

Abstract
Silica nanoparticles (SiO2 NPs) have potential utility in controlled release. Despite significant research in this area, there is a gap in the understanding of the correlation between SiO2 NP physicochemical properties on the one hand and their degradation in solutions, in cells, and in vivo on the other. Here, we fabricated SiO2 NPs with variations in size, porosity, density, and composition: 100 nm Stöber, 100 and 500 nm mesoporous, 100 nm disulfide-based mesoporous, and 100 nm disulfide-based hollow mesoporous. Degradation profiles over 28 days were investigated in simulated biological fluids and deionized water. Results show Meso 100, and 500 nanoparticles degraded faster at higher pH values. Results from macrophages indicate Meso 100 nanoparticles showed the highest degradation amount (~3.8%). Cytotoxicity evaluation of the particles in Human Aortal Endothelial Cells (HAECs) shows concentration-dependent toxicity for the particles. Results from CD-1 mice show ~53% of Meso 100 nanoparticles (25 mg kg−1) degraded and were detected in urine after seven days. It was shown nanoparticle porosity and composition as well as pH and ionic strength of the medium play the predominant roles for degradation of SiO2 NPs. Based on histological evaluations, at the injected doses investigated, the particles did not show toxicity.
Keywords: silica nanoparticles, physicochemical properties, degradation, toxicity, biodistribution, clearance
Introduction
Silicon dioxide is “generally regarded as safe (GRAS)” ingredient by the United States Food and Drug Administration (US-FDA) [1, 2]. Silica nanoparticles (SiO2 NPs) can provide for robust delivery systems due to the ease of synthesis and scale-up, tunable stability, porosity, surface area, size and size distribution, entrapment efficiency, core composition, density and the ability for surface functionalization [3-5]. These particles are widely investigated for the delivery of bioactive and imaging agents [6-8]. Their safety as a function of physicochemical properties has been assessed [9-14]. SiO2 NPs can be porous or nonporous in structure with variations in size, shape, porosity, degree of condensation, and zeta potential [12, 15]. Incorporation of degradable moieties such as disulfide, tetrasulfide, and carbamate can make these structures susceptible to controlled degradation by intracellular or extracellular stimuli [16-20]. Also, pH-sensitive and enzyme-responsive SiO2 NPs have been designed for use in drug delivery [21, 22]. Selective etching techniques can be used to fabricate hollow structures with considerable loading capacity [23-25].
Despite significant advances in SiO2 NP research, to the best of our knowledge, a comparative and systematic study on degradation profiles of SiO2 NPs in different biological fluids, in cells, and in vivo as a function of time and particle physicochemical properties has not been thoroughly conducted. A key concern is a gap in understanding between the degradation of SiO2 NPs in vitro, in cells, and in vivo. Hence, a comprehensive analysis of degradation as well as the biological fate of SiO2 NPs and their degradation products is needed. Such study will provide valuable baseline data for tailor-making SiO2 NPs for delivery of bioactive and imaging agents and give us information for the effect of different parameters on degradation, biodistribution, and clearance of the particles. In vitro and in vivo degradation studies of nonporous and mesoporous SiO2 NPs have been reported in several papers [26-31]. For example, it was demonstrated that nanoparticle shape and geometry can alter the degradation rate of SiO2 NPs [28]. Also, it was reported that particle’s degree of condensation could influence the degradation rate of SiO2 NPs and highly-condensed mesoporous SiO2 NPs slowly degrade in simulated body fluid (SBF) over several weeks [32]. It was demonstrated that surface functionalization of the particles using poly(ethylene glycol) (PEG) slows the degradation rate of mesoporous particles [31]. For in vivo investigation of degradation, silicon (Si) detected in urine and feces was reported as the degradation products. It was shown that size did not play a significant role in the degradation of mesoporous SiO2 NPs in mice [26].
To fill this gap, there are several points to consider. First, intact particles should be fabricated as uniform as possible and thoroughly characterized such that one can minimize the influence of particle polydispersity and exclude the results for which characterization is suboptimal. Second, there is a need to accurately measure the degradation profiles of SiO2 NPs, the amount of released Si, and the mechanisms of degradation. Third, it is crucial to systematically correlate nanoparticle physicochemical properties such as size, porosity, density, shape, and composition with degradation. Fourth, the pH and composition of the degradation media need to be carefully considered.
To address these issues, herein, we fabricated a series of well-characterized SiO2 NPs with low polydispersity and different physicochemical properties such as size, porosity, density, and composition. Degradation of the synthesized particles was then evaluated as a function of time and particle physicochemical characteristics. The same particles were utilized for all in vitro, in macrophages, and in vivo experiments. The particles were evaluated for their cytotoxicity in RAW 264.7 macrophages and Human Aortic Endothelial Cells (HAECs). In vitro degradation profiles were measured over a 28-day period in simulated biological fluids such as simulated gastric fluid (SGF) pH 1.2, simulated lysosomal fluid (SLF) pH 4.5, simulated intestinal fluid (SIF) pH 6.5, simulated body fluid (SBF) pH 7.4, and in deionized (DI) water pH 6.5. The degradation of disulfide-based particles was also investigated in SBF containing glutathione (GSH) as a reducing agent to evaluate the influence of intracellular GSH on degradation profile. Degradation of the nanoparticles was further analyzed in RAW 264.7 macrophages. The in vivo degradation, biodistribution, and toxicity of selected nanoparticles were investigated upon intravenous injection in CD-1 mouse (Scheme 1).
Scheme 1.
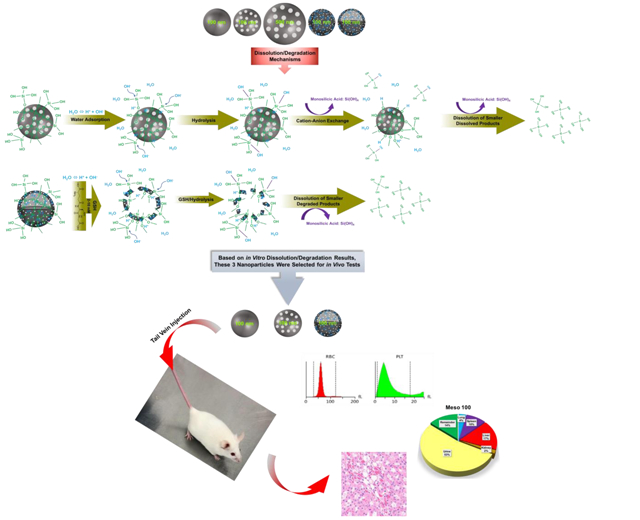
Schematic representation of in vitro and in vivo studies. The top panel illustrates the fabricated nanoparticles with variations in size, porosity, density, and composition (from left to right): Stöber 100, Meso 100, Meso 500, Disulfide Meso 100, and Disulfide Hollow 100 nanoparticles. The middle panel indicates degradation mechanisms. For regular SiO2 NPs, hydroxyl ions (OH−) attacking Si-O bonds are forming an unstable pentavalent structure which can be broken by attachment of other ions. In disulfide-based SiO2 NPs, a similar process is involved in addition to degradation via disulfide breakage in a redox environment containing GSH as a reducing agent which has high intracellular concentrations (2-10 mM) [33-35]. Release of monosilicic acid Si(OH)4 is the product of these processes. The bottom panel depicts the in vivo studies conducted in CD-1 mice. Three nanoparticles (Stöber 100, Meso 100, and Disulfide Hollow 100) were selected based on in vitro results. Particles were tested for in vivo degradation, toxicity, and biodistribution.
Materials and Methods
Materials.
The following chemicals were obtained from Sigma-Aldrich, Inc. (St. Louis, MO, USA): Silicic acid (Si(OH)4, 99.9%), simulated intestinal fluid (SIF) without enzyme, pepsin, cetyltrimethylammonium bromide (CTAB, ≥99.0%), Triton™ X-100, fetal bovine serum (FBS), triethylamine (TEA, ≥99.0%), tetraethyl orthosilicate (TEOS, ≥99.0% GC), and glutathione (GSH). Simulated gastric fluid (SGF) without pepsin was purchased from RICCA® Chemical Company (Arlington, Texas, USA). Simulated lysosomal fluid (SLF; human liver lysosomes) was obtained from XenoTech (Kansas City, KS, USA). Cell lysis buffer (ab152163) was obtained from Abcam (Cambridge, MA, USA). 10% Neutral buffered formalin, Roswell Park Memorial Institute-1640 (RPMI-1640) medium, hydrochloric acid ACS Grade BDH (36.5-38.0%), and phosphate-buffered saline (PBS) biotechnology grade tablets were received from VWR (Radnor, PA, USA). TrypLE™, Vascular Cell Basal Medium (Medium 200), Low Serum Growth Supplement (LSGS), and BD Vacutainer® Blood Collecting Tubes (Heparin- and K2 EDTA-coated) were purchased from Thermo Fisher Scientific (Grand Island, NY, USA). SALJET (Sterile 0.9% W/V Sodium Chloride) was purchased from Winchester Laboratories LLC (New York, NY, USA). Histosette I (Tissue Processing/Embedding Cassettes with LID, Pink) was received from Simport (Beloeil, QC, Canada). Sodium carbonate (Na2CO3, anhydrous, granular, ≥99.5%) was received from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). Ammonium hydroxide (NH4OH, 28.0-30.0% as NH3) was obtained from EMD Millipore Corporation (Billerica, MA, USA). Trypan Blue Stain 0.4% was obtained from Invitrogen (Carlsbad, CA, USA). Sodium Chloride High Purity Grade (NaCl, 99.9%) was purchased from AMRESCO® (Solon, OH, USA). Bis[3-(triethoxysilyl)propyl] disulfide (BTESPD, 90.0%) was purchased from Gelest, Inc. (Morrisville, PA, USA). RAW 264.7 macrophages (ATCC® TIB-71™) and Human Aortic Endothelial Cells (HAECs; ATCC® PCS-100-011™) were received from American Type Culture Collection (ATCC, Manassas, VA, USA). CCK-8 cytotoxicity assay kit was received from Dojindo (Rockville, MD, USA). EHT5 LH LYSE and EHT5 DIFF LYSE, GOT/AST and Pre-Surgical S-Panel (containing TP, ALP, GLU, GPT/ALT, and BUN) DRI-CHEM Kits were obtained from Heska Corporation (Loveland, CO, USA). 1.5 mL FUJI Plain Tubes (designed for FUJI DRI-CHEM Analyzer) was received from FUJIFILM Corporation (Minato-ku, Tokyo, Japan). Limulus Amebocyte Lysate (LAL) QCL-1000™ kit was purchased from Lonza (Walkersville, MD, USA). CD-1 female mice were obtained from Charles River Laboratories (San Diego, CA, USA). All materials were used as received without further purification.
Nanoparticle Synthesis
Stöber 100.
Nonporous Stöber SiO2 NPs were synthesized as follows [24]: 180 mmol of DI water, 200 mmol of ammonium hydroxide, and 1700 mmol of absolute ethanol were mixed in a 250-mL flask under stirring rate of 400 RPM for 10 min. Then, 18 mmol of TEOS was added dropwise, and the reaction was left under stirring for 24 h at room temperature. The synthesized NPs were precipitated by centrifugation using Sorvall® RC-5B Refrigerated Superspeed Centrifuge (Du Pont Instruments Ltd., Wilmington, DE, USA) at 15,000 RPM for 20 min, washed thoroughly with DI water and ethanol 95%, and stored in DI water for further use.
Meso 100.
300 mg of CTAB was dissolved in 100 mL of DI water at 60 °C. After the solution was cooled to room temperature, 2.8 mL of ammonium hydroxide was added, and the mixture was kept under stirring at 400 RPM for 30 min. Then, 1 mL of TEOS was added dropwise, and the mixture was kept under constant stirring for 4 h. The product was collected by centrifugation at 15,000 RPM for 20 min. As-synthesized nanoparticles were suspended in acidic ethanol (1 mL of HCl 36.5% in 30 mL of absolute ethanol) and heated at 80 °C under reflux for 6 h to remove the surfactant. The acidic ethanol washing step was repeated twice. Complete removal of surfactant was confirmed by Fourier-Transform Infrared (FT-IR) spectroscopy [12].
Meso 500.
CTAB (2 gr) was dissolved in the mixture of 700 mL of DI water and 50 mL of ammonium hydroxide. The mixture was stirred at room temperature for 1 h to allow the surfactant form aligned hexagonal micelles. TEOS was added, and the mixture was kept under constant stirring at 300 RPM for 4 h. The obtained nanoparticles were washed twice with DI water/ethanol 95% followed by centrifugation, and particles were suspended in acidic ethanol (1 mL of HCl 36.5% in 30 mL of absolute ethanol) and heated at 80 °C under reflux for 6 h to remove the surfactant. The acidic ethanol washing step was repeated twice.
Disulfide Meso 100.
For fabricating these nanoparticles, 55 mL of DI water, 5 mL of absolute ethanol, 125 mg of CTAB, and 425 μL of sodium hydroxide (2 M) were mixed in a 100-mL round bottom flask at 80 °C under stirring at 600 RPM for 1 h. Stirring rate was increased to 1400 RPM and BTESPD/TEOS with the molar ratio of 1:5 was added simultaneously. Then, the reaction was kept under constant stirring for 6 h. The obtained nanoparticles were washed with water and ethanol 95% for several times. CTAB was removed by suspending the nanoparticles in acidic ethanol (1 mL of HCl 36.5% in 30 mL of absolute ethanol) and the suspension was kept under reflux at 80 °C for another 6 h. The acidic ethanol washing step was repeated twice [16].
Disulfide Hollow 100.
First, Stöber core NPs were prepared as mentioned above and stored in 50 mL of DI water for use in the second step (stock Stöber SiO2 NPs). Second, the fabricated Stöber SiO2 NPs were coated with surfactant-based mesoporous shell-forming core-shell SiO2 NPs. In this step, 1400 mmol of DI water, 65 mmol of absolute ethanol, 0.25 mmol of TEA, 0.18 mmol of CTAB, and 10 mL of the stock Stöber SiO2 NPs were mixed in a 100-mL round bottom flask at 80 °C under the stirring rate of 400 RPM for 1 h. For preparing GSH-responsive shell, stirring rate was increased to 1400 RPM, and 0.1 mmol of BTESPD and 0.44 mmol of TEOS were added simultaneously. The reaction was kept under rotation for 4 h. Then, the obtained particles were precipitated by centrifugation at 15,000 RPM for 20 min and washed with ethanol 95%.
Third, the hollow particles were obtained via etching using a high concentration of sodium carbonate. The etching occurs due to structural and compositional differences between the core and the shell in mesoporous-coated Stöber SiO2 NPs. For synthesizing Disulfide Hollow 100 nanoparticles, 18 mmol of sodium carbonate was dissolved in 10 mL of DI water in a 100-mL round bottom flask at 50 °C under the stirring rate of 600 RPM for 1 h. The fabricated mesoporous-coated Stöber SiO2 NPs were dispersed in 10 mL of DI water, and sonicated for 1 h. Then, stirring rate was increased to 1200 RPM, and the NP suspension was added to sodium carbonate aqueous solution, and the reaction was left under stirring for another 12 h. The product was washed thrice with water/ethanol 95%, suspended in acidic ethanol (1 mL of HCl 36.5% in 30 mL of absolute ethanol), and heated to 80 °C under reflux for 6 h to remove the surfactant. The acidic ethanol washing step was repeated twice [24].
Physicochemical Characterization of Nanoparticles.
Size and morphology of the nanoparticles were analyzed by electron microscopy methods. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images were acquired by FEI Tecnai™ 12 transmission electron microscope (Hillsboro, OR, USA) operating at 120 kV and FEI Quanta 600F scanning electron microscope (Hillsboro, OR, USA) operating at 20 kV, respectively. Scanning transmission electron microscopy (STEM) images of the nanoparticles were obtained on JEOL JEM-2800 (Akishima, Tokyo, Japan) scanning transmission electron microscope with dual-energy dispersive X-ray spectrometer (EDS) detectors at an electron beam energy of 200 kV. Sample preparation was done by drop-casting the nanoparticles on a carbon-coated TEM grid. Hydrodynamic diameter and zeta potential were determined by dynamic light scattering (DLS) in a Malvern Instruments Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK). Measurements were performed in triplicate. Thermogravimetric analyses (TGA) were conducted using a TA Instruments hi-res TGA 2950 Thermogravimetric Analyzer (New Castle, DE, USA). All TGA experiments were conducted under a N2 atmosphere from 35 to 800 °C at a heating rate of 20 °C/min. Nitrogen adsorption-desorption isotherm analyses were performed at −196 °C on a Micromeritics ASAP 2020 (Norcross, GA, USA) for measuring surface area and pore size. All samples were dried at 100 °C overnight before analysis. Pore volume and pore size distributions were acquired from an adsorption branch by using the Barrett-Joyner-Halenda (BJH) method. The Brunauer–Emmett–Teller (BET) specific surface areas were measured via adsorption data at P/P0= 0.05–0.20. X-ray diffraction (XRD) patterns of all nanoparticles were investigated on a Bruker D2 Phaser X-ray diffractometer (Bruker AXS, Madison, WI, USA) using Cu Kα radiation (λ = 0.1542 nm) at 45 kV and 40 mA. The XRD spectra were recorded at a scanning speed of 0.01 deg/s, with a step size of 0.02° in a 2-theta scattering angle and a range of 2-8. X-ray photoelectron spectroscopy (XPS) analyses of the nanoparticles were conducted by Axis Ultra DLD instrument (Kratos Analytical, Manchester, UK). For analyses, the samples were mounted on a C tape and pumped overnight in the load lock before introduction into the analysis chamber. A mono Al source was employed. Survey scans were collected with a pass energy of 160 eV, step size of 1 eV, and dwell time of 200 ms. High-resolution region scans were obtained with a pass energy of 40 eV, 0.1 eV step size, and 400 ms live time. Data were processed using CASA XPS software. Fourier-Transform Infrared (FT-IR) spectra were obtained on a Varian 3100 FT-IR Excalibur Series Spectrometer (Randolph, MA, USA) with attenuated total reflectance mode to confirm complete removal of the surfactant (CTAB). For Limulus Amebocyte Lysate (LAL) test, LAL Chromogenic Assay Kit was used according to the manufacturer’s protocol to measure the free soluble endotoxin levels in all fabricated particles.
In Vitro Degradation.
Fabricated particles with a concentration of 100 μg mL−1 (total volume of 3.5 mL) were placed in simulated gastric fluid (SGF) pH 1.2, simulated lysosomal fluid (SLF; human liver lysosomes) pH 4.5, deionized (DI) water pH 6.5, simulated intestinal fluid (SIF) pH 6.5, and simulated body fluid (SBF) pH 7.4. To evaluate the degradation of disulfide-based nanoparticles (Disulfide Meso 100 and Disulfide Hollow 100), these particles were placed in SBF pH 7.4 containing 10 mM of glutathione (GSH) as a reductant. Samples were prepared in triplicate and incubated in a multi-therm shaker under constant rotation at 120 RPM. At specific time points: 0, 0.25, 1, 3, 7, 14, and 28 days, 100 μL of each sample was taken for TEM and Inductively Coupled Plasma Mass Spectrometry (ICP-MS) analyses. For ICP-MS, the samples were added to 0.5 mL centrifugal filter tubes (Millipore Ultrafree®-MC with a cut-off size of 100 kDa), centrifuged at 13,000 RPM for 20 min, and the collected supernatants were submitted for silicon (Si) measurement.
Cytotoxicity Assays.
Cytotoxicity of the intact nanoparticles was tested in Human Aortic Endothelial Cells (HAECs) as a model normal human cell line. Cells were passaged three times using Medium 200 containing low serum growth supplement (LSGS), seeded onto 96-well plates with the density of 8,000 cells per well, and incubated for 24 h at 37 °C in 5% CO2. Next, the cells were treated using different concentrations of nanoparticles in media (12.5, 25, 50, 100, 200, and 400 μg mL−1). Wells with media and 0.01% V/V Triton™ X-100 (without nanoparticles) were used as negative and positive controls, respectively. The cells were then incubated for 24 h, the media was aspirated, and the cells were washed twice with PBS. Cell viability was measured using the CCK-8 Cytotoxicity Assay Kit according to an established protocol and absorbance was measured at 450 nm with SpectraMax® M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Assays were performed at least in triplicate.
Cytotoxicity of the particles was measured in RAW 264.7 macrophages. Cells were seeded onto 6-well plates containing RPMI + 10% FBS with the density of 100,000 cells per well and allowed to grow for 24 h at 37 °C in 5% CO2 After 24 h, the cells were washed with PBS. Fresh media containing 10% FBS was then added with varying nanoparticle concentrations (20, 40, and 80 μg mL−1). These concentrations were chosen based on our previous studies [12]. Wells with media and Triton™ X-100 (without nanoparticles) were utilized as negative and positive controls, respectively. The cells were then incubated for another 72 h with nanoparticles (72 h was chosen because at least 90% confluency was achieved at this time point with 100,000 cells per well density of seeding cells), the media was aspirated, and the cells were washed twice with PBS. Cell viability was evaluated using the CCK-8 Cytotoxicity Assay Kit mentioned above. LC20 of each nanoparticle was then measured for further use in degradation study in RAW 264.7 macrophages.
Cytotoxicity of free silicic acid [Si(OH)4] was also investigated in RAW 264.7 macrophages after incubation for 24 h using a concentration range of 0.0625 to 400 μg mL−1. Cell viability was measured using the CCK-8 Kit, and assays were conducted at least in triplicate.
Intracellular Degradation of SiO2 NPs.
100,000 cells per well were seeded in 6-well plates containing RPMI + 10% FBS and incubated for 24 h at 37 °C in 5% CO2. Based on the CCK-8 results, cells were treated with LC20 of each nanoparticle and incubated for another 72 h. After 72 h, cells were washed with PBS and lysed with cell lysis buffer according to the manufacturer’s protocol. The lysed cells were then centrifuged at 1500 RPM for 8 min. The supernatants were collected and submitted for Si measurement via ICP-MS. As a control, nanoparticles were incubated in RPMI medium containing 10% FBS (without cells) for 72 h and proceeded with the same conditions.
Animal Studies.
Female CD-I mice (6-8 weeks old) were purchased from Charles River Laboratories, housed in a group of 4 animals in standard cages and were acclimated to the animal facility for two weeks. All animals had free access to food and water and were subject to 12 h light and dark cycles. Then, mice were randomly assigned to treatment groups (N=3 mice per group), transferred to individually housed metabolic cages (Nalgene®) and kept for one week before experimental procedures. Particles were injected to CD-1 mice via the lateral tail vein with Stöber 100, Meso 100, and Disulfide Hollow 100 nanoparticles in total volume of 150 μL of sterile normal saline (0.9% w/v NaCl) at the dose of 25 mg per weight of the animal. Control mice received 150 μL of sterile normal saline under the same condition. The individual behavioral reaction to single-dose administration of SiO2 NPs was assessed daily up to 7 days. Mice were weighed daily from day 0 to day 7. Urine and fecal samples were collected at each time point (6, 12, 24, 48, 72, 120, and 168 h) in separate 50-mL polypropylene tubes and submitted for Si measurement via ICP-MS. On day 7, animals were euthanized using 5% Isoflurane (Fluriso™, USP). Hematological and blood chemical marker levels were recorded. Vital organs (liver, lung, spleen, and kidney) were isolated, weighted post-necropsy, preserved in 10% formalin at −80 °C for further histopathological examination and Si measurement. All animal experiments were performed in compliance with the University of Utah Institutional Animal Care and Use Committee (IACUC).
Sample Preparation for Measurement of Silicon Content In Vivo.
Since Si is most stable in hydrofluoric acid, a two-step procedure was used for digesting urine, feces, and organs. First, samples were initially dried and treated with hot concentrated nitric acid, and then a mixture of 0.1% HF and 5% HCl was used to solubilize Si.
Urine.
Thawed samples were transferred into 50 mL polypropylene vials and dried in a graphite block digester at 105 °C. Next, 2.5 mL of HNO3 (concentrated, trace metal grade) was added, refluxed at 95 °C for 2 h, and dried at 105 °C. After cooling, the residues were treated with 1 mL of 0.1% HF (trace metal grade), 5% HCl (trace metal grade), and 10 ppb Gallium (Ga) as an internal standard. Vials were kept under stirring overnight. Then, the digests were transferred into polystyrene autosampler tubes for injection into ICP-MS for Si measurement.
Feces.
Thawed samples were transferred into 50 mL polypropylene vials, mixed with 30 mL of HNO3, and kept at room temperature for 1 week. The suspension was homogenized by mixing, 1 mL was poured into a 50-mL polypropylene vial, weighted, and then dried at 105 °C. 2.5 mL of HNO3 was added, the samples were refluxed at 95 °C for 2 h, and dried at 105 °C.
After cooling, the residues were treated with 2 mL of 0.1% HF and 5% HCl, and 10 ppb Ga as an internal standard. Then, the digests were transferred into polystyrene autosampler tubes for injection into ICP-MS for Si measurement.
Organs.
Thawed samples were transferred into 50 mL polypropylene vials, mixed with 5 mL of HNO3 and kept at room temperature overnight. Then, the samples were refluxed at 95 °C for 2 h and dried at 105 °C. After cooling, the residues were treated with 2 mL of 0.1% HF and 5% HCl, and 10 ppb Ga as an internal standard. Then, the digests were transferred into polystyrene autosampler tubes for injection into ICP-MS for Si measurement.
ICP-MS Preparation.
Samples were run in a quadrupole ICP-MS (Agilent 7500ce, Santa Clara, CA, USA) using a PTFE spray chamber, sapphire injector, platinum cones, and a syringe-FAST introduction system (ESI, Omaha, NE, USA). The instrument was tuned for Si (mass 28) to minimize the background signal obtained for 0.1% HF and 5% HCl to approximately 100 Kcps. Typical sensitivities were over 1 Mcps per ppm of Si. A calibration curve containing 0, 0.25, 0.5, 1, 5, and 25 ppm of Si was run with samples. Ga 10 ppb was added to calibration curve, blanks, and samples. Sets of 4 samples and 2 blanks were run after the calibration curve. Limit of detection was calculated as three times the standard deviation of the background and was typically below 0.1 ppm.
Complete Blood Count and Blood Chemistry Analyses.
Blood was withdrawn from the heart tissue immediately following euthanasia or animal death. The collected blood was stored in heparin- and EDTA-coated blood collection tubes. The complete blood counts (CBC) were measured within 4 h post-collection with HT5 CBC-DIFF Instrument (Heska, Loveland, CO, USA). Plasma was obtained from centrifugation of blood samples at 3500 RPM for 15 min, frozen at −80 °C and the analyses were conducted within 24 h of blood collection using a DRI-CHEM veterinary blood chemistry analyzer (Heska, Loveland, CO, USA). In CBC analysis, significant hematology markers from the whole blood were evaluated such as red blood cells (RBC), hemoglobin, platelets, and white blood cells (WBC) including neutrophils, basophils, eosinophils, monocytes, and lymphocytes. In blood chemistry analysis, the following markers were measured: blood urea nitrogen (BUN), total protein, glucose, alkaline phosphatase (ALP), alanine aminotransferase (ALT), and aspartate aminotransferase (AST).
Histopathological Examination.
Isolated liver, spleen, lung, and kidneys were fixed in 10% formalin. The tissues were processed using the Tissue-Tek VIP 6 Vacuum Infiltration Processor and embedded in molten paraffin wax using the Tissue-Tek Embedding Center. The tissues were then sliced on Leica RM 655 Rotary Microtome, and placed onto glass slides. The slides were stained with hematoxylin and eosin (H&E) using the Tissue-Tek Prisma Automated Slide Stainer. The histopathological interpretation was performed by an animal pathologist (Dr. Lawrence D. McGill) who was unaware of the treatment modalities of each animal. Slides were scanned at 20X by Aperio digital pathology slide scanner and analyzed via Aperio ImageScope software.
Nanoparticle Tissue Uptake by TEM In Vivo.
The liver was isolated from the mouse 7 days after nanoparticle injection and immediately placed in a fixative solution pH 7.45 containing 2.5% glutaraldehyde, 1% paraformaldehyde, 0.1 M of sodium cacodylate containing 4% sucrose and 8 mM of calcium chloride. The tissues were then processed by the following steps: samples were rinsed with 0.1 M of sodium cacodylate buffer at room temperature for 10 min and post-fixed in 2% osmium tetroxide and 0.1 M of sodium cacodylate buffer for 1 h. Osmium tetroxide was removed, and samples were rinsed in water at room temperature for 5 min and stained en bloc with saturated Aqueous Uranyl Acetate for 1 h. Samples were then dehydrated through a graded series of ethanol: 50%, 70%, 95%, and 100% at room temperature for 5 min for each change and transitioned to absolute Acetone for 5 min. Afterward, samples were infiltrated through increasing ratios of Plastic: Acetone, such as 1:1 at room temperature for 1 h, and then 3:1 for overnight. The next day, the tissues were infiltrated three times in 100% plastic with 1 h of rotation and 1 h of vacuum for each step. Molds were prepared and tissues placed in appropriate molds with labels and placed in an oven at 60-70 °C overnight. Blocks were removed from the molds before cutting. One block was hand-trimmed with a double edge razor blade. Next, 0.5 μm of block face sections were cut with glass knives on a Leica EM UC6 ultramicrotome. Once areas of interest were found, the block-face was trimmed, and a diamond knife was used to cut 80 nm sections. Sections were placed on 200 mesh copper grids and then contrasted at room temperature for 10 min with saturated aqueous Uranyl Acetate. Sections were examined on FEI TECNAI T-12 TEM operated at 120 kV, and images were generated on a Gatan Ultrascan 1000.
Statistical Analysis.
Data are expressed as mean ± standard deviations (SD) for at least three separate experiments. The difference between multiple study groups was analyzed by Analysis of Variance (ANOVA) followed by a Tukey’s or Dunnett’s post-hoc tests. The difference between the study groups was considered significant when Pvalue< 0.05.
Results and Discussion
Synthesis and Characterization of Nanoparticles.
Five SiO2 NPs were fabricated with variations in diameter (100 nm vs. 500 nm), porosity (nonporous vs. mesoporous), density (dense vs. hollow), and composition (TEOS vs. TEOS/BTESPD). These particles were named Stöber 100, Meso 100, Meso 500, Disulfide Meso 100, and Disulfide Hollow 100. Stöber 100 nanoparticles were synthesized based on a reported sol-gel method [24]. Meso 100 and Meso 500 particles were fabricated using surfactant (CTAB)-based techniques [13, 36]. Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles were fabricated by modified hydrolysis and polycondensation of TEOS and BTESPD precursors containing …Si-O-Si-C-C-C-S-S-C-C-C-Si-O-Si… (precursors’ structures are illustrated in Fig. 1A and B) [16, 24]. Structural and compositional difference-based (between the inner core and the outer shell) selective etching strategy was utilized for the preparation of these lower density hollow particles [24]. All the synthesized particles were carefully characterized using different techniques. Size, size distribution, and morphology of the particles were evaluated using transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. 1). Electron microscopy images confirmed the fabrication of uniform particles with average diameters of Stöber 100 (105 ± 5 nm), Meso 100 (125 ± 15 nm), Meso 500 (475 ± 45 nm), Disulfide Meso 100 (105 ± 10 nm), and Disulfide Hollow 100 (130 ± 5 nm). Fig. 1M and N show STEM images for Disulfide Meso 100 and Disulfide Hollow 100 particles revealing the homogeneous distribution of sulfur (S) within these particles.
Figure 1.

Structure of the precursors used for fabrication of the nanoparticles; A: tetraethyl orthosilicate (TEOS) and B: bis[3-(triethoxysilyl)propyl] disulfide (BTESPD). C-G: TEM images of (C) Stöber 100, (D) Meso 100, (E) Meso 500, (F) Disulfide Meso 100, and (G) Disulfide Hollow 100 nanoparticles indicating the formation of particles with narrow size distribution. C1-G1 insets: TEM images of the particles under higher magnifications. H-L: SEM images displaying size, shape, and surface roughness of the nanoparticles. M and N: STEM images showing atomic density in Disulfide Meso 100 and Disulfide Hollow 100 particles. In these particles, sulfur (S) has a homogeneous distribution [16, 24].
Mean hydrodynamic diameters measured in DI water and surface area of the particles are summarized in Table 1. Free soluble endotoxin levels were evaluated using the LAL chromogenic assay. Endotoxin is the lipopolysaccharide (O-antigen) component of the outer membrane of gram-negative bacteria [37]. The presence of free endotoxin should be accurately monitored for in vitro and in vivo experiments since it can cause unwanted inflammatory adverse effects [38]. For a 20 gr mouse, the maximum endotoxin level considered safe after IV injection of nanoparticles is 0.1 EU administered over a 1 h period [39, 40]. The results indicate this level was in an acceptable range for the nanoparticles injected to mice with an average of 30 gr used in this study (Table 1).
Table 1.
Characterization of nanoparticles in terms of size, surface area, average pore diameter, and endotoxin level a.
| Nanoparticle | Diameter (TEM; nm) |
Diameter (DLS; nm) |
Surface Area (m2 g −1) |
Pore Diameter (nm) |
Endotoxin Level (EU mL−1) |
|---|---|---|---|---|---|
| Stöber 100 | 105 ± 5 | 134 ± 15 | 37 ± 0.3 | 0 | 0.126 ± 0.02 |
| Meso 100 | 125 ± 15 | 188 ± 30 | 1027 ± 29 | 2.9 ± 0.5 | 0.103 ± 0.05 |
| Meso 500 | 475 ± 45 | 810 ± 23 | 950 ± 23 [36] | 2.9 ± 0.7 [36] | 0.02 ± 0.01 |
| Disulfide Meso 100 | 105 ± 10 | 140 ± 8 | 342 ± 8 | 2 ± 0.8 | 0.04 ± 0.02 |
| Disulfide Hollow 100 | 130 ± 5 | 178 ± 15 | 495 ± 8 | 2.3 ± 0.5 | 0.174 ± 0.06 |
Data are mean ± SD (N=3).
Fig. 2 illustrates the physiochemical characterization of the synthesized nanoparticles. Fig. 2A indicates nitrogen adsorption-desorption isotherm plots. Meso 100, Meso 500, Disulfide Meso 100, and Disulfide Hollow 100 nanoparticles follow similar patterns which are related to type IV isotherm according to IUPAC classification. A hysteresis loop was observed in the isotherm plot of Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles attributed to capillary condensation in the mesopores at high relative pressures (P/P0). Stöber 100 nanoparticles follow type III isotherm recognized for nonporous nanoparticle.
Figure 2.

Characterization of the fabricated nanoparticles. A: NPs nitrogen adsorption-desorption isotherms indicating type IV for mesoporous particles and type III for nonporous dense Stöber 100 particles. B: pore size distribution confirming the presence of 2-3 nm pores in mesoporous particles. C: TGA graphs showing more weight loss in disulfide-based particles due to the existence of …Si-O-Si-C-C-C-S-S-C-C-C-Si-O-Si… hydrophobic backbone structure. D: XRD plots of the particles confirming the presence of Bragg peaks in Meso 100 and Meso 500 nanoparticles and the absence of these peaks in Disulfide Meso 100 and Disulfide Hollow 100 particles. E: XPS spectra for Disulfide Meso 100 (inset) and Disulfide Hollow 100 particles revealing the incorporation of BTESPD precursor in these particles during the fabrication step. Purple and green arrows show the presence of carbon and sulfur atoms, respectively. F: FT-IR analyses for CTAB and Meso 100 particles which confirms the absence of typical CTAB peaks around 3000 cm−1 in washed Meso 100 nanoparticle.
Fig. 2B indicates the pore size distribution in the fabricated nanoparticles. Thermogravimetric analysis (TGA) graphs in Fig. 2C reveals weight loss percentages of these nanoparticles when the temperature is increased up to 800 °C. Based on the structure of bis[3-(triethoxysilyl)propyl] disulfide precursor used in the fabrication of Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles, …Si-O-Si-C-C-C-S-S-C-C-C-Si-O-Si… is embedded in these particles. This leads to more weight loss of these nanoparticles at temperatures higher than 400 °C due to the burning of the carbon-based hydrophobic moiety in comparison to regular nonporous and mesoporous particles.
As illustrated in Fig. 2C, Disulfide Hollow 100 particles exhibit less weight loss than Disulfide Meso 100 particles which are related to the lower density of this nanoparticle due to the presence of a hollow cavity in its structure (Fig. 1G). In Fig. 2D, X-ray diffraction (XRD) graphs are illustrated confirming the presence of aligned and well-ordered pore structure in Meso 100 and Meso 500 nanoparticles due to the observation of Bragg peaks and disordered pore structure in Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles. The latter phenomenon is often observed in periodic mesoporous organosilica NPs (PMO NPs) in which long bissilylated precursor (here BTESPD) is utilized in the preparation step with no typical Bragg peaks observed in the corresponding XRD graphs [41].
X-ray photoelectron spectroscopy (XPS) plots in Fig. 2E, confirm the incorporation of disulfide-based precursor in Disulfide Meso 100 and Disulfide Hollow 100 particles due to the presence of sulfur and carbon peaks. XPS atomic percentages for the synthesized particles are summarized in Table S1. Complete removal of CTAB surfactant was shown in Fig. 2F in which typical CTAB peaks around 3000 cm−1 do not exist in Fourier-Transform Infrared (FT-IR) spectrum of the washed nanoparticles.
In Vitro Degradation.
Degradation of the nanoparticles (100 μg mL−1, the total volume of 3.5 mL) was studied in simulated biological fluids with different pH values at 37 °C under constant rotation. These fluids were SGF pH 1.2 ± 0.1, SLF pH 4.5 ± 0.1, DI water pH 6.5 ± 0.1, SIF pH 6.5 ± 0.1, and SBF pH 7.4 ± 0.2. SBF containing 10 mM of GSH was also applied mimicking intracellular microenvironment to evaluate the influence of S-S bonds on degradation profile of Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles. GSH is a peptide composed of three amino acids, namely glycine, L-glutamic acid, and L-cysteine. L-cysteine residue has free S-H (thiol) group. GSH has a high intracellular concentration (2-10 mM), which can accelerate intracellular degradation of S-S bonds [42, 43]. Extracellular concentration of GSH is 2–20 μM, which is 100–1000 times lower than intracellular concentrations [44]. The exact composition of the media is described in Table S2.
Several factors may influence the degradation of SiO2 NPs. These include, but are not limited to nanoparticle size, morphology, porosity, pore diameter, surface area, density, -Si-O-Si-condensation degree, composition, zeta potential, concentration, condensation degree, surface functionality, in vivo route of administration, and composition of degradation media including in this case GSH concentration. Table 2 summarizes zeta potential values of the synthesized particles in the studied media. All particles have higher zeta potential values (conversion of silanol groups to silanolate; Si-OH to Si-O−) in DI water due to the absence of ions. Ionic strength and pH can reduce Debye length by screening surface charges, which can result in lower zeta potential values [45-47]. The particles had zeta potential values around zero in SGF, which is attributed to the presence of a high concentration of proton (H+) ions in this medium shielding the surface charges. Also, point of zero charge (PZC) for silicon dioxide is 2-3 meaning that at pH values around 2-3, the net charge of SiO2 NPs is ca. zero which confirms our findings in SGF [48-50].
Table 2.
Zeta potential values in mV for the nanoparticles in five different media a.
| Nanoparticle | SGF pH 1.2 |
SLF pH 4.5 |
DI Water pH 6.5 |
SIF pH 6.5 |
SBF pH 7.4 |
|---|---|---|---|---|---|
| Stöber 100 | 3 ± 3 | −11 ± 3 | −43 ± 1 | −17 ± 1 | −19 ± 2 |
| Meso 100 | 3 ± 2 | −2 ± 1 | −39 ± 2 | −7 ± 1 | −13 ± 1 |
| Meso 500 | −2 ± 2 | −5 ± 2 | −29 ± 1 | −8 ± 0.5 | −13 ± 1 |
| Disulfide Meso 100 | 2 ± 2 | −5 ± 1 | −28 ± 2 | −2 ± 0.2 | −6 ± 0.2 |
| Disulfide Hollow 100 | 0 ± 0.5 | −10 ± 2 | −24 ± 1 | −20 ± 3 | −16 ± 1 |
Data are mean ± SD (N=3).
Fig. S1 illustrates the amount of released Si from the nanoparticles over 28 days in different degradation media. Variations in pH play the predominant role in the degradation process where a higher Si content is released at higher pH values, and degradation dramatically increases under alkaline conditions such as pH 7.4. As shown in Scheme 1, sequential hydration occurs in which the water molecules adsorb onto the siloxane and silanol networks and hydroxyl (OH−) ions present in aqueous solution attack electropositive silicon atoms (nucleophilic attack) in silanol groups forming unstable pentavalent structures. When another hydroxyl group attacks this intermediate complex, monosilicic acid [Si(OH)4] (a.k.a. silicon hydroxide) releases in the medium by the ion-exchange reaction. Monosilicic acid is the soluble form of silica with the pKa of ca. 9.8 (weak acid) containing silicon tetrahedrally coordinated to four hydroxyl groups [51]. This molecule is nontoxic at specific concentrations and can be transferred through the tissues, enter blood vessels, and eventually be excreted and cleared from the body through the urinary system due to its small size (<5.5 nm) [52]. The degradation kinetics of SiO2 NPs (RDegradation) is dependent on the concentration of three surface groups: i) protonated (SiOH2+), ii) neutral (SiOH), and iii) deprotonated silanols (SiO−) [53].
✓ m and p are reaction orders
✓ k(H+), k(H2O), and k(OH−) are rate constants
Also, anions such as Cl−, PO43−, SO42−, and taurocholate can affect the degradation process by enhancing deprotonation of silanol groups and hydrolysis of Si-O-Si bonds, which is catalyzed by the nucleophilic attack. Also, protons (H+) and other cations (Na+, K+, Ca2+, and Mg2+) can interact with electronegative oxygen atoms and weaken Si-O-Si bonds due to the formation of electrostatic clouds around these bonds and facilitate the degradation process [54]. In SGF, we have a high concentration of H+ and Cl−, but low pH value of this medium around PZC overcomes the ionic concentration.
To study the cytotoxicity of the degradation products of the nanoparticles, we investigated the cytotoxicity of free silicic acid [Si(OH)4] in RAW 264.7 macrophages (Fig. S1F). RAW 264.7 macrophages were chosen due to their high phagocytic activity. Over a wide range of concentrations from 0.0625 to 400 μg mL−1, no cytotoxicity was observed after incubation of these cells with silicic acid for 24 h.
SiO2 is an acidic oxide which can react with basic solutions. SiO2 dissolves in alkaline solutions and can form salts with bases (forming charged species) which weaken the backbone structure of Si-O-Si [55-57]. SiO2 NPs cannot react with DI water quickly due to the difficulty of breaking up the strong covalent Si-O-Si bonds (with average bond length of 1.64 angstroms and bond dissociation energy variations from 452 KJ mol−1 to 799 KJ mol−1 based on other adjacent groups) [58-61]. However, when the surface area of the particles is higher such as the case for Meso 100 and Meso 500 nanoparticles, more than 80% degradation is observed in SLF, SIF, and SBF after 28-days. In Fig. 3, degradation percentages of SiO2 NPs over time are shown based on the amount of Si that was added to each at the beginning of the experiments.
Figure 3.

Degradation profiles of SiO2 NPs in different media; A: SGF, B: SLF, C: DI water, D: SIF, and E: SBF. Results suggest that Mesoporous particles with higher surface area and porous structure, degrade faster than nonporous and disulfide-based counterparts. Due to the hydrophobic structure of disulfide-based particles, no difference was observed for degradation rates in the presence and absence of GSH (shown in E). Effect of pH and ionic strength on the degradation of SiO2 NPs; F: Stöber 100 and G: Meso 100 nanoparticles (** means there is statistically significant difference at each time point in SBF). Stöber 100 particles degrade very slowly in SGF, SLF, and DI water while much faster degradation was observed in SIF and SBF. Meso 100 nanoparticles degrade up to 100% in SLF, SIF, and SBF while in SGF and DI water maximum degradation is 6% and 71%, respectively. H: cytotoxicity of SiO2 NPs in HAECs after incubation for 24 h. Results indicate concentration-dependent toxicity for all the particles with the highest toxicity observed for Meso 100 and Meso 500 nanoparticles at dosages equal or more than 200 μg mL−1. Data are mean ± SD (N=3). ** means statistically significant difference (Pvalue< 0.05) was observed for degradation percentages between Meso 100 and other nanoparticles at day 28. ANOVA followed by a Tukey’s post-hoc test was applied for statistical analyses.
As demonstrated in Fig. 3A, maximum ca. 8.7% of the particles degraded after 28 days at pH 1.2. Fig. 3B shows that Meso 100 and Meso 500 nanoparticles degrade up to ca. 100% in SLF while the maximum degradation for Stöber 100 nanoparticles is ca. 7% which implies the role of nanoparticle’s porosity and surface area; Stöber 100 (37 m2 g−1), Meso 100 (1027 m2 g−1), and Meso 500 (950 m2 g−1). For Disulfide Hollow 100 and Disulfide Meso 100 nanoparticles, the percentages were ca. 59% and 19%, respectively. As shown, Disulfide Hollow 100 nanoparticles showed ca. 3.1 times higher degradation (Pvalue< 0.05) than Disulfide Meso 100 particles. This could be attributed to their thin shell thickness (~10-15 nm) which can be broken easily in the medium, particularly in a redox environment. In DI water (Fig. 3C), Meso 100 and Meso 500 particles degraded up to 71% and 55%, respectively after 28 days. However, the other three particles did not degrade to the same extent (ca. 17% for Disulfide Hollow 100 nanoparticles).
Fig. 3D indicates the importance of medium composition in the degradation of the nanoparticles. As illustrated, with the same pH as DI water, all particles degraded to a higher degree in SIF after 28 days, up to 46% (Stöber 100), 100% (Meso 100), 87% (Meso 500), 55% (Disulfide Meso 100), and 79% (Disulfide Hollow 100). In SBF (Fig. 3E), there is a synergistic effect of high ionic strength, and basic condition (Table S2). The highest degradation occurs in SBF for all the particles. In the first 6 hours, the degradation of the particles was as follows: Stöber 100 (6%), Meso 100 (51%), Meso 500 (41%), Disulfide Meso 100 (13%), and Disulfide Hollow 100 (34%). These values are higher than the observed degradation in all the other media at the same time point. It is noteworthy that the presence of 10 mM GSH does not affect the degradation rate of disulfide-based particles probably due to the hydrophobic backbone of BTESPD precursor containing …-C-C-C-S-S-C-C-C-… which does not exist in the TEOS precursor. Therefore, even when these particles are exposed to a reductive environment, and the disulfide bonds degrade and the particles break into smaller fragments. Still these fragments do not degrade faster since they probably repel water molecules because of their hydrophobic nature. Keeping this in mind, these nanocarriers can be designed with controlled degradation rates by utilizing different ratios of precursors in the fabrication process. The disulfide-containing particles can then release the active agent in a sustained manner desirable for specific delivery applications. Between Meso 100 and Meso 500 particles, we observed modest differences in degradation rates of the particles, as shown in Fig. 3B-E. Hence, it seems that particle size has a moderate influence on in vitro degradation kinetics of SiO2 NPs and smaller particles (Meso 100) with higher surface area (Table 1) degrade faster than larger particles (Meso 500). These results are consistent with previous observations for degradation of mesoporous SiO2 NPs with variations in size in simulated body fluid at 37 °C [62]. Comparing Stöber 100 and Meso 100 nanoparticles, differences in degradation kinetics were statistically significant, which confirms that an increase in porosity and surface area (37 and 1027 m2 g−1, respectively) increases the degradation rates. Comparing Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles, the differences in degradation kinetics were negligible. This suggests that in the range studied, particle density does not seem to play a significant role in the degradation of these particles. Between Meso 100 and Disulfide Meso 100 particles, there were significant differences in degradation kinetics in various media due to the hydrophobic nature of the disulfide-containing precursor as discussed earlier.
To elucidate the influence of pH and ionic strength, the degradation kinetics of Stöber 100 and Meso 100 nanoparticles (as examples of nonporous and mesoporous particles, respectively) was investigated in different media as demonstrated in Fig. 3F and Fig. 3G, respectively. Both particles degraded in a pH-, ionic concentration-, and time-dependent fashion. Stöber 100 nanoparticles exhibited statistically (Pvalue< 0.05) higher degradation in SIF and SBF at 28 days (up to 46% and 64%, respectively) while maximum degradation in SGF, SLF, and DI water was ca. 10% (very slow degradation kinetics). For Meso 100 nanoparticles, the effect of pH and ionic strength was even more pronounced, and up to 100% degradation was observed in SLF, SIF, and SBF while maximum degradation was ca. 6% and 71% in SGF and DI water, respectively. In general, in SBF, due to high ionic concentration of this medium (Table S2 indicates the composition of SBF), higher degradation was observed for all the nanoparticles (Fig. 3).
Cytotoxicity of these nanoparticles was evaluated in Human Aortic Endothelial Cells (HAECs; Fig. 3H). Toxicity of SiO2 NPs can occur by the generation of reactive oxygen species (ROS) via silanol groups which initiate intracellular oxidative stress, or hydrogen bonding and electrostatic interactions between SiO2 NPs group (SiOH/SiO−) and proteins or phospholipids existing in cell membranes. Also, ROS generation might lead to DNA damage and abnormal expressions of genes (genotoxicity). SiO2 NPs can also induce the formation of nuclear inclusions and protein aggregates inside the cells leading to cytotoxicity. HAECs were chosen as an alternative human cell line to investigate the effect of nanoparticle size, porosity, density, and composition on the cytotoxicity profile. After incubation of these cells with nanoparticles for 24 h, concentration-dependent toxicity was observed for all the particles. Results show that with concentrations equal or more than 200 μg mL−1, Meso 100 and Meso 500 particles killed ca. 90% of the cells. When the cells were treated with 12.5 μg mL−1 of nanoparticles, no toxicity was observed except for Stöber 100 nanoparticles in which only 78% of the cells were found viable after 24 h.
For observing degradation process, TEM images were taken for selected nanoparticles (based on the results shown in Fig. 3, Stöber 100, Meso 100, and Disulfide Hollow 100 particles were chosen) in SGF, DI water, and SIF over 28 days. The nonporous dense Stöber 100 particles undergo surface (external) degradation (Fig. 4A). In contrast, Fig. 4B suggests that Meso 100 particles degrade both in surface and in bulk due to the porous scaffold. Consistent with Fig. 3 degradation data, Stöber 100 particles lose their mass integrity and shape and degrade much faster in SIF than in DI water or SGF. In DI water (Fig. 4A), few particles degrade within 28 days (as indicated by red arrows). For Meso 100 particles, degradation of the particles start from the very first hours of incubation, especially in SIF. After immersion of these particles in SIF and DI water, some large pores were formed in particles by altering the structure of the ordered mesopores which accelerates the degradation kinetics of these mesoporous particles. After 48 hours, many of Meso 100 particles aggregated and formed large irregularly shaped clusters. Disulfide Hollow 100 particles seem to collapse or rupture and disintegrate into smaller particles especially in SIF and SBF with 10 mM of GSH while in SGF the particles remain almost intact even after 28 days (Fig. 4C). This disintegration of hollow particles is due to their shell thickness. We have previously demonstrated that the diameter of the shell and the size of the interior hollow cavity are tunable based on reaction conditions [24] and the thinner the shell is, the easier it breaks after immersion in an aqueous environment. Images of the nanoparticles incubated in SBF and SLF are shown in Fig. S2. Since SBF has high ionic concentration, and SLF contains human liver lysosome homogenates, the degraded particles were hard to find in these media and TEM images taken were not explanatory (Fig. S2).
Figure 4.
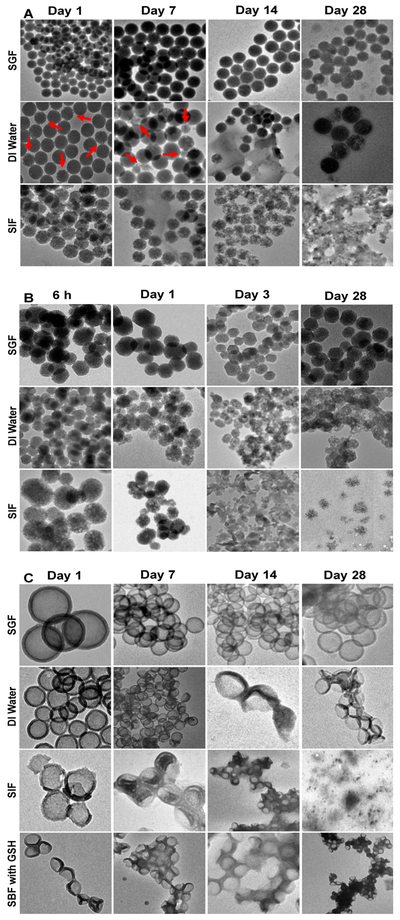
Degradation images of the nanoparticles taken by TEM after immersion in SGF, DI water, and SIF over 28 days (from left to right). A: Stöber 100, B: Meso 100, and C: Disulfide Hollow 100 nanoparticles. The latter was also investigated in SBF with 10 mM of GSH. Stöber 100 nanoparticles underwent surface degradation while Meso 100 nanoparticles underwent surface and bulk degradation. Disulfide Hollow 100 particles tend to collapse or rupture and disintegrate into smaller fragments. Images are in accordance with the observed degradation results with this order for all the particles: SIF>DI water>SGF (> means quicker degradation). Since the fabricated nanoparticles behave differently in simulated dissolution media and some particles like Meso 100 degraded much faster than their counterparts, the time points indicated in this figure are specific to each nanoparticle.
Intracellular Degradation of SiO2 NPs.
For investigating intracellular degradation of these nanoparticles, RAW 264.7 macrophages were selected. As shown in Fig. 5A, Meso 100 particles cause toxic effects even at concentrations less than 80 μg mL−1 in which 67%, 50%, and 21% cell viability was observed after the cells were treated with 20, 40, and 80 μg mL−1 of Meso 100 particles, respectively. However, more than 70% of cell viability was observed with other particles used under the same concentrations. Next, using particles’ LC20 (Stöber 100: 40 μg mL−1, Meso 100: 10 μg mL−1, Meso 500: 40 μg mL−1, Disulfide Meso 100: 20 μg mL−1, and Disulfide Hollow 100: 20 μg mL−1), intracellular degradation was measured in macrophages. After 72 h, macrophages were lysed using lysis buffer, and the supernatant Si content was measured by ICP-MS. As illustrated in Fig. 5B, Meso 100 particles have the highest degradation rate (ca. 3.8%). The percent degradation values for Stöber 100 and Meso 500 nanoparticles were ca. 0.3% and 2%, respectively. The intracellular degradation percentages were ca. 1.2% and 0.4% for Disulfide Meso 100 and Disulfide Hollow 100 nanoparticles, respectively. The results indicate that for intracellular degradation of SiO2 NPs, size (Meso 100 vs. Meso 500), porosity and surface area (Stöber 100 vs. Meso 100), density (Disulfide Meso 100 vs. Disulfide Hollow 100), and composition (Meso 100 and Disulfide Meso 100) play significant roles. As a control, intact nanoparticles were incubated in RPMI medium containing 10% FBS (without cells). No degradation was observed after incubation of the particles for 72 h with this media as measured by ICP-MS.
Figure 5.

A: Cytotoxicity of SiO2 NPs in RAW 264.7 macrophages after incubation for 72 h with 20, 40, and 80 μg ML−1 of nanoparticles. LC20 values for the particles were 40, 10, 40, 20, and 20 μg mL−1 for Stöber 100, Meso 100, Meso 500, Disulfide Meso 100, and Disulfide Hollow 100 nanoparticles, respectively. B: Degradation percentages of the particles. The macrophages were treated with particles LC20 and incubated for 72 h. The cells were lysed, and Si content in the supernatant was measured using ICP-MS. As shown, Meso 100 particles have the highest degradation rate (3.8%) while Stöber 100 nanoparticles have the lowest (0.3%). Data are mean ± SD (N=6). **means statistically significant differences (Pvalue< 0.05) were observed for degradation percentages between Meso 100 and other nanoparticles. ANOVA followed by a Tukey’s post-hoc test was applied for statistical analyses.
In Vivo Degradation and Biodistribution.
For investigating the in vivo degradation and biodistribution, Stöber 100, Meso 100, and Disulfide Hollow 100 nanoparticles were selected based on the in vitro results. Since less significant differences were observed in the degradation rates between Meso 100 and Meso 500 nanoparticles (in comparison to other nanoparticles), Meso 500 nanoparticles were excluded for in vivo studies. Particles at the dose of 25 mg kg−1 were tail vein injected to immunocompetent CD-1 female mice housed individually in metabolic cages for 7 days post-injection. Control mice were injected with 0.9% normal saline. Urine samples were collected in separate tubes at each time point for each mouse. No mortality or sign of morbidity was observed for mice for 7 days. Fig. 6A indicates the cumulative percentages of Si measured in urine samples. The glomerular capillary wall has three different layers: fenestrated endothelium, glomerular basement membrane (basal lamina, which is negatively charged), and podocyte extensions. The cut-off size for these layers is ~5.5 nm [63, 64]. Based on kidney glomerulus anatomy, particles should degrade or become fragmented to the sizes less than ~5 nm, and pass through these layers to be filtered and excreted in the urine. As illustrated in Fig. 6A, after 24 h, renal excretion of Disulfide Hollow 100 nanoparticles is ca. 25.9% while this value is ca. 11.6% and 21.7% for Stöber 100 and Meso 100 particles, respectively. After 7 days, Meso 100 particles have the highest degradation in mice with 53% of the degradation products containing Si detected in urine while for Stöber 100 and Disulfide Hollow 100 nanoparticles, Si excretion was 27% and 39%, respectively. Fig. 6B-D illustrates degradation, and biodistribution of these particles after 7 days. Highly perfused organs were selected for these experiments, including liver, spleen, lung, and kidney. As shown, all three particles accumulated in the liver and spleen more than lung and kidney. It is noteworthy that in organs, Si can be found in the form of soluble degradation products, and as intact or broken nanoparticles while in the urine Si should be in its soluble degradation form or in the form of nanoparticle fragments that are less than ~5 nm. As shown in Fig. 6B, for Stöber 100 nanoparticles, the Si content was: 27% (urine), 25% (liver), 16% (spleen), 4% (lung), 2% (kidney), and 26% (the rest of the body). For Meso 100 nanoparticles (Fig. 6C), the Si content was: 53% (urine), 17% (liver), 10% (spleen), 4% (lung), 2% (kidney), and 14% (the rest of the body). As indicated in Fig. 6D, these values for Disulfide Hollow 100 nanoparticles were: 39% (urine), 19% (liver), 13% (spleen), 3% (lung), 3% (kidney), and 23% (rest of the body). Regarding fecal and blood analyses after 7 days, no Si was found in fecal samples and only ca. 0.2% of Si was found in the blood of the mice treated with Disulfide Hollow 100 nanoparticles. Results indicate that porosity, surface area, and composition play significant roles for degradation and bio-elimination of these SiO2 NPs in vivo since in mice treated with Meso 100 nanoparticles ca. 2 times more Si was found in urine than mice treated with Stöber 100 nanoparticles. Also, for the particles studied, density does not seem to play a substantial influence on biodistribution of these particles in CD-1 mice since regardless of their density, all of the particles followed a similar pattern of biodistribution.
Figure 6.

In vivo investigation of degradation and biodistribution of Stöber 100, Meso 100, and Disulfide Hollow 100 nanoparticles. Particles were IV injected to CD-1 female mice and housed individually in metabolic cages for 7 days post-injection. Control mice were injected with 0.9% normal saline. A: Percentages of cumulative Si detected in urine samples. As indicated, Meso 100 nanoparticles have the highest degradation in mice with 53% detected in urine after 168 h while for Stöber 100 and Disulfide Hollow 100 particles it is 27% and 39%, respectively. Biodistribution of the administered SiO2 NPs measured 7 days post-injection; B: Stöber 100, C: Meso 100, and D: Disulfide Hollow 100 nanoparticles. Results show that particles are distributed mostly to liver and spleen and a lesser degree in lung and kidney. In Fig. 6B-D, the numbers are rounded to the nearest integer. Data are mean ± SD (N=3). ** means statistically significant difference (Pvalue< 0.05) was observed for urinary excretion of silicon between Meso 100 and other nanoparticles at day 7. ANOVA followed by a Tukey’s post-hoc test was applied for statistical analyses.
Complete Blood Count and Blood Chemistry Analyses.
We further evaluated potential hematotoxicity of the nanoparticles at 25 mg kg−1 dose injected. Mice blood was collected on day 7 after euthanasia and used for complete blood count (CBC) and blood chemistry analyses. For CBC analysis, significant markers such as white blood cells (WBC; a.k.a. leukocytes which include neutrophils, basophils, eosinophils, monocytes, and lymphocyte), red blood cells (RBC), hemoglobin (Hb), and platelet and for blood chemistry, blood urea nitrogen (BUN), total protein, glucose, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were investigated. The total protein level was measured as a marker of immune reactions. Renal function was examined by BUN, and liver function was investigated mainly through AST, ALT, and ALP. As shown in Table 3, for the period studied, some alterations of these indices were observed for nanoparticle treated groups. It seems that hollow particles have a less overall negative effect on CBC and blood chemistry comparing to Meso 100 and Stöber 100 counterparts, especially on WBC and platelet counts. No immune or inflammatory responses were observed in the treated mice as the total protein remained in the normal range, which was further confirmed by histopathological evaluation.
Table 3.
Complete blood count and blood chemistry analyses upon IV injections a.
| Treated Mice Dose |
Control | Stöber 100 | Meso 100 | Disulfide Hollow 100 |
|---|---|---|---|---|
| 0.9% Normal Saline |
25 mg kg−1 | 25 mg kg−1 | 25 mg kg−1 | |
| ❖ Complete Blood Count | ||||
| ✓ White Blood Cell (103/μL) | 1.35 ± 0.30** | 2.12 ± 1.52** | 2.07 ± 0.88** | 1.63 ± 0.56 |
| ▪ Neutrophil (103/μL) | 0.28 ± 0.07 | 0.31 ± 0.11 | 0.37 ± 0.16 | 0.28 ± 0.08 |
| ▪ Basophil (103/μL) | 0.02** | 0.02 ± 0.01 | 0.04 ± 0.06 | 0.007 ± 0.004** |
| ▪ Eosinophil (103/μL) | 0.06 ± 0.02** | 0.11 ± 0.08** | 0.22 ± 0.09** | 0.05 ± 0.01 |
| ▪ Monocyte (103/μL) | 0.02 ± 0.01** | 0.07 ± 0.04 | 0.13 ± 0.03** | 0.07 ± 0.01 |
| ▪ Lymphocyte (103/μL) | 0.96 ± 0.50 | 1.59 ± 0.47** | 1.29 ± 0.33 | 1.22 ± 0.49 |
| ✓ Reb Blood Cell (106/μL) | 8.39 ± 0.21 | 7.67 ± 2.39 | 7.98 ± 0.27 | 8.77 ± 0.50 |
| ✓ Hemoglobin (g/dL) | 14.82 ± 0.10 | 13.43 ± 4.24 | 14.66 ± 0.37 | 15.02 ± 0.64 |
| ✓ Platelet (103/μL) | 895 ± 128** | 295.44 ± 28.56** | 262.87 ± 48.32** | 843.14 ± 76.18 |
| ❖ Blood Chemistry | ||||
| ✓ Blood Urea Nitrogen (mg/dL) | 18.93 ± 3.30 | 21.45 ± 3.46 | 21 ± 0.70 | 19.3 ± 4.55 |
| ✓ Total Protein (g/dL) | 5.33 ± 0.32 | 4.75 ± 0.21 | 5.23 ± 0.20 | 5.3 ± 0.36 |
| ✓ Glucose (mg/dL) | 225.66 ± 24.68 | 312.5 ± 60.10 | 258 ± 29.05 | 233 ± 61.73 |
| ✓ ALT (GPT; IU/L) | 24.33 ± 5.50** | 55.5 ± 13.06** | 23.66 ± 9.29** | 21 ± 3.60 |
| ✓ AST (GOT; IU/L) | 101 ± 36.76 | 123.5 ± 14.84 | 94 ± 5.65 | 67.33 ± 8.14 |
| ✓ ALP (IU/L) | 62 ± 1 | 71 | 76.33 ± 16.07 | 71.66 ± 11.50 |
Data are mean ± SD (N=3).
means statistically significant difference (Pvalue< 0.05) was observed between nanoparticle-injected mice and the control group. ANOVA followed by a Dunnett’s post-hoc test was applied for statistical analyses.
Histopathological Examination, In Vivo Tissue Uptake, and Growth Charts.
To further gain insight into the toxicity of these particles in liver, spleen, lung, and kidney, samples were processed using paraffin wax, sliced, and stained with hematoxylin and eosin (H&E). As shown in Fig. 7A, all the isolated tissues looked the same as the control group, and no toxicity was observed in these organs based on histological observations. Also, no granuloma, lymphocytic infiltration, sinusoid, hyperemia, necrosis, and degeneration of the tissues were observed in mice treated with these nanoparticles. More H&E stained histology images are shown in Fig. S3-6. For observing the engulfed nanoparticles in tissues, the liver was chosen as it was shown in Fig. 6B-D that particles are taken up more by this organ. As indicated in Fig. 7C-E, particles are taken up in a group (Meso 100 nanoparticles: Fig. 7D) or as individual particles (Stöber 100: Fig. 7C and Disulfide Hollow 100: Fig. 7E). Control group treated with 0.9% normal saline is shown in Fig. 7B.
Figure 7.

A: Light microscopy analysis for representative sections of liver, spleen, lung, and kidney obtained from control and nanoparticle treated groups after 7 days post-injection at the dose of 25 mg kg−1. The slides were H&E stained. As illustrated, no toxicity, including lymphocytic infiltration, inflammation, necrosis, and degeneration of the tissues, was observed. TEM images of the particles taken up by the liver tissues treated with B: 0.9% normal saline (control group), C: Stöber 100, D: Meso 100, and E: Disulfide Hollow 100 nanoparticles. As demonstrated, nanoparticles are entrapped by liver cells as a group of particles (Meso 100) or as a single particle (Stöber 100 and Disulfide Hollow 100). F: Animal growth chart measured daily. No significant body weight changes were observed in control and nanoparticle treated groups. Mice growth data are mean ± SD (N=3).
Animals were observed daily. No clinical toxicity or behavioral changes were observed during the treatment period, and no significant fluctuations in body weight were seen in control, and nanoparticle treated groups (Fig. 7F).
Overall, the results of this study indicate that SiO2 NP porosity and composition, and the pH and ionic strength of the environment play the predominant roles for the degradation of SiO2 NPs. As observed, regular mesoporous particles (Meso 100 and Meso 500) exhibited faster degradation and degraded in all the studied media, which could be attributed to the high surface area of these particles. In SBF with 10 mM of GSH, the degradation of the disulfide-based degradable mesoporous particles was slower than their regular mesoporous counterparts due to the hydrophobic nature of the precursors. Also, the influence of the pH of the degradation media is prominent as the degradation rates of all the particles considerably increased by increasing the pH, especially to pH values equal or above 4.5. Degradation studies conducted in RAW 264.7 macrophages and CD-1 mice, re-confirm the effect of porosity on degradation. In vivo results indicate, in the range studied, particle porosity and density do not have drastic impact on the biodistribution of the particles.
These results have implications for controlled delivery using SiO2 NPs. For example, it is possible to increase loading capacity and tune degradation rates based on the desired location and rate of drug release as well as the route of delivery. Based on our investigation of SiO2 NP degradation in different simulated biological fluids, these particles degrade very slowly in gastric fluid over 28 days, whereas the degradation rates are much higher in intestinal fluid. These results suggest that if these particles are used for oral drug delivery, they can bypass the gastric environment as the gastric residence time in fasted and fed states is about 1-3 h and these nanostructures do not degrade or release the cargo (or degrade and release to very negligible amounts due to diffusion) in this period of time in acidic pH of the stomach. When the particles reach to the small intestine (with the residence time of ~9-12 h) and large intestine (with the residence time up to ~72 h), they can release their cargo for localized or systemic purposes. The pH value of the large intestine varies from 4 to 7, which is also similar to the pH values used in this study. Thus, with appropriate design criteria, SiO2 NPs can be fabricated for utility in oral delivery where the nanocarrier degrades over time, releases the cargo, and the degradation products are excreted from the body through urine or feces.
As shown in Fig. 3, the degradation profiles of disulfide-based degradable SiO2 NPs follow a much slower pattern which makes these particles a promising candidate for sustained intracellular delivery. When the particles are taken up by the cells, they are entrapped in the endolysosomal compartments with pH values around 4-5. Additionally, in lysosomes, intrinsic concentrations of cysteine amino acid, a reducing agent, are quite high, which in turn can enhance the degradation process [65]. Also, if the particles escape from the endolysosomal compartments and enter the cytosol, they can be degraded in the cytosol (approximately pH 7.4) due to the presence of high concentrations of GSH which can reduce disulfide bonds (2-10 mM) [66]. Thus, in both microenvironments (either lysosome or cytosol), slow degradation and cargo release are expected. In our previous studies, we have indicated that ca. 10% w/w of a model drug can be loaded in GSH-sensitive hollow mesoporous SiO2 NPs [24]. Therefore, for example, if we want to target the liver which was shown here to have the highest accumulation of the nanoparticles, using the same dosing regimen of 25 mg kg−1, ca. 16 μg of the free drug can be delivered to the liver cells [67, 68].
Based on our in vivo results, the injected nanocarriers at the dose of 25 mg kg−1 did not cause toxic effects in CD-1 mice over 7 days. However, the long-term toxicity and fate of the intact nanoparticles, as well as the toxicity and fate of the degradation products need further investigations. In particular, further research is necessary to understand the effect of silsesquioxane fragments used in degradable organosilica nanoparticles on the biological microenvironment as these products may not completely degrade. As the core composition of these nanoparticles is different, the long-term degradation rate should be explored to understand how long it takes for each type of nanoparticle to degrade and be excreted from the body.
Conclusion
In summary, five SiO2 NPs were fabricated with differences in size, porosity, density, and composition and tested for in vitro and in vivo degradation, biodistribution, toxicity, and clearance. Results indicate that particle porosity and composition play predominant roles on these processes while for the particles studied density did not have a substantial influence. Faster degradation rates were observed for smaller particles with higher surface areas. Additionally, higher degradation of the particles was observed at pH values equal to or greater than 4.5. Meso 100 nanoparticles had the highest degradation in RAW 264.7 macrophages and CD-1 mice. Disulfide-based degradable particles underwent both hydrolysis and disulfide reduction degradation in the reductive environment and disintegrated into smaller fragments. These particles did not exhibit higher degradation rates compared to mesoporous nanoparticles composed of TEOS due to the hydrophobic nature of the precursor used in the synthesis of disulfide-containing systems. Results from in vivo studies in CD-1 mice indicate that most of the injected particles (25 mg kg−1) biodistributed in the liver and cleared from the body via kidneys after 7 days. Toxicity of the particles in RAW 264.7 macrophages and Human Aortic Endothelial Cells (HAECs) show concentration-dependence for all the studied particles. Based on the degradation rates measured in different biological fluids as well as organ residence time, it seems that oral and intracellular deliveries can be achieved by tailoring the porosity, density, and core composition of these particles. These future studies should be built on previous work that has demonstrated the application of SiO2 NPs in oral delivery [28, 69-71]. Findings from this study provide additional design parameters for SiO2 NPs in controlled delivery applications.
Supplementary Material
Highlights.
Silica nanoparticles degrade over time in simulated biological fluids
Faster degradation is observed at higher pH values
Nanoparticle porosity and composition play the predominant roles for degradation
pH and ionic strength of the media influence degradation
Acknowledgments.
We acknowledge financial support from the National Institute of Environmental Health Sciences of the NIH (R01ES024681) and the University of Utah Nanotechnology Training Program Fellowship (Seyyed Pouya Hadipour Moghaddam). This work made use of the University of Utah shared facilities of the Micron Microscopy Suite, and the University of Utah USTAR shared facilities supported in part by the MRSEC Program of the NSF under Award No. DMR-1121252. We would also like to acknowledge Dr. Lawrence D. McGill for his suggestions and interpretations on histological sample evaluation, Venkata Yellepeddi for assisting in mouse tail vein injections, and Mostafa Yazdimamaghani for technical advice on the fabrication of Meso 500 SiO2 NPs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].March D, Overview of Food Ingredients, Additives & Colors, 21 (2013) 455–463. [Google Scholar]
- [2].Zhou Y, Quan G, Wu Q, Zhang X, Niu B, Wu B, Huang Y, Pan X, Wu C, Mesoporous Silica Nanoparticles for Drug and Gene Delivery, APSB 8(2) (2018) 165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yamamoto E, Kuroda K, Preparation and Controllability of Mesoporous Silica Nanoparticles, Enzymes 44 (2018) 1–10. [DOI] [PubMed] [Google Scholar]
- [4].Yazdimamaghani M, Barber ZB, Hadipour Moghaddam SP, Ghandehari H, Influence of Silica Nanoparticle Density and Flow Conditions on Sedimentation, Cell Uptake, and Cytotoxicity, Mol. Pharm. 15(6) (2018) 2372–2383. [DOI] [PubMed] [Google Scholar]
- [5].Patel K, Angelos S, Dichtel WR, Coskun A, Yang Y, Zink JI, Stoddart JF, Enzyme-Responsive Snap-Top Covered Silica Nanocontainers, JACS 130(8) (2008) 2382–2383. [DOI] [PubMed] [Google Scholar]
- [6].Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI, Mesoporous Silica Nanoparticles in Biomedical Applications, Chem. Soc. Rev. 41(7) (2012) 2590–2605. [DOI] [PubMed] [Google Scholar]
- [7].Meng H, Xue M, Xia T, Zhao Y-L, Tamanoi F, Stoddart JF, Zink JI, Nel A, Autonomous In Vitro Anticancer Drug Release from Mesoporous Silica Nanoparticles by pH-Sensitive Nanovalves, JACS 132 (2010) 12690– 12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lu J, Liong M, Li Z, Zink JI, Tamanoi F, Biocompatibility, Biodistribution, and Drug Delivery Efficiency of Mesoporous Silica Nanoparticles for Cancer Therapy in Animals, JACS 6 (2010) 1794–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee JE, Lee N, Kim T, Kim J, Hyeon T, Multifunctional Mesoporous Silica Nanocomposite Nanoparticles for Theranostic Applications, Acc. Chem. Res. 44 (2011) 893–902. [DOI] [PubMed] [Google Scholar]
- [10].Mohammadpour R, Yazdimamaghani M, Cheney DL, Jedrzkiewicz J, Ghandehari H, Subchronic Toxicity of Silica Nanoparticles As a Function of Size and Porosity, J. Control. Release 304 (2019) 216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nel A, Meng H, An Engineered Approach to Pancreatic Cancer Using Mesoporous Silica Nanocarriers and Immune Perturbation, Nanomedicine: NBM 14 (2018) 1856–1857. [Google Scholar]
- [12].Yu T, Malugin A, Ghandehari H, Impact of Silica Nanoparticle Design on Cellular Toxicity and Hemolytic Activity, ACS Nano 5 (2011) 5717–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu T, Greish K, McGill LD, Ray A, Ghandehari H, Influence of Geometry, Porosity, and Surface Characteristics of Silica Nanoparticles on Acute Toxicity: Their Vasculature Effect and Tolerance Threshold, ACS Nano 6 (2012) 2289–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Greish K, Thiagarajan G, Herd H, Price R, Bauer H, Hubbard D, Burckle A, Sadekar S, Yu T, Anwar A, Ray A, Ghandehari H, Size and Surface Charge Significantly Influence the Toxicity of Silica and Dendritic Nanoparticles, Nanotoxicology 6(7) (2012) 713–723. [DOI] [PubMed] [Google Scholar]
- [15].Huang X, Teng X, Chen D, Tang F, He J, The Effect of the Shape of Mesoporous Silica Nanoparticles on Cellular Uptake and Cell Function, Biomaterials 31 (2010) 438–448. [DOI] [PubMed] [Google Scholar]
- [16].Hadipour Moghaddam SP, Saikia J, Yazdimamaghani M, Ghandehari H, Redox-Responsive Polysulfide-Based Biodegradable Organosilica Nanoparticles for Delivery of Bioactive Agents, ACS Appl. Mater. Interfaces 9 (2017) 21133–21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maggini L, Cabrera I, Ruiz-Carretero A, Prasetyanto EA, Robinet E, De Cola L, Breakable Mesoporous Silica Nanoparticles for Targeted Drug Delivery, Nanoscale 8 (2016) 7240–7247. [DOI] [PubMed] [Google Scholar]
- [18].Gao Z, Hadipour Moghaddam SP, Ghandehari H, Zharov I, Synthesis of Water-Degradable Silica Nanoparticles from Carbamate-Containing Bridged Silsesquioxane Precursor, RSC Adv. 8 (2018) 4914–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Croissant J, Cattoën X, Man MWC, Gallud A, Raehm L, Trens P, Maynadier M, Durand J, Biodegradable Ethylene-Bis (Propyl) Disulfide-Based Periodic Mesoporous Organosilica Nanorods and Nanospheres for Efficient In-Vitro Drug Delivery, Adv. Mater. 26 (2014)6174–6180. [DOI] [PubMed] [Google Scholar]
- [20].Croissant J, Fatieiev Y, Khashab N, Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles, Adv. Mater. 9 (2017) 1–51. [DOI] [PubMed] [Google Scholar]
- [21].Fatieiev Y, Croissant J, Julfakyan K, Deng L, Anjum D, Gurinov A, Khashab N, Enzymatically Degradable Hybrid Organic–Inorganic Bridged Silsesquioxane Nanoparticles for In Vitro Imaging, Nanoscale 7 (2015) 15046–15050. [DOI] [PubMed] [Google Scholar]
- [22].He Y, Zeng B, Liang S, Long M, Xu H, Synthesis of pH-Responsive Biodegradable Mesoporous Silica–Calcium Phosphate Hybrid Nanoparticles as A High Potential Drug Carrier, ACS Appl. Mater. Interfaces 9 (2017) 44402–44409. [DOI] [PubMed] [Google Scholar]
- [23].Chen F, Hong H, Shi S, Goel S, Valdovinos HF, Hernandez R, Theuer CP, Barnhart TE, Cai W, Engineering of Hollow Mesoporous Silica Nanoparticles for Remarkably Enhanced Tumor Active Targeting Efficacy, Sci. Rep. 4 (2014) 5080–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hadipour Moghaddam SP, Yazdimamaghani M, Ghandehari H, Glutathione-Sensitive Hollow Mesoporous Silica Nanoparticles for Controlled Drug Delivery, J. Control. Release 282 (2018) 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Du L, Liao S, Khatib H, Stoddart J, Zink J, Controlled-Access Hollow Mechanized Silica Nanocontainers, JACS 131 (2009) 15136–15142. [DOI] [PubMed] [Google Scholar]
- [26].He Q, Zhang Z, Gao F, Li Y, Shi J, In Vivo Biodistribution and Urinary Excretion of Mesoporous Silica Nanoparticles: Effects of Particle Size and PEGylation, Small 7 (2011) 271–280. [DOI] [PubMed] [Google Scholar]
- [27].Cho M, Cho W-S, Choi M, Kim SJ, Han BS, Kim SH, Kim HO, Sheen YY, Jeong J, The Impact of Size on Tissue Distribution and Elimination by Single Intravenous Injection of Silica Nanoparticles, Toxicol. Lett. 189 (2009) 177–183. [DOI] [PubMed] [Google Scholar]
- [28].Li L, Liu T, Fu C, Tan L, Meng X, Liu H, Biodistribution, Excretion, and Toxicity of Mesoporous Silica Nanoparticles After Oral Administration Depend on Their Shape, Nanomedicine: NBM 11 (2015) 1915–1924. [DOI] [PubMed] [Google Scholar]
- [29].Yamada H, Urata C, Aoyama Y, Osada S, Yamauchi Y, Kuroda K, Preparation of Colloidal Mesoporous Silica Nanoparticles with Different Diameters and Their Unique Degradation Behavior in Static Aqueous Systems, Chem. Mater. 24 (2012) 1462– 1471. [Google Scholar]
- [30].Shen D, Yang J, Li X, Zhou L, Zhang R, Li W, Chen L, Wang R, Zhang F, Zhao D, Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres, Nano Lett. 14 (2014) 923–932. [DOI] [PubMed] [Google Scholar]
- [31].Cauda V, Schlossbauer A, Bein T, Biodegradation Study of Colloidal Mesoporous Silica Nanoparticles: Effect of Surface Functionalization with Organosilanes and Poly (ethylene glycol), Micropor Micropor Mat. 132 (2010) 60–71. [Google Scholar]
- [32].He Q, Shi J, Zhu M, Chen Y, Chen F, The Three-Stage In Vitro Degradation Behavior of Mesoporous Silica in Simulated Body Fluid, Micropor Micropor Mat. 131(1-3) (2010) 314–320. [Google Scholar]
- [33].Montero D, Tachibana C, Rahr Winther J, Appenzeller-Herzog C, Intracellular Glutathione Pools are Heterogeneously Concentrated, Redox Biol. 1(1) (2013) 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Circu ML, Aw TY, Glutathione and Modulation of Cell Apoptosis, Biochim Biophys Acta 1823 (2012) 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Forman HJ, Zhang H, Rinna A, Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis, Mol. Aspects Med. 30 (2009) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Saikia J, Yazdimamaghani M, Hadipour Moghaddam SP, Ghandehari H, Differential Protein Adsorption and Cellular Uptake of Silica Nanoparticles Based on Size and Porosity, ACS Appl. Mater. Interfaces 8 (2016) 34820–34832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Smulders S, Kaiser JP, Zuin S, Van Landuyt KL, Golanski L, Vanoirbeek J, Wick P, Hoet P, Contamination of Nanoparticles by Endotoxin: Evaluation of Different Test Methods, Particle and Fibre Toxicol. 9 (2012) 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li Y, Boraschi D, Endotoxin Contamination: A Key Element in the Interpretation of Nanosafety Studies, Nanomedicine 11 (2016) 269–287. [DOI] [PubMed] [Google Scholar]
- [39].Dobrovolskaia M, Preclinical Immunotoxicity Studies of Nanotechnology-Formulated Drugs: Challenges, Considerations and Strategy, J. Control. Release 220 (2015) 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Williams KL, Endotoxins: Pyrogens, LAL Testing and Depyrogenation, CRC Press; 2007 [Google Scholar]
- [41].Van Der Voort P, Esquivel D, De Canck E, Goethals F, Van Driessche I, Romero-Salguero F, Periodic Mesoporous Organosilicas: From Simple to Complex Bridges; A Comprehensive Overview of Functions, Morphologies and Applications, Chem. Soc. Rev. 42 (2013) 3913–3955. [DOI] [PubMed] [Google Scholar]
- [42].Wardman P, Dennis MF, Stratford MR, White J, Extracellular: Intracellular and Subcellular Concentration Gradients of Thiols, Int. J. Radiat. Oncol. Biol. Phys. 22 (1992) 751–754. [DOI] [PubMed] [Google Scholar]
- [43].Montero D, Tachibana C, Winther JR, Appenzeller-Herzog C, Intracellular Glutathione Pools Are Heterogeneously Concentrated, Redox Biol. 1 (2013) 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND, Glutathione Metabolism and Its Implications for Health, J. Nutr. 134(3) (2004) 489–492. [DOI] [PubMed] [Google Scholar]
- [45].Pfeiffer C, Rehbock C, Hühn D, Carrillo-Carrion C, de Aberasturi DJ, Merk V, Barcikowski S, Parak WJ, Interaction of Colloidal Nanoparticles with Their Local Environment: the (ionic) Nanoenvironment Around Nanoparticles is Different from Bulk and Determines the Physicochemical Properties of the Nanoparticles, J. Royal Soc. 11 (2013) 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kobayashi M, Juillerat F, Galletto P, Bowen P, Borkovec M, Aggregation and Charging of Colloidal Silica Particles: Effect of Particle Size, Langmuir 21(13) (2005) 5761–5769. [DOI] [PubMed] [Google Scholar]
- [47].Singh G, Song L, Experimental Correlations of pH and Ionic Strength Effects on the Colloidal Fouling Potential of Silica Nanoparticles in Crossflow Ultrafiltration, J. Membrane Sci. 303(1)(2007)112–118. [Google Scholar]
- [48].Phanichphant S, Nakaruk A, Channel D, Photocatalytic Activity of the Binary Composite CeO2/SiO2 For Degradation of Dye, Appl. Surf. Sci. 387 (2016) 214–220. [Google Scholar]
- [49].Nguyen QX, Belgard TG, Taylor JJ, Murthy VS, Halas NJ, Wong MS, Water-Phase Synthesis of Cationic Silica/Polyamine Nanoparticles, Chem. Mater. 8 (2012) 1426–1433. [Google Scholar]
- [50].Cloarec J-P, Chevalier C, Genest J, Beauvais J, Chamas H, Chevolot Y, Baron T, Souifi A, pH Driven Addressing of Silicon Nanowires onto Si3N4/SiO2 Micro-Patterned Surfaces, Nanotechnology 27 (2016) 295602–295610. [DOI] [PubMed] [Google Scholar]
- [51].Belton DJ, Deschaume O, Perry C, An Overview of The Fundamentals of The Chemistry of Silica with Relevance to Biosilicification and Technological Advances, FEBS J. 279 (2012) 1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Popplewell J, King S, Day J, Ackrill P, Fifield L, Cresswell R, Di Tada M, Liu K, Kinetics of Uptake and Elimination of Silicic Acid by a Human Subject: A Novel Application of 32Si and Accelerator Mass Spectrometry, J. Inorg. Biochem. 69 (1998) 177–180. [DOI] [PubMed] [Google Scholar]
- [53].Rimola A, Costa D, Sodupe M, Lambert J, Ugliengo P, Silica Surface Features and Their Role in The Adsorption of Biomolecules: Computational Modeling and Experiments, Chem. Rev. 113 (2013)4216–4313. [DOI] [PubMed] [Google Scholar]
- [54].Braun K, Pochert A, Beck M, Fiedler R, Gruber J, Linden M, Dissolution Kinetics of Mesoporous Silica Nanoparticles in Different Simulated Body Fluids, JSST 79 (2016) 319–327. [Google Scholar]
- [55].Iler RK, Chemistry of Silica Solubility: Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry of Silica 24 (1979). [Google Scholar]
- [56].Rimstidt JD, Barnes H, The Kinetics of Silica-Water Reactions, Geochim. Cosmochim. Acta 44(1980) 1683–1699. [Google Scholar]
- [57].Monk DJ, Soane DS, Howe R, Hydrofluoric Acid Etching of Silicon Dioxide Sacrificial Layers Experimental Observations, J. Electrochem. Soc. 141 (1994) 264–269. [Google Scholar]
- [58].Weinhold F, West R, The Nature of the Silicon–Oxygen Bond, Organometallics 30(21) (2011)5815–5824. [Google Scholar]
- [59].Luo Y, Kerr J, Bond Dissociation Energies, CRC Handbook of Chemistry and Physics, 89 (2012) 70–92. [Google Scholar]
- [60].Lu W, Wang C, Nguyen V, Schmidt M, Gordon MS, Ho K, Structures and Fragmentations of Small Silicon Oxide Clusters by Ab Initio Calculations, J. Phys. Chem. 107 (2003)6936–6943. [Google Scholar]
- [61].Hildenbrand DL, Murad E, Dissociation Energy and Ionization Potential of Silicon Monoxide, J. Chem. Phys. 51 (1969) 807–811. [Google Scholar]
- [62].Chen G, Teng Z, Su X, Liu Y, Lu G, Unique Biological Degradation Behavior of Stöber Mesoporous Silica Nanoparticles from Their Interiors to Their Exteriors, J. Biomed. Nanotechnol. 11 (2015) 722–729. [DOI] [PubMed] [Google Scholar]
- [63].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV, Renal Clearance of Quantum Dots, Nat. Biotechnol. 25 (2007) 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Longmire M, Choyke PL, Kobayashi H, Clearance Properties of Nano-sized Particles and Molecules as Imaging Agents: Considerations and Caveats, Nanomedicine 3 (2008) 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Greene AA, Marcusson E, Morell G, Schneider J, Characterization of The Lysosomal Cystine Transport System in Mouse L-929 Fibroblasts, J. Biol. Chem. 265 (1990) 9888–9895. [PubMed] [Google Scholar]
- [66].Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z, Glutathione-Responsive Nanovehicles as a Promising Platform for Targeted Intracellular Drug and Gene Delivery, J. Control. Release 152 (2011) 2–12. [DOI] [PubMed] [Google Scholar]
- [67].Wu L, Wu M, Zeng Y, Zhang D, Zheng A, Liu X, Liu J, Multifunctional PEG Modified DOX Loaded Mesoporous Silica Nanoparticle-CuS Nanohybrids as Photo- Thermal Agent and Thermal-Triggered Drug Release Vehicle for Hepatocellular Carcinoma Treatment, Nanotechnology 26 (2014) 1242–1249. [DOI] [PubMed] [Google Scholar]
- [68].Xie M, Xu Y, Shen H, Shen S, Ge Y, Xie J, Negatively Charge Functionalized Mesoporous Silica Nanoparticles as Drug Vehicles Targeting Hepatocellular Carcinoma, Int. J. Pharm. 474 (2014) 223–231. [DOI] [PubMed] [Google Scholar]
- [69].Mura S, Nicolas J, Couvreur P, Stimuli-Responsive Nanocarriers for Drug Delivery, Nat. Mater. 12 (2013)991. [DOI] [PubMed] [Google Scholar]
- [70].Florek J, Caillard R, Kleitz F, Evaluation of Mesoporous Silica Nanoparticles for Oral Drug Delivery-Current Status and Perspective of MSNs Drug Carriers, Nanoscale 9 (2017) 15252–15277. [DOI] [PubMed] [Google Scholar]
- [71].Lee CH, Lo LW, Mou CY, Yang CS, Synthesis and Characterization of Positive-Charge Functionalized Mesoporous Silica Nanoparticles for Oral Drug Delivery of an Anti-Inflammatory Drug, Adv. Funct. Mater. 18 (2008) 3283–3292. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.