

Abstract
We investigated the Central Nervous System (CNS) and skeletal muscle tissue from A woman was clinically diagnosed with amyotrophic lateral sclerosis (ALS) at the age of 22. Neuropathologic evaluation showed upper and lower motor neuron loss, corticospinal tract degeneration and skeletal muscle denervation. Analysis of the patient's Deoxyribonucleic acid (DNA) revealed a AGT>GGT change resulting in an S375G substitution in the C‐terminal region of TDP‐43. This variant was previously reported as being benign. Considering the early onset and severity of the disease in this patient, we tested the effects of this genetic variant on TDP‐43 localization, pre‐mRNA splicing activity and toxicity, in parallel with the effects on known neighboring disease‐associated mutations. In cell lines, expressed in culture, S375G TDP‐43 appeared to be more significantly localized in the nucleus and to exert higher toxicity than wild‐type TDP‐43. Strikingly, a phosphomimic mutant at the same residue (S375E) showed a strong tendency to accumulate in the cytoplasm, especially under stress conditions, and molecular dynamics simulations suggest that phosphorylation of this residue can disrupt TDP‐43 intermolecular interactions. The results of the current study highlight the importance of phosphorylation and regulation of TDP‐43 nuclear‐cytoplasmic shuttling/redistribution, in relation to the pathogenetic mechanisms involved in different forms of ALS.
Keywords: ALS, FTLD, mRNA splicing, neurodegeneration, phosphorylation, TARDBP mutations, TDP‐43 mutations
Introduction
Recent studies have uncovered many mutations in the genes associated with the ALS/FTD spectrum 1, 2, 3, 4. The study of these mutations has been very useful to establish a firm causative link between the proteins aggregating in the Central Nervous System (CNS) of the patients and the occurrence of disease. An important causative link between aggregation of specific proteins and disease has also been established for nuclear protein TDP‐43, that is responsible for the formation of characteristic aggregates in the neurons of patients affected by ALS/FTD, but whose pathophysiological relevance was initially controversial 5, 6, 7. In 2008, following the identification of TARDBP mutations, that segregate with disease, the role of the protein TDP‐43 in the pathogenesis of amyotrophic lateral sclerosis (ALS) was firmly established 8, 9, 10. To date, more than 50 TARBDP variants, associated with genetically determined ALS, have been reported 11, 12. Pathogenic TDP‐43 mutations occur mainly in familial and sporadic forms of ALS but have also been detected in cases of Frontotemporal Dementia (FTD) 11, 12, 13, 14, 15, 16, 17, 18, 19.
The low‐complexity domain C‐terminal region of TDP‐43 appears to be the most affected by genetic variants. This region spans residues 262, at the end of RRM2 domain, to residue 414 at the protein's C‐terminus, and is known to contain several subdomains, including the hydrophobic segments, Q/N‐rich and Gly‐rich. Similarly to what happens for other hnRNPs, the C‐terminus of TDP‐43 principally mediates protein–protein interactions allowing TDP‐43 to play a role in many cellular processes, especially alternative splicing regulation 20. In addition, several in vitro and in vivo studies have shown that these mutations in TARDBP may impact, to a variable degree, the basic biochemical properties of TDP‐43, such as protein half‐life, aggregation potential, stress granule recruitment or altered intracellular localization 11. Furthermore, new focus has been also directed toward measuring the possible impact of TDP‐43 mutations on the alteration of the non aqueous liquid microdroplets formed by this protein 21, 22 or alterations in the axonal transport and viscosity of TDP‐43‐containing RNP granules in neurons 23, 24, 25, 26.
RNA processing, one of the best characterized functional properties of TDP‐43 has often been reported to be a target of mutations 27. In particular, using murine model systems, engineered to overexpress the well‐known Q331K and M337V mutations, it has been shown that splicing alterations associated with TDP‐43 mutations do occur in vivo 28. Compared to mice expressing wild‐type TDP‐43, the transgenic mice displayed aberrant splicing alterations for many genes. The Q331K mice show unique splicing changes resulting in gain‐of‐function or loss‐of‐function that depend on the pre‐mRNA substrate 28. Consistent with these data, a cognitive dysfunction and a paucity of parvalbumin interneurons, due to alterations in the TDP‐43 autoregulation process, has recently been demonstrated in a Q331K knock‐in murine model 29. On the other hand, splicing‐affecting N‐ethyl‐N‐nitrosourea ENU‐derived mutations in the mouse TARDBP gene showed that an in vivo gain‐of‐function, due to a single mutation (M323K), correlates with adult‐onset neuromuscular phenotype characterized by motor neuron loss and neurodegenerative changes 30.
Nonetheless, the impact of human pathogenic TARDBP mutations on the age at onset of disease and its progression is still not determined. In fact, patients harboring these mutations do not show marked differences related to age at onset or disease progression from the vast majority of patients (>95%) who do not carry any change in the TDP‐43 sequence. This has been observed also for the cohort of more than one‐hundred A382T‐mutation carriers, living on the island of Sardinia 31. Generally, these people do not develop any particular feature that can distinguish them from non‐carrier ALS patients, the only possible exception being a younger age at onset when the mutation coexists with the C9ORF72 repeat expansion 32.
The study of TARDBP mutations is important for several reasons. First, they might point to both the most important protein regions and the molecular pathways leading to ALS even in patients with aggregated wild‐type TDP‐43 14. Second, regardless of the possible effects of mutations at the protein level, the presence of nucleotide changes in the mRNA coding region allows mutant mRNA expression to be specifically targeted without affecting the wild‐type transcript 33, 34.
Aim of this study, was to characterize the TDP‐43 S375G variant, that was identified in a case of early onset ALS.
The finding of a genetic variant, associated with an early onset form of ALS, made us reconsider its biological significance, since this substitution (previously described during the screening of a large cohort of familial and sporadic ALS patients) was interpreted as a variant of unclear pathological significance (Ensembl: rs766196255) 35. In keeping with the initial classification, the prediction of its potential pathogenic effects using the PolyPhen and SIFT programs reported in ALSdb (variant: 1‐11082589‐A‐G) suggested that this variant is “benign” and “tolerated,” respectively 36. We now provide detailed functional and structural analyses of the S375G substitution. The present findings strongly suggest that a revision of the characteristics of this TDP‐43 variant is necessary. In fact, the current data support the concept that the S375 residue is a potential phosphorylation site and that its removal by the S375G mutation can alter the regulation of the TDP‐43 nuclear‐cytoplasmic localization and possibly lead to a disease condition.
Materials and Methods
Patient and family history
The proband was followed over a period of 4 years. During the patient's clinical evaluation, testing of Deoxyribonucleic acid (DNA) from a blood sample was carried out at a commercial laboratory. Sequence alterations in SOD1, ANG, FIG4 and FUS genes were not identified; however, a change of unknown significance was found in the TARDBP gene.
Both parents of the proband are healthy and without clinical history of neurological disease. The patient's older siblings are healthy. One of the patient's parents had a cousin who was diagnosed with ALS after age 50. The other parent had a great uncle who was clinically diagnosed with ALS. Autopsies were not performed. A grandmother was reported to have died with late onset Alzheimer disease.
A detailed clinical history of the patient is reported in the Supporting Information.
Neuropathology
The brain and spinal cord were removed 24 h after death. The right hemi‐brain was sectioned fresh. Coronal and transverse slabs were frozen and stored at −80°C. The left hemi‐brain was fixed in 10% buffered formalin solution. A detailed description of the macroscopic and light microscopy examination of the CNS of the patient is reported in the Supporting Information.
Neurohistology
For the neurohistopathologic and immunohistochemical studies, formalin‐fixed tissue blocks from multiple areas of the central nervous system were processed. The selected areas included middle frontal, primary motor, cingulate, temporal, insular, parietal and occipital cortex, hippocampus, caudate nucleus, putamen, globus pallidus, amygdala, thalamus, cerebellar cortex, dentate nucleus, midbrain, pons, medulla, cervical, thoracic, lumbar and sacral levels of the spinal cord. Five‐micrometer‐thick sections were obtained from formalin‐fixed, paraffin‐embedded tissue and stained with hematoxylin and eosin with Luxol Fast Blue.
Skeletal muscle
Skeletal muscle samples were obtained from the quadriceps and the biceps muscles. Five‐micrometer‐thick sections were prepared from formalin‐fixed, paraffin‐embedded tissue and stained with hematoxylin and eosin. Findings related to the muscle changes are reported in the Supporting Information.
Immunohistochemistry
Antibodies to phosphorylated TDP‐43 (pS409/410, monoclonal, Cosmo Bio, Tokyo, Japan), nonphosphorylated TDP‐43 (polyclonal, Proteintech Group, Chicago, IL, USA), FUS (polyclonal, Pierce Biotechnology, Rockford, IL, USA), β‐amyloid (6F/3D, monoclonal, DAKO Carpinteria, CA, USA), tau (PHF‐1, monoclonal, courtesy of Peter Davies) and α‐synuclein (4D6, monoclonal, Covance, Emeryville, CA, USA) were used to evaluate sections of the brain and spinal cord. Antibodies to myosin heavy chain slow and myosin heavy chain fast (monoclonal, Novocastra, Leica, Newcastle Upon Tyne, United Kingdom) were used to evaluate sections of skeletal muscle.
Immunocytochemistry
HeLa cells plated in p35 dishes with cover glasses inside (Corning‐Costar, Corning, NY, USA) were transfected with the constructs of interest. After 24 h, they were fixed with 2 mL of 3.2% p‐formaldehyde/PBS solution (32% Paraformaldehyde aqueous solution, Electron Microscopy Science) for 2 h at room temperature. After three washings with 2 mL of PBS each, the slides were permeabilized with 2 mL of 0.3% Triton/PBS for 5 minutes on ice. Then, cells were washed 3 times with 2 mL of PBS, blocked with 1.5 mL of 2% BSA/PBS for 20 minutes at RT, and incubated with the primary antibody overnight at 4°C. The day after, slides were washed and incubated with secondary antibody in 2% BSA/PBS for 1 h at room temperature in a dark humidified chamber. After this step, slides were incubated face down on a drop of Vectashield with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Vector Labs, Burlingame, CA, USA). Fluorescence was analyzed and quantified using a Nikon Eclipse C1 confocal microscope using (60x oil object). Qualitative and quantitative analyses were performed using ImageJ 37. For the quantitative analysis, the regions of interest were selected and their fluorescence signals were measured (in arbitrary units). The average ratio between the cytoplasmic and nuclear signal, for each condition, was plotted in a grouped graph with standard deviation and number of analyzed samples using GraphPad software (GraphPad Software, La Jolla, CA, USA).
DNA extraction and genetic analysis
Permission from the next of kin was received for DNA analysis. Genomic DNA was extracted from frozen brain tissue using standard protocols 38. The amplified products were examined by agarose gel electrophoresis. Remaining product was treated with ExoSAP‐IT (USB, Cleveland, OH, USA) then asymmetrically amplified using the DTCS Quick Start Kit (Beckman Coulter, Fullerton, CA, USA). Products were analyzed on a CEQ 8000 GeXP Genetic Analysis System (Beckman Coulter). The resulting DNA sequences were compared to the known sequences (www.ncib.nlm.nih.gov).
Mutant TDP‐43 production and add‐back assay
Using specific sets of primers (sequence available upon request) the S375G, S375E, G376D and N378D mutations were inserted in the previously described si‐resistant plasmid expressing wild‐type TDP‐43 39. Briefly, in order to maximize TDP‐43 silencing efficiency, HeLa cells were plated at 30% of confluence (day 0) and two rounds of TDP‐43 siRNA transfections were carried out on days 1 and 2, according to the procedure already described 40. On day 2 afternoon, co‐transfection was performed with 1 μg of pFLAG‐fusion protein expression vector and 0.5 μg of the CFTR C155T reporter minigene 40. On day 3, cells were harvested, and total RNA was extracted with EuroGold Trifast (Euroclone, Milan, Italy). Reverse transcription was performed using M‐MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. PCR with DNA Polymerase (New England Biolabs, Ipswich, MA, USA) using minigene‐specific primers was carried out for 35 amplification cycles (95°C for 45 s, 54°C for 45 s, 72°C for 45 s). The PCR products were analyzed using QIAxcel DNA Screening Kit capillary electrophoresis (Qiagen, Hilden, Germany). Expression levels of the added‐back TDP‐43 proteins were monitored through western blotting, using a commercially available rabbit polyclonal TDP‐43 antibody (Proteintech, Chicago, IL, USA, catalog number 10782‐2‐AP). Endogenous tubulin was used as a loading control, using a mouse monoclonal antibody produced in‐house.
Real time quantitative PCR
Total RNA was extracted from brain samples with miRNeasy Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. Generation of cDNA for Real Time PCR experiments (qPCR) was performed using Moloney murine leukemia virus (MMLV) Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and exameric random primers, according to the manufacturer's protocol. Real time PCRs were performed on a CFX96 real time PCR detection system with iQ SYBR Green Supermix (Bio‐Rad, Hercules, CA, USA). Real time PCR conditions were an initial temperature of 95°C for 2 minutes (activation of Hot Start Polymerase), and 40 cycles of 95°C for 15 s and 55°C for 30 s. PCR products were subsequently analyzed by a melting curve analysis. Relative TDP‐43 expression (Forward primer: 5′‐atctggtgtatgttgtcaactatcc‐3′; Reverse primer:5′‐gaacttctccaaaggtactaaaatactc‐3′), determined by real time RT‐PCR, was normalized to the expression of the housekeeper GAPDH (Forward primer: 5′‐aaggtgaaggtcggagtcaa‐3′;Reverse primer: 5′‐aatgaaggggtcattgatgg‐3′) and using the ΔCt method as described in detail by Livak and Schmittgen 41. The results represent the average and standard deviation of three independent experiments, each with two technical replicates.
LDH cytotoxicity assay
The LDH levels were measured using the kit CytoTox96 Non‐Radioactive Cytotoxicity assay (Promega, Madison, WI, USA) according to manufacturer's instructions. Briefly, HeLa cells (6 × 104) were plated in 24‐well plates in 500 μL of culture medium (D‐MEM). After 24 h, the cells were transfected with 0.1 μg of plasmid expressing TDP‐43 WT or the following point mutations: S375G; G376D; N378D or the truncation mutation Y374X, which lacks the last 40 C‐terminal residues. After 24, 48 and 72 h of transfection, the activity of LDH enzyme release by the cells was assayed by measuring the absorbance at 490 nm (485 nm) in a plate reader (EnVision Multilabel Reader – Waltham, MA, USA) within 1 h after addition of the stop solution.
Statistical analysis
An unpaired t‐test or a multiple comparison Anova test with Bonferroni correction were carried out using GraphPad software (GraphPad Software, La Jolla, CA, USA). The statistical test performed in each experiment is reported in caption.
Molecular modeling and molecular dynamics simulations
Molecular Dynamics simulations were performed on two sets of different structures: the experimental X‐ray structure corresponding to the segment 370‐375, with sequence GNNSYS in its amyloidal form which was reported very recently by Eisenberg's laboratory 42, and an analogous molecular model in which the sequence was shifted one residue (i.e., 371–376, corresponding to the segment with sequence NNSYSG). The first model (GNNSYS) was downloaded from the PDB (5wia), and the structure of NNSYSG was modeled using ZipperDB 43. This second model was found to be more appropriate to conduct analyses on the S375G mutant, as explained in the Results section. Accordingly, we used the mutation tool implemented in PyMOL to generate S375G and S375E variants from the NNSYSG fibrillar assembly.
In all simulations, all the above structures were explicitly solvated using TIP3P water molecules 44, pre‐equilibrated at 1 atm and 300 K, and then individually submitted to 50‐ns molecular dynamics simulations. All these calculations were carried out using the GROMACS package 45, 46, using the amber99sb‐ildn force field 47, and the set of parameters reported in Mompeán et al, 48 which was shown to satisfactorily describe fibrillar assemblies very similar to the ones we study here. Snapshots from all the MD trajectories were extracted as PDB files every nanosecond in order to measure Cα–Cα distances between the different β‐strands throughout the simulations, which serve as an indicator of the structural integrity of the distinct assemblies over time.
Results
Patient characteristics
A woman was diagnosed with ALS at the age of 22 and died at 26. ALS in distant relatives was documented. The Supporting Information includes the clinical history of the patient as well as the description of the neuropathologic findings in the patient. Of particular interest was the observation that the patient carried a S375G change in the TARDBP gene (Supplementary Figure S1). No C9ORF72 expansion was identified.
TDP‐43 neuropathology
In the primary motor cortex, numerous neurons and glial cells contained intracytoplasmic inclusions, that were immunolabeled using TDP‐43 antibodies. The upper and lower cortical layers were both involved (Figure 1A,B). Fine TDP‐43 immunopositive threads were observed in the neuropil (Figure 2A). Neurons containing TDP‐43 inclusions were more numerous than glial cells. Astrocytes and oligodendroglial cells were involved. In the subcortical white matter, adjacent to the primary motor cortex, numerous oligodendroglial cells contained TDP‐43 immunopositive intracytoplasmic inclusions (Figure 2B). A much lower involvement was observed in other cortical areas. In the subcortical gray nuclei, the putamen was more affected than caudate nucleus, globus pallidus, thalamus and subthalamic nucleus (Figure 2C,D).
Figure 1.
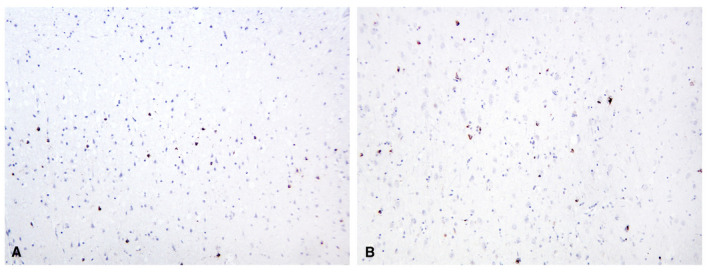
pTDP‐43 immunohistochemical staining of brain and spinal cord. Overview of the primary motor cortex stained with pTDP‐43 showing density and distribution of immunoreactive neurons and glia. A. Upper cortical layers. B. Lower cortical layers (pTDP‐43, 10x original magnification).
Figure 2.
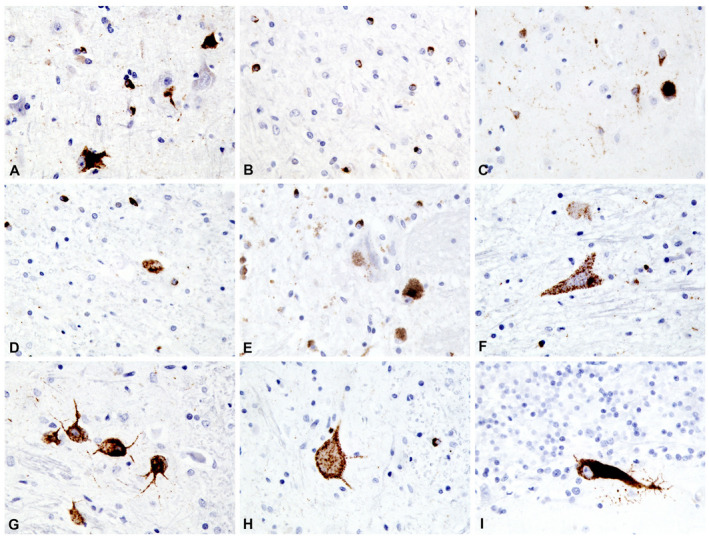
Higher power view of CNS areas stained with pTDP‐43. A. Primary motor cortex. B. White matter adjacent to primary motor cortex. C. Putamen. D. Subthalamic nucleus. E. Substantia nigra. F. Hypoglossal nucleus. G. Inferior olivary nucleus. H. Lumbar spinal cord, anterior horn. I. Cerebellum, vermis (pTDP‐43, 40x original magnification).
In the cerebellum, rare Purkinje cells had TDP‐43 immunopositive cytoplasm (Figure 2I). In the brain stem, most involved were the nuclei of the hypoglossus, vagus nerve and inferior olive. The substantia nigra and the tectal nuclei were less involved (Figure 2E,F, and G). In the spinal cord, TDP‐43 inclusions were present in numerous oligodendroglial cells but less frequently seen in neurons (Figure 2H).
FUS‐immunoreactive cytoplasmic inclusions were occasionally seen in neurons and glia in the primary motor cortex, putamen, subthalamic nucleus, red nucleus, inferior olivary nucleus and spinal cord neurons (results not shown.) The neuronal inclusions have a compact appearance. No immunoreactivity to beta‐amyoid, tau or alpha‐synuclein was seen in the central nervous system.
Genetic analysis
DNA analysis of frozen brain tissue by direct sequencing revealed an AGT>GGT point mutation in the TARDBP gene (Supplementary Figure S1). This mutation is predicted to lead to the substitution of serine for glycine at residue 375 (S375G). A C9ORF72 expansion was not identified.
PhosphoTDP‐43 immunohistochemical expression in the S375G patient brain compared to total TDP‐43 expression levels
Based on the availability of numerous fixed and frozen CNS regions sampled from this ALS case, it was possible to compare the expression levels of pTDP‐43 (Figure 3A) in neurons/glia with total TDP‐43 mRNA expression (Figure 3B). The aim was to determine whether pTDP‐43 expression levels in ALS correlated with total TDP‐43 expression levels.
Figure 3.
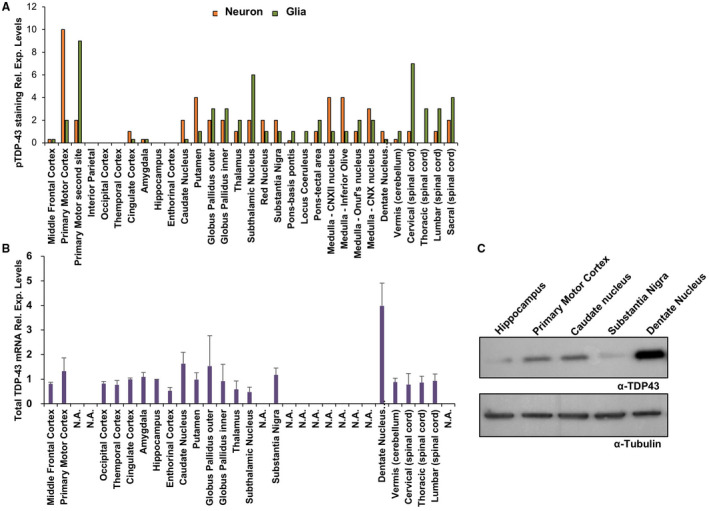
Comparison of TDP‐43 and pTDP‐43 expression patterns in the S375G patient brain. A. Levels of pTDP‐43 expression in the neurons and glia of different brain regions. After a low power scan, three noncontiguous microscopic fields were examined at 40x magnification using an Olympus BX41 brightfield microscope and averaged for the count. A nucleus had to be recognized for the count. B. Total TDP‐43 mRNA expression in different brain regions of ALS S375G patient as measured by RT‐qPCR. Three independent experiments were analyzed and both relative expression and standard deviations are shown for each region. The brain regions that were not available in sufficient amounts for RNA extraction are reported as not available (N.A.). C. Western blot analysis of patient samples that were in sufficient quantity to extract proteins. The expression levels of total TDP‐43 were determined using the Proteintech antibody. Tubulin was used as a loading control.
As shown in Figure 3, this comparison reveals marked differences in the analyzed brain regions. As expected, most of the pTDP‐43 staining was concentrated in the areas affected by the disease, such as the Primary Motor Cortex and the Spinal Cord 49, 50. Interestingly, pTDP‐43 pathology was also prominently detected in the glia of the Subthalamic nucleus, although previous reports found no evidence of pTDP‐43 signal in this area 50, 51. Also of interest was the absence of pTDP‐43 pathology in the hippocampus, where TDP‐43 pathology has been reported by several groups 52. In contrast, TDP‐43 total mRNA expression levels were rather constant in all the analyzed regions with few exceptions, especially in the dentate nucleus (cerebellum) where expression levels were considerably higher than in the other tissues. These mRNA expression data were confirmed by western blot analysis that showed an increased expression of TDP‐43 in this region (Figure 3C). Compared to normal brains, it is interesting to note that the TDP‐43 expression pattern found in the S375G carrier is apparently similar to normal TDP‐43 expression profiles in healthy brains as reported by two different databases of gene expression profiling: the Genotype‐Tissue Expression pilot analysis 53, 54 and BioGPS 55. In both databases, in fact, the levels of TDP‐43 mRNA seemed to be comparable in most of the analyzed brain regions with a slight increase only in the cerebellar area.
Intracellular localization of S375G and of the adjacent G376D, N378D and Y374X TDP‐43 disease‐associated mutants
Based on the results described above, we investigated whether the S375G substitution could induce alterations in the intracellular localization of TDP‐43 in comparison with that of the wild‐type protein or with respect to the localization of TDP‐43, occurring in association with disease‐associated variants located closely to S375G. In particular, we focused on the G376D and N378D substitutions, previously described, but not well characterized, in a various studies 11.
The subcellular distribution of all the TARDBP mutants was investigated in HeLa cell lines. It is shown that endogenous wild‐type TDP‐43 is predominantly nuclear, with a lower abundance in the cytoplasm, due to the continuous shuttling of this protein between the two compartments (Figure 4A and Supplementary Figure S2) 56. While the G376D and N378D substitutions did not significantly affect the distribution of TDP‐43 which remained prominently nuclear, in the S375G variant the distribution of TDP‐43 was significantly more nuclear than in any other mutants. As for the wild‐type protein, only 10% of it was present in the cytoplasm (Figure 4A and Supplementary Figure S2). This result was confirmed by nucleo‐cytoplasmic fractionation followed by western blot against the wild‐type and S375G mutant proteins (Figure 4C).
Figure 4.
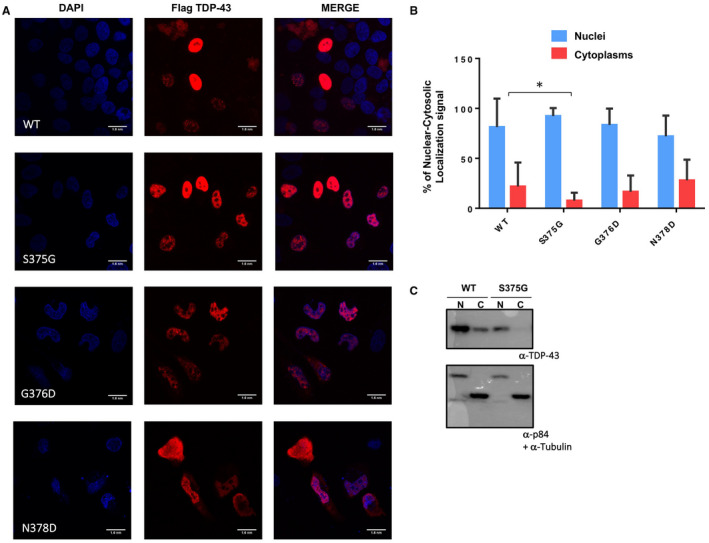
Intracellular localization of TDP‐43 mutants. A. Immunofluorescence of transiently transfected WT Flag‐TDP‐43 and S375G, G376D and N378D Flag‐TDP‐43 in HeLa cells. The overexpressed proteins were visualized using anti‐Flag polyclonal antibody. The slides were analyzed with 60X oil objective and the acquired fields mesure100 nm/pixel. Scale bar = 10 μm. B. Quantification of nuclear cytoplasmic staining intensity for WT flag‐TDP‐43, S375G, G376D and N378D. Unpaired t‐test was performed for statistical analysis (*P < 0.05). C. Nucleo‐cytoplasmic (N/C) distribution of transiently transfected HeLa cells with flag‐tagged wild‐type TDP‐43 and flag‐tagged S375G mutant. The detection of p84 and tubulin was performed as control for nuclear/cytoplasmic contamination of the two fractions.
In view of this difference, we experimentally tested the splicing functionality of the mutants using an add‐back assay previously set up to study the TDP‐43 structure–splicing function correlation 39. This assay is based on a minigene system carrying CFTR exon 9, that includes a mutation in a splicing enhancer element within its sequence to obtain a 50/50 ratio of exon inclusion/skipping (Supplementary Figure S3, lane 1). This method provides an optimal condition to monitor whether a change in TDP‐43 structure can result in either a loss‐of‐function (less exon 9 skipping) or a gain‐of‐function effect (more exon 9 skipping). In this system, when endogenous TDP‐43 is removed from the cells by siRNA treatment, the levels of CFTR exon recognition substantially increase to almost 80% (Supplementary Figure S3, lane 2). Conversely, splicing inhibition can be fully rescued following expression of a si‐resistant wild‐type TDP‐43 protein (Supplementary Figure S3, lane 3) but not when a si‐resistant F4L TDP‐43 mutant, that cannot bind RNA due to mutations in the RRM1 and RRM2 domains is expressed at comparable levels (Supplementary Figure S3, lane 4). Then, we tested the effect of si‐resistant S375G, G376D and N378D TDP‐43 variants. In all these mutants, however, we observed that exon 9 splicing was inhibited similarly to the si‐resistant wild‐type TDP‐43 (Supplementary Figure S3, compare lane 3 with 5‐7).
In ALS patients carrying TARDBP mutations, in addition to missense mutations, the Y374X is the only truncation mutation described to date 11. The functional significance of this mutation is not known, except for the fact that it has been described to impair binding of TDP‐43 to the PPI/CypA factor 57. Although this type of mutation cannot be compared to missense mutations, we also investigated the effects of Y374X both with regard to its nuclear localization and to its functional impact (in add‐back splicing assays). The results showed that the localization of TDP‐43, associated with Y374X, is comparable to that of wild‐type TDP‐43 (i.e., predominantly nuclear) and that the absence of the last 40 residues does not alter significantly its splicing ability (Supplementary Figure S4).
Cytotoxicity of TDP‐43 mutants
Next, we examined whether the S375G, G376D, N378D and Y374X substitutions can increase cellular toxicity, as several mutations have been shown to be toxic when expressed in various animal models of ALS, from yeast to zebrafish 58, 59 and patient‐derived iPS cell lines 60. To perform this analysis, HeLa cells were transiently transfected with the flagged TDP‐43 vector, either wild type or carrying all these substitutions. In all transfected cells, western blot analysis against total TDP‐43 showed that the flagged wild‐type and mutant proteins were expressed at similar levels 48 h post‐transfection (Figure 5D). Note that as it lacks 40 residues the Y374X truncation mutant has the same electrophoretic mobility of the unflagged endogenous protein. The medium was collected after 24, 48 and 72 h and lactate dehydrogenase (LDH) levels were measured for each time point. Importantly, compared to cells transfected with wild‐type TDP‐43 the LDH levels after 24 hrs were not different in any of the mutants (Figure 5A). On the other hand, the expression of the S375G, G376D and Y374X mutants caused a significant citotoxicity after 48 h (Figure 5B) and the N378D mutant showed a toxic effect after 72 h (Figure 5C).
Figure 5.
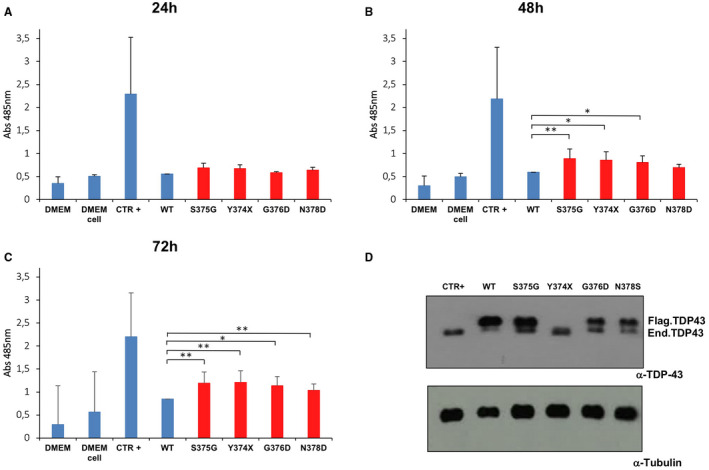
Analysis of TDP‐43 mutants’ toxicity. A. The release of LDH into the media was used as an indicator of cell toxicity. LDH levels were measured 24 h A, 48 h B and 72 h C after cells transfection with the indicated constructs. Data from three separate experiments were analyzed by multiple comparison 1‐way ANOVA, followed by Bonferroni's posthoc analysis (**P < 0.001). D. Western blot analysis of flagged TDP‐43 protein expression after 48 h to show comparable expression of all mutants in our cell line. The signal for the flagged‐Y374X mutant co‐migrates with the signal for the endogenous TDP‐43 due to its shorter size.
Functional effects of mimicking post‐translational phosphorylation at the S375 position
Previous in vitro studies have shown Serine‐375 phosphorylation by CK1 61. Furthermore, a recent mass spectrometry analysis of TDP‐43 post‐translational modifications in two brains of ALS patient has highlighted the phosphorylation of this residue in both cases 62. Therefore, the replacement of serine with a glycine should prevent the phosphorylation at position 375. To test the potential effects of phosphorylation at this position, we prepared a phosphomimic mutant of TDP‐43 (S375E) and analyzed TDP‐43 immunolocalization inside cells.
As shown in Figure 6A, immunohistochemistry analysis showed that, contrary to what happens with the S375G variant, the localization of the S375E mutant protein is not confined to the nucleus, but is present in the cytoplasm. The toxicity of the S375E substitution was comparable to that of the S375G variant (Figure 6B). Add‐back experiments carried out to tentatively correlate mislocalization with CFTR exon 9 splicing alteration showed that there was no significant difference from wild‐type TDP‐43 (Figure 6C). This result suggests that the amount of TDP‐43 persisting in the nucleus is sufficient to drive correct splicing of the transfected minigene (at least within the limits of this kind of assay).
Figure 6.
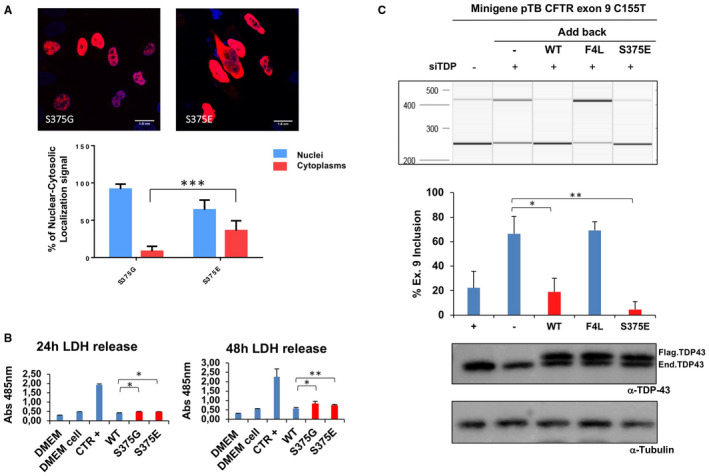
Immunofluorescence of transiently transfected Flag‐tagged S375G and S375E mutants in HeLa cells. A. The overexpressed proteins were visualized using anti‐Flag polyclonal antibody in a 100 nm/pixel field. Scale bar = 10 μm. The graph in the lower panel reports the immunofluorescence intensity ratio between cytoplasms and nuclei of transiently transfected S375G and S375E. B. Analysis of S375G and S375E mutants’ toxicity. The LDH levels released in the media were measured at 24 h and 48 h after cells transfection with the indicated constructs. Data from three separate experiments were analyzed by multiple comparison one‐way ANOVA, followed by Bonferroni's posthoc analysis (**P < 0.001). C. QIAxcel analysis of CFTR exon 9 inclusion upon expression of wild‐type and S375E variants. The pTB CFTR exon 9 C155T reporter minigene was transfected in HeLa cells following the knockdown of the endogenous TDP‐43 protein. After RT‐PCR amplification with minigene‐specific primers, the levels of CFTR exon 9 inclusion (Ex 9+) were measured in each sample using the QIAxcel system (upper panel). The changes in CFTR exon 9 inclusion relatively to added‐back wild‐type TDP‐43 (WT) were then measured following the addition of siRNA‐resistant wild‐type TDP‐43 (WT), an RNA‐binding impaired mutant (F4L) and the S375E variant (middle panel). The two lower panels show western blots carried out using a polyclonal against TDP‐43 and tubulin to check for silencing efficiency of the endogenous protein (enTDP) and comparable transgene expression (flagTDP). Results from three biological triplicates (three individual transfection experiments) are reported in the graph, below with standard deviations and p‐values.
Treatment with Arsenite has been reported to efficiently induce the inclusion of TDP‐43 in stress granules 63, 64 and this event seems to be a major feature toward aberrant aggregation of this protein in patient brains 65. Therefore, considering the higher propensity of S375E to be localized in the cytoplasm, it was interesting to see whether this change could be enhanced by Arsenite treatment. As shown in Supplementary Figure S5, the S375E was even more pronoucedly localized in the cytoplasm following Arsenite treatment, while S375G seemed to remain mostly unaffected and still predominantly localized in the cytoplasm.
Molecular modeling analysis of the effects of the pathological S375G and phosphomimetic S375E substitutions on the propensity of β sheet formation in this TDP‐43 segment
To gain insights, at a near‐atomic level, about the effects of S375G and S375E on TDP‐43 structure, we employed Molecular Dynamics (MD) simulations to study the structural integrity of the WT sequence with respect to the S375G mutant in the corresponding amyloid‐like assemblies. To this end, we first selected two segments containing the WT target region; namely, G370‐NNSYS375 and N371‐NSYSG376. Although a crystallographic structure has very recently been reported for fibrils with the former sequence (G370‐NNSYS375) 42, S375 lies right at the edge, at the C‐terminal position of the sequence and therefore, the impact of the S375G substitution is difficult to ascertain in the context of this structure. The investigators, that solved this structure, used the ZipperDB server 43 to identify the self‐assembly properties of the G370‐NNSYS375 segment; thus, we used the same tool to generate a model for N371‐NSYSG376, in which the target residue 375 does not lie at the edge of the sequence. Interestingly, we found that the predicted G370‐NNSYS375 assembly differs significantly from the experimental structure. In contrast, we found that side chains in the predicted model for N371‐NSYSG376 display orientations very similar to those observed in the crystallographic structure of G370‐NNSYS375 (Supplementary Figure S6), which strongly suggests that similar stabilizing interactions are present. Should a fibrillar assembly made up by N371‐NSYSG376 display a similar structural stability as the experimental structure of G370‐NNSYS375, the former would be a more reliable model to analyze the impact of S375G because the mutation would not occur at the edge residue. To test this premise, we performed MD analyses of both the experimental G370‐NNSYS375 and modeled N371‐NSYSG376 structures. As expected from the similar side chain orientations, both display a remarkable structural stability (Supplementary Figure S7). Therefore, we selected the N371‐NSYSG376 model to study the impact of S375G on TDP‐43 assembly.
Next, we performed MD simulations on N371‐NSYGG376, that is, the S375G variant. Figure 7 shows the structures corresponding to the beginning (time 0) and the end (time 50 ns) of the simulations, as well as Cα–Cα intra‐β‐sheet distances measured throughout the course of the simulations. The results indicate that both WT and S375G exhibit high structural stability, which we interpret as the ability to S375G to maintain amyloid‐like assemblies similar to those of the WT sequence.
Figure 7.

Structural stability of WT, S375G and S375E. A. Snapshots from MD trajectories taken at 0 (green structures) and after 50 ns (cyan) of simulation. WT and S375G remain assembled during the simulation time. In contrast, the phosphomimetic variant S375E disassembled and does not overlap with the initial conformation (B. Upper panel) Inter‐β‐strands, intra‐β‐sheet measurements from Cα to Cα between stacked residues. Edge (first and sixth) residues are excluded from the analysis. Yellow, dotted lines represent Cα–Cα distances between the upper and central strand. Red lines indicate the analogous distances between the central and lower strands. (B. Lower panel) Each of these distances was measured at each data point of the simulation, and they were all averaged and plotted. Whereas, WT and S375G inter‐strand distances remain constant over time, that of S375E increases as a result of disassembly.
In contrast, the assembly formed by the N371‐NSYGE376 variant dissociated (Figure 7C). We attributed its instability to a strong electrostatic repulsion induced by juxtaposed Glu side chains. In the RIPK1–RIPK3 functional amyloid, this type of mutation in the amyloid core destabilizes the structure and prevents amyloid assembly 66. In FUS fibrils, phosphorylation has been shown to disrupt liquid droplet formation 67. These results in aggregate support the idea that phosphorylation of S375 might be a key aspect of TDP‐43 assembly and nuclear localization.
Identification of other phosphomimic mutants near Serine 375 that might also contribute to affect the nuclear cytoplasmic localization of TDP‐43
In order to determine whether the above results reveal a peculiar feature of Serine 375 or also of other Serine residues in the C‐terminal domain of TDP‐43, we looked at the prevalent distribution of phosphorylation sites described in the TDP‐43 C‐terminal domain. As shown in Supplementary Table S1, phosphorylation has been consistently observed to occur for a series of Serine residues at positions S242, S305, S387–389–393–395 and S404 in addition to S375 61, 62 (Figure 8A). As shown in Figure 8B,C, introduction of a phosphomimic mutation in these sites caused a significant reduction of the nuclear localization for S375E and especially for the S387E–389E–393E–395E (S387–395E) TDP‐43 mutants, as quantified in Figure 8D. This result suggests that the potential phosphorylation events in the region surrounding the residue serine 375 could be critical for the control of TDP‐43 intracellular redistribution.
Figure 8.
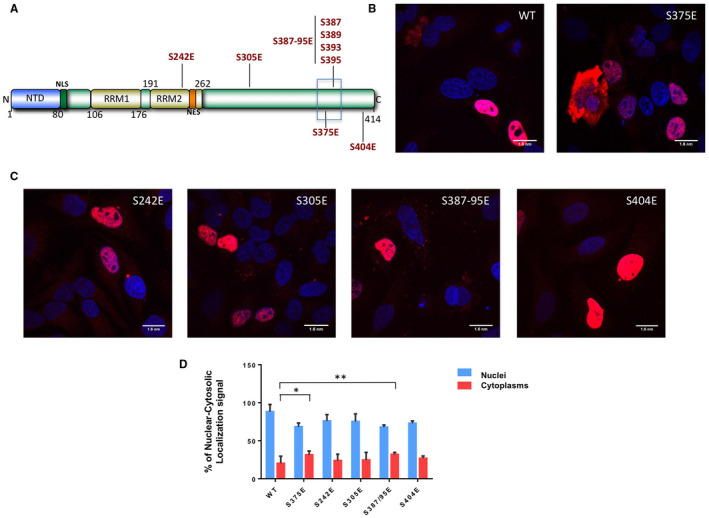
Intracellular localization of TDP‐43 mutants. A. Schematic diagram of wild‐type TDP‐43 showing the nuclear localization signal (NLS), nuclear export signal (NES), RNA Recognition Motifs (RRMs) and the phosphomimic mutations introduced in the various potential phosphorylation sites at positions S242, S305, S375, S387–389–393–395 and S404. B–C. Immunofluorescence of transiently transfected WT Flag‐TDP‐43 and S242E, S305E, S375E, S387E–389E–393E–395E (S387–395E) and S404E flag‐tagged TDP‐43 mutants in HeLa cells. The overexpressed proteins were visualized using anti‐Flag polyclonal antibody in a 100 nm/pixel field. Scale bar = 10 μm. D. Shows a quantification of nuclear‐cytoplasmic staining intensity for all these mutants. Statistical analysis was performed using multiple comparison 1‐way Anova test with Bonferroni's correction (*P < 0.05; **P < 0.001).
Discussion
Genetically determined ALS may occur in association with mutations in the TARDBP gene. We have studied clinically and neuropathologically a case of early onset ALS in a Caucasian woman and found a AGT>GGT change in TARDBP resulting in a S375G substitution in the C‐terminal region of TDP‐43. The potential role of this mutation in the pathogenesis of ALS was investigated through the analysis of TDP‐43 neuropathology and experiments of genetic engineering.
TDP‐43 intracytoplasmic inclusions revealed the aberrant localization of the protein in neurons and glia in multiple anatomical regions of the CNS. It was also noted that in spite of the fact that FUS gene mutations were not found in the genomic DNA of the ALS patient, both neurons and glia occasionally contained intracytoplasmic inclusions of FUS, another RNA‐binding protein. Such finding has also been previously reported in association with mutant TARDBP 68.
Although relatively rare, TARDBP mutations provide important conditions that allow to understand the potential pathogenetic links between TDP‐43 and neurodegenerative disease 11, 12, 13, 14, 15, 16, 17. Furthermore, there is a growing number of published intronic and exonic variants in TARDBP sequences, whose significance is currently unknown (http://www.ensembl.org). Clarifying the role of these variants in relation to disease may have important consequences for clinical counseling purposes. Recently, a few studies have started addressing the issue of comparing TDP‐43 variants of uncertain significance such as A90V with well‐characterized disease‐associated mutations in order to better evaluate whether these changes might be considered a low‐ or high‐risk factor for developing TDP‐43 proteinopathy 69. In addition to A90V, there are also several other variants (i.e., S375G) that are currently listed in ALS databases as tolerated/benign. However, the discovery of this variation in a very early onset case of ALS suggested that this missense change might actually represent a pathological mutation. From a biological point of view, this variation is very interesting as it has the potential to remove a possible phosphorylation site from the C‐terminus of the protein.
As recently reviewed, phosphorylation is one of the major posttranslational modifications of TDP‐43 that has been described to affect protein function both in healthy and disease conditions 70. Indeed, aberrant TDP‐43 phosphorylation at residues S409/410 is one of the major distinguishing features of TDP‐43 pathology 49, 71, 72, 73, 74. Regarding this issue, one important conclusion that can be drawn from our study is that there is no correlation between the abundance of TDP‐43 protein expression in different brain regions and the pathological S409/410 pTDP‐43 immunoreactivity. Moreover, new evidence suggests that in physiological conditions TDP‐43 phosphorylation can contribute to change its RNA‐binding properties following heat shock 75 and can profoundly affect its oligomerization within cells 76. In our work, we also show that a S375E phosphomimetic displays altered cellular properties and toxicity, raising the possibility that reversible phosphorylation at this position may play an important role in regulating TDP‐43 self‐association through its C‐terminal domain. It should be noticed that no increased aggregation of S375E was apparent from our results but further studies are needed to better address this issue.
Whereas, intrinsically disordered proteins are typical rich in charged residues, TDP‐43's C‐terminal domain is remarkably poor; it only contains only one Asp, two Glu, two Lys, five Arg and one His residues; with a net charge of about +4 at pH 7.4. This scarcity of charged residues makes the C‐terminal domain very sensitive to solution conditions. Even slight changes in pH dramatically reduce electrostatic repulsion producing soluble to aggregative phase changes 77. In keeping with this view, alternations in temperature, salt concentration and RNA concentration induce TDP‐43 CTD microdroplet formation: that is, reversible liquid phase/phase separation 21, 78. This process is mediated by interactions contributed by the partially populated hydrophobic helix (formed by residues 320–340) and a series of aromatic residues; namely: F276, F283, F289, F315, F317, W334, Y374, W385, F401 and W412, scattered throughout the CTD 78. In addition, RNA‐binding has been recently shown to modulate microdroplet formation by TDP‐43 and other hnRNP proteins; moderate RNA concentrations, such as those found in the cytoplasm, promote liquid microdroplet formation, whereas higher concentrations, akin to those present in the nucleus, lead to microdroplet dissolution 79. The formation of these microdroplets is a physiological event, as it enables TDP‐43 and its associated mRNAs, to bind to stress granules under certain conditions. However, an excess tendency to form microdroplets could be detrimental considering their proposed role in the amyloid formation of the protein FUS/TLS 80, a protein whose functions and domain structure show similarities to TDP‐43 and whose aggregation has also been linked to ALS and FTD.
Bearing all these considerations in mind, the S375G mutation and the loss of this phosphorylation site could affect these processes in different ways. Very recently, the X‐ray crystal structures of amyloid‐like structures formed by several short segments of TDP‐43's C‐terminal domain have been reported, including one segment which contains S375 42. Our molecular dynamics simulations reported here show that phosphorylation of S375 would strongly destabilize this amyloid‐like structure and produce its dissociation. However, it is quite possible that this segment is not the one which forms the pathologically relevant amyloid. In fact, our previous findings put the spotlight on the Q/N‐rich segment composed of residues 341–367 81, the residues which follow the hydrophobic helix formed by residues 320–340. A peptide corresponding to the segment is capable of forming an amyloid‐like structure in vitro 82, 83. Like TDP‐43, Huntingtin 84 and the Androgen Receptor 85 also have hydrophobic helices followed by Q/(N)‐rich segments which are implicated in pathological amyloid formation.
Taken together, the S375G mutation could affect amyloid formation by the 341–367 segment in at least two different ways. First, phosphorylation would add two negative charges at pH 7.4, which would lower the net charge from +4 to +2, thus potentially reducing the electrostatic repulsion and favoring self‐association processes (either microdroplet formation or aggregation). Secondly, this additional negative charge could potentially weaken the interaction with RNA, which might lead to less microdroplet formation in the cytoplasm where the RNA concentration is lower. Considering that S375 is adjacent to Y374, which favors microdroplet formation, these effects could be magnified. Although the relative importance of these distinct effects is still unresolved, it is clear that the balance between aqueous phases vs. distinct microdroplet phases is finely regulated and that mutations such as S375G that upset this balance could lead to disease.
In conclusion, our work raises the important need for a future comprehensive analysis of many seemingly benign genetic variations, which are increasingly uncovered by the numerous studies, that are directed to population screening in the area of neurodegenerative diseases.
Funding
This work was supported by the International Scientific Co‐operation Agreement between Italy and Israel (SCREENCELLS4ALS, Ministero Affari Esteri, MAE, Italy) and SAF‐2016‐76678‐C2‐2‐R (DVL). BG is supported by grant P30 AG010133. KN is supported by grant P30 AG035982.
Supporting information
Figure S1. In the patient, DNA analysis identified a novel S375G (AGT>GGT) change in a mutational hot spot in the TARDBP gene. This change is predicted to lead to posttranslational modifications due to the potential loss of a phosphorylation site. No C9orf72 expansion was detected.
Figure S2. This graph shows the number of counted transfected cells (16) and the TDP‐43 protein localization between nuclei and cytoplasm for the WT pFlagSiR plasmid; for the S375G mutation, and for the G376D and N378D mutations.
Figure S3. A. QIAxcel gel analysis of CFTR exon 9 inclusion upon expression of TDP‐43 variants. The pTB CFTR exon 9 C155T reporter minigene was transfected in HeLa cells following the knockdown of the endogenous TDP‐43 protein. The changes in CFTR exon 9 inclusion relative to added‐back TDP‐43 WT were then measured following the addition of siRNA‐resistant wild‐type TDP‐43 (WT), an RNA‐binding impaired mutant (F4L) and the mutant variants. After RT‐PCR amplification using minigene‐specific primers, the levels of CFTR exon 9 inclusion (Ex 9+) were measured in each sample by QIAxcel. B. Results from three biological triplicates (three individual transfection experiments) are reported in the graph, below with standard deviations and p‐values. C. The two lower panels show western blots using a polyclonal against TDP‐43 and tubulin to check for silencing efficiency of the endogenous protein (enTDP) and comparable transgene expression (FlagTDP).
Figure S4. A. Immunofluorescence of transiently transfected WT Flag‐TDP‐43 and Y374X Flag‐TDP‐43 in HeLa cells. The overexpressed proteins were visualized using anti‐Flag polyclonal antibody Scale bar = 10 µm. B. Agarose gel analysis of CFTR exon 9 inclusion upon expression of these two variants. The change in CFTR exon 9 inclusion relative to added‐back TDP‐43 WT were then measured following the addition of siRNA‐resistant wild type TDP‐43 (WT), an RNA‐binding impaired mutant (F4L) and the Y374X (Y374X) variant. Image analysis was performed using ImageJ. C. Results from three biological triplicates (three individual transfection experiments) are reported in the graph, below with standard deviations and P‐values.
Figure S5. This graph shows the TDP‐43 protein localization between nuclei and cytoplasm for the WT TDP‐43 protein, the S375G and the S375E phosphomimic mutants following Arsenite stress (+Ars).
Figure S6. A. Predicted structure for G370NNSYS375 using ZipperDB. All Ser residues are exposed to the solvent (upper left and lower right, Ser in stick representation), whereas Asn residues meet at the inter‐sheet interface (right, Asn in stick representation). B. Experimental X‐ray structure of G370NNSYS375. Side chain orientations are drastically different from the predictions shown in panel A: serine residues meet at the inter‐sheet interface (left figure, Ser in stick representation), and are also on the edges, where they face juxtaposed Asn residues (right figure, both Ser and Asn in stick representation). C. Predicted structure of N371NSYSG376 using ZipperDB. The structure of this segment resembles that of the experimental amyloid shown in B: serines meet at the inter‐sheet region and also interact with Asn residues at the edge of the motif.
Figure S7. Structural stability of the G370NNSYS375 experimental structure A and the N371NSYSG376 working model B. Snapshots corresponding to the initial (left: time 0, in green) and final (middle: 50 ns, in cyan) time points of independent MD simulations are shown. Superposition of the structures at both times is shown to emphasize the stability (right). As illustrated in Figure S6, these two structures display very similar side chain orientations, which strongly encourages the use of N371NSYSG376 to model mutations at S375.
Table S1. This Table reports the TDP‐43 phosphorylation sites as determined by in vitro phosphorylation assay and by mass spectrometric analysis of TDP‐43 derived from two patient aggregates (ALS P1 and P2) as reported in two previous studies (1, 2). Phosphorylation has been consistently observed to occur for a series of Serine residues at positions S242, S305, S387–389–393–395 and S404 in addition to S375 (highligthed in yellow).
Acknowledgments
Images in this paper were generated in the Optical Microscopy Center of the University of Trieste at the Life Sciences Department, funded as detailed at http://www.units.it/confocal. We wish to thank Cynthia Shaddy‐Gouvion and Marsha Danley for technical assistance. We are grateful to the patient and her family for their commitment to the advancing of new knowledge about ALS.
Contributor Information
Kathy Newell, Email: knewell@kumc.edu.
Emanuele Buratti, Email: buratti@icgeb.org.
References
- 1. Quintana‐Murci L (2016) Understanding rare and common diseases in the context of human evolution. Genome Biol 17:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zufiria M, Gil‐Bea FJ, Fernandez‐Torron R, Poza JJ, Munoz‐Blanco JL, Rojas‐Garcia R et al (2016) ALS: a bucket of genes, environment, metabolism and unknown ingredients. Prog Neurobiol 142:104–129. [DOI] [PubMed] [Google Scholar]
- 3. Mann DMA, Snowden JS (2017) Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathol 27:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kapeli K, Martinez FJ, Yeo GW (2017) Genetic mutations in RNA‐binding proteins and their roles in ALS. Hum Genet 136:1193–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 6. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 7. Rothstein JD (2007) TDP‐43 in amyotrophic lateral sclerosis: pathophysiology or patho‐babel? Ann Neurol 61:382–384. [DOI] [PubMed] [Google Scholar]
- 8. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 9. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D et al (2008) TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buratti E (2015) Functional significance of TDP‐43 mutations in disease. Adv Genet 91:1–53. [DOI] [PubMed] [Google Scholar]
- 12. Harrison AF, Shorter J (2017) RNA‐binding proteins with prion‐like domains in health and disease. Biochem J 474:1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banks GT, Kuta A, Isaacs AM, Fisher EM (2008) TDP‐43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome 19:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pesiridis GS, Lee VM, Trojanowski JQ (2009) Mutations in TDP‐43 link glycine‐rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18:R156–R162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barmada SJ, Finkbeiner S (2010) Pathogenic TARDBP mutations in amyotrophic lateral sclerosis and frontotemporal dementia: disease‐associated pathways. Rev Neurosci 21:251–272. [DOI] [PubMed] [Google Scholar]
- 16. Gendron TF, Rademakers R, Petrucelli L (2013) TARDBP mutation analysis in TDP‐43 proteinopathies and deciphering the toxicity of mutant TDP‐43. J Alzheimer's Dis: JAD 33(Suppl. 1):S35–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lattante S, Rouleau GA, Kabashi E (2013) TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat 34:812–826. [DOI] [PubMed] [Google Scholar]
- 18. Caroppo P, Camuzat A, Guillot‐Noel L, Thomas‐Anterion C, Couratier P, Wong TH et al (2016) Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol Genet 2:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ et al (2009) TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord 24:1843–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Budini M, Baralle FE, Buratti E (2014) Targeting TDP‐43 in neurodegenerative diseases. Expert Opin Ther Targets 18:617–632. [DOI] [PubMed] [Google Scholar]
- 21. Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by alpha‐helical structure in the TDP‐43 low‐complexity C‐terminal domain. Structure 24:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt HB, Rohatgi R (2016) In vivo formation of vacuolated multi‐phase compartments lacking membranes. Cell Rep 16:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gopal PP, Nirschl JJ, Klinman E, Holzbaur EL (2017) Amyotrophic lateral sclerosis‐linked mutations increase the viscosity of liquid‐like TDP‐43 RNP granules in neurons. Proc Natl Acad Sci U S A 114:E2466–E2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fallini C, Bassell GJ, Rossoll W (2012) The ALS disease protein TDP‐43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet 21:3703–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowden HA, Dormann D (2016) Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem 138(Suppl. 1):112–133. [DOI] [PubMed] [Google Scholar]
- 26. Liu‐Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF et al (2014) ALS‐linked mutations enlarge TDP‐43‐enriched neuronal RNA granules in the dendritic arbor. J Neurosci 34:4167–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ratti A, Buratti E (2016) Physiological functions and pathobiology of TDP‐43 and FUS/TLS proteins. J Neurochem 138(Suppl. 1):95–111. [DOI] [PubMed] [Google Scholar]
- 28. Arnold ES, Ling SC, Huelga SC, Lagier‐Tourenne C, Polymenidou M, Ditsworth D et al (2013) ALS‐linked TDP‐43 mutations produce aberrant RNA splicing and adult‐onset motor neuron disease without aggregation or loss of nuclear TDP‐43. Proc Natl Acad Sci U S A 110:E736–E745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM et al (2018) TDP‐43 gains function due to perturbed autoregulation in a TARDBP knock‐in mouse model of ALS‐FTD. Nat Neurosci 21:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H et al (2018) Mice with endogenous TDP‐43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J 37:e98684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orru S, Manolakos E, Orru N, Kokotas H, Mascia V, Carcassi C, Petersen MB (2012) High frequency of the TARDBP p.Ala382Thr mutation in Sardinian patients with amyotrophic lateral sclerosis. Clin Genet 81:172–178. [DOI] [PubMed] [Google Scholar]
- 32. Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G et al (2014) Genetic architecture of ALS in Sardinia. Neurobiol Aging 35:2882 e7–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishimura AL, Shum C, Scotter EL, Abdelgany A, Sardone V, Wright J et al (2014) Allele‐specific knockdown of ALS‐associated mutant TDP‐43 in neural stem cells derived from induced pluripotent stem cells. PLoS One 9:e91269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhandare VV, Ramaswamy A (2016) Identification of possible siRNA molecules for TDP43 mutants causing amyotrophic lateral sclerosis: in silico design and molecular dynamics study. Comput Biol Chem 61:97–108. [DOI] [PubMed] [Google Scholar]
- 35. Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM et al (2015) Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 77:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to imageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murrell J, Farlow M, Ghetti B, Benson MD (1991) A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 254:97–99. [DOI] [PubMed] [Google Scholar]
- 39. D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM et al (2009) Functional mapping of the interaction between TDP‐43 and hnRNP A2 in vivo . Nucleic Acids Res 37:4116–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ayala YM, Pagani F, Baralle FE (2006) TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett 580:1339–1344. [DOI] [PubMed] [Google Scholar]
- 41. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 42. Guenther EL, Cao Q, Trinh H, Lu J, Sawaya MR, Cascio D et al (2018) Atomic structures of TDP‐43 LCD segments and insights into reversible or pathogenic aggregation. Nat Struct Mol Biol 25:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldschmidt L, Teng PK, Riek R, Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci U S A 107:3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:726–735. [Google Scholar]
- 45. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447. [DOI] [PubMed] [Google Scholar]
- 46. Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. Software X 1:19–25. [Google Scholar]
- 47. Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO et al (2010) Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mompean M, Gonzalez C, Lomba E, Laurents DV (2014)Combining classical MD and QM calculations to elucidate complex system nucleation: a twisted, three‐stranded, parallel beta‐sheet seeds amyloid fibril conception. J Phys Chem B 118:7312–7316. [DOI] [PubMed] [Google Scholar]
- 49. Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y et al (2009) Phosphorylation of S409/410 of TDP‐43 is a consistent feature in all sporadic and familial forms of TDP‐43 proteinopathies. Acta Neuropathol 117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013)Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gomez‐Pinedo U, Villar‐Quiles RN, Galan L, Matias‐Guiu JA, Benito‐Martin MS, Guerrero‐Sola A et al (2016) Immununochemical markers of the amyloid cascade in the hippocampus in motor neuron diseases. Front Neurol 7:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Geser F, Brandmeir NJ, Kwong LK, Martinez‐Lage M, Elman L, McCluskey L et al (2008) Evidence of multisystem disorder in whole‐brain map of pathological TDP‐43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. [DOI] [PubMed] [Google Scholar]
- 53. Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M et al (2015) Human genomics. The human transcriptome across tissues and individuals. Science 348:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Consortium GT (2015) Human genomics. The Genotype‐Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI (2016) BioGPS: building your own mash‐up of gene annotations and expression profiles. Nucleic Acids Res 44:D313–D316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E et al (2008) Structural determinants of the cellular localization and shuttling of TDP‐43. J Cell Sci 121(Pt 22):3778–3785. [DOI] [PubMed] [Google Scholar]
- 57. Lauranzano E, Pozzi S, Pasetto L, Stucchi R, Massignan T, Paolella K et al (2015) Peptidylprolyl isomerase A governs TARDBP function and assembly in heterogeneous nuclear ribonucleoprotein complexes. Brain 138(Pt 4):974–991. [DOI] [PubMed] [Google Scholar]
- 58. Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD (2009) TDP‐43 is intrinsically aggregation‐prone, and amyotrophic lateral sclerosis‐linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284:20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P et al (2010)Gain and loss of function of ALS‐related mutations of TARDBP (TDP‐43) cause motor deficits in vivo . Hum Mol Genet 19:671–683. [DOI] [PubMed] [Google Scholar]
- 60. Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M et al (2012)Mutant induced pluripotent stem cell lines recapitulate aspects of TDP‐43 proteinopathies and reveal cell‐specific vulnerability. Proc Natl Acad Sci U S A 109:5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kametani F, Nonaka T, Suzuki T, Arai T, Dohmae N, Akiyama H et al (2009) Identification of casein kinase‐1 phosphorylation sites on TDP‐43. Biochem Biophys Res Commun 382:405–409. [DOI] [PubMed] [Google Scholar]
- 62. Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y et al (2016) Mass spectrometric analysis of accumulated TDP‐43 in amyotrophic lateral sclerosis brains. Sci Rep 6:23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E et al (2009) TDP‐43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111:1051–1061. [DOI] [PubMed] [Google Scholar]
- 64. Liu‐Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T et al (2010) Tar DNA binding protein‐43 (TDP‐43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W et al (2011) TAR DNA‐binding protein 43 (TDP‐43) regulates stress granule dynamics via differential regulation of G3BP and TIA‐1. Hum Mol Genet 20:1400–1410. [DOI] [PubMed] [Google Scholar]
- 66. Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G et al (2018) The structure of the necrosome RIPK1‐RIPK3 core, a human hetero‐amyloid signaling complex. Cell 173:1244–1253 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL et al (2017) Structure of FUS protein fibrils and its relevance to self‐assembly and phase separation of low‐complexity domains. Cell 171:615–627 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K et al (2010) FUS‐immunoreactive inclusions are a common feature in sporadic and non‐SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wobst HJ, Wesolowski SS, Chadchankar J, Delsing L, Jacobsen S, Mukherjee J et al (2017) Cytoplasmic relocalization of TAR DNA‐binding protein 43 is not sufficient to reproduce cellular pathologies associated with ALS in vitro . Front Mol Neurosci 10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Buratti E (2018) TDP‐43 post‐translational modifications in health and disease. Expert Opin Ther Targets 22:279–293. [DOI] [PubMed] [Google Scholar]
- 71. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nonaka T, Arai T, Buratti E, Baralle FE, Akiyama H, Hasegawa M (2009) Phosphorylated and ubiquitinated TDP‐43 pathological inclusions in ALS and FTLD‐U are recapitulated in SH‐SY5Y cells. FEBS Lett 583:394–400. [DOI] [PubMed] [Google Scholar]
- 73. Takeuchi R, Tada M, Shiga A, Toyoshima Y, Konno T, Sato T et al (2016) Heterogeneity of cerebral TDP‐43 pathology in sporadic amyotrophic lateral sclerosis: evidence for clinico‐pathologic subtypes. Acta Neuropathol Commun 4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mackenzie IR, Neumann M (2017) Reappraisal of TDP‐43 pathology in FTLD‐U subtypes. Acta Neuropathol 134:79–96. [DOI] [PubMed] [Google Scholar]
- 75. Li W, Reeb AN, Lin B, Subramanian P, Fey EE, Knoverek CR et al (2017) Heat shock‐induced phosphorylation of TAR DNA‐binding protein 43 (TDP‐43) by MAPK/ERK kinase regulates TDP‐43 function. J Biol Chem 292:5089–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang A, Conicella AE, Schmidt HB, Martin EW, Rhoads SN, Reeb AN et al (2018) A single N‐terminal phosphomimic disrupts TDP‐43 polymerization, phase separation, and RNA splicing. EMBO J 37:e97452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mompean M, Chakrabartty A, Buratti E, Laurents DV (2016) Electrostatic repulsion governs TDP‐43 C‐terminal domain aggregation. PLoS Biol 14:e1002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li HR, Chiang WC, Chou PC, Wang WJ, Huang JR (2018) TAR DNA‐binding protein 43 (TDP‐43) liquid‐liquid phase separation is mediated by just a few aromatic residues. J Biol Chem 293:6090–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I et al (2018) RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science 360:918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY et al (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. [DOI] [PubMed] [Google Scholar]
- 81. Budini M, Buratti E, Stuani C, Guarnaccia C, Romano V, De Conti L et al (2012) Cellular model of TAR DNA‐binding protein 43 (TDP‐43) aggregation based on its C‐terminal Gln/Asn‐rich region. J Biol Chem 287:7512–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mompean M, Buratti E, Guarnaccia C, Brito RM, Chakrabartty A, Baralle FE et al (2014) Structural characterization of the minimal segment of TDP‐43 competent for aggregation. Arch Biochem Biophys 545C:53–62. [DOI] [PubMed] [Google Scholar]
- 83. Mompean M, Hervas R, Xu Y, Tran TH, Guarnaccia C, Buratti E et al (2015) Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP‐43. J Phys Chem Lett 6:2608–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pandey NK, Isas JM, Rawat A, Lee RV, Langen J, Pandey P et al (2018) The 17‐residue‐long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J Biol Chem 293:2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eftekharzadeh B, Piai A, Chiesa G, Mungianu D, Garcia J, Pierattelli R et al (2016) Sequence context influences the structure and aggregation behavior of a PolyQ tract. Biophys J 110:2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. In the patient, DNA analysis identified a novel S375G (AGT>GGT) change in a mutational hot spot in the TARDBP gene. This change is predicted to lead to posttranslational modifications due to the potential loss of a phosphorylation site. No C9orf72 expansion was detected.
Figure S2. This graph shows the number of counted transfected cells (16) and the TDP‐43 protein localization between nuclei and cytoplasm for the WT pFlagSiR plasmid; for the S375G mutation, and for the G376D and N378D mutations.
Figure S3. A. QIAxcel gel analysis of CFTR exon 9 inclusion upon expression of TDP‐43 variants. The pTB CFTR exon 9 C155T reporter minigene was transfected in HeLa cells following the knockdown of the endogenous TDP‐43 protein. The changes in CFTR exon 9 inclusion relative to added‐back TDP‐43 WT were then measured following the addition of siRNA‐resistant wild‐type TDP‐43 (WT), an RNA‐binding impaired mutant (F4L) and the mutant variants. After RT‐PCR amplification using minigene‐specific primers, the levels of CFTR exon 9 inclusion (Ex 9+) were measured in each sample by QIAxcel. B. Results from three biological triplicates (three individual transfection experiments) are reported in the graph, below with standard deviations and p‐values. C. The two lower panels show western blots using a polyclonal against TDP‐43 and tubulin to check for silencing efficiency of the endogenous protein (enTDP) and comparable transgene expression (FlagTDP).
Figure S4. A. Immunofluorescence of transiently transfected WT Flag‐TDP‐43 and Y374X Flag‐TDP‐43 in HeLa cells. The overexpressed proteins were visualized using anti‐Flag polyclonal antibody Scale bar = 10 µm. B. Agarose gel analysis of CFTR exon 9 inclusion upon expression of these two variants. The change in CFTR exon 9 inclusion relative to added‐back TDP‐43 WT were then measured following the addition of siRNA‐resistant wild type TDP‐43 (WT), an RNA‐binding impaired mutant (F4L) and the Y374X (Y374X) variant. Image analysis was performed using ImageJ. C. Results from three biological triplicates (three individual transfection experiments) are reported in the graph, below with standard deviations and P‐values.
Figure S5. This graph shows the TDP‐43 protein localization between nuclei and cytoplasm for the WT TDP‐43 protein, the S375G and the S375E phosphomimic mutants following Arsenite stress (+Ars).
Figure S6. A. Predicted structure for G370NNSYS375 using ZipperDB. All Ser residues are exposed to the solvent (upper left and lower right, Ser in stick representation), whereas Asn residues meet at the inter‐sheet interface (right, Asn in stick representation). B. Experimental X‐ray structure of G370NNSYS375. Side chain orientations are drastically different from the predictions shown in panel A: serine residues meet at the inter‐sheet interface (left figure, Ser in stick representation), and are also on the edges, where they face juxtaposed Asn residues (right figure, both Ser and Asn in stick representation). C. Predicted structure of N371NSYSG376 using ZipperDB. The structure of this segment resembles that of the experimental amyloid shown in B: serines meet at the inter‐sheet region and also interact with Asn residues at the edge of the motif.
Figure S7. Structural stability of the G370NNSYS375 experimental structure A and the N371NSYSG376 working model B. Snapshots corresponding to the initial (left: time 0, in green) and final (middle: 50 ns, in cyan) time points of independent MD simulations are shown. Superposition of the structures at both times is shown to emphasize the stability (right). As illustrated in Figure S6, these two structures display very similar side chain orientations, which strongly encourages the use of N371NSYSG376 to model mutations at S375.
Table S1. This Table reports the TDP‐43 phosphorylation sites as determined by in vitro phosphorylation assay and by mass spectrometric analysis of TDP‐43 derived from two patient aggregates (ALS P1 and P2) as reported in two previous studies (1, 2). Phosphorylation has been consistently observed to occur for a series of Serine residues at positions S242, S305, S387–389–393–395 and S404 in addition to S375 (highligthed in yellow).