

Abstract
Antibodies that specifically bind polyethylene glycol (PEG), i.e. anti-PEG antibodies (APA), are associated with reduced efficacy and increased risk of serious adverse events for several PEGylated therapeutics. Here, we explored the concept of using free PEG molecules to saturate circulating APA. Surprisingly, we found that 40 kDa free PEG effectively restored the prolonged circulation of PEGylated liposomes in the presence of high titers of pre-existing APA for at least 48 hours in mice. In contrast, lower molecular weight free PEG (≤ 10 kDa) failed to restore circulation beyond a few hours. These in vivo results were consistent with estimates from a minimal physiologically based pharmacokinetic model. Importantly, the infusion of free PEG appeared to be safe in mice previously sensitized by injection of PEGylated liposomes, and free PEG did not elicit excess APA production even in mice with pre-existing adaptive immunity against PEG. Our results support further investigation of high molecular weight free PEG as a potential method to control and overcome high titers of APA, restoring the prolonged circulation of PEGylated liposomes and possibly other PEGylated therapeutics.
Keywords: polyethylene glycol antibodies, anti-PEG antibodies, PEGylation, polyethylene glycol, antidrug antibodies, drug delivery, accelerated blood clearance, nanoparticles
Introduction
The ability of polyethylene glycol (PEG) to improve the pharmacokinetic profile of various protein and nanoparticle therapeutics has led to its popular use in a wide variety of drug products [1–3]. Indeed, PEGylation of drug molecules increases the aqueous solubility of hydrophobic drugs, improving colloidal stability and reducing aggregation. PEGylation also increases hydrodynamic size, reducing renal clearance of otherwise small therapeutics [4]. As PEG chains are highly flexible, they can sterically inhibit interactions with immune system blood components, such as opsonins and degradative enzymes [4]. This frequently reduces the immunogenicity and antigenicity compared to the underlying therapeutic drugs or particles [5]. These various “stealth” effects of PEG grafting enable less frequent dosing to patients, increased safety, and improved therapeutic outcomes.
Although PEGylation is intended to increase the half-life of drugs, the repeated injection of PEGylated liposomes and selected PEGylated proteins can cause subsequent injections to be rapidly eliminated from circulation, a phenomenon called the accelerated blood clearance (ABC) effect [6–8]. Similarly, the administration of PEGylated microbubbles (an ultrasound contrast enhancing agent) also led to rapid clearance of subsequent doses of microbubbles in rats [9]. This ABC effect occurs even in mice lacking T cells, but does not occur in mice without spleens, suggesting that APA-mediated ABC occurs as a T-independent response, likely mediated by marginal zone B cells in the spleen [10, 11]. Recent rodent studies have suggested that the immunogenicity of the conjugated protein or nanoparticle is correlated with the titer of anti-PEG antibodies (APA) that are induced [12]. The association of ABC with APA has now been extensively documented for a number of PEGylated therapeutics in human studies [13–18]. In a study of PEG-asparaginase, roughly one-third of treated patients were found to be APA-positive by serology and the presence of APA was associated with poor therapeutic efficacy [15]. Similarly, about 40% of patients with chronic refractory gout that were on a pegloticase regimen developed APA that led to rapid clearance of the enzyme from the blood [13, 14]. In addition to infusion reactions, APA have been associated with serious adverse events, including anaphylactic responses that contributed to discontinuation of the use of the PEGylated RNA aptamer pegnivacogin in phase III clinical trials [16, 17].
With the large number of PEGylated therapeutics that are approved or in clinical development and the high costs of bringing alternative treatments to the market, there is a need to restore the safe and effective use of existing PEGylated therapeutics in patients who produce high levels of APA. Here, we decided to test a simple yet non-obvious approach: using an infusion of free PEG molecules to bind and saturate circulating APA, which hypothetically provides a window for the efficacious use of PEGylated therapeutics. The duration and extent to which free PEG can saturate circulating APA depends on both the amount and the MW of the infused PEG. Lower MW PEG (~2-10 kDa) is frequently used in PEGylating proteins and drug carriers and would be expected to limit the formation of large immune complexes relative to higher MW free PEG [19]. However, high MW PEG undergoes slower renal clearance, and thus offers the possibility of extending the window of effective APA saturation [20, 21]. We hypothesized that, with a substantial molar excess of infused free PEG, APA could be effectively saturated while minimizing the formation of immune complexes (even with very high MW PEG). Here, we showed that infusion of 40 kDa PEG effectively restored the prolonged circulation of PEGylated liposomes without causing toxicity or excess stimulation of humoral anti-PEG responses in mice.
Methods
Minimal physiologically based pharmacokinetic model
We previously developed a minimal physiologically based pharmacokinetic (mPBPK) model for the purpose of predicting the interactions between APA and PEGylated entities in both mice and humans [22]. Briefly, we used MATLAB to simulate the behavior of individual APA and PEGylated drug molecules by assigning them association and dissociation constants, volumes of diffusion, rates of transfer between plasma and interstitial fluid, and rates of elimination in bound and unbound forms, all of which were taken from published literature or independent experiments. We adapted the model to demonstrate the effects of administering a dose of free PEG (of varying MW and amounts) into mice with defined APA concentrations that can change over time. The primary model output is a prediction of the kinetics and clearance of a subsequently-infused PEGylated drug. All model parameters and assumptions were identical to our previously published methods, except for the addition of free PEG, for which circulation kinetics were simplified as single-compartment behavior and were adapted from McSweeney et al, 2018 [21].
Mouse model of PEGylated liposomal doxorubicin (PLD) clearance
Adult BALB/c mice (18-22 weeks, female, Charles River) were passively immunized with APA by the intravenous administration of mouse anti-PEG IgG1 (Silver Lake Research, CH2076, lot K0868) 24 hours prior to the intravenous administration of PLD (3 mg/kg, Doxil®, Janssen Products, LP). The administration of 30 μg of APA resulted in a plasma concentration of about 7 μg/mL at the time of PLD injection. In test groups, free PEG was administered intravenously (2,200 mg/kg for PEG 10 kDa, 550 mg/kg for PEG 20 kDa and 40 kDa) to mice 0.5 hours prior to the administration of PLD (3 mg/kg). Doses of PEG were administered and analyzed in mg/kg instead of μmole/kg in order to normalize for the number of repeating ethylene oxide backbone units regardless of PEG chain length. When technically possible, free PEG and PLD were administered into contralateral tail veins. At time points of 0.083, 3, 6, 24, 48, and 96 hours following PLD administration, mice were anesthetized using ketamine (100 mg/kg, i.p.) and medetomidine hydrochloride (1 mg/kg, i.p.), and sacrificed via cardiac puncture (into sodium heparin vacutainers) and cervical dislocation. The blood, liver, spleen, and lungs were collected for doxorubicin quantification.
PLD Quantification
The methods used for sample collection, preparation, and analysis of encapsulated and released doxorubicin in plasma and total doxorubicin in tissues after administration of PLD have been previously described [23–26]. Briefly, blood samples were collected in sodium heparin tubes at 0.083, 3, 6, 24, 48, and 96 hr after the administration of PLD. Blood was centrifuged at 1,500g at 4°C for 5 minutes to obtain plasma. Encapsulated and released doxorubicin in plasma were separated using solid phase separation. Tissues were flash frozen using liquid nitrogen and were stored at −80°C until processing. Upon processing, tissues were thawed, weighed, and diluted in a 1:3 ratio with phosphate buffered saline prior to homogenizing with a Precellys 24 bead mill homogenizer (Omni International Inc, Kennesaw,GA). Samples were further processed by addition of 800 μL extraction solution (acetonitrile with 100 ng/mL daunorubicin internal standard) to 200 μL of plasma or tissue homogenate. The samples were vortexed for 10 minutes and then centrifuged at 10,000 g for 10 minutes at 4°C. The supernatant was removed to a clean tube, evaporated to dryness under nitrogen, and reconstituted in 150 μL of 15% acetonitrile in water plus 0.1% formic acid. The samples were vortexed, transferred to autosampler vials, and analyzed by high-performance liquid chromatography with fluorescence detection (HPLC-FL) set to excitation wavelength 490 nm / emission wavelength 590 nm. The HPLC-FL technique had a quantitative range of 10 – 3,000 ng/mL for total doxorubicin in tissues and released doxorubicin in plasma and 300 – 30,000 ng/mL for encapsulated doxorubicin in plasma. Samples that returned a concentration above the quantitative limit were diluted to fall within the quantitative range.
Blood pressure measurement and cardiac rhythm analysis
We continuously measured blood pressure in BALB/c mice (female, age ranging from 2.5 months to 8 months) that were administered free PEG 40 kDa or PBS. Some mice were vaccinated against PEG four weeks prior to this study by i.v. administration of empty PEGylated liposomes (0.1 μmole lipid per kg bodyweight). APA responses were confirmed in these mice via competition ELISA. Blood pressure was measured using a pressure-volume measurement system (AVD 500, Scisense). To do this, mice were anesthetized with 100 mg/kg ketamine and 15 mg/kg xylazine, administered intraperitoneally. Nair was used to remove fur from the anterior neck. The right carotid artery was dissected and ligated to prevent blood flow. A nick was made in the carotid artery inferior to the ligation, and a pressure transducer was inserted to the level of the innominate artery, near the aortic arch. Arterial blood pressure was measured continuously. Baseline pressure measurements were recorded for approximately one minute prior to the intravenous injection of PEG 40 kDa at a dose of 550 mg/kg in PBS in approximately 150 uL total volume, and pressure was recorded for 20 minutes. Pulse rate was determined by the frequency of the arterial pressure change. Treatment allocation of free PEG versus PBS was concealed to the researchers administering injections and performing surgery.
Histopathology and Clinical Laboratory Analysis
8-week old female BALB/c mice (Charles River) were administered free PEG 40 kDa (550mg/kg) or PBS (equal volume) weekly for six weeks. Mice were sensitized against PEG by i.v. injection of 0.1 μmole/kg of lipids in the form of PEGylated liposome. Mice were given weekly injections of free PEG. At the end of the study, mice were sacrificed, and the liver, kidneys, and blood were collected.
Organs were cut in half and one half was flash frozen in OCT media using liquid nitrogen. The other half was fixed in 10% neutral buffered formalin for one week at room temperature for routine histology. Tissue frozen in OCT was sectioned using a cryostat at a thickness of 10 μm. Formalin-fixed specimens were then processed, embedded in paraffin wax, sectioned at 4 microns thick, and stained by hematoxylin and eosin (H&E) or Periodic acid-Schiff (PAS). Blood was collected by cardiac puncture into EDTA tubes for whole blood or serum separator tubes for serum testing. Urine was collected by scruffing the animal and gently stimulating the bladder or, if necessary, using a syringe to remove urine from the bladder following sacrifice. Clinical blood chemistry testing, including metabolic panels, and urine analyte measurements were performed on the Alfa Wasserman Vet Axcel using manufacturer-supported analyte assays. Complete blood counts (CBCs) were performed on the IDEXX Procyte Dx hematology analyzer utilizing murine species-specific parameters. Clinical chemistry and histology were analyzed by a partially-blinded board certified veterinary pathologist. Toxicity studies were pre-registered at preclinicaltrials.eu (pre-clinical Registry ID: PCTE0000119).
Anti-PEG Antibody Quantification
APA concentrations in blood were measured as previously described [22]. Briefly, whole blood, collected from submandibular bleed, was allowed to coagulate at room temperature for 15 minutes and then centrifuged at 2,000g for 15 minutes to generate serum. Untreated half-area 96-well Costar plates (Corning #3695) were coated with 50 μl of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-methoxy PEG-5kDa (DSPE-PEG; Nanocs, New York, NY, USA) at a concentration of 50 μg/mL in PBS overnight at 4°C. The plates were blocked with 5% w/v milk in DPBS for 1 hour at room temperature. Plasma samples were diluted 50-fold in 1% milk in DPBS and added to the plate, 50 μL per well. The samples were plated in both the presence and absence of free PEG 8 kDa (final concentration 10 mg/mL) as competition, incubated at 4°C overnight, and subsequently washed with DPBS. A standard curve was generated using mouse anti-PEG IgG1 (Silver Lake Research, CH2076, lot K0868). Secondary antibodies were goat anti-mouse IgG-HRP (1:5,000 dilution, Santa Cruz, sc-2005, lot #D1816), with 1 step Ultra TMB (ThermoFisher) as substrate. The HRP reaction was quenched with 2N sulfuric acid, and the absorbance was read at 450 nm (signal) and 570 nm (background) using a Spectramax M2 plate reader (Molecular Devices). All washes were with DPBS without TWEEN (which contains PEG moieties). Sample absorbances were corrected for competition results to adjust for the effect of non-specific binding by non-APA antibodies and analyzed using a 5-parameter logistic regression.
Results
High MW free PEG rescues the pharmacokinetics of PEG-drugs in mice with APA
We and others have previously established the use of PEGylated liposomal doxorubicin (PLD) as a model PEGylated drug to investigate the clearance of PEGylated drugs in mice passively immunized with APA, due to the ease of quantifying the fluorescent doxorubicin in plasma and tissues as well as the ability to accurately control APA levels [22, 27]. Utilizing this same model, we first investigated the effectiveness with which different MW free PEGs could restore the prolonged circulation of PLD in the presence of APA. Specifically, we first intravenously injected mice with APA, then injected PBS or free PEG of various MW 23.5 hours later, and finally administered PLD 0.5 hours later (Figure 1A, schematic). In all data figures, the time at which PLD was administered is referred to as t = 0 hr.
Figure 1: High MW free PEG rescues the pharmacokinetics of PEG-drugs in mice with APA.
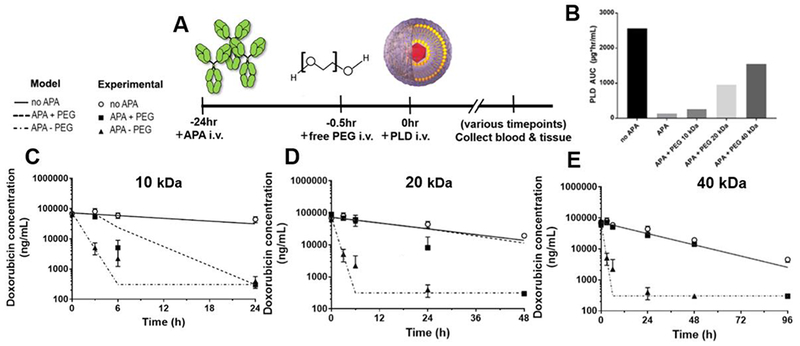
(A) We developed a mouse model of APA-mediated ABC by administering defined amounts of APA to naïve mice i.v., followed 24 h later by i.v. injection of PLD ± free PEG. (B) The AUC0-96h of PLD in mice that received the various sizes of free PEG polymer. (C–E) The PK of PLD was measured in naïve mice (open circle) as well as passively immunized mice receiving either i.v. PBS (filled triangle) or various free PEG interventions (filled square), including (C) 10 kDa (2200 mg/kg), (D) 20 kDa (550 mg/kg), and (E) 40 kDa (550 mg/kg) free PEG 30 min prior to PLD administration. Lines depict PK predictions from mPBPK model. For comparison, converting the above mg/kg doses of PEG 10 kDa, 20 kDa, and 40 kDa to molar doses equals 220 μmole/kg, 27.5 μmole/kg, and 13.75 μmole/kg, respectively.
We first evaluated 10 kDa PEG, which is within the common MW range used in PEGylated drugs. We found that 10 kDa PEG (at a dose of 2,200 mg/kg) provided an exceedingly transient reversal of the effects of APA, only through the first 3 hours-post injection (Figure 1C). At APA concentrations of 7 μg/mL, the pre-infusion of 10 kDa free PEG increased the mean plasma encapsulated doxorubicin concentrations ~10-fold at the 3-hour time point, from ~5,000 ng/mL in saline-treated controls to ~53,000 ng/mL in the free PEG-treated arm. By the 6-hour timepoint, however, the measured doxorubicin concentrations in PEG-treated mice were only 2.5-fold higher, at ~5,000 ng/mL vs. ~2,000 ng/mL in the saline control group. For comparison, control mice that did not receive APA still had ~59,000 ng/mL doxorubicin in the plasma at the 6-hour time point. These in vivo data were generally consistent with projections from our mPBPK model, which predicted that through the 3-hour timepoint, APA-positive animals treated with free PEG 10 kDa would retain ~6-fold greater PLD than PBS control. The limited duration of its effect was most likely due to rapid renal clearance of free PEG, since 10 kDa PEG has a terminal half-life of ~18 minutes in BALB/cCrSlc mice [21]. Thus, in the 3 hours between t=3 hr and t=6 hr, the amount of PEG 10 kDa remaining in circulation would be reduced by ~50-fold to an amount that was inadequate for sustained competitive inhibition of APA.
We next evaluated 20 kDa and 40 kDa free PEG (each at a dose of 550 mg/kg), which possess markedly longer elimination half-lives of ~3 hours and ~15 hours in mice, respectively [21]. Consistent with their prolonged circulation, both 20 and 40 kDa free PEG provided much longer effective saturation of high titers of APA, effectively restoring PLD’s circulation through 24 and 48 hours, respectively (Figure 1D and 1E). Over these durations, PLD concentrations in APA-positive, free PEG-treated mice were indistinguishable from control mice without APA. The use of higher MW free PEG increasingly improved the total systemic exposure of PLD in mice: APA-positive mice that received free PEG 10 kDa, 20 kDa, and 40 kDa had 2x, 7x, and 11x greater AUC relative to saline control (Figure 1B). Importantly, the administration of 40 kDa free PEG recovered ~60% of the PLD AUC0-96hr compared to mice with no APA at all (Figure 1B). This is likely to be an underestimate, since we did not test the plasma concentration between 48 hours (at which point the PEG-treated group was equal to controls) and 96 hours (at which point PEG-treated mice had eliminated all PLD). This enhanced systemic exposure would be expected to be of particular importance for therapeutics whose primary mechanism of action is to degrade specific compounds found in the blood, such as pegloticase, pegaspargase, pegarginase, etc.
Free PEG reduces the APA-mediated hepatic accumulation of PEG-liposomes
In animals with no APA, PEGylated liposomes and polymeric particles typically exhibited a tendency toward hepatic accumulation, with the spleen as the second leading site of accumulation [22, 28, 29]. We previously found that mice with high levels of APA exhibited greatly increased hepatic but otherwise reduced splenic and pulmonary distribution [22], whereas some have observed APA directing the clearance of PLD toward the spleen [27]. To investigate whether free PEG alters the biodistribution of PLD in mice with APA, we next measured total doxorubicin concentrations in the liver, spleen and lungs of mice (Figure 2). Free PEG appeared to reduce the hepatic accumulation of PLD, with increasing MW free PEG possibly resulting in less hepatic accumulation (Supplemental Figure 1 A and B). Indeed, mice treated with 20 kDa or 40 kDa free PEG had approximately 40% and 78% reduction in liver concentration of doxorubicin, at their respective terminal time points, relative to mice treated with saline (Supplemental 1). Mice treated with 40 kDa free PEG also appeared to accumulate less doxorubicin in the spleen and lung, relative to mice treated with 20 kDa free PEG (Supplemental 1C). To confirm that the PK-prolonging effect of free PEG was due to saturation of APA, and not nonspecific saturation of phagocytosis, we compared the PK of a dose of PLD in naïve mice that were pre-treated with either PBS or free PEG. We saw no appreciable difference in PK between these two groups, suggesting that free PEG acts specifically on APA (Supplemental 2).
Figure 2: Free PEG-treated mice delay PLD accumulation in liver.
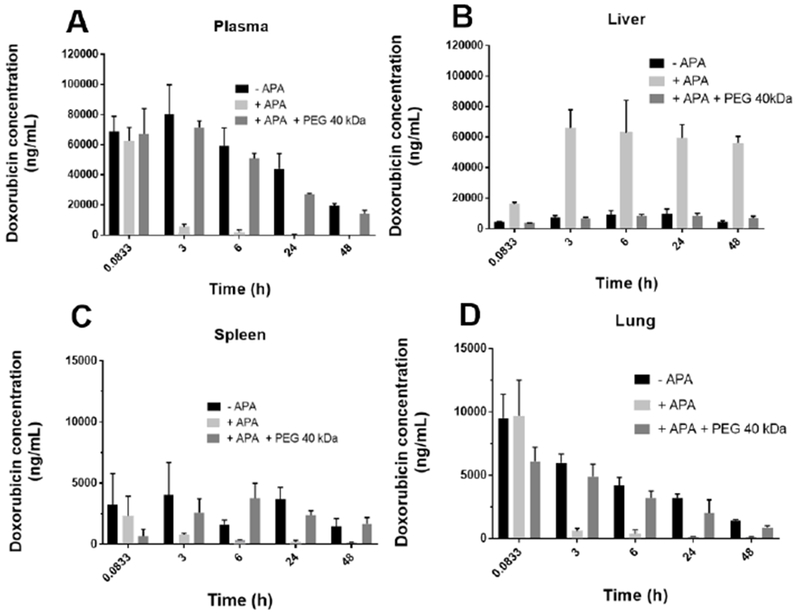
(A) The plasma concentration of encapsulated PLD was determined in naïve mice (black bars) as well as passively immunized mice treated with either free PEG (dark grey bars) or PBS (light grey bars). Tissue concentrations of total doxorubicin (encapsulated and released) in homogenate of the (C) liver, (D) spleen, and (E) lung.
Free PEG administration to PEG-sensitized mice does not result in apparent systemic, cardiac, hepatic, or renal toxicity
There is significant concern whether introducing free PEG in the presence of high titers of APA would result in PEG/APA immune complexes that could trigger significant renal and systemic toxicities. For example, in systemic lupus erythematosus and several other autoimmune diseases, the deposition of immune complexes in the kidneys leads to inflammation and glomerular disease [30]. Thus, we next sought to determine whether free PEG may cause any identifiable pathologies in mice sensitized to PEG and possessing substantial APA titers, induced via intravenous injection of PEGylated liposomes. Given that the primary route of elimination of free PEG is renal, we first conducted a series of tests to assess potential damage to the kidneys. In particular, we tested for evidence of membranous glomerular nephropathy, i.e. immune complex deposition in the functional space of renal excretion which could lead to glomerular inflammation and dysfunction.
Our mouse model to assess the toxicity of 40 kDa free PEG was generated by sensitizing mice against PEG via i.v. injection of PEGylated liposomes (after which the mice develop immunity to PEG and produce endogenous APA). We then administered free PEG 40 kDa weekly through week 6 to these PEG-sensitized mice, which had ~10 μg/mL APA at the time of the first dose of free PEG. One week after the final dose of free PEG, mice were sacrificed, and their kidneys were processed using standard histochemical methods and analyzed by a board-certified veterinary pathologist. H&E and PAS stains of the kidneys and liver of treated mice showed no discernable differences compared to saline-treated control mice (Figure 3). We also performed immunofluorescence staining on the liver and kidneys of the same mice to detect the presence of immune complex deposits. We observed no apparent difference in the deposition of PEG, IgG, IgM, or complement protein C3 in the renal glomerular spaces or livers of mice that received repeated injections of free PEG or PBS (Figure 3B and D, and Supplemental 4). Taken together, these results suggest minimal risk of immune complex-mediated disease in mice.
Figure 3: Repeat dosing of High MW free PEG does not cause immune complex deposition or apparent tissue damage.
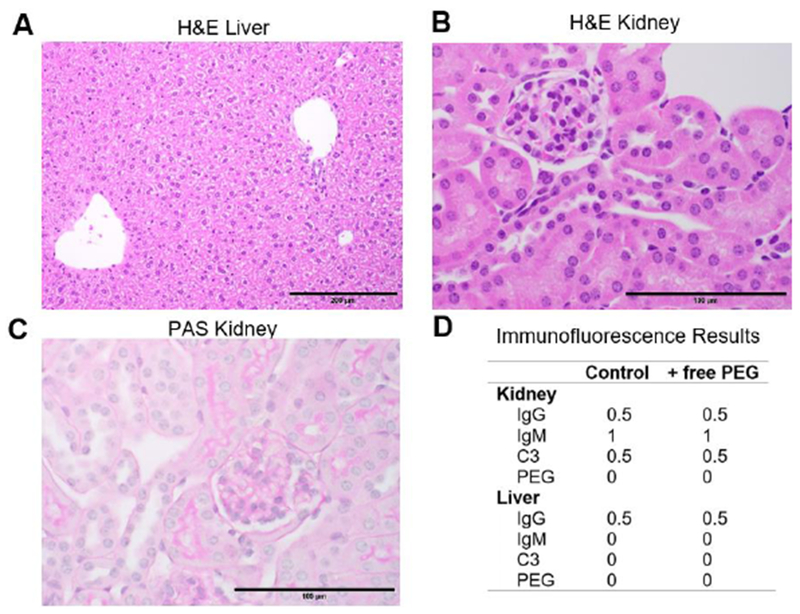
PEG-sensitized mice were given six weekly injections of 40 kDa free PEG. (A and C) Routine H&E staining of liver (200X magnification) and kidney (600X). Tissue histomorphology is within normal limits. (A) Regionally, hepatocytes in the liver display mild vacuolar change consistent with normal glycogen storage. (B) H&E of the kidney suggests that renal tubular epithelium occasionally display attenuated cells, consistent with post-mortem change, and lack vacuolation. (C) PAS staining of kidney (600X) highlights thin, positive-staining basement membranes of the glomerulus and renal tubules. Note that in the inner lumen of renal tubules there is irregular PAS-positive staining of PAS-positive secretions that are not basement membranes. (D) Summary immunofluorescence (IF) averages for PEG-sensitized mice that received either PBS or free PEG for six weeks. See complete panel of representative IF images in Supplemental 4.
To gain further insights into the potential toxicities resulting from using free PEG in PEG-sensitized mice, we collected blood and urine samples and performed a complete blood count as well as serum tests. We again found no differences between free-PEG treated vs. control mice (Tables 1 and 2 for PEG 20 kDa and 40 kDa, respectively). Importantly, there was no evidence of increased protein excretion to urine (measured by the ratio of total urine protein to creatinine, Supplemental 3), changes in leukocyte counts, or changes in serum creatinine or BUN. There was a slightly lower mean corpuscular volume in mice treated with free PEG (Supplemental 3). This was measured alongside a statistically-insignificant decrease in red blood cell count, hemoglobin, and hematocrit and an increased reticulocyte count. These results suggest a mild, subclinical (i.e. unlikely to have symptomatic manifestations), microcytic anemia. Given the statistical uncertainty, further studies will be required to determine if this is a real effect. Since PEGylated proteins have previously been shown to accumulate in the liver, we also measured serum albumin concentration, as well as AST and ALT, as potential markers of hepatic pathology. We found no evidence of free PEG-induced liver pathology through these systemic markers or through histopathologic analysis.
Table 1: Administration of 20 kDa free PEG does not affect clinical blood chemistry or urinalysis results.
PEG 20 kDa (in mice injected with APA)
Naïve mice were given exogenous APA (30 μg per mouse) or PBS, followed 24hr later by an injection of 20 kDa free PEG (550 mg/kg). Mice were given one more injection of PBS or PEG at day 7 and were sacrificed at day 14. Terminal blood was collected via cardiac puncture. Complete blood counts were performed within 60 minutes of sample collection, and serum/urine tests were performed according to manufacturer recommendations.
| PBS + PBS (n=10) | APA + PEG (n=10) | p-value | ||
|---|---|---|---|---|
| Complete Blood Count | ||||
| Red blood cell(M/μL) | 10.8 | 10.6 | 0.08 | |
| Hemoglobin (g/dL) | 16.6 | 16.3 | 0.12 | |
| Hematocrit (%) | 48.7 | 47.7 | 0.15 | |
| MCV (fL) | 44.5 | 45.1 | 0.56 | |
| Platelet (K/μL) | 114.4 | 113.4 | 0.96 | |
| Reticulocyte (K/μL) | 457.7 | 477.4 | 0.54 | |
| WBC (K/uL) | 6.3 | 6.8 | 0.46 | |
| Neutrophil # (K/μL) | 1.3 | 1.4 | 0.56 | |
| Lymphocyte # (K/μL) | 4.5 | 5.0 | 0.39 | |
| Serum | ||||
| Creatinine (mg/dL) | 0.38 | 0.39 | 0.91 | |
| Albumin (g/dL) | 2.9 | 3.0 | 0.68 | |
| BUN (mg/dL) | 22.1 | 21.8 | 0.83 | |
| Urine | ||||
| Creatinine (mg/dL) | 65.2 | 64.5 | 0.92 | |
| Total protein (mg/dL) | 203.1 | 161.3 | 0.10 |
Table 2: Repeat dosing of 40 kDa free PEG does not affect clinical blood chemistry or urinalysis results.
PEG 40 kDa (in vaccinated mice)
Sensitized mice were given six weekly injections of 40 kDa free PEG (550 mg/kg) or PBS, and urine and terminal blood were collected. Terminal blood was collected via cardiac puncture. Complete blood counts were performed within 60 minutes of sample collection, and serum/urine tests were performed according to manufacturer recommendations.
| vax + PBS (n=5) | vax + PEG (n=9) | p-value | ||
|---|---|---|---|---|
| Complete Blood Count | ||||
| Red blood cell(M/μL) | 10.3 | 10.2 | 0.7 | |
| Hemoglobin (g/dL) | 16.3 | 15.6 | 0.17 | |
| Hematocrit (%) | 49.7 | 47.6 | 0.14 | |
| MCV (fL) | 48.1 | 46.5 | 0.01 | |
| Platelet (K/μL) | 77 | 131.22 | 0.46 | |
| Reticulocyte (K/μL) | 446.16 | 523.97 | 0.43 | |
| WBC (K/μL) | 4.82 | 4.62 | 0.76 | |
| Neutrophil # (K/μL) | 0.9 | 1.03 | 0.70 | |
| Lymphocyte # (K/μL) | 3.4 | 3.38 | 0.97 | |
| Serum | ||||
| Creatinine (mq/dL) | 0.1 | 0.1 | 0.86 | |
| Total protein (g/dL) | 4.9 | 5.0 | 0.22 | |
| BUN (mg/dL) | 19.2 | 20.8 | 0.03 | |
| ALT (U/L) | 31.8 | 32.3 | 0.98 | |
| AST (U/L) | 179.8 | 103.1 | 0.07 | |
| Urine (n=8PEG, 5 PBS) | ||||
| Creatinine (mg/dL) | 70.7 | 54.1 | 0.157 | |
| Total protein (mg/dL) | 200.6 | 93.6 | 0.090 |
The injection of free PEG 20 kDa as a low volume resuscitation (LVR) solution has recently shown efficacy in preclinical models of hypovolemic shock within 20 minutes of administration [31, 32]. Given that we were administering free PEG to normotensive animals, we sought to determine whether treatment with free PEG would cause an osmotically-driven increase in the blood pressure of treated animals. We administered free PEG or PBS to mice, half of which had been previously sensitized against PEG via i.v. injection of PEGylated liposomes, and continuously measured their arterial blood pressure for a period of 20 minutes (the time frame in which PEG-LVR solutions were reported to be active). We found no measurable change in the blood pressure or heart rate of these mice (Figure 4A and B).
Figure 4: The administration of free PEG to sensitized or naïve mice does not acutely affect blood pressure or heart rate.
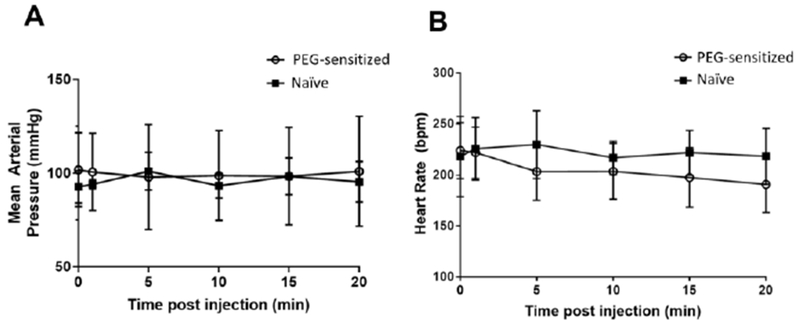
Sensitized mice received empty PEGylated liposomes (0.1 μmole lipid / kg weight, i.v.) on Day 0, and serum APA was confirmed by ELISA. PEG-sensitized and naïve mice were anesthetized, and a transducer was placed into the carotid artery to continuously measure (A) arterial blood pressure and (B) heart rate. Baseline values were first recorded for one minute (shown in figure as the t=0 timepoint), followed by dosing of 40 kDa free PEG (550 mg/kg, i.v.). For each time point, the MAP and HR values were averaged over a 30-second collection interval (e.g. MAP or HR at t = 5 min is the average from 4:45-5:15). The 0 minute timepoint represents average values over the 30 seconds prior to PEG injection.
Free PEG does not cause runaway production of APA upon chronic dosing to sensitized mice
The common paradigm in immunology is that antigen-specific B cells would respond to the introduction of their corresponding antigens with the secretion of more antigen-specific antibodies. These additional antibodies are meant to neutralize harmful foreign entities, but unfortunately can trigger uncontrolled hypersensitive reactions to therapeutics. To determine if free PEG induces elevated APA production, we administered free PEG 40 kDa (550 mg/kg) or PBS to PEG-sensitized mice weekly, with a blood sample taken immediately prior to each weekly PEG injection as well as at the end of the study to quantify APA levels. Much to our surprise, the repeated administration of free PEG did not cause APA concentrations to increase over time (Figure 5). Indeed, mice that received weekly free PEG injections possessed APA concentrations that were indistinguishable from those receiving weekly injections of saline.
Figure 5: Repeated free PEG injection does not cause runaway APA production in sensitized mice.
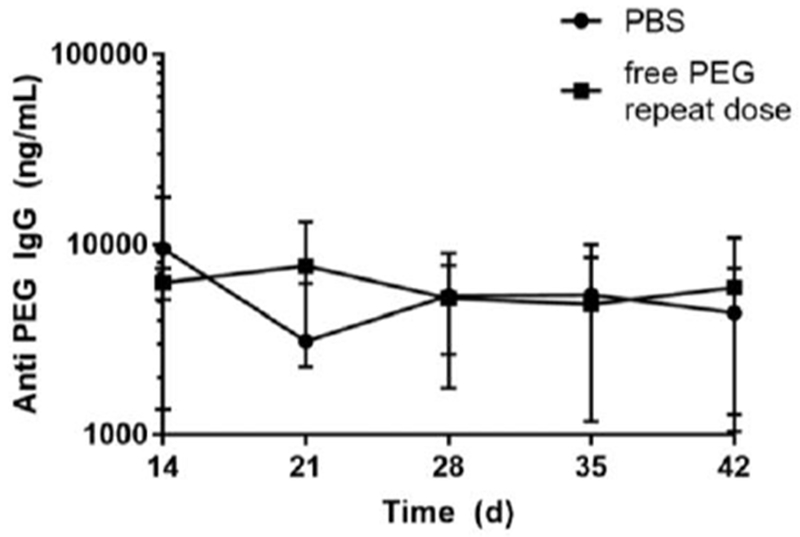
Mice were first sensitized against PEG on Day 0 by the i.v. administration of empty PEGylated liposomes (0.1 μmole lipid / kg weight) and then given injections of PBS or free PEG 40 kDa (550 mg/kg, i.v.) on days 14, 21, 28, and 35. Serum was collected before each weekly infusion for APA measurements.
Discussion
Numerous clinical studies have shown that APA, if not addressed, can result in loss of efficacy or serious adverse events in sensitized patients [17, 33, 34]. There is currently no method to restore the use of PEGylated medication in patients with high APA titers. Thus, the only available recourse is to discontinue treatment with the affected PEGylated therapeutic and switch to an alternative PEG-free therapy. However, for some drugs such as pegloticase, the last-line therapy for treatment-refractory chronic tophaceous gout, no alternative treatment is available. Here, we showed that an exceptionally simple and readily translatable approach – administrating high MW free PEG – can safely and effectively restore the prolonged circulation and efficacy of PEGylated medicines in animals with high APA titers. Importantly, by only minimally stimulating APA-specific B cells, it is likely that free PEG infusion could be used as a repeated intervention, possibly administered prior to each dose of a PEGylated therapeutic in patients with high APA titers. When we apply the principles of allometric scaling from mice to humans to the pharmacokinetics of free PEG, the administration of high MW free PEG to suppress free circulating APA is predicted to last for at least 7 days. This makes infusion with high MW free PEG a potentially practical strategy for control over APA, compatible with use of PEGylated therapeutics that require weekly or biweekly dosing.
It remains surprising that free PEG only modestly stimulated the production of additional APA, given that other PEGylated entities, including both PEGylated proteins and PEG-liposomes, typically potently stimulate APA production. Since elevated antibody production is elicited by activation of B-cells due to crosslinking of surface B-cell receptors (BCR) [35], we speculate that free PEG must be inherently less efficient at crosslinking multiple BCRs on APA-secreting B-cells relative to the PEGylated therapeutic counterparts. When PEG chains are grafted onto substrates such as proteins or liposomes, they present PEG motifs with relatively fixed spatial orientation at a high local concentration. In other words, distinct BCRs could be effectively crosslinked despite binding to different PEG chains as long as those PEG chains are anchored to the same protein or liposome. The resulting BCR crosslinking could then activate T-independent APA production. In contrast, when PEG is not anchored to a substrate (e.g. particle or protein), the disperse, flexible nature of free PEG chains in solution is less likely to crosslink multiple B cell receptors on the surface of APA-specific B cells, particularly when the ratio of free PEG to APA-BCR is high. Indeed, repeated dosing of free PEG did not result in runaway APA production, and the corresponding titer of induced APA is low enough to be managed with therapeutic free PEG.
This approach of using free PEG to saturate circulating antibodies differs from other strategies currently used to address unwanted immune responses. In autoimmune disease, current standards of care often rely upon non-specific immune downregulation, e.g. through the use of glucocorticoids [36] or anti-inflammatory cytokines, which have the potential to increase susceptibility to infection [37]. Glucocorticoid use has been found to be associated with significantly increased risk of pneumonia (hazard ratio of up to 2.3 in patients on glucocorticoids), and other infections [38, 39]. As a separate strategy, in the field of food allergy, patients who have unwanted hypersensitive responses are increasingly treated with oral antigen as an immunomodulatory strategy [40]. In peanut allergy, for example, this approach has been shown to be safe and effective in reducing allergic responses to subsequent peanut challenge [40, 41]. However, such strategies that aim to shift the isotype of the humoral response do not present an ideal solution to ABC caused by APA. Specifically, although isotype switching may help reduce the risk of anaphylaxis following treatment with a therapeutic, the presence of APA would likely still quickly clear the PEGylated therapeutic, leading to loss of efficacy. In contrast, the use of free PEG directly blocks APA from binding to PEGylated drugs, regardless of the isotype of APA present. A particular advantage to free-PEG infusion is the antigen-specific control over the humoral response, rather than broad immune suppression.
In our animal model of APA-induced accelerated clearance of PEGylated liposomes, we administered anti-PEG IgG and then characterized the pharmacokinetics of subsequently-injected PLD. Many T-independent antigens do not elicit antibody class switching, resulting in primarily an IgM-dominant response. However, many other T-independent antigens, such as bacterial capsular polysaccharides (with highly repetitive structure similar to PEG), can elicit antibody class switching to IgG as well as the generation of memory responses [42, 43]. Further, clinical studies of pegnivacogin and pegloticase in humans have suggested that anti-PEG IgG is significantly associated with adverse events to PEGylated drugs, with titer-dependent risks [16]. Specifically, patients who had severe reactions to pegnivacogin had approximately 6-fold more anti-PEG IgG than patients who experienced no reaction [16]. It was found that patients who had anti-PEG IgG at a titer three times higher than the assay detection limit were about three times more likely to experience a severe allergic reaction than were patients who had anti-PEG IgG titers that were only one time higher than the assay detection limit. Finally, it was previously shown that humans typically exhibit a IgG-dominant APA response [44]. Altogether, these clinical realities informed our decision to focus on anti-PEG IgG as our model for PEG sensitivity in PK studies, which indeed resulted in rapid ABC. For our efficacy studies, we chose to administer exogenous APA instead of vaccinating the mice for several reasons. First, the concentration of APA in sensitized mice induced by i.v. exposure to PEGylated liposomes is both widely variable and extremely high [6, 45], reaching levels that exceed titers measured to date in the general population [44, 46]. By utilizing mice passively immunized against PEG, we were able to better ensure both the relevance of our findings and also greatly reduce the number of animals needed in the study. Now that we have established a baseline level of efficacy of free PEG in suppressing the APA response, future studies will further investigate free PEG’s capacity to saturate an actively-induced APA response achieved using clinically relevant PEGylated drugs such as pegloticase. Separately, we recently verified the ability of free PEG to saturate APA in an actively immunized model, preventing ABC in rats that were sensitized against PEGylated microbubbles used as ultrasound contrast agents [9]. Finally, for our toxicity studies, we first elicited adaptive anti-PEG immunity by i.v. injection of PEGylated liposomes. We then administered weekly doses of free PEG to these sensitized mice to simulate chronic dosing in sensitized patients.
PEG is generally considered safe, inert, and non-immunogenic, and has been approved by the U.S. Food and Drug Administration (FDA) as a Generally Recognized as Safe (GRAS) substance [2]. PEGs of varying molecular weight are also available as USP-grade pharmaceutical excipients, and have been routinely included in various drug products. However, since PEG is frequently included in formulations at lower dosages than our current study, we investigated whether a high dose of high MW (40 kDa) free PEG would cause pathology in naïve or sensitized animals. PEG’s toxicity is thought to result from end group metabolism by alcohol dehydrogenase, a process that can be treated with the administration of fomepizole or ethanol, competitive inhibitors of the alcohol dehydrogenase enzyme [47, 48]. Since the toxic metabolic byproducts are a result of reactions of the end groups of PEG, we would expect that larger MW species of PEG would be less toxic than shorter PEG species at the same mass dose. Indeed, we found no organ-specific or systemic evidence of toxicity associated with the repeated administration of free PEG in mice that had been previously administered PEGylated liposomes to generate an APA response.
We believe that the unchanged terminal biodistribution profile of PLD in mice with APA is the result of free PEG only temporarily binding to APA rather than clearing them from circulation as APA/PEG complexes. As such, once the plasma concentration of free PEG drops below its effective threshold, the APA in the plasma is once again able to bind to PLD and affect its rapid clearance to the liver. We found that the efficacy of free PEG was maintained at 3, 24, and 48 hours, but lost by 6, 48, and 96 hours for PEG 10, 20, and 40 kDa, respectively. Based upon the initial dose of PEG given and the expected half-life of each PEG size, the amount of free PEG remaining in circulation for each type of PEG would be roughly 1, 2, and 60 mg/kg for PEGs 10, 20, and 40 kDa, respectively, at the timepoint where control over APA was maintained, but dropped to 0.003, 0.01, and 6.5 mg/kg at the time at which control over APA was lost. These results suggest that the threshold amount of free PEG needed to maintain control over single-digit μg/mL APA is somewhere in the range of 1-60 mg/kg, varying by MW.
Although our mPBPK model predicted that higher MW PEG molecules would be more effective than lower MW PEG in prolonging APA saturation, the estimated duration of effect differed from that measured in vivo. Whereas the model predicted that PEG 20 kDa and 40 kDa could effectively saturate the circulating APA for ~48 hours and ~96 hours respectively, the actual durations of effect were ~24 hours and ~48 hours, respectively (Figure 1). We believe these differences are likely attributed to two main factors. First, the estimated effective circulation half-life of free PEG from literature may differ from the actual circulation half-life in our mice. Second, the movement of free PEG out of the plasma compartment and into the peripheral fluids could result in a reduced concentration of PEG in plasma. This suggests our mPBPK model would benefit from more complex compartment analysis with respect to both the movement of free PEG and PEGylated drug.
We wish to point out that as part of FDA’s Summary Basis for Regulatory Action of REBINYN, Novo Nordisk’s treatment for hemophilia that consisted of Factor IX linked to 40kDa PEG, the document discussed extensive preclinical safety studies of 40 kDA PEG administered via weekly intravenous injections in healthy cynomolgus monkeys and Wistar rats, at doses that were 5- to 100-fold in excess of the clinical dose [49]. The majority of animals evaluated in the toxicity studies remained healthy until their scheduled sacrifice time point and had no overt signs of toxicity (e.g., irregularities in heart rate, body weight, food consumption, etc.). These studies did, however, find an accumulation of free PEG in tissues of the body, and the FDA expressed uncertainty over the physiological and clinical implications. However, despite PEG accumulation, the lack of danger signals from other toxicity assays led to a recommendation of approval for the drug product. Accumulation (and subsequent elimination) of free PEG has been extensively documented [50]. Indeed, a 2013 survey found that PEG vacuolization had been documented histologically for 10 out of 17 PEGylated therapeutics on the market [50]. Notably, PEG accumulation has not been associated with any pathological effect. It is not known whether the accumulation and vacuolization of PEG in tissues poses any safety risk [50]. The primary route of elimination for free PEG is renal, and the extreme flexibility of PEG means that even high MW molecules can pass through the glomerular filter, albeit at a slower rate [21, 50]. Future studies of the use of free PEG to saturate APA should be complemented by further long-term safety studies of the extent of PEG vacuolization in other organs and any possible pathologic risk. The excellent safety profile observed to date offers promise that the administration of free PEG could provide a safe strategy to combat APA that can be quickly advanced into the clinical setting.
We previously found that the majority of people actually possess pre-existing APA, with detectable levels of anti-PEG IgM or IgG in ~70% of the general population, underscoring the potential for rapid immune stimulation upon treatment with certain PEGylated therapeutics [44]. With an increased number of PEGylated therapeutics gaining FDA approval, the clinical significance of APA will likely increase markedly over time. This will be especially important for patients on a drug regimen containing multiple different PEGylated therapeutics; even if one PEGylated drug does not elicit high APA titers, its efficacy and safety could be impacted due to APA elicited by another PEGylated drug. It will be important for clinical care teams to be aware of the possible risks posed by APA. A recent survey of physicians from specialties in which PEG-drug use is routine found that although 83% of providers surveyed regularly prescribed at least one PEGylated drug, only 22% were aware of APA [51]. Developing a safe and effective method to allow the use of PEGylated drugs in sensitive patients could allow for more effective control of a range of pathologies for which treatment with a PEGylated drug is standard course. We believe infusion with high MW free PEG is a simple yet promising solution to this problem. Our current findings must next be validated in larger animal models, particularly those that afford sensitive detection of hypersensitive, hemodynamic, or other toxicological responses.
Data Availability Statement:
Raw data for the pharmacokinetics and biodistribution of PEGylated liposomal doxorubicin in naïve and vaccinated mice ± free PEG are publicly available at data.mendeley.com/datasets/37mxbmdjx9/1 [52].
Supplementary Material
Acknowledgements:
Animal Studies were performed with the help of the UNC Lineberger Animal Studies Core Facility at the University of North Carolina at Chapel Hill. The UNC Lineberger Animal Studies Core is supported in part by an NCI Center Core Support Grant (CA16086) to the UNC Lineberger Comprehensive Cancer Center. Animal histopathology and clinical services were performed by the Animal Histopathology & Laboratory Medicine Core at the University of North Carolina, which is supported in part by an NCI Center Core Support Grant (5P30CA016086-41) to the UNC Lineberger Comprehensive Cancer Center.
Funding: This work was supported by a National Science Foundation Graduate Research Fellowship (DGE-1650116, MDM), The David and Lucile Packard Foundation (2013-39274, SKL), National Institutes of Health (R01 HL141934, SKL, WCZ, and R35 GM119661, YC), and a UNC Research Opportunities Initiative grant in Pharmacoengineering (SKL). MGF was supported in part from the National Science Foundation DMS-1462992, DMS-1412844, DMS-1517274, DMS-1664645.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: McSweeney MD and Lai SK are inventors on Intellectual Property (IP) related to this research. This IP has not been licensed.
References
- 1.Veronese FM and Pasut G, PEGylation, successful approach to drug delivery. Drug Discov Today, 2005. 10(21): p. 1451–8. [DOI] [PubMed] [Google Scholar]
- 2.Milla P, Dosio F, and Cattel L, PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab, 2012. 13(1): p. 105–19. [DOI] [PubMed] [Google Scholar]
- 3.Jokerst JV, et al. , Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond), 2011. 6(4): p. 715–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veronese FM and Mero A, The impact of PEGylation on biological therapies. BioDrugs, 2008. 22(5): p. 315–29. [DOI] [PubMed] [Google Scholar]
- 5.Gefen T, et al. , The impact of PEGylation on protein immunogenicity. Int Immunopharmacol, 2013. 15(2): p. 254–9. [DOI] [PubMed] [Google Scholar]
- 6.Mima Y, et al. , Anti-PEG IgM Is a Major Contributor to the Accelerated Blood Clearance of Polyethylene Glycol-Conjugated Protein. Mol Pharm, 2015. 12(7): p. 2429–35. [DOI] [PubMed] [Google Scholar]
- 7.Richter AW and Akerblom E, Antibodies against polyethylene glycol produced in animals by immunization with monomethoxy polyethylene glycol modified proteins. Int Arch Allergy Appl Immunol, 1983. 70(2): p. 124–31. [DOI] [PubMed] [Google Scholar]
- 8.Ishida T, et al. , Accelerated blood clearance of PEGylated liposomes upon repeated injections: effect of doxorubicin-encapsulation and high-dose first injection. J Control Release, 2006. 115(3): p. 251–8. [DOI] [PubMed] [Google Scholar]
- 9.Fix SM, et al. , Accelerated Clearance of Ultrasound Contrast Agents Containing Polyethylene Glycol is Associated with the Generation of Anti-Polyethylene Glycol Antibodies. Ultrasound Med Biol, 2018. 44(6): p. 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishida T, et al. , PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J Control Release, 2007. 122(3): p. 349–55. [DOI] [PubMed] [Google Scholar]
- 11.Ishida T, et al. , Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release, 2006. 115(3): p. 243–50. [DOI] [PubMed] [Google Scholar]
- 12.Li B, et al. , Revealing the Immunogenic Risk of Polymers. Angewandte Chemie International Edition, 2018. 0(0). [DOI] [PubMed] [Google Scholar]
- 13.Lipsky PE, et al. , Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther, 2014. 16(2): p. R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershfield MS, et al. , Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Research & Therapy, 2014. 16(2): p. R63–R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong JK, et al. , Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer, 2007. 110(1): p. 103–11. [DOI] [PubMed] [Google Scholar]
- 16.Ganson NJ, et al. , Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol, 2016. 137(5): p. 1610–1613. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Povsic TJ, et al. , Pre-existing anti-PEG antibodies are associated with severe immediate allergic reactions to pegnivacogin, a PEGylated aptamer. Journal of Allergy and Clinical Immunology, 2016. 138(6): p. 1712–1715. [DOI] [PubMed] [Google Scholar]
- 18.Garay RP, et al. , Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Deliv, 2012. 9(11): p. 1319–23. [DOI] [PubMed] [Google Scholar]
- 19.Rojko JL, et al. , Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical-related immune complexes: review and case studies. Toxicol Pathol, 2014. 42(4): p. 725–64. [DOI] [PubMed] [Google Scholar]
- 20.Longley CB, et al. , Biodistribution and excretion of radiolabeled 40 kDa polyethylene glycol following intravenous administration in mice. J Pharm Sci, 2013. 102(7): p. 2362–70. [DOI] [PubMed] [Google Scholar]
- 21.Yamaoka T, Tabata Y, and Ikada Y, Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci, 1994. 83(4): p. 601–6. [DOI] [PubMed] [Google Scholar]
- 22.McSweeney MD, et al. , A minimal physiologically based pharmacokinetic model that predicts anti-PEG IgG-mediated clearance of PEGylated drugs in human and mouse. Journal of Controlled Release, 2018. 284: p. 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabizon A, Shiota R, and Papahadjopoulos D, Pharmacokinetics and tissue distribution of doxorubicin encapsulated in stable liposomes with long circulation times. J Natl Cancer Inst, 1989. 81(19): p. 1484–8. [DOI] [PubMed] [Google Scholar]
- 24.Amselem S, Gabizon A, and Barenholz Y, Optimization and upscaling of doxorubicin-containing liposomes for clinical use. J Pharm Sci, 1990. 79(12): p. 1045–52. [DOI] [PubMed] [Google Scholar]
- 25.Zamboni WC, et al. , Plasma, Tumor, and Tissue Disposition of STEALTH Liposomal CKD-602 (S-CKD602) and Nonliposomal CKD-602 in Mice Bearing A375 Human Melanoma Xenografts. Clinical Cancer Research, 2007. 13(23): p. 7217. [DOI] [PubMed] [Google Scholar]
- 26.Anders CK, et al. , Pharmacokinetics and efficacy of PEGylated liposomal doxorubicin in an intracranial model of breast cancer. PLoS One, 2013. 8(5): p. e61359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh YC, et al. , Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes. Theranostics, 2018. 8(11): p. 3164–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Awasthi VD, et al. , Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. International Journal of Pharmaceutics, 2003. 253(1): p. 121–132. [DOI] [PubMed] [Google Scholar]
- 29.Li Y-P, et al. , PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. Journal of Controlled Release, 2001. 71(2): p. 203–211. [DOI] [PubMed] [Google Scholar]
- 30.Toong C, Adelstein S, and Phan TG, Clearing the complexity: immune complexes and their treatment in lupus nephritis. International Journal of Nephrology and Renovascular Disease, 2011. 4: p. 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plant V, et al. , Low-Volume Resuscitation for Hemorrhagic Shock: Understanding the Mechanism of PEG-20k. J Pharmacol Exp Ther, 2017. 361(2): p. 334–340. [DOI] [PubMed] [Google Scholar]
- 32.Plant V, et al. , Low-volume resuscitation using polyethylene glycol-20k in a preclinical porcine model of hemorrhagic shock. J Trauma Acute Care Surg, 2016. 81(6): p. 1056–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, et al. , Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Control Release, 2016. 244(Pt B): p. 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Q and Lai SK, Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol, 2015. 7(5): p. 655–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fagarasan S and Honjo T, T-Independent Immune Response: New Aspects of B Cell Biology. Science, 2000. 290(5489): p. 89. [DOI] [PubMed] [Google Scholar]
- 36.Coutinho AE and Chapman KE, The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol, 2011. 335(1): p. 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingrid Loma RH, Multiple Sclerosis: Pathogenesis and Treatment. Current Neuropharmacology, 2011. 9(3): p. 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youssef J, Novosad SA, and Winthrop KL, Infection Risk and Safety of Corticosteroid Use. Rheumatic diseases clinics of North America, 2016. 42(1): p. 157–x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolfe F, Caplan L, and Michaud K, Treatment for rheumatoid arthritis and the risk of hospitalization for pneumonia: associations with prednisone, disease-modifying antirheumatic drugs, and anti-tumor necrosis factor therapy. Arthritis Rheum, 2006. 54(2): p. 628–34. [DOI] [PubMed] [Google Scholar]
- 40.Vickery BP, et al. , Early oral immunotherapy in peanut-allergic preschool children is safe and highly effective. J Allergy Clin Immunol, 2017. 139(1): p. 173–181. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vickery BP, et al. , Sustained unresponsiveness to peanut in subjects who have completed peanut oral immunotherapy. J Allergy Clin Immunol, 2014. 133(2): p. 468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Defrance T, Taillardet M, and Genestier L, T cell-independent B cell memory. Curr Opin Immunol, 2011. 23(3): p. 330–6. [DOI] [PubMed] [Google Scholar]
- 43.Obukhanych TV and Nussenzweig MC, T-independent type II immune responses generate memory B cells. The Journal of experimental medicine, 2006. 203(2): p. 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Q, et al. , Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal Chem, 2016. 88(23): p. 11804–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichihara M, et al. , Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats. Pharmaceutics, 2011. 3(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen B-M, et al. , Measurement of Pre-Existing IgG and IgM Antibodies against Polyethylene Glycol in Healthy Individuals. Analytical Chemistry, 2016. 88(21): p. 10661–10666. [DOI] [PubMed] [Google Scholar]
- 47.Webster R, et al. , PEG and PEG conjugates toxicity: towards an understanding of the toxicity of PEG and its relevance to PEGylated biologicals, in PEGylated Protein Drugs: Basic Science and Clinical Applications, Veronese FM, Editor. 2009, Birkhäuser Basel: Basel; p. 127–146. [Google Scholar]
- 48.Brent J, et al. , Fomepizole for the treatment of ethylene glycol poisoning. Methylpyrazole for Toxic Alcohols Study Group. N Engl J Med, 1999. 340(11): p. 832–8. [DOI] [PubMed] [Google Scholar]
- 49.FDA, Summary Basis for Regulatory Action for REBINYN, (N9-GP, GlycoPEGylated rFIX) 2017.
- 50.Ivens IA, et al. , PEGylated Biopharmaceuticals: Current Experience and Considerations for Nonclinical Development. Toxicologic Pathology, 2015. 43(7): p. 959–983. [DOI] [PubMed] [Google Scholar]
- 51.McSweeney MD, et al. , Physician Awareness of Immune Responses to Polyethylene Glycol-Drug Conjugates. Clinical and Translational Science, 2018. 11(2): p. 162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McSweeney MD, (raw data) Overcoming anti-PEG antibody mediated accelerated blood clearance of PEGylated liposomes by pre-infusion with high molecular weight free PEG. 10.17632/37mxbmdjx9.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for the pharmacokinetics and biodistribution of PEGylated liposomal doxorubicin in naïve and vaccinated mice ± free PEG are publicly available at data.mendeley.com/datasets/37mxbmdjx9/1 [52].