were treated with 200 mg per kg gefitinib or DMSO vehicle via oral gavage for 5 days (n=5–6 per group). Mice were sacrificed 6 hours after the final dose and livers were analyzed for (B) EGFR phosphorylation and (D) SMAD5 phosphorylation levels by immunoblot and chemiluminescence quantitation and (C) Hamp mRNA by qRT-PCR. (E-F) Primary hepatocytes were isolated from 7-week-old Smad15;Alb-Cre+ and littermate Cre- control mice. Hepatocyte Hamp mRNA was analyzed by qRT-PCR in cells treated with (E) 100 nM LDN-193189 in 2% 2-hydroxypropyl-β-cyclodextrin or (F) 20 ng/ml mouse recombinant EGF or vehicle alone for 24 hours. Data from 4 (E) or 3 (F) independent experiments, each performed in triplicate, are shown. Transcripts were normalized to Rpl19 and the average of male Smad15;Alb-Cre- or vehicle-treated Cre- control mice were set to 1. Data are presented in scatter plots with mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 relative to female mice or vehicle-treated control mice of the same genotype or as otherwise noted by Student’s t test.
EGF fails to suppress hepcidin in Smad15;Alb-Cre+ hepatocytes
Although our data suggest that EGFR signaling is not involved in testosterone-mediated hepcidin suppression, EGF itself has been demonstrated to suppress hepcidin expression (17). To determine whether hepcidin suppression by EGF requires BMP R-SMAD signaling, we tested whether EGF reduced Hamp expression in Smad15;Alb-Cre+ primary hepatocytes that have a residual capacity for hepcidin suppression (Fig. 2D), which we confirmed in vitro by BMP type I receptor inhibitor, LDN-193189 (21), treatment (Fig. 4E). Whereas EGF suppressed Hamp mRNA in Cre- primary hepatocytes, it did not suppress Hamp mRNA in Smad15;Alb-Cre+ hepatocytes (Fig. 4F). Collectively, these data provide functional evidence that SMAD1/5 signaling is required for hepcidin suppression by EGF, at least in vitro.
LPS induces hepcidin production in Smad158;Alb-Cre+ mice
IL-6 plays a key role in hepcidin induction by inflammation (12). To investigate whether IL-6-mediated hepcidin induction requires BMP R-SMADs, we tested the effects of LPS injection in Smad158;Alb-Cre+ mice versus Cre- controls. LPS significantly induced endogenous liver Il6 mRNA in both genotypes after 6 hours (Fig. 5A). In Cre- mice, LPS also increased liver Smad1 and Smad5, but suppressed Smad8 mRNA (Fig. 5B–C). Liver Hamp mRNA and serum hepcidin were significantly increased by LPS in both genotypes (Fig. 5D–E). The fold-induction in Hamp mRNA was similar or greater in Cre+ compared to Cre- mice, although the fold-induction in serum hepcidin was less, and lower baseline hepcidin resulted in lower peak hepcidin levels in Cre+ mice. Since LPS also induces inflammatory cytokines other than IL-6, we also measured Hamp mRNA in primary hepatocytes treated with IL-6 and found similar results (Fig. 5F). These data suggest that IL-6 still induces hepcidin in the absence of BMP R-SMADs, although R-SMADs do influence the absolute levels of hepcidin achieved by altering the basal setpoint.
Fig. 5. Inflammation significantly increases hepcidin production in Smad158;Alb-Cre+ mice.

Male and female Smad158;Alb-Cre+ and littermate Cre- control mice (n=6–7 per group) at 6 weeks of age were injected with PBS or LPS (1 mg per kg body weight) and sacrificed after 6 hours. Livers and serum were collected to determine (A) Il6, (B) Smad1/5, (C) Smad8 (D) Hamp mRNA expression by qRT-PCR, and (E) serum hepcidin protein levels by ELISA. (F) Primary hepatocytes isolated from 6-week-old Smad158;Alb-Cre+ and littermate Cre- control mice (n=4 per group) were treated with 2 ng/ml IL-6 for 6 hours and Hamp mRNA levels were determined. Data from 4 independent experiments, each performed in triplicate, are shown. Transcripts were normalized to Rpl19 and the average of PBS-treated Cre- control mice were set to 1. Data are presented as scatter plots with mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 relative to PBS-treated controls of the same genotype by Student’s t test. Fold-change relative to PBS treatment for each genotype are reported in panels D-F.
Smad1/5/8;Alb-Cre+ mice exhibit liver tissue injury and fibrosis at 8 weeks
Tissue iron accumulation in human hemochromatosis patients leads to injury and organ dysfunction (3). Although mouse models of hemochromatosis phenocopy the iron overload seen in humans, most mouse models do not develop tissue injury and fibrosis (31–34). The severity of liver iron accumulation in Smad158;Alb-Cre+ mice led us to examine for evidence of liver damage and fibrosis. Eight-week-old male and female Smad158;Alb-Cre+ mice exhibited a significant increase in serum ALT (3–6x) and total bilirubin compared to Cre- controls, consistent with liver injury (Fig. 6A–B). Liver malondialdehyde was also increased, indicative of oxidative stress and lipid peroxidation (Fig. 6C). Histologic analysis (Fig. 6D–E, Fig. S9–10) showed scattered apoptotic hepatocytes, confirmed by TUNEL staining, and numerous ceroid-laden macrophages (which have phagocytosed dead hepatocytes), predominantly in the centrilobular (zone 3) region. Hepatocyte mitotic figures were also present, indicative of response to injury. Occasional foci of mononuclear inflammation were present in zone 3, but overall inflammation was mild. These changes were not observed in Cre- controls. Prussian blue staining showed marked hepatocytic iron deposition as well as positive staining of ceroid-laden macrophages in zone 3 in Smad158;Alb-Cre+ mice, but not Cre- mice. Since there was no visible iron staining in sinusoidal Kupffer cells, the iron in ceroid-laden macrophages likely reflects iron originally deposited in damaged, phagocytosed hepatocytes. Picrosirius red staining showed sinusoidal/pericellular fibrosis in zone 3 in the Smad158;Alb-Cre+ mice (5–6x higher than Cre- mice), indicating chronic injury to hepatocytes. Additional indicators of liver fibrosis, collagen type I alpha 1 (Col1a1) mRNA and hydroxyproline, were also increased in Smad158;Alb-Cre+ mice compared to Cre- controls (Fig. 6F). Collectively, the histopathologic findings in Smad158;Alb-Cre+ mice reveal hepatocytic iron deposition, accompanied by active and chronic hepatocyte injury in a predominantly zone 3 distribution. In contrast to the liver, we did not find biochemical or histologic evidence of injury in the pancreas or heart of Smad158;Alb-Cre+ mice, despite iron loading (Fig. S11–12).
Fig. 6. Smad158;Alb-Cre+ mice develop liver injury and fibrosis at 8 weeks of age.
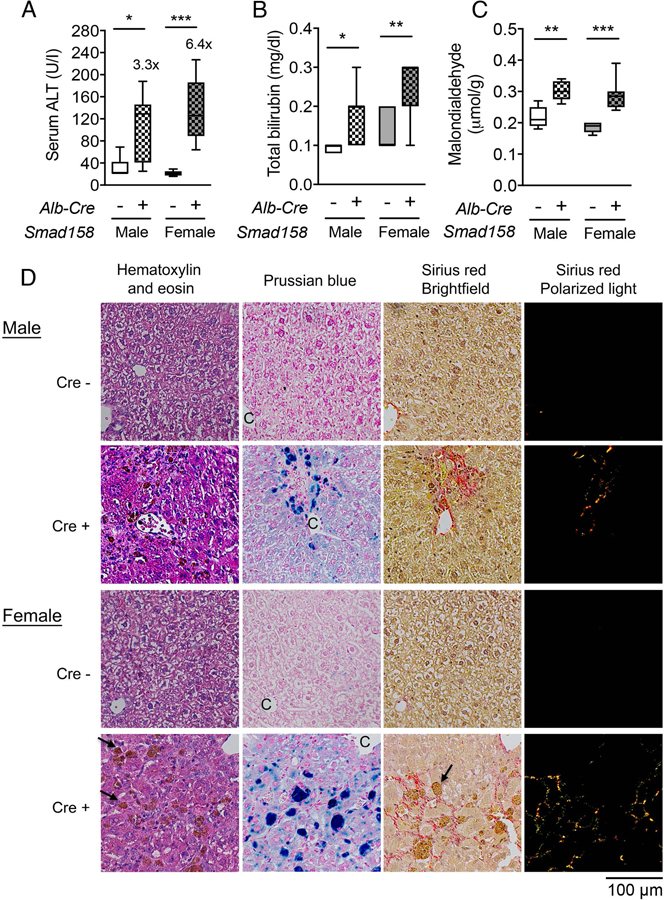
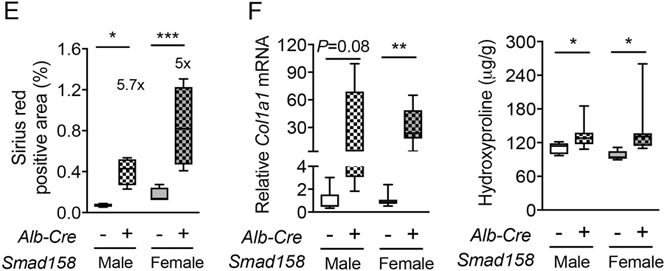
Male and female Smad158;Alb-Cre+ and littermate Cre- control mice were sacrificed at 8 weeks of age (n=6–8 per group). Serum was collected to measure (A) alanine aminotransferase (ALT) and (B) total bilirubin. (C, F) Liver malondialdehyde, hydroxyproline, and Col1a1 relative to Rpl19 mRNA levels were quantified by colorimetric assays or qRT-PCR. (D) Liver sections from a subset of mice (n=4 per group) were stained with hematoxylin and eosin, picrosirius red for collagen formation, or Perls’ Prussian blue for tissue iron. Branches of the central vein (C) and ceroid-laden macrophages (arrows) are indicated. Images were taken under the brightfield and/or polarized light, and representative images are shown. (E) The sirius red positive area was quantitated by dividing total area calculated in polarized light (after subtraction of major veins) by total area calculated under the brightfield (n=4 per group). Data are presented as box plots with min to max whiskers. For qRT-PCR, the average of male Cre- control mice was set to 1. *P < 0.05, **P < 0.01, ***P < 0.001 relative to their respective Cre- controls by Student’s t test.
Hepatic iron loading and SMAD1/5/8 deficiency both contribute to liver injury and fibrosis in Smad158;Alb-Cre+ mice
To investigate the cause(s) of liver injury and fibrosis in Smad158;Alb-Cre+ mice, we examined liver injury parameters and histology in 8-week-old Smad15;Alb-Cre+ mice versus Cre- controls. Serum ALT was 3-fold higher in Smad15;Alb-Cre+ females compared with Cre- controls with some features of liver injury by histologic analysis, although picrosirius red quantitation and Col1a1 mRNA levels were not significantly increased due to large animal-to-animal variability (Fig. S13). Serum ALT, liver Col1a1 mRNA, and picrosirus red were significantly increased in both male and female Smad15;Alb-Cre+ mice fed a high iron diet (Fig. S13). Thus, Smad8 deletion per se is not required for liver injury.
Notably, the degree of liver injury and fibrosis in Cre+ mice (Fig. 6, Fig. S13) paralleled the degree of liver iron loading, which was most severe in Smad158:Alb-Cre+ females, followed by Smad158:Alb-Cre+ males and Smad15:Alb-Cre+ females, followed by Smad15:Alb-Cre+ males (Fig. 2C). Moreover, serum ALT was only modestly increased in females and Col1a1 mRNA was not increased in 5-week-old Smad158;Alb-Cre+ mice whose livers were less iron loaded (Fig. S6) than 8-week-old Smad158;Alb-Cre+ mice. To confirm whether iron loading plays a pathogenic role in liver injury and fibrosis, we evaluated Smad158;Alb-Cre+ mice and Cre- controls maintained on a low iron diet between 4 and 8 weeks of age to minimize liver iron loading. Although serum and liver iron levels were still higher in low iron diet Smad158;Alb-Cre+ mice compared with Cre- controls (Fig. 7A–C) in the context of hepcidin deficiency (Fig. 7D), liver iron levels were much lower in Smad158;Alb-Cre+ mice on a low iron diet (~200 µg/g, Fig. 7C) compared with a house diet (~2200 µg/g, Fig 2C). Importantly, serum ALT and liver Col1a1 mRNA were not increased in low iron diet Smad158;Alb-Cre+ mice compared to Cre- mice (Fig. 7E–F).
Fig. 7. A low iron diet prevents liver injury and fibrosis in Smad158;Alb-Cre+ mice.
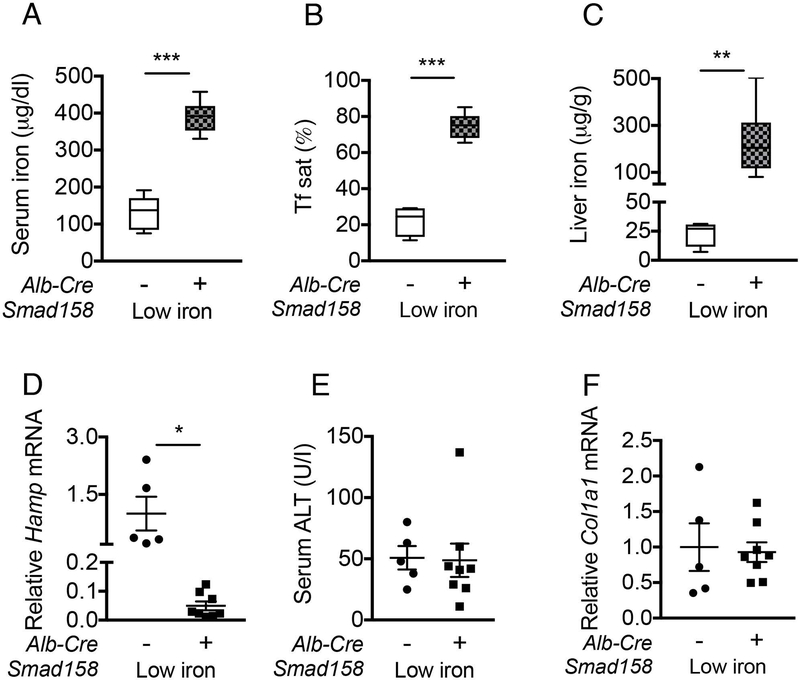
Four-week-old male and female Smad158;Alb-Cre+ and littermate Cre- control mice were treated with a low iron diet for 4 weeks (n=5–8 per group). At 8 weeks of age, serum and livers were collected to determine (A) serum iron, (B) serum Tf sat, (C) liver iron, and (E) serum ALT. (D) Liver Hamp and (F) liver Col1a1 mRNA levels were measured by qRT-PCR. Transcript levels were normalized to Rpl19, and the average of Cre- control mice was set to 1. Data are presented as scatter plots with mean ± SEM or box plots with min to max whiskers. *P < 0.05, **P < 0.01, ***P < 0.001 relative to Cre- control mice on a low iron diet by Student’s t test.
To explore whether a deficiency in SMAD1/5/8 signaling also contributes to liver injury and fibrosis, 3-week-old Cre- control mice received a high iron diet for 5 weeks to induce serum and liver iron loading to similar levels as Smad158;Alb-Cre+ mice on a house diet (Fig. 8A). As expected, Hamp mRNA was increased in Cre- mice fed a high iron diet, compared to both Cre- and Cre+ mice on a house diet (Fig. 8B). Interestingly, despite similar liver iron loading, serum ALT, liver Col1a1 mRNA, liver picrosirius red staining, and other histologic evidence of hepatocyte injury were significantly reduced in high iron diet Cre- females compared to house diet Smad158;Alb-Cre+ females, with similar trends seen in males (Fig. 8C–F). Collectively, these data suggest that hepatocyte Smad1/5/8 ablation contributes to the development of liver injury and fibrosis in response to iron loading.
Fig. 8. Hepatocyte ablation of Smad1/5/8 worsens iron-induced liver injury and fibrosis in mice.

Three-week-old male and female Smad15;Alb-Cre- mice were fed a high iron (2% carbonyl iron) diet for 5 weeks (Cre- +Fe, n=4–8 per group). At 8 weeks of age, tissues were collected to measure (A) serum iron and liver iron and (C) serum ALT. (B) Liver Hamp and (D) Col1a1 relative to Rpl19 mRNA levels were measured by qRT-PCR. Data are presented as box plots with min to max whiskers. Results from Smad158;Alb-Cre+ mice on a house diet from Figure 2 and Figure 6 were replotted for comparison (TKO). The dotted lines represent the average of all Cre- mice on a house diet from Figure 2 and 6 for reference. For qRT-PCR, results are normalized to the average of male Smad158;Alb-Cre- control mice on a house diet, which was set to 1. #P < 0.05, ##P < 0.01, ###P < 0.001 relative to Cre- mice on a house diet by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001 relative to sex-matched TKO mice by Student’s t test. (E) In a subset of mice (n=4 per group), liver sections were stained with hematoxylin and eosin, picrosirius red for collagen formation, or Perls’ Prussian blue for tissue iron. Images were taken under the brightfield and/or polarized light, and representative images are shown. (F) The sirius red positive area was quantitated by dividing total area calculated in polarized light (after subtraction of major veins) by total area calculated under the brightfield (n=4 per group). Data in panel F are presented and analyzed as in panels A-D.
DISCUSSION
BMP R-SMADs are major transcriptional regulators of hepcidin to govern systemic iron homeostasis (4); however, the relative contribution of SMAD1, SMAD5, and SMAD8 has been uncertain. We recently demonstrated that SMAD1 and SMAD5 have critical, but redundant and dose-dependent roles in hepcidin and iron homeostasis regulation since hepatocyte ablation of both Smad1 and Smad5 in mice resulted in hepcidin deficiency and iron overload, whereas ablation of either Smad1 or Smad5 reduced basal hepcidin levels, but only marginally impacted iron- and BMP-dependent hepcidin induction (27). Here, we demonstrated that unlike SMAD1 or SMAD5, SMAD8 is dispensable for basal hepcidin expression since hepcidin levels were unchanged in Smad8fl/fl;Alb-Cre+ mice. Although liver Smad8 expression was regulated by dietary iron, Smad8fl/fl;Alb-Cre+ mice responded appropriately to a dietary iron challenge. This dispensability of SMAD8 in the presence of SMAD1 and SMAD5 may be due to the lower expression of Smad8 compared with Smad1 and Smad5 in mouse hepatocytes. SMAD8 was also reported to have reduced transcriptional activity compared with SMAD1 and SMAD5 (28). Although not required for hepcidin regulation in the presence of SMAD1 and SMAD5, SMAD8 does have a critical role in hepcidin transcription in the absence of SMAD1 and SMAD5, since triple-knockout Smad158;Alb-Cre+ mice had lower hepcidin expression than double-knockout Smad15;Alb-Cre+ mice. Moreover, lower hepcidin in Smad158;Alb-Cre+ mice was associated with increased iron overload, including the development of extrahepatic iron loading. A similar pattern of lower hepcidin leading to extrahepatic iron overload has been observed in other mouse hemochromatosis models, attributed to a greater or faster accumulation of non-transferrin-bound iron that overwhelms the liver’s uptake capacity (5, 15).
The Smad158;Alb-Cre+ mice provide a novel model to study the mechanisms of liver injury and fibrosis in hemochromatosis. In contrast to humans (3), liver injury and fibrosis are not reported in most mouse hemochromatosis models (31–34). For example, no liver injury or fibrosis were reported in mice lacking Hfe or Hjv, even after a high iron diet (31, 34). Hamp knockout mice developed ALT elevations only after receiving a high iron diet for 5 months and liver fibrosis was not detected until 12 months (32). Hfe/Tfr2 double-mutant mice were reported to develop ALT elevation and portal fibrosis, but the changes were relatively modest (80% increase in ALT, 3.5-fold increase in collagen) at 11 weeks (33). There was a brief mention of liver injury and inflammation in 8-month-old hepatocyte Smad4 knockout mice, but this was poorly characterized (20). In the present study, 8-week-old Smad158;Alb-Cre+ mice were found to have 3–6-fold higher ALT levels, with a histopathologic correlate of apoptotic hepatocytes, ceroid-laden macrophages, and increased collagen formation compared to Cre- controls. It is difficult to quantitatively compare the Smad158;Alb-Cre+ mice and other hemochromatosis models because they are on different background strains, which influence iron loading severity (35), and some models are not fully characterized. However, our data suggest that at least 2 factors contribute to the pathogenesis of liver injury and fibrosis in Smad158;Alb-Cre+ mice. Iron overload is required since liver injury and fibrosis were prevented by a low iron diet. SMAD1/5/8 deficiency also plays a pathogenic role since Cre- mice with similar iron overload as Smad158;Alb-Cre+ were protected from liver injury and fibrosis. This was not specifically a consequence of Smad8 deficiency since Smad15;Alb-Cre+ mice also exhibited some degree of liver injury. Notably, HFE, TFR2, and HJV mutations are also associated with reduced hepatic SMAD1/5/8 signaling (8–11, 36), raising the hypothesis that deficient SMAD1/5/8 signaling could contribute to the pathophysiology of liver disease in human patients with hereditary hemochromatosis. There were some differences between the pattern of hepatocyte injury and fibrosis in Smad158;Cre+ mice, which was predominantly centrilobular (zone 3), compared with human hemochromatosis patients, where injury and fibrosis typically begin in the portal/periportal region (zone 1) before progressing to bridging zone 1-zone 3 fibrosis and cirrhosis. The explanation for these apparent differences will require additional investigation.
Our study helps to clarify the role of BMP-SMAD1/5/8 signaling in hepcidin induction by inflammation. It was previously proposed that SMAD1/5/8 signaling is required for hepcidin induction by LPS/IL-6 via crosstalk at the receptor and/or intracellular levels since LPS/IL-6 still induced hepcidin in Bmp6−/− or Hjv−/− mice (37, 38), but failed to induce hepcidin in hepatocyte Alk3 or Smad4 knockout mice (20, 23). However, we demonstrated here that in the absence of hepatocyte Smad1/5/8, hepcidin was still induced by LPS in vivo and IL-6 in vitro. These data demonstrate that BMP R-SMADs are not required for hepcidin induction by LPS/IL-6. The apparent lack of hepcidin induction by IL-6 in the Alk3 and Smad4 mice may be due to differences in experimental protocol. Alternatively, there could be signals governed by SMAD1/5/8-independent, but ALK3- or SMAD4-dependent, pathways. For example, ALK3 can activate non-SMAD pathways and TGF-beta and Activin ligands require SMAD4 but utilize SMAD2/3 rather than SMAD1/5/8 (39). Although our study was limited by the use of conditional knockout models, we previously demonstrated >99% recombination efficiency with the Alb-Cre (5) and both the Alk3 and Smad4 mice have the same limitation (20, 23). Notably, although LPS and IL-6 increased hepcidin in Smad1/5/8-deficient hepatocytes, basal hepcidin levels were dramatically lower resulting in lower peak hepcidin levels. Thus, although not required for hepcidin induction by LPS/IL-6, SMAD1/5/8 does influence final hepcidin levels achieved by altering the basal setpoint. These data are consistent with the hepcidin lowering effects reported for BMP-SMAD1/5/8 inhibitors in animal models of anemia of inflammation (21, 22).
The Smad158;Alb-Cre+ mice have also enabled new insights into the mechanisms of hepcidin regulation by testosterone and EGF. Multiple groups demonstrated that the hepcidin suppressive effects of testosterone account for lower hepcidin expression in male compared with female mice (15, 16), and this is proposed to account for the greater extrahepatic iron overload seen in Bmp6 (5, 15), Hjv (26), and Bmp2 (6) global or conditional knockout mice. Here, we demonstrated that hepcidin expression was not lower and extrahepatic iron loading was not increased in male compared with female Smad15;Alb-Cre+ or Smad158;Alb-Cre+ mice, providing the first in vivo evidence of a crucial functional role for BMP R-SMADs in hepcidin regulation by testosterone. Interestingly, Hamp expression was lower in Smad15;Alb-Cre+ females compared with males, which may be attributed to the hepcidin suppressive effects of estrogen that acts directly via an estrogen response element on the hepcidin promoter (13, 14). Notably, our data did not corroborate one proposed model that testosterone suppresses hepcidin by enhancing EGFR signaling (15), since EGFR inhibition did not reverse hepcidin suppression in male mice in our study. Our data are consistent with another proposed model that testosterone sequesters SMAD1 and SMAD4 with the androgen receptor to reduce their association with the hepcidin promoter (16), although this requires further validation in vivo. These findings may help shed light on some of the sex differences in iron loading and liver disease progression in hemochromatosis patients.
Our data also provide the first functional evidence of a critical role for BMP R-SMADs in hepcidin suppression by EGF. These findings are consistent with a prior study demonstrating that EGF reduces nuclear localization of BMP R-SMADs in cell culture (17). Another growth factor, HGF, was also reported to increase expression of the SMAD co-repressor TG-interacting factor (17). Future studies will be needed to understand the relative contribution of these proposed mechanisms in vivo. These studies may have implications for developing new therapies for chronic liver diseases, where hepcidin suppression by EGF has been postulated to contribute to hepcidin deficiency and iron loading, which correlates with poorer outcomes (17, 40).
In summary, Smad158;Alb-Cre+ mice are a new model of hemochromatosis that establish the redundant, but critical, role for all BMP R-SMADs in hepcidin and iron homeostasis regulation. This model reveals the crucial role of BMP R-SMADs in hepcidin suppression by EGF and the sexual dimorphism reported in some mouse models of hemochromatosis, which is independent of EGFR signaling. Additionally, although BMP R-SMADs contribute to elevated hepcidin levels in the context of inflammation, this is mainly as a consequence of reducing basal hepcidin levels rather than impacting the IL-6 response. Finally, our data suggest a functional role for SMAD1/5/8 deficiency in liver injury and fibrosis in the context of iron overload.
Supplementary Material
Acknowledgments
Financial Support
This work was supported by NIH grant RO1-DK087727 to JLB. CW is supported in part by a Cooley’s Anemia Foundation Research Fellowship.
List of Abbreviations
- EGF
epidermal growth factor
- TFR2
transferrin receptor 2
- HJV
hemojuvelin
- BMP
bone morphogenetic protein
- R-SMADs
receptor-activated SMAD transcription factors
- HGF
hepatocyte growth factor
- IL-6
interleukin-6
- EGFR
epidermal growth factor receptor
- LPS
lipopolysaccharides
Footnotes
Disclosures
JLB has ownership interest in Ferrumax Pharmaceuticals and has received consulting fees from Keryx Biopharmaceuticals and Disc Medicine. All other authors have nothing to declare.
REFERENCES
- 1.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090–2093. [DOI] [PubMed] [Google Scholar]
- 2.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003;33:21–22. [DOI] [PubMed] [Google Scholar]
- 3.Brissot P, Pietrangelo A, Adams PC, de Graaff B, McLaren CE, Loreal O. Haemochromatosis. Nat Rev Dis Primers 2018;4:18016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang CY, Babitt JL. Liver iron sensing and body iron homeostasis. Blood 2018. [DOI] [PMC free article] [PubMed]
- 5.Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2016. [DOI] [PMC free article] [PubMed]
- 6.Canali S, Wang CY, Zumbrennen-Bullough KB, Bayer A, Babitt JL. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am J Hematol 2017;92:1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch PS, Olsavszky V, Ulbrich F, Sticht C, Demory A, Leibing T, et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2016. [DOI] [PMC free article] [PubMed]
- 8.Corradini E, Garuti C, Montosi G, Ventura P, Andriopoulos B, Jr., Lin HY, et al. Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis. Gastroenterology 2009;137:1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009;50:1992–2000. [DOI] [PubMed] [Google Scholar]
- 10.Ryan JD, Ryan E, Fabre A, Lawless MW, Crowe J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology 2010;52:1266–1273. [DOI] [PubMed] [Google Scholar]
- 11.Corradini E, Rozier M, Meynard D, Odhiambo A, Lin HY, Feng Q, et al. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 2011;141:1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell 2017;168:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou Y, Zhang S, Wang L, Li J, Qu G, He J, et al. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene 2012;511:398–403. [DOI] [PubMed] [Google Scholar]
- 14.Yang Q, Jian J, Katz S, Abramson SB, Huang X. 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 2012;153:3170–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latour C, Kautz L, Besson-Fournier C, Island ML, Canonne-Hergaux F, Loreal O, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 2014;59:683–694. [DOI] [PubMed] [Google Scholar]
- 16.Guo W, Bachman E, Li M, Roy CN, Blusztajn J, Wong S, et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 2013;12:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology 2012;56:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, Roth MP, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012;120:431–439. [DOI] [PubMed] [Google Scholar]
- 19.Canali S, Core AB, Zumbrennen-Bullough KB, Merkulova M, Wang CY, Schneyer AL, et al. Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice. Endocrinology 2016;157:1146–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005;2:399–409. [DOI] [PubMed] [Google Scholar]
- 21.Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011;118:4977–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinbicker AU, Sachidanandan C, Vonner AJ, Yusuf RZ, Deng DY, Lai CS, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011;117:4915–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayeur C, Lohmeyer LK, Leyton P, Kao SM, Pappas AE, Kolodziej SA, et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 2014;123:2261–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besson-Fournier C, Gineste A, Latour C, Gourbeyre O, Meynard D, Martin P, et al. Hepcidin upregulation by inflammation is independent of Smad1/5/8 signaling by activin B. Blood 2017;129:533–536. [DOI] [PubMed] [Google Scholar]
- 25.Verga Falzacappa MV, Casanovas G, Hentze MW, Muckenthaler MU. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J Mol Med (Berl) 2008;86:531–540. [DOI] [PubMed] [Google Scholar]
- 26.Latour C, Besson-Fournier C, Gourbeyre O, Meynard D, Roth MP, Coppin H. Deletion of BMP6 worsens the phenotype of HJV-deficient mice and attenuates hepcidin levels reached after LPS challenge. Blood 2017;130:2339–2343. [DOI] [PubMed] [Google Scholar]
- 27.Wang CY, Core AB, Canali S, Zumbrennen-Bullough KB, Ozer S, Umans L, et al. Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice. Blood 2017;130:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep 2014;4:7596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold SJ, Maretto S, Islam A, Bikoff EK, Robertson EJ. Dose-dependent Smad1, Smad5 and Smad8 signaling in the early mouse embryo. Dev Biol 2006;296:104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood 2008;112:1503–1509. [DOI] [PubMed] [Google Scholar]
- 31.Lebeau A, Frank J, Biesalski HK, Weiss G, Srai SK, Simpson RJ, et al. Long-term sequelae of HFE deletion in C57BL/6 × 129/O1a mice, an animal model for hereditary haemochromatosis. Eur J Clin Invest 2002;32:603–612. [DOI] [PubMed] [Google Scholar]
- 32.Lunova M, Goehring C, Kuscuoglu D, Mueller K, Chen Y, Walther P, et al. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J Hepatol 2014;61:633–641. [DOI] [PubMed] [Google Scholar]
- 33.Delima RD, Chua AC, Tirnitz-Parker JE, Gan EK, Croft KD, Graham RM, et al. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron-induced liver injury in mice. Hepatology 2012;56:585–593. [DOI] [PubMed] [Google Scholar]
- 34.Padda RS, Gkouvatsos K, Guido M, Mui J, Vali H, Pantopoulos K. A high-fat diet modulates iron metabolism but does not promote liver fibrosis in hemochromatotic Hjv(−)/(−) mice. Am J Physiol Gastrointest Liver Physiol 2015;308:G251–261. [DOI] [PubMed] [Google Scholar]
- 35.Fleming RE, Holden CC, Tomatsu S, Waheed A, Brunt EM, Britton RS, et al. Mouse strain differences determine severity of iron accumulation in Hfe knockout model of hereditary hemochromatosis. Proc Natl Acad Sci U S A 2001;98:2707–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006;38:531–539. [DOI] [PubMed] [Google Scholar]
- 37.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009;41:478–481. [DOI] [PubMed] [Google Scholar]
- 38.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 2005;115:2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem 2010;147:35–51. [DOI] [PubMed] [Google Scholar]
- 40.Fargion S, Valenti L, Fracanzani AL. Beyond hereditary hemochromatosis: new insights into the relationship between iron overload and chronic liver diseases. Dig Liver Dis 2011;43:89–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.