

Abstract
Recent studies indicate that 4-hydroxy-trans-2-nonenal (HNE), a major oxidative stress triggered lipid peroxidation-derived aldehyde, plays a critical role in the pathophysiology of various human pathologies including metabolic syndrome, diabetes, cardiovascular, neurological, immunological, and age-related diseases and various types of cancer. HNE is the most abundant and toxic α, β-unsaturated aldehyde formed during the peroxidation of polyunsaturated fatty acids in a series of free radical-mediated reactions. The presence of an aldehyde group at C1, a double bond between C2 and C3 and a hydroxyl group at C4 makes HNE a highly reactive molecule. These strong reactive electrophilic groups favor the formation of HNE adducts with cellular macromolecules such as proteins and nucleic acids leading to the regulation of various cell signaling pathways and processes involved in cell proliferation, differentiation, and apoptosis. Many studies suggest that the cell-specific intracellular concentrations of HNE dictate the anti-oxidative and pro-inflammatory activities of this important molecule. In this review, we focused on how HNE could alter multiple anti-oxidative defense pathways and pro-inflammatory cytotoxic pathways by interacting with various cell-signaling intermediates.
1. Oxidative Stress and Lipid Peroxidation
Free radicals are regularly generated in aerobes because of normal respiration processes and the activity of cellular antioxidant defense machinery maintains a balance of the free radicals utilizing a variety of antioxidant enzymes in the cells. A balanced redox homeostasis is necessary for the maintenance of normal cellular processes in aerobes [1]. Under oxidative stress conditions, disruption of cellular redox homeostasis leads to an imbalance between reactive oxygen species (ROS) generation and their elimination by antioxidant enzymes. The major reasons for the redox imbalance could be the overproduction of free radical species or the inability of the cellular antioxidant defense machinery to eliminate or sequester the free radicals generated in the body. Free radicals such as superoxide anion (O2˙ˉ), hydroxyl radical (OH˙), nitric oxide (NO˙), peroxyl radical (LOO˙) and non-radical oxidants such as hydrogen peroxide (H2O2), peroxynitrite (ONOOˉ), hypochlorous acid (HOCl), nitrous acid (HNO2), and singlet oxygen (1O2), are the most commonly generated ROS in the cells and act as initiators of oxidative damage contributing to pathophysiology of multiple disease complications. Cells upon exposure to external oxidants such as xenobiotics, environmental pollutants, UV radiation, carcinogens and allergens, and internally formed oxidants in the body such as cytokines, growth factors, and chemokines could lead to altered cellular metabolic processes leading to the production of ROS. The activity of enzymes such as NADPH oxidase, xanthine oxidase, and auto-oxidation of glucose can generate ROS under different oxidant stimuli. In addition, mitochondrial oxidative phosphorylation that usually participates in the cellular respiration to generate energy in the cells can also contribute to the generation of free radicals (Figure 1). Apart from inducing damage to cellular macromolecules and dysregulation of cellular homeostasis, the free radicals formed during metabolic processes also act as secondary signaling intermediates and regulate various oxidative and anti-oxidative signaling pathways [2]. Antioxidant peptides such as glutathione (GSH) play an important role in detoxification of ROS. GSH is the most essential regulator of cellular redox homeostasis as it metabolizes or scavenges a number of free radicals or free radical- generated products in the cells. The ratio of GSH (reduced)/GSSG (oxidized) is a key indicator of cellular redox potential. Apart from GSH, enzymes such as glutathione-S-transferases (GSTs), plays a very important role in the maintenance of redox balance in cells. GSTs play a significant role in detoxification of various xenobiotics as well as endogenous toxic products generated in the cells by conjugation with GSH, facilitating further metabolism or detoxification by multiple other antioxidant detoxification and defense pathways [3]. Further, antioxidant enzyme superoxide dismutase (SOD) catalyzes the dismutation of superoxide free radical (O2˙ˉ), into hydrogen peroxide and oxygen. The hydrogen peroxide is further catalyzed by catalase into oxygen and water [4]. The activity of these antioxidant enzymes controls the free radical content in the body. However, during pathological complications, alterations in various antioxidant defense pathways along with increased ROS production imbalances cellular homeostasis leading to tissue damage and dysfunction.
Figure 1.

Schematic figure showing the ROS -induced formation of HNE via lipid peroxidation and its metabolism by various antioxidative enzymes.
Though ROS play an important role in regulating various physiological functions such as the elimination of pathogenic bacteria by immune cells, maintenance of vascular tone, cardiovascular functions, cell proliferation, and differentiation, a disturbance of ROS homeostasis leads to the development of various pathological complications. Several studies have shown that oxidative stress generated by ROS plays a critical role in multiple pathologies including various types of cancers, inflammatory disorders, neurodegenerative diseases, and cardiovascular complications [5, 6]. Apart from the spontaneous direct effect on DNA, RNA, amino acids, proteins and lipids, the secondary products generated by ROS -mediated reactions significantly damage various macromolecules and propagate their deleterious effects in the cells. Most importantly, even though the ROS generated in cells are short-lived, the secondary products generated by ROS are comparatively stable and further acts as important mediators of cellular signaling. The secondary ROS generated products maintain and propagate the effect of ROS long after ROS generation. These secondary toxic products could migrate to distant sites, which are far from their site of origin and can induce tissue damage and organ dysfunction in multiple sites, thus exerting multifactorial side effects. One such effect of ROS is the oxidative damage to the membrane lipids, which is termed as lipid peroxidation. Membrane phospholipids such as poly-unsaturated fatty acids (PUFA) are the major targets of lipid peroxidation induced by ROS. Lipid peroxidation-derived products such as HNE (4-hydroxy-trans-2-nonenal), acrolein and malondialdehyde (MDA) are more stable; have a longer half-life than ROS themselves and have attained considerable attention in the recent past as important mediators of oxidative stress-induced pathological complications [7].
During oxidative stress, lipid peroxidation occurs via three major steps, (a) initiation, (b) propagation and (c) termination. In the initial initiation step, the free radicals (e.g. OH˙) attack PUFA and generates lipid radicals (L˙). The removal of hydrogen atoms from the lipids leads to the reduction of ROS into water and generation of lipid radicals. During propagation step, the unstable lipid radicals react with oxygen leading to the generation of lipid peroxyl radicals (LOO˙) and additional lipid radicals (L˙), which further reacts in a chain reaction and new lipid radicals and lipid peroxides are generated. The presence of metal ions such as iron (Fe2+) and copper (Cu2+) is shown to accelerate the propagation reaction. In the final termination step, hydroperoxides are generated by the reaction of peroxyl radicals with vitamin E (α-tocopherol). The lipid peroxides and lipid radicals react with each other to generate more stable non-radical products during the termination of the lipid peroxidation process (Figure 1). The β-oxidation of lipid peroxides leads to the generation of various toxic lipid aldehydes such as alkanals, alkenals, hydroxyalkenals, and alkadienes [8, 9]. The formation of lipid aldehydes such as MDA and HNE is often considered as the most common toxic end products of lipid peroxidation and generally used as indicators of oxidative damage in the cells and tissues. Further, the α,β-unsaturated hydroxyalkenal, HNE has been shown to be the most abundant and toxic lipid peroxidation end product generated during lipid peroxidation [8, 9]. HNE with its highly reactive electrophilic groups can interact with cellular proteins, GSH, nucleic acids and can cause cytotoxicity or genotoxicity (Figure 1). Recent studies have postulated that HNE is a biomarker of oxidative stress-induced pathological complications and correlated its damaging effects to various human diseases such as cancer, neurodegenerative, inflammatory and autoimmune diseases, various metabolic diseases, and mitochondrial dysfunction [8, 10–15]. Besides its reactivity with macromolecules, HNE can also alter the membrane potential, electron transport and ion imbalance in the cells leading to neurological disorders [16, 17]. Further, HNE also plays an important role in cell survival and death by signaling through different pathways mediated by caspase3, Bax, Bcl2, death receptors, multiple kinases and transcription factors such as Nrf2, AP-1 and NF-κB (Figure 2) [18–21]. The effect of HNE on the cellular macromolecules is dependent on the type of tissue and concentration of HNE, with specificities for proteins containing cysteine, histidine and lysine residues [8, 9, 22]. The detoxification and metabolism of HNE is also differential in different tissues [23]. In the succeeding section, we have discussed the reactivity of HNE with different macromolecules and its metabolism.
Figure 2.

HNE is a pleiotropic signaling molecule: Depending on the concentration and duration of exposure, HNE induces multiple signaling pathways in cells by regulating various signaling intermediates.
2. Reactivity and Metabolism of HNE
HNE has three functional groups 1) carbonyl group on C1 (C=O), 2) a double bond between C2 and C3 (C=C) and 3) hydroxyl group on C4 (which facilitates C=C polarization and cyclization reactions), which makes HNE a highly reactive aldehyde product of lipid peroxidation process (Figure 1). HNE can react with different macromolecules such as proteins, phospholipids, nucleic acids, and glutathione (GSH). Membrane-bound entities and proteins containing an abundance of cysteine, lysine and histidine residues and phospholipids such as phosphatidyl-ethanolamine are preferred targets for adduct formation by HNE. Electrophilic sites of HNE leads to Schiff base formation between an amino group and the carbonyl group at C1 and Michael addition of thiol or amino compounds at C3 [9, 24, 25]. The reactivity of HNE towards amino acids has been shown to be in the order of cysteine>histidine>lysine [26–28]. First, the amino acids undergo Michael addition at the C=C double bond of HNE. Michael addition to the C=C bond confers rotational freedom at the C2-C3 bond, which facilitates secondary reactions of primary amines with the carbonyl group of HNE to form Schiff bases [29]. Although cysteine is the most preferred amino acid for reactivity with HNE, HNE-histidine adducts have been shown to be more stable as compared to cysteine- and lysine-HNE adducts [28, 30]. Further, HNE-induced covalent modification of nucleophilic residues of amino acids regulate protein activation/inactivation, which could alter cellular signaling pathways. Specifically, important cellular signaling pathways controlling apoptosis, cell cycle, oxidative and nitrosative stress-associated pathways are reported to be significantly affected by HNE leading to cellular toxicities [31–33]. HNE also inhibits the proteasomes such as 20S proteasome and hence impairs the cellular proteasomal degradation of damaged or modified protein subunits generated subsequent to oxidative stress [34]. Chaperone activities specifically mediated by HSP 72 and HSP 90 have been shown to be modified by HNE adduct formation [35, 36]. HSP 90 function has been reported to be modified by the modification of Cys-572 residue by HNE, which has important pathological implications in alcoholic liver diseases (ALD) [36, 37]. HNE can directly interact with guanosine bases in DNA to form 1,N-2-propano-deoxyguanosine with an ability to form 1.2 ± 0.5 adducts/107 nucleotides [38]. Etheno-DNA (ɛ-DNA) adducts have been shown to be generated by the reaction of HNE with nucleotides in DNA that could lead to mutational changes in DNA, increasing the susceptibility to cancerous transformation in cells [39]. Formation of HNE-DNA adducts indicate the genotoxic and mutagenic effects induced by lipid peroxidation [40]. HNE-DNA adduct formation hampers DNA repair mechanism in the cells. Studies have shown that HNE inhibits nucleotide excision repair in DNA damage-induced either by UV radiation or carcinogens such as benzo[a]pyrene diol epoxide (BDPE); a major environmental pollutant and component of cigarette smoke [41, 42]. Thus, these studies provide evidence that HNE is a multifactorial effector of signaling and cellular functions in the body.
HNE is short-lived in the cells with a half-life of less than 2 min and is immediately conjugated with other macromolecules or metabolized by various antioxidant enzymes [43]. Based on cellular antioxidant defense capacity and cell types, multiple studies have demonstrated varying concentrations and half-life of HNE in vitro and in vivo [44]. Major cellular metabolic pathways which play an important role in the detoxification of HNE are: (a) alcohol dehydrogenase (ADH) or aldose reductase (AR, AKR1B1), which reduces HNE to 1,4-dihydroxy-2-nonene (DHN), (b) aldehyde dehydrogenase (ALDH), which oxidizes HNE to 4-hydroxy-2-nonanoic acid (HNA) and (c) glutathione-S-transferases (GSTs), which catalyze the conjugation of HNE to GSH forming GS-HNE, which is then transported out of the cell in an ATP- dependent manner by various drug transporters such as MRP1, MRP2, and RLIP76 (Figure 1) [8, 45–47]. HNE has the ability to induce modifications in enzymes involved in cellular detoxification such as GSTs [48], glutathione reductase (GR) [49], either by Michael adduct formation or by covalent modifications of amino acid residues in the proteins. Interestingly, the cellular GSH levels control the concentration of HNE in cells and on the other hand, HNE has been shown to regulate the expression of enzymes responsible for GSH synthesis. Exposure of cells to HNE has been shown to increase γ-glutamate cysteine ligase (GCL) activity. GCL catalyzes the rate-limiting step in GSH biosynthesis and hence is important for the maintenance of cellular GSH levels. Further, HNE has also been shown to induce the transcriptional activation of γ-glutamyl cysteine synthase, thus contributing to enhanced GSH biosynthesis [50]. HNE can conjugate with GSH spontaneously or by the conjugation reactions catalyzed by the specific GST isozymes [51, 52]. GSTA4-4 and GST5.8 have been shown to specifically participate in the GSH-conjugation with HNE [53, 54]. Studies have shown that overexpression of HNE metabolizing GST (mGSTA4-4, hGSTA4-4, or hGST5.8) confers protection to cells against oxidative injury [55]. Exposure of hepatoma cells to exogenous HNE leads to the formation of major metabolites of HNE: glutathione-HNE (GS-HNE) indicating the importance of HNE-GSH conjugation as a major metabolic route for its detoxification [47]. Apart from GST, studies have also demonstrated that aldose reductase (AR; AKR1B1) further metabolizes GS-HNE to GS-DHN and aldehyde dehydrogenase (ALDH) isozymes metabolize GS-HNE to GS-HNA [54, 56]. Several studies have demonstrated that aldo-keto reductases are an important class of enzymes upregulated by oxidative stress or exposure to HNE [57]. Up-regulation of AR has been shown to play an important role in HNE detoxification in HepG2 cells. Burczynski et al., have demonstrated that HNE generated in the cells induces its own metabolism and detoxification by up-regulating the aldo-keto reductase isozyme AKR1C1 [58]. Both HNE and its GSH metabolite, GS-HNE, have been shown to be reduced by AR to 1,4-dihydroxy-2-nonene (DHN) and GS-DHN, respectively [59–62]. Further, AR reduces GSH conjugates of aldehydes (e.g. GS-HNE and GS-acrolein) more effectively as compared to their parent aldehydes (e.g. HNE and acrolein) indicating that AR has a specific binding site for glutathionylated aldehydes [63, 64]. Indeed, Singh et al., have crystallized AR bound GS-analogue and found that AR has specific GS-aldehyde binding domain [65]. These studies demonstrate that catalytic activity mediated by AR is a major step in the detoxification of lipid peroxidation-derived aldehydes such as HNE and their glutathione conjugates. Indeed multiple studies modulating AR activity have shown to exert therapeutic effects in various oxidative stress-induced inflammatory pathologies including cancer [56, 66–68].
HNE-mediated regulation of signaling pathways is pleiotropic and the signaling pathways activated or inhibited by HNE depends on the concentration of HNE and the type of cells used in the study. In cell culture studies, HNE concentration greater than 10 μM has been reported to induce apoptosis whereas sub-lethal dose ≤5 μM induces cell proliferation. Concentration-dependent activation of various pro-inflammatory and anti-oxidative pathways by HNE is shown to regulate multiple kinases and transcription factors important for disease pathology (Figure 2). In certain pathologies, a very high concentration of HNE has been observed. Elevated plasma levels of HNE (~100 μM) have been reported in children with systemic lupus erythematosus [69] and in the liver tissues of mouse models of experimental alcoholic liver disease [70, 71]. However, it is still unclear how the cells cope with such high levels of HNE in vivo and the deleterious effect exerted by such high concentrations of HNE in various human diseases. With the increasing evidence of the importance of ROS and lipid peroxidation-derived aldehydes in different human pathologies, and advancement in analytical techniques to detect and identify the specific role of free radical intermediates in various diseases [72], studies on the roles of lipid peroxidation-derived aldehydes in modulating cellular signaling pathways has attained a considerable attention in the recent years. In the following section, we have briefed some of the important findings on the role of HNE in mediating anti - and pro-inflammatory signaling pathways.
3. Regulation of Anti-oxidative Pathways by HNE
NFE2-related nuclear factor 2 (Nrf2) is a master transcription factor regulating the expression of genes involved in the anti-oxidative and anti-inflammatory pathways [73]. Various exogenous stimuli such as xenobiotics, flavonoids, and antioxidants activate Nrf2 nuclear translocation, which then binds to its consensus antioxidant response elements (ARE) and induces the transcription of anti-oxidative genes. Under the basal conditions, Nrf2 is sequestered in the cytoplasm by its association with KEAP1, which keeps the complex inactive. The Nrf2-KEAP1 complex is ubiquitylated and subsequently, the association of Nrf2-KEAP1 complex with Cul3 induces proteasomal degradation. However, under conditions of oxidative stress or antioxidant stimuli, phosphorylation of Nrf2 triggers its dissociation from KEAP1. The active Nrf2 is then translocated to the nucleus and binds to its consensus ARE to activate respective antioxidant defense genes [74, 75]. (Figure 3).
Figure 3.

Role of HNE on Nrf2-mediated anti-oxidative signaling: Nrf2 is a master regulator of various anti-oxidative and anti-inflammatory pathways in the cells. Under basal conditions, Nrf2 is bound to KEAP-1 and remains inactive. KEAP-1 is an adaptor protein and association of KEAP-1 with Cul3, promotes proteasomal degradation of the Nrf2-KEAP1 complex. However, when activated by an external stimulus, (a) phosphorylation and dissociation of Nrf2 occurs from the Nrf2-Keap1 complex leading to nuclear translocation of Nrf2 and transcription of target genes. (b) HNE could promote Nrf2 activation either by modifying the C151 residues in KEAP-1 or by promoting Nrf2 phosphorylation directly. Apart from directly affecting Nrf2 and KEAP-1, various upstream kinases such as p38MAPK, PKC and ERK can also be modulated by HNE, which further initiates the activation of Nrf2.
Activation of the KEAP1-Nrf2 signaling pathway has been shown to be a major approach for HNE-induced cellular antioxidant defense. HNE is electrophilic in nature and hence activates the EpRE/ARE response elements. The highly reactive cysteine residues in KEAP1 makes it a preferred target for electrophilic attack by HNE [76]. Specifically, HNE has been shown to modify the cysteine amino acid residue at C151 in the BTB domain of KEAP1 leading to the dissociation of KEAP1 from the Nrf2-KEAP1 complex [76]. Three distinct cysteine amino acid-based sensors in KEAP1 have been reported to be recognized by carbonyl groups of HNE [77]. Apart from directly modulating KEAP1-Nrf2 interaction, HNE can also modulate various upstream protein kinases that are required for the phosphorylation Nrf2. Among these, PKC [78], PI3K [79] p38-MAPK, and ERK [80] have been shown to be important cellular protein kinases whose activities are specifically regulated by HNE. Although the precise mechanism of how these kinases modulate HNE-induced Nrf2 activation is not known, it is hypothesized that the adduct formation of protein kinases with HNE could alter the physiological functions of these protein kinases and favor phosphorylation of Nrf2 at specific sites (Figure 3).
Activation of Nrf2 either directly or indirectly acts as an important transcriptional regulator of various Phase -II detoxifying enzymes conferring protection to cells against oxidative damage [81]. Some important Phase- II detoxification enzymes activated by HNE include aldo-keto reductases (AKRs), γ-glutamylcysteine ligase (GCL), glutathione peroxidase (GPX), glutathione-S-transferase (GST), NADPH quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), thioredoxin (Trx), thioredoxin reductase (TrxR), and drug transporter proteins such as MRPs, Pgp1 and RLIP76. All the Phase-II detoxification enzymes have been shown to have specific ARE consensus sequences for Nrf2 binding in response to oxidative stress [82]. It is important that under oxidative stress, the major cellular detoxification pathways are activated in a concerted mechanism as a defense response to maintain the cellular redox balance homeostasis to confer protection against oxidative injury.
Several studies have demonstrated that the activation of Nrf2 plays an important role in the detoxification of xenobiotics, including lipid peroxidation products such as HNE [83, 84]. Miller et al., have shown that the intraperitoneal injection of Nrf2-ARE activators such as sulforaphane or carnosic acid protects cerebral cortical mitochondria from HNE-induced toxicity. Further, treatment with sulforaphane or carnosic acid up-regulated the mRNA levels of antioxidant enzymes such as HO-1 and prevented HNE-induced disruption of mitochondrial respiration [85]. HNE has also been shown to induce the expression of antioxidant enzymes AKR1C1, GSTA4, and HO-1 in HeLa cells, which play an important role in the protection against oxidative stress-induced toxicity [86]. Using siRNA-mediated silencing of Nrf2, this study has provided strong evidence that Nrf2 plays an important role in exerting protection against HNE-induced toxicity in HeLa cells [86]. Exposure to low concentrations of HNE (<5 μM) is likely to induce an elevated anti-oxidant response, which prepares the cells to cope with oxidative insults. Incubation of macrophages and vascular smooth muscle cells with HNE has been shown to activate Nrf2 and protect cells from oxidative injury during vascular complications. Abundant formation of lipid peroxidation products such as HNE has been reported in atherosclerotic plaques and hence identifying the signaling pathways regulated by HNE in vascular cells is of immense importance in atherosclerosis [87]. Gargiulo et al., have shown that the exposure of monocytes to HNE induces various inflammatory cytokines such as IL-1β, IL-8, and TNF-α which regulate atherosclerotic plaque stability [88, 89]. Incubation of HNE with murine peritoneal macrophages isolated from Nrf2 knockout mouse models did not show increased expression of HO-1, Prx1, and A170 antioxidant proteins, indicating the significance of Nrf2 in HNE-induced protective effects [88]. HNE-induced upregulation of CD36, a major scavenger receptor, has been shown to exert protective functions during atherosclerosis and oxLDL-induced oxidative damage in an Nrf2 dependent manner [90]. Studies have demonstrated that multiple antioxidant response elements and oxLDL uptake receptors are HNE targets, which play an important role in atherosclerosis progression, the anti-oxidant response in endothelial and vascular cells and atheroprotective functions [91, 92]. In 661 W retinal ganglion cells, apart from upregulating the nuclear translocation and Nrf2-ARE transcription activity, treatment with 5 μM HNE induced the expression of antioxidant genes such as Trx, TrxR, and HO-1, exerting protective functions. Similarly, the up-regulation of antioxidant genes by HNE conferred protection against H2O2-induced apoptosis in 661 W cells [93]. This study also demonstrated that siRNA-mediated silencing of Nrf2 failed to confer protection induced by HNE against H2O2-induced cell damage [93]. However, the signaling mechanisms activated by HNE are elusive. As we have already discussed in the preceding section that the concentration of HNE used in a study could act as an important factor to properly decipher the signaling mechanisms activated or inhibited by HNE (Figure 2). In studies using primary cultures of human optic nerve head astrocytes, it is observed that HNE concentrations greater than 50 μM induce apoptosis and significantly decrease the cellular GSH levels [94]. Treatment with a lesser concentration of HNE (< 25 μM) also leads to a significant decrease in cellular GSH levels after 1 h and 3 h of treatment. However, when the cells were allowed to recover for 24 h without HNE after 1 h or 3 h treatment with 25 μM HNE, significant restoration of GSH levels have been observed [94]. In this study, real-time PCR gene expression analysis of mRNA also showed a significant increase in the levels of AKR1C1, GSTA4 and GCLC mRNA along-with increase in transcription factor Nrf2 and c-Fos. Thus, the concentration and time of exposure to HNE are important in mediating HNE-induced antioxidant responses, which prepares the cells for protection against oxidant-induced damage [94]. It is already clear that strategies such as using natural or synthesized antioxidants to activate or augment Nrf2 mediated cellular antioxidant defense pathways exert protective functions against oxidative stress injury by multiple oxidants including lipid peroxidation products [86, 95]. Thus, HNE-mediated activation of Nrf2 may be an important step in regulating the HNE-induced antioxidant response in cells and tissues.
Studies to decipher the molecular mechanisms of HNE-mediated antioxidant defense response has provided evidence that apart from Nrf2-mediated regulation of antioxidant response, HNE has the ability to directly regulate the transcriptional activity of other antioxidant enzymes responsible for GSH biosynthesis. HNE regulates GSH levels by regulating the rate-limiting step of GSH biosynthesis by modifying Cys553 of the catalytic subunit (~73 kDa GCLC) and Cys35 of the modulatory subunit (~31 kDa, GCLM) of glutamate-cysteine ligase (GCL) [96]. GCL is a heterodimeric holoenzyme complex consisting of a 73 kDa catalytic subunit (GCLC) and a 31-kDa modifier subunit, which catalyzes the first and rate-limiting step of GSH biosynthesis [97]. Further, HNE induces the promoter activity of γ-glutamyl transpeptidase (GGT) by interacting with an electrophile response element (EpRe) in the proximal region of GGT promoter region 5 (GP5). GGT plays an important role in glutathione homeostasis and metabolism of GSH and hence increasing the transcriptional activity of GGT confers protection against oxidative injury. Moreover, HNE has been shown to be involved in Nrf2 binding to the GP5 EpRE complex [98]. Apart from directly inducing GGT promoter activity, HNE could also activate various upstream MAPK kinases such as ERK and p38-MAPK, which are involved in the activation of GGT. The transcriptional regulation of EpRE by p38-MAPK and ERK has been shown to induce GGT mRNA transcription, which regulates antioxidant response and cellular homeostasis [99]. MAPKs could act as important components in the regulation of Nrf2 activity induced by HNE as they have been reported to directly induce phosphorylation of Nrf2 leading to its activation [80]. Further, the transcription factor AP1 has been shown to be involved in the HNE-induced increase in the glutamate cysteine ligase (GCL) expression by regulating antioxidant response [100, 101]. These studies have provided evidence that multiple signaling pathways co-operate to induce antioxidant defense response induced by HNE. Low concentrations of HNE has been shown to induce the expression of cytoprotective genes such as thioredoxin reductase 1 (TrxR1) in a Nrf2/ERK/Akt-dependent pathway in PC12 cells [102]. Further, the expression of TrxR1 has been shown to induce an adaptive response and enhances cellular tolerance to oxidative insult. The HNE-induced ERK and AKT signaling pathways may directly or indirectly affect Nrf2 expression in PC12 cells [102]. HNE-mediated activation of ERK also plays an important role in the regulation of HO-1 expression induced by HNE. In rat pulmonary epithelial cells, HO-1 expression is reported to be translationally regulated by HNE-mediated phosphorylation of ERK [103].
ROS are important for HNE production; ROS-induced lipid peroxidation is a major source of HNE generation in cells [7, 9]. Furthermore, studies have provided evidence that HNE itself could induce ROS generation in cells; the HNE-induced ROS further propagates the effects of HNE-mediated antioxidant defense mechanisms in cells [104]. On the other hand, a few studies showed that ROS could play a bystander effect on HNE-induced cell signaling. Studies on PC12 cell lines provide evidence that HNE-induced cell damage was a result of HNE-induced modification of cellular GSH or modification in cysteine and histidine residues in proteins rather than the direct effect of ROS alone. HNE-induced apoptosis, intercellular Ca+2 accumulation, caspase activation and cellular quiescence were not ameliorated by scavenging the ROS generated in cells [105]. Thus, protein modifications, rather than ROS generation in HNE exposed cells seem to be a major regulator of HNE-mediated cell signaling [105]. Several other pathways such as the unfolded protein response- (UPR) and ER stress-associated signaling pathways have been shown to be affected by HNE [105]. In the same study, they have demonstrated that HNE induces ER stress by regulating ATF4 expression which regulates the unfolded protein response (UPR). A significant increase in UPR target genes such as GRP78 and CHOP has been observed in PC12 cells treated with HNE [105]. Thus, HNE by interacting with various protein kinases such as MAPK and AKT, transcription factors and AREs regulates the Nrf2-mediated antioxidant pathways in the cells. Furthermore, exposure of cells to low concentrations of HNE prepares the cells to withstand further oxidative injury, whereas a high concentration activates the apoptotic pathways leading to cellular toxicity.
4. Regulation of Pro-Inflammatory Pathways by HNE
Besides the regulation of Nrf2-mediated anti-oxidant pathways, HNE has been shown to regulate various pro-inflammatory pathways mediated by NF-κB and AP1. The activation of these pathways leads to the expression of multiple cytokines, chemokines and growth factors responsible for inflammatory response and various disease pathologies. NF-κB; a major redox-sensitive transcription factor activated during oxidative stress conditions, is a key regulator of cell viability and death [106]. Various studies have provided evidence of the myriad and elusive role of HNE as a critical regulator of NF-κB-mediated inflammatory signaling pathways in cells. NF-κB is a heterodimeric transcription factor with five subunits consisting of p50, p52, p65, c-Rel, and RelB. In the inactive state, the NF-κB complex is localized in the cytoplasm due to its association with IκBα. Upon stimulation by exogenous stimuli, NF-κB is phosphorylated and translocated to the nucleus where it binds to its consensus DNA sequence element and induces transcription of target genes. The IκB kinases (IKK) play an important role in the activation of NF-κB by inducing phosphorylation on serine residues of IκBα, which leads to dissociation of IκBα from NF-κB, leading to NF-κB activation and nuclear translocation. After dissociation, IκBα is polyubiquitinylated and degraded by the proteasome [106].
HNE can induce either activation or inhibition of NF-κB depending on the cell type and concentration (Figure 4). Although the mechanisms are not clearly understood, it has been shown in many studies that exposure to a high concentration of HNE inhibits the activation of NF-κB [107]. In fact, oxidative stress-induced HNE and NF-κB activation are positively correlated with the increased inflammatory response in many studies [108–113].
Figure 4.

Role of HNE on NF-κB-mediated pro-inflammatory signaling: During oxidative stress conditions ROS activate a series of protein kinases such as IKK which phosphorylates IkBα leading to dissociation of the inactive complex of NF-κB-IκBα. The active NF-κB is translocated to the nucleus and binds to its consensus sequences to induce the transcription of target genes. HNE exerts pleiotropic effects on NF-κB; (a) the low concentrations of HNE (<5 μM) could activate NF-κB whereas (b) high concentrations (>5 μM) of HNE could inhibit NF-κB. HNE could modulate NF-κB activity by interacting with upstream protein kinases and can directly modify NF-κB subunits.
It has been demonstrated in THP1 cells that HNE prevents phosphorylation of IκBα, which is necessary for NF-κB activation. Specifically, pre-treatment of THP1 cells with 25 μM HNE resulted in the reduction of LPS-induced phosphorylation in Ser-32 of IκBα, thus preventing the activation of NF-κB by HNE [114]. Besides LPS, HNE also inhibits phosphorylation of IκBα induced by IL-1β and Phorbol 12-myristate 13-acetate (PMA) in monocytes [114]. Studies have further demonstrated that HNE could directly regulate the activity of IKK Kinase by forming covalent adducts with the IKK complex. IKK-induced phosphorylation IκBα is necessary for NF-κB activation [115]. Studies using H1299 and Jurkat T-cells have shown that pre-treatment with HNE blocks TPA- or ionomycin-induced activation of NF-κB [115]. HNE could also regulate NF-κB by directly forming adducts with important signaling intermediates. In a model of long-term hepatic injury following alcohol exposure to rodents, HNE-IκBα adduct formation has been demonstrated to inhibit NF-κB, which is independent of IKK phosphorylation [116]. Inhibition of NF-κB by HNE exerts anti-inflammatory effects. HNE-induced inhibition of NF-κB prevents LPS-induced production of IL-6 in rat kupffer cells [117]. HNE-IKK adduct formation and suppression of IκBα phosphorylation could lead to the inhibition of NF-κB. HNE has been shown to prevent IL-1β-induced nuclear translocation of p65 and NF-κB-DNA binding [118]. Similarly, in rat cortical neuronal cells, exposure to HNE prevented the NF-κB-DNA binding activity [119].
On the other hand, many studies have provided substantial evidence that HNE activates NF-κB (Figure 4). Exposure of cells to low concentrations of HNE (1 μM) induces the activation of NF-κB in vascular smooth muscle cells [120]. The same concentration of HNE has been shown to induce phosphorylation and DNA-binding of NF-κB [120]. Similarly, another study demonstrates that exposure of vascular smooth muscle cells to HNE increases ROS production and activation of NF-κB/AKT signaling pathway leading to cell proliferation [121]. 5-Lipoxygenase (5-LOX) plays an important role in inflammation and atherosclerotic plaque formation. HNE has been shown to induce 5-LOX mRNA expression in murine macrophages by signaling through NF-κB, p38MAPK and ERK [122]. Similarly, exposure of murine macrophages to LPS increased ROS production and subsequently increased lipid peroxidation products [123]. Results from this study also demonstrate that HNE is an important mediator of LPS-induced inflammation by increasing the release of inflammatory cytokines such as TNF-α, IL-1β, IL-6, and MCP-1 [123]. HNE-mediated increase in the NF-κB activity has been shown to be involved in inflammatory signaling in rheumatoid arthritis. Treatment of synovial cells with 5 μM HNE resulted in a time-dependent increase in IL-1β, IL-6, and TNFα [124]. Exposure to a higher concentration of HNE (50 μM), led to a transient increase in IL-1β, IL-6, and TNF-α, with the maximum increase observed after 1 h. However, the levels of inflammatory cytokines gradually decreased after 6 and 12 h. Increase in Cox2 expression, an important inflammatory mediator was also observed in the same study [124]. Further, HNE has been reported to induce endothelial dysfunction, by increasing the production of NF-κB-mediated expression of IL-8, ICAM, and impairment of endothelial barrier function [125].
Although the literature provides conflicting data on the role of HNE in inhibiting or activating NF-κB based on its concentration, it is clear that exposure of cells to HNE induces a state of oxidative stress imbalance in the cells. Exposure to HNE leads to depletion of cellular GSH [126] and studies have provided evidence that depletion of GSH leads to activation of NF-κB [127]. HNE has also been reported to induce p47phox-mediated NADPH oxidase activity in murine macrophages contributing to oxidative stress [128]. Although the involvement of HNE-induced ROS in HNE-mediated signaling is controversial, studies have shown that HNE-induced mitochondrial damage in cells is primarily due to ROS-generation, which is also a major contributor of vascular damage induced by HNE [129]. HNE-induced oxidative stress has been shown to be an inducer of endothelial dysfunction. Several studies have demonstrated that treatment with antioxidants such as NAC and mercaptopropionyl glycine, which inhibits oxidative stress, attenuated the HNE-induced loss of endothelial cell function [130, 131]. These studies highlight that HNE-induced oxidative stress as a major player in HNE-induced endothelial cell damage and dysfunction.
Apart from directly regulating NF-κB, multiple lines of evidence suggest that HNE induces NF-κB -mediated pro-inflammatory signaling by regulating the activation of major protein kinases such as PKC, p38-MAPK, and JNK [132]. PKC is an important kinase in mediating HNE signaling. Treatment of RAW 264.7 macrophages with HNE decreases phorbol 12-myristate 13-acetate (PMA)-induced ROS production in a dose- and time-dependent manner by interacting with PKC [133]. Further, MAPK kinases are also reported to play an important role in HNE-induced endothelial dysfunction. In a study using human macrophages, induction of Cox2 expression was observed upon exposure to HNE. In this study, the authors did not observe any change in the expression of iNOS or NF-κB but p38-MAPK was found to be an important regulator of HNE-induced inflammatory signaling in macrophages [87]. HNE also alters the balance of calcium ions in cells, which is important for signal transduction mediated by PKC and PI3K isozymes. In human chondrocytes, treatment with HNE induced the HNE-IKK adduct formation leading to inhibition of nuclear translocation of p65 and DNA-binding. Surprisingly, data from the same study show that HNE induces Cox2 and PGE2 in chondrocytes independent of NF-κB [118].
Activator protein 1 (AP-1) transcription factor is an important regulator of cellular signaling pathways controlling differentiation, proliferation, and apoptosis. HNE is well known to alter AP-1 transcriptional activity and affects various cellular processes. In rat cortical neurons, HNE-induced AP-1 nuclear translocation and DNA-binding plays an important role in neuronal calcium homeostasis and mitochondrial dysfunction [119]. Furthermore, the regulation of AP-1 activity by HNE has been shown to induce vascular smooth muscle cell proliferation leading to vascular complications [134].
HNE has been shown to be involved in the regulation of both intrinsic and extrinsic apoptotic signaling pathways in cells [19]. Exposure of SK-N-BE neuroblastoma cell lines to HNE induces the expression of p53, p73, p63, p21 and Bax leading to apoptotic cell death [135]. HNE has also been reported to induce p53-mediated apoptosis in the retinal pigment epithelial cells. Using knockout mouse models of GSTA4-4, Sharma et al., have demonstrated that HNE induces p53-mediated apoptotic signals via p21, Bax, and caspase 3. Inhibiting the cells ability to detoxify HNE leads to HNE-induced phosphorylation and nuclear translocation of p53 and induction of apoptosis [136]. In addition to HNE-induced p53 expression, HNE-mediated cytochrome C release has been shown to be necessary for HNE-induced apoptosis in macrophages [137].
HNE also induces the activation and phosphorylation of Src kinases. Mass spectrophotometric analysis of HNE-Src interaction showed that HNE interacts with His236, Cys241 and Cys248 residues of Src. Activation of Src by HNE leads to the expression of inflammatory mediator Cox2 and transcription factor AP-1, via activation of p38MAPK, JNK and ERK1/2 [138]. The HNE-Src adduct formation plays an important role in pro-inflammatory signaling in aged kidneys [139]. HNE also plays an important role in age-related oxidative stress. Higher plasma levels of HNE has been reported in obese individuals [140]. HNE-induced TNF-α expression in adipocytes was regulated by HNE mediated transcriptional regulation of ETS1 transcription factors [140]. HNE induced inflammation and Cox-2 production in many studies has been reported to be mediated by p38-MAPK mediated signaling and activation of ATF-2/CREB-1, JNK, c-JUN and AP-1 [20]. Many other transcription factors have been reported to play an important role in HNE-induced vascular endothelial cell dysfunction [141]. Transcription factors such as ATF3 and ATF4, have been reported to play an important role in HNE- induced ER stress in endothelial cells. The siRNA mediated ATF4 deletion prevented HNE-induced monocyte adhesion and IL-8 production and exerted protective functions against HNE-induced toxicity in endothelial cells [126].
HNE also plays an important role in cancer-associated inflammation and cancerous progression as evidenced by recent reports highlighting the importance of HNE-mediated pro-inflammatory signaling in cancer [142]. HNE is an important second messenger molecule modulating cellular signaling pathways in cancer and mediates cancer cell proliferation, apoptosis, and antioxidant defense [44]. HNE either by extrinsic or intrinsic mechanisms has been reported to facilitate cancerous progression [9, 13, 41, 44]. Co-administration of HNE accelerated DSS-induced colitis in mice. Along with the increase in DSS-induced colitis, a significant increase in the expression of pro-inflammatory genes such as IL-6, TLR4, Cox2 along-with infiltration of CD45+ and CD45+F4/80+ immune cells were observed in HNE + DSS treated mice compared to DSS-alone. Treatment with HNE also impairs intestinal barrier function by reducing the colonic expression of occludin protein. Loss of barrier function leads to an increase in the bacterial LPS in the circulation. Further, by using TLR4 null mouse models they have demonstrated that HNE mediates inflammatory signaling in DSS-induced colitis by signaling through TLR4 [143]. Apart from this, analyzing transcript levels of genes altered by HNE in human colorectal carcinoma cells have provided evidence that exposure to HNE induces an alteration in multiple signaling pathways related to antioxidant response, ER stress, apoptosis and cell cycle regulation in a time- and dose-dependent manner [144]. Apart from cancer progression, studies also provide evidence that HNE plays an important role in inducing apoptosis in colon carcinoma cells. It has been demonstrated that HNE-induced apoptosis in colon carcinoma cells is mediated by signaling through HSF-1. siRNA-mediated inhibition of HSF-1 prevented HNE-induced cleavage of PARP and caspase 3. Overexpression of Bcl-xl attenuated HNE-induced apoptosis in colon carcinoma cells [145]. HNE also plays an central role in the pathogenesis of inflammatory lung pathologies such as COPD. High levels of HNE-modified proteins have been observed in lung tissues from COPD patients with a positive correlation to inflammation and expression of inflammatory mediators [146]. Recent studies have also demonstrated the importance of HNE in neuronal inflammation and maintenance of neuronal cells. Studies using animal models suggest that HNE reduces neuronal intracellular calcium (Ca+2) in CSF and leads to the loss of motor neurons. Calcium levels were also altered in the surviving neurons with no observable morphological alterations. Thus, HNE induces a prominent change in ionic equilibrium in neuronal cells implicating its importance in neurodegenerative diseases [147, 148]. The importance of HNE and lipid peroxidation products in neuronal biology and neuroinflammation is attaining considerable attention in recent years [148, 149]. HNE induces the expression of Cox2, PGE2 and IL-6 expression and inhibition of HNE is a beneficial target to attenuate inflammation during osteoarthritis [150, 151]. Apart from the evidence provided above on the role of HNE in inflammatory pathologies, HNE also plays an important role in the pathogenesis and progression of other inflammatory diseases such as cataract, AMD and COPD [152]. Thus, these studies provide evidence that HNE is an important mediator of various pro-and anti-inflammatory signaling pathways in different cell types (Figure 5) and understanding the cellular signaling pathways regulated by lipid peroxidation-derived aldehydes such as HNE will provide insights into the mechanism of disease and open avenues for the development of new therapeutic strategies in the future.
Figure 5.
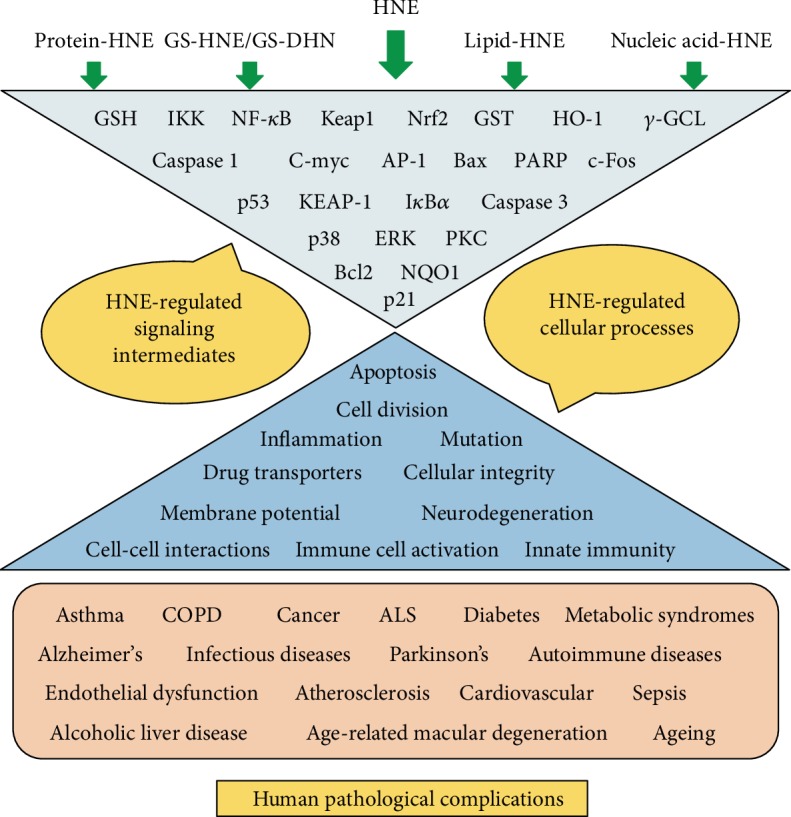
Central role of HNE and its metabolites in the regulation of various signaling pathways leading to various human disease pathologies. A summary of important signaling intermediates and cellular processes affect by HNE and its metabolites which can contribute to the pathophysiology of various human disorders are listed.
5. Conclusions and Future Perspectives
Recent studies have shown that depending upon the cell type, concentration and adduct formation with macromolecules, HNE could dictate cells to undergo proliferation or death. Interaction of HNE with cellular GSH has been shown to be a major metabolic route for its detoxification as well as intervening the cellular signaling pathways. Recent studies also suggest that HNE and HNE-protein adducts could act as biomarkers of various disease processes. Importantly, HNE metabolizing enzymes such as AR, ALDH1, and GSTs could regulate various anti-oxidative and pro-inflammatory pathways by generating HNE metabolites such as GS-HNE and GS-DHN which can further act as secondary signaling molecules. These studies opened new dimensions to understand the significance of HNE as well as other lipid aldehydes formed during lipid peroxidation and to understand the significance of aldehyde metabolizing enzymes in the pathophysiology of various diseases. These studies have provided a redox link between lipid aldehyde formation with oxidative stress and immune and inflammatory responses. Most importantly, we have shown that AR that reduces HNE, acrolein and other lipid aldehydes mediate oxidative stress signals initiated by various oxidants such as allergens, bacterial toxins, hyperglycemia, cytokines, and growth factors [56]. In fact, inhibition of AR has been shown to prevent several inflammatory complications including colon cancer, asthma, sepsis, uveitis, and cardiomyopathy [67, 68]. Thus, the specific use of antioxidants or synthetic inhibitors of HNE metabolizing enzymes that control the intracellular levels of HNE and its GSH-metabolites could be developed as potential therapeutic drug targets. It is a challenge for future research to clearly understand the complex interactions of HNE with various cellular proteins and how the protein-HNE adducts regulate various physiological processes. Further, it is still not clear how HNE commands cell signaling pathways based on a specific cell or tissue type. A better understanding of HNE-adduct formation with cellular macromolecules and identification of new HNE-adducts as potential biomarkers for various human pathologies will help to understand the pathophysiology of disease progression. In addition, identification of new immune and inflammatory response pathways regulated by HNE and its metabolites could help to understand the tissue and organ damage and dysfunction. Further, the use of novel transgenic approaches, metabolomics and next-generation sequencing (NGS) could ease the identification of critical signaling pathways intervened by HNE and its metabolites.
Acknowledgments
Supported by funding from NIH/NIDDK grant DK104786.
Abbreviations
- ADH:
Alcohol dehydrogenase
- AKR1B1:
Aldo-keto reductase family 1 member B1
- ALD:
Alcoholic liver disease
- ALDH:
Aldehyde dehydrogenase
- AMD:
Age-related macular degeneration
- AP-1:
Activator protein-1
- AR:
Aldose reductase
- ARE:
Antioxidant response element
- ATF4:
Activating transcription factor 4
- COPD:
Chronic obstructive pulmonary disease
- Cox-2:
Cyclooxygenase 2
- CREB:
cAMP response element-binding protein
- CSF:
Cerebrospinal fluid
- Cul3:
Cullin3
- DHN:
1,4-dihydroxy-2-nonene
- DNA:
Deoxyribonucleic acid
- DSS:
Dextran sulfate sodium
- EpRE:
Electrophile responsive element
- ERK:
Extracellular signal-regulated kinase
- ETS1:
ETS proto-oncogene1
- GGT:
γ-Glutamyl transpeptidase
- GPX:
Glutathione peroxidase
- GRP-78:
Glucose regulated protein 78
- GSH:
Glutathione
- GSSG:
Glutathione disulfide
- GST:
Glutathione-S-transferase
- H2O2:
Hydrogen peroxide
- HNA:
4-hydroxy-2-nonanoic acid
- HNE:
4-hydroxy-trans-2-nonenal
- HO-1:
Heme oxygenase-1
- HSF-1:
Heat shock factor-1
- ICAM-1:
Intercellular adhesion molecule -1
- IKK:
I-kappaB kinase
- IL-6:
Interleukin-6
- iNOS:
Inducible nitric oxide synthase
- IκBα:
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor- alpha
- KEAP-1:
Kelch like ECH associated protein 1
- LPS:
Lipopolysaccharide
- MCP-1:
Monocyte chemoattractant protein-1
- MDA:
Malondialdehyde
- MRP:
Multidrug resistance-associated protein
- NAC:
N-acetylcysteine
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NGS:
Next-generation sequencing
- NF-κB:
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NQO1:
NADPH quinone oxidoreductase 1
- Nrf2:
NFE2-related nuclear factor 2
- OxLDL:
Oxidized low-density lipoprotein
- p38MAPK:
p38 mitogen activated protein kinase
- PARP:
Poly ADP ribose polymerase
- PGE2:
Prostaglandin E2
- Pgp1:
ATP binding cassette subfamily B member 1 (ABCB1)
- PKC:
Protein kinase C
- PMA:
Phorbol myristate acetate
- Prx1:
Peroxiredoxin1
- PUFA:
Polyunsaturated fatty acid
- RLIP76:
RalA binding protein 1(RALBP1)
- ROS:
Reactive oxygen species
- SOD:
Superoxide dismutase
- TLR-4:
Toll-like receptor 4
- TNF-α:
Tumor necrosis factor α
- TPA:
Tissue plasminogen activator
- Trx:
Thioredoxin
- UPR:
Unfolded protein response
- γ-GCL:
γ-Glutamate cysteine ligase.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Davies K. J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50(4):279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 2.Bae Y. S., Oh H., Rhee S. G., Yoo Y. D. Regulation of reactive oxygen species generation in cell signaling. Molecules and Cells. 2011;32(6):491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aquilano K., Baldelli S., Ciriolo M. R. Glutathione: new roles in redox signaling for an old antioxidant. Frontiers in Pharmacology. 2014;5:196–196. doi: 10.3389/fphar.2014.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants & Redox Signaling. 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ray P. D., Huang B.-W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular Signalling. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pham-Huy L. A., He H., Pham-Huy C. Free radicals, antioxidants in disease and health. International Journal of Biomedical Science. 2008;4(2):89–96. [PMC free article] [PubMed] [Google Scholar]
- 7.Guéraud F., Atalay M., Bresgen N., et al. Chemistry and biochemistry of lipid peroxidation products. Free Radical Research. 2010;44(10):1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- 8.Mol M., Regazzoni L., Altomare A., et al. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: methodological aspects and biological consequences. Free Radical Biology & Medicine. 2017;111:328–344. doi: 10.1016/j.freeradbiomed.2017.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Poli G., Schaur R. J., Siems W. G., Leonarduzzi G. 4-Hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Medicinal Research Reviews. 2008;28(4):569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 10.Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Molecular Aspects of Medicine. 2003;24(4-5):281–291. doi: 10.1016/S0098-2997(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 11.Milkovic L., Cipak Gasparovic A., Zarkovic N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radical Research. 2015;49(7):850–860. doi: 10.3109/10715762.2014.999056. [DOI] [PubMed] [Google Scholar]
- 12.Barrera G., Pizzimenti S., Ciamporcero E. S., et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxidants & Redox Signaling. 2015;22(18):1681–1702. doi: 10.1089/ars.2014.6166. [DOI] [PubMed] [Google Scholar]
- 13.Zarkovic K., Jakovcevic A., Zarkovic N. Contribution of the HNE-immunohistochemistry to modern pathological concepts of major human diseases. Free Radical Biology & Medicine. 2017;111:110–126. doi: 10.1016/j.freeradbiomed.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Castro J. P., Jung T., Grune T., Siems W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radical Biology & Medicine. 2017;111:309–315. doi: 10.1016/j.freeradbiomed.2016.10.497. [DOI] [PubMed] [Google Scholar]
- 15.Sottero B., Rossin D., Poli G., Biasi F. Lipid oxidation products in the pathogenesis of inflammation-related gut diseases. Current Medicinal Chemistry. 2018;25(11):1311–1326. doi: 10.2174/0929867324666170619104105. [DOI] [PubMed] [Google Scholar]
- 16.Fleuranceau-Morel P., Barrier L., Fauconneau B., Piriou A., Huguet F. Origin of 4-hydroxynonenal incubation-induced inhibition of dopamine transporter and Na+/K+ adenosine triphosphate in rat striatal synaptosomes. Neuroscience Letters. 1999;277(2):91–94. doi: 10.1016/S0304-3940(99)00652-7. [DOI] [PubMed] [Google Scholar]
- 17.Subramaniam R., Roediger F., Jordan B., et al. The Lipid Peroxidation Product, 4-Hydroxy-2-trans-Nonenal, Alters the Conformation of Cortical Synaptosomal Membrane Proteins. Journal of Neurochemistry. 1997;69(3):1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhary P., Sharma R., Sharma A., et al. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry. 2010;49(29):6263–6275. doi: 10.1021/bi100517x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dalleau S., Baradat M., Guéraud F., Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death and Differentiation. 2013;20(12):1615–1630. doi: 10.1038/cdd.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kutuk O., Basaga H. Apoptosis signalling by 4-hydroxynonenal: a role for JNK–c-Jun/AP-1 pathway. Redox Report. 2007;12(1-2):30–34. doi: 10.1179/135100007X162329. [DOI] [PubMed] [Google Scholar]
- 21.Feng H., Stockwell B. R. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biology. 2018;16(5) doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H., Forman H. J. Signaling by 4-hydroxy-2-nonenal: exposure protocols, target selectivity and degradation. Archives of Biochemistry and Biophysics. 2017;617:145–154. doi: 10.1016/j.abb.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng R., Dragomir A. C., Mishin V., et al. Differential metabolism of 4-hydroxynonenal in liver, lung and brain of mice and rats. Toxicology and Applied Pharmacology. 2014;279(1):43–52. doi: 10.1016/j.taap.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poli G., Schaur R. J. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life. 2000;50(4):315–321. doi: 10.1080/15216540051081092. [DOI] [PubMed] [Google Scholar]
- 25.Siems W., Grune T. Intracellular metabolism of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24(4-5):167–175. doi: 10.1016/S0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 26.Doorn J. A., Petersen D. R. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chemical Research in Toxicology. 2002;15(11):1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 27.Doorn J. A., Petersen D. R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chemico-Biological Interactions. 2003;143-144:93–100. doi: 10.1016/S0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 28.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Progress in Lipid Research. 2003;42(4):318–343. doi: 10.1016/S0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 29.Schaur R. J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24(4-5):149–159. doi: 10.1016/S0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto M., Sibata T., Wasada H., Toyokuni S., Uchida K. Structural basis of protein-bound endogenous aldehydes. Chemical and immunochemical characterizations of configurational isomers of a 4-hydroxy-2-nonenal-histidine adduct. The Journal of Biological Chemistry. 2003;278(7):5044–5051. doi: 10.1074/jbc.M210129200. [DOI] [PubMed] [Google Scholar]
- 31.Stewart B. J., Doorn J. A., Petersen D. R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chemical Research in Toxicology. 2007;20(8):1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 32.Sampey B. P., Carbone D. L., Doorn J. A., Drechsel D. A., Petersen D. R. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Molecular Pharmacology. 2007;71(3):871–883. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 33.Nakashima I., Liu W., Akhand A. A., et al. 4-hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Molecular Aspects of Medicine. 2003;24(4-5):231–238. doi: 10.1016/S0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 34.Ferrington D. A., Kapphahn R. J. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Letters. 2004;578(3):217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Carbone D. L., Doorn J. A., Kiebler Z., Sampey B. P., Petersen D. R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chemical Research in Toxicology. 2004;17(11):1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connor R. E., Marnett L. J., Liebler D. C. Protein-selective capture to analyze electrophile adduction of hsp90 by 4-hydroxynonenal. Chemical Research in Toxicology. 2011;24(8):1275–1282. doi: 10.1021/tx200157t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carbone D. L., Doorn J. A., Kiebler Z., Ickes B. R., Petersen D. R. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. The Journal of Pharmacology and Experimental Therapeutics. 2005;315(1):8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 38.Schuler D., Budiawan, Eder E. Development of a 32P-Postlabeling Method for the Detection of 1,N2-Propanodeoxyguanosine Adducts of 2-Hexenal in Vivo. Chemical Research in Toxicology. 1999;12(4):335–340. doi: 10.1021/tx980225b. [DOI] [PubMed] [Google Scholar]
- 39.Nair J., Barbin A., Velic I., Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutation Research. 1999;424(1-2):59–69. doi: 10.1016/S0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 40.Karlhuber G. M., Bauer H. C., Eckl P. M. Cytotoxic and genotoxic effects of 4-hydroxynonenal in cerebral endothelial cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 1997;381(2):209–216. doi: 10.1016/S0027-5107(97)00170-X. [DOI] [PubMed] [Google Scholar]
- 41.Feng Z., Hu W., Tang M.-S. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(23):8598–8602. doi: 10.1073/pnas.0402794101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blair I. A. DNA adducts with lipid peroxidation products. The Journal of Biological Chemistry. 2008;283(23):15545–15549. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siems W. G., Zollner H., Grune T., Esterbauer H. Metabolic fate of 4-hydroxynonenal in hepatocytes: 1,4-dihydroxynonene is not the main product. Journal of Lipid Research. 1997;38(3):612–622. [PubMed] [Google Scholar]
- 44.Gueraud F. 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free Radical Biology & Medicine. 2017;111:196–208. doi: 10.1016/j.freeradbiomed.2016.12.025. [DOI] [PubMed] [Google Scholar]
- 45.Sharma R., Yang Y., Sharma A., et al. Mechanisms and physiological significance of the transport of the glutathione conjugate of 4-hydroxynonenal in human lens epithelial cells. Investigative Ophthalmology & Visual Science. 2003;44(8):3438–3449. doi: 10.1167/iovs.03-0051. [DOI] [PubMed] [Google Scholar]
- 46.Awasthi S., Singhal S. S., Sharma R., Zimniak P., Awasthi Y. C. Transport of glutathione conjugates and chemotherapeutic drugs by RLIP76 (RALBP1): A novel link between G-protein and tyrosine kinase signaling and drug resistance. International Journal of Cancer. 2003;106(5):635–646. doi: 10.1002/ijc.11260. [DOI] [PubMed] [Google Scholar]
- 47.Tjalkens R. B., Cook L. W., Petersen D. R. Formation and export of the glutathione conjugate of 4-hydroxy-2, 3-E-nonenal (4-HNE) in hepatoma cells. Archives of Biochemistry and Biophysics. 1999;361(1):113–119. doi: 10.1006/abbi.1998.0946. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell A. E., Morin D., Lame M. W., Jones A. D. Purification, mass spectrometric characterization, and covalent modification of murine glutathione S-transferases. Chemical Research in Toxicology. 1995;8(8):1054–1062. doi: 10.1021/tx00050a009. [DOI] [PubMed] [Google Scholar]
- 49.Vander Jagt D. L., Hunsaker L. A., Vander Jagt T. J., et al. Inactivation of glutathione reductase by 4-hydroxynonenal and other endogenous aldehydes. Biochemical Pharmacology. 1997;53(8):1133–1140. doi: 10.1016/S0006-2952(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 50.Liu R.-M., Gao L., Choi J., Forman H. J. γ-Glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. American Journal of Physiology-Lung Cellular and Molecular Physiology. 1998;275(5):L861–L869. doi: 10.1152/ajplung.1998.275.5.l861. [DOI] [PubMed] [Google Scholar]
- 51.Berhane K., Widersten M., Engstrom A., Kozarich J. W., Mannervik B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(4):1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartley D. P., Ruth J. A., Petersen D. R. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Archives of Biochemistry and Biophysics. 1995;316(1):197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- 53.Balogh L. M., Atkins W. M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metabolism Reviews. 2011;43(2):165–178. doi: 10.3109/03602532.2011.558092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Awasthi Y. C., Ramana K. V., Chaudhary P., Srivastava S. K., Awasthi S. Regulatory roles of glutathione-S-transferases and 4-hydroxynonenal in stress-mediated signaling and toxicity. Free Radical Biology and Medicine. 2017;111:235–243. doi: 10.1016/j.freeradbiomed.2016.10.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Awasthi Y. C., Sharma R., Cheng J. Z., et al. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Molecular Aspects of Medicine. 2003;24(4-5):219–230. doi: 10.1016/S0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 56.Ramana K. V. ALDOSE REDUCTASE: new insights for an old enzyme. Biomolecular Concepts. 2011;2(1-2):103–114. doi: 10.1515/BMC.2011.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spycher S., Tabataba-Vakili S., O'Donnell V. B., Palomba L., Azzi A. 4-hydroxy-2, 3-trans-nonenal induces transcription and expression of aldose reductase. Biochemical and Biophysical Research Communications. 1996;226(2):512–516. doi: 10.1006/bbrc.1996.1386. [DOI] [PubMed] [Google Scholar]
- 58.Burczynski M. E., Sridhar G. R., Palackal N. T., Penning T. M. The reactive oxygen species--and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. The Journal of Biological Chemistry. 2001;276(4):2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- 59.Choudhary S., Srivastava S., Xiao T., Andley U. P., Srivastava S. K., Ansari N. H. Metabolism of lipid derived aldehyde, 4-hydroxynonenal in human lens epithelial cells and rat lens. Investigative Ophthalmology & Visual Science. 2003;44(6):2675–2682. doi: 10.1167/iovs.02-0965. [DOI] [PubMed] [Google Scholar]
- 60.Srivastava S., Dixit B. L., Cai J., et al. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radical Biology & Medicine. 2000;29(7):642–651. doi: 10.1016/S0891-5849(00)00351-8. [DOI] [PubMed] [Google Scholar]
- 61.Srivastava S., Liu S. Q., Conklin D. J., Zacarias A., Srivastava S. K., Bhatnagar A. Involvement of aldose reductase in the metabolism of atherogenic aldehydes. Chemico-Biological Interactions. 2001;130-132(1-3):563–571. doi: 10.1016/S0009-2797(00)00299-4. [DOI] [PubMed] [Google Scholar]
- 62.Srivastava S., Chandra A., Wang L. F., et al. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. The Journal of Biological Chemistry. 1998;273(18):10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava S., Watowich S. J., Petrash J. M., Srivastava S. K., Bhatnagar A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry. 1999;38(1):42–54. doi: 10.1021/bi981794l. [DOI] [PubMed] [Google Scholar]
- 64.Dixit B. L., Balendiran G. K., Watowich S. J., et al. Kinetic and structural characterization of the glutathione-binding site of aldose reductase. The Journal of Biological Chemistry. 2000;275(28):21587–21595. doi: 10.1074/jbc.M909235199. [DOI] [PubMed] [Google Scholar]
- 65.Singh R., White M. A., Ramana K. V., et al. Structure of a glutathione conjugate bound to the active site of aldose reductase. Proteins: Structure, Function, and Bioinformatics. 2006;64(1):101–110. doi: 10.1002/prot.20988. [DOI] [PubMed] [Google Scholar]
- 66.Yadav U. C., Ramana K. V. Regulation of NF-κB-Induced Inflammatory Signaling by Lipid Peroxidation-Derived Aldehydes. Oxidative Medicine and Cellular Longevity. 2013;2013 doi: 10.1155/2013/690545.690545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramana K. V., Srivastava S. K. Aldose reductase: a novel therapeutic target for inflammatory pathologies. The International Journal of Biochemistry & Cell Biology. 2010;42(1):17–20. doi: 10.1016/j.biocel.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tammali R., Srivastava S. K., Ramana K. V. Targeting aldose reductase for the treatment of cancer. Current Cancer Drug Targets. 2011;11(5):560–571. doi: 10.2174/156800911795655958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grune T., Michel P., Sitte N., et al. Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Radical Biology and Medicine. 1997;23(3):357–360. doi: 10.1016/S0891-5849(96)00586-2. [DOI] [PubMed] [Google Scholar]
- 70.Kamimura S., Gaal K., Britton R. S., Bacon B. R., Triadafilopoulos G., Tsukamoto H. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: association of lipid peroxidation with liver fibrogenesis. Hepatology. 1992;16(2):448–453. doi: 10.1002/hep.1840160225. [DOI] [PubMed] [Google Scholar]
- 71.Tsukamoto H., Horne W., Kamimura S., et al. Experimental liver cirrhosis induced by alcohol and iron. The Journal of Clinical Investigation. 1995;96(1):620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spickett C. M. The lipid peroxidation product 4-hydroxy-2-nonenal: advances in chemistry and analysis. Redox Biology. 2013;1(1):145–152. doi: 10.1016/j.redox.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Motohashi H., Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends in Molecular Medicine. 2004;10(11):549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 74.Suzuki T., Yamamoto M. Molecular basis of the Keap1–Nrf2 system. Free Radical Biology and Medicine. 2015;88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 75.Kobayashi A., Kang M. I., Watai Y., et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Molecular and Cellular Biology. 2006;26(1):221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McMahon M., Lamont D. J., Beattie K. A., Hayes J. D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(44):18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Numazawa S., Ishikawa M., Yoshida A., Tanaka S., Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. American Journal of Physiology. Cell Physiology. 2003;285(2):C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 79.Chen J., Wang L., Chen Y., Sternberg P., Cai J. Phosphatidylinositol 3 kinase pathway and 4-hydroxy-2-nonenal-induced oxidative injury in the RPE. Investigative Ophthalmology & Visual Science. 2009;50(2):936–942. doi: 10.1167/iovs.08-2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun Z., Huang Z., Zhang D. D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4(8):e6588–e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang M., An C., Gao Y., Leak R. K., Chen J., Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Progress in Neurobiology. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen T., Nioi P., Pickett C. B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of Biological Chemistry. 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology. 2013;53(1):401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singhal S. S., Singh S. P., Singhal P., Horne D., Singhal J., Awasthi S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicology and Applied Pharmacology. 2015;289(3):361–370. doi: 10.1016/j.taap.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller D. M., Singh I. N., Wang J. A., Hall E. D. Administration of the Nrf2–ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radical Biology & Medicine. 2013;57:1–9. doi: 10.1016/j.freeradbiomed.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang Y., Li W., Kong A.-N. T. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell & Bioscience. 2012;2(1):40–40. doi: 10.1186/2045-3701-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumagai T., Matsukawa N., Kaneko Y., Kusumi Y., Mitsumata M., Uchida K. A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. The Journal of Biological Chemistry. 2004;279(46):48389–48396. doi: 10.1074/jbc.M409935200. [DOI] [PubMed] [Google Scholar]
- 88.Gargiulo S., Gamba P., Testa G., et al. Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell. 2015;14(4):569–581. doi: 10.1111/acel.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gargiulo S., Testa G., Gamba P., Staurenghi E., Poli G., Leonarduzzi G. Oxysterols and 4-hydroxy-2-nonenal contribute to atherosclerotic plaque destabilization. Free Radical Biology and Medicine. 2017;111:140–150. doi: 10.1016/j.freeradbiomed.2016.12.037. [DOI] [PubMed] [Google Scholar]
- 90.Ishii T., Itoh K., Ruiz E., et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circulation Research. 2004;94(5):609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 91.Siow R. C., Ishii T., Mann G. E. Modulation of antioxidant gene expression by 4-hydroxynonenal: atheroprotective role of the Nrf2/ARE transcription pathway. Redox Report. 2007;12(1-2):11–15. doi: 10.1179/135100007X162167. [DOI] [PubMed] [Google Scholar]
- 92.Negre-Salvayre A., Garoby-Salom S., Swiader A., Rouahi M., Pucelle M., Salvayre R. Proatherogenic effects of 4-hydroxynonenal. Free Radical Biology & Medicine. 2017;111:127–139. doi: 10.1016/j.freeradbiomed.2016.12.038. [DOI] [PubMed] [Google Scholar]
- 93.Tanito M., Agbaga M. P., Anderson R. E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radical Biology & Medicine. 2007;42(12):1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 94.Malone P. E., Hernandez M. R. 4-Hydroxynonenal, a product of oxidative stress, leads to an antioxidant response in optic nerve head astrocytes. Experimental Eye Research. 2007;84(3):444–454. doi: 10.1016/j.exer.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang Y., Li W., Su Z. Y., Kong A. N. T. The complexity of the Nrf2 pathway: beyond the antioxidant response. The Journal of Nutritional Biochemistry. 2015;26(12):1401–1413. doi: 10.1016/j.jnutbio.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Backos D. S., Fritz K. S., Roede J. R., Petersen D. R., Franklin C. C. Posttranslational modification and regulation of glutamate-cysteine ligase by the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal. Free Radical Biology and Medicine. 2011;50(1):14–26. doi: 10.1016/j.freeradbiomed.2010.10.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griffith O. W., Mulcahy R. T. The Enzymes of Glutathione Synthesis: γ-Glutamylcysteine Synthetase. Advances in Enzymology and Related Areas of Molecular Biology. 1999;73:209–267. doi: 10.1002/9780470123195.ch7. [DOI] [PubMed] [Google Scholar]
- 98.Zhang H., Liu H., Dickinson D. A., et al. γ-Glutamyl transpeptidase is induced by 4-hydroxynonenal via EpRE/Nrf2 signaling in rat epithelial type II cells. Free Radical Biology & Medicine. 2006;40(8):1281–1292. doi: 10.1016/j.freeradbiomed.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang H., Liu H., Iles K. E., et al. 4-Hydroxynonenal induces rat γ-Glutamyl transpeptidase through mitogen-activated protein kinase-mediated electrophile response element/nuclear factor erythroid 2-related factor 2 signaling. American Journal of Respiratory Cell and Molecular Biology. 2006;34(2):174–181. doi: 10.1165/rcmb.2005-0280OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dickinson D. A., Iles K. E., Watanabe N., et al. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radical Biology & Medicine. 2002;33(7):974–987. doi: 10.1016/S0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 101.Iles K. E., Liu R. M. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radical Biology & Medicine. 2005;38(5):547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 102.Chen Z. H., Saito Y., Yoshida Y., Sekine A., Noguchi N., Niki E. 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via activation of Nrf2. The Journal of Biological Chemistry. 2005;280(51):41921–41927. doi: 10.1074/jbc.M508556200. [DOI] [PubMed] [Google Scholar]
- 103.Iles K. E., Dickinson D. A., Wigley A. F., Welty N. E., Blank V., Forman H. J. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radical Biology & Medicine. 2005;39(3):355–364. doi: 10.1016/j.freeradbiomed.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Raza H., John A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicology and Applied Pharmacology. 2006;216(2):309–318. doi: 10.1016/j.taap.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 105.Lin M. H., Yen J. H., Weng C. Y., Wang L., Ha C. L., Wu M. J. Lipid peroxidation end product 4-hydroxy-trans-2-nonenal triggers unfolded protein response and heme oxygenase-1 expression in PC12 cells: roles of ROS and MAPK pathways. Toxicology. 2014;315:24–37. doi: 10.1016/j.tox.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 106.Morgan M. J., Liu Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Park S., Sung B., Jang E. J., et al. Inhibitory action of salicylideneamino-2-thiophenol on NF-κB signaling cascade and cyclooxygenase-2 in HNE-treated endothelial cells. Archives of Pharmacal Research. 2013;36(7):880–889. doi: 10.1007/s12272-013-0116-4. [DOI] [PubMed] [Google Scholar]
- 108.Selcuk M. Y., Aygen B., Dogukan A., et al. Chromium picolinate and chromium histidinate protects against renal dysfunction by modulation of NF-κB pathway in high-fat diet fed and Streptozotocin-induced diabetic rats. Nutrition & Metabolism. 2012;9(1):p. 30. doi: 10.1186/1743-7075-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Valacchi G., Sticozzi C., Belmonte G., et al. Vitamin Ccompound mixtures prevent ozone-induced oxidative damage in human keratinocytes as initial assessment of pollution protection. PLoS One. 2015;10(8) doi: 10.1371/journal.pone.0131097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Potočnjak I., Škoda M., Pernjak-Pugel E., Peršić M. P., Domitrović R. Oral administration of oleuropein attenuates cisplatin-induced acute renal injury in mice through inhibition of ERK signaling. Molecular Nutrition & Food Research. 2016;60(3):530–541. doi: 10.1002/mnfr.201500409. [DOI] [PubMed] [Google Scholar]
- 111.Venkatachalam K., Prabhu S. D., Reddy V. S., Boylston W. H., Valente A. J., Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. The Journal of Biological Chemistry. 2009;284(12):7853–7865. doi: 10.1074/jbc.M808824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sonowal H., Pal P., Shukla K., Saxena A., Srivastava S. K., Ramana K. V. Aldose reductase inhibitor, fidarestat prevents doxorubicin-induced endothelial cell death and dysfunction. Biochemical Pharmacology. 2018;150:181–190. doi: 10.1016/j.bcp.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Timucin A. C., Basaga H. Pro-apoptotic effects of lipid oxidation products: HNE at the crossroads of NF-κB pathway and anti-apoptotic Bcl-2. Free Radical Biology and Medicine. 2017;111:209–218. doi: 10.1016/j.freeradbiomed.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 114.Page S., Fischer C., Baumgartner B., et al. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. The Journal of Biological Chemistry. 1999;274(17):11611–11618. doi: 10.1074/jbc.274.17.11611. [DOI] [PubMed] [Google Scholar]
- 115.Ji C., Kozak K. R., Marnett L. J. IκB Kinase, a Molecular Target for Inhibition by 4-Hydroxy-2-nonenal. The Journal of Biological Chemistry. 2001;276(21):18223–18228. doi: 10.1074/jbc.M101266200. [DOI] [PubMed] [Google Scholar]
- 116.Dou X., Li S., Wang Z., et al. Inhibition of NF-κB activation by 4-Hydroxynonenal contributes to liver injury in a mouse model of alcoholic liver disease. The American Journal of Pathology. 2012;181(5):1702–1710. doi: 10.1016/j.ajpath.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Luckey S. W., Taylor M., Sampey B. P., Scheinman R. I., Petersen D. R. 4-Hydroxynonenal Decreases Interleukin-6 Expression and Protein Production in Primary Rat Kupffer Cells by Inhibiting Nuclear Factor-κB Activation. The Journal of Pharmacology and Experimental Therapeutics. 2002;302(1):296–303. doi: 10.1124/jpet.102.033522. [DOI] [PubMed] [Google Scholar]
- 118.Vaillancourt F., Morquette B., Shi Q., et al. Differential regulation of cyclooxygenase-2 and inducible nitric oxide synthase by 4-hydroxynonenal in human osteoarthritic chondrocytes through ATF-2/CREB-1 transactivation and concomitant inhibition of NF-κB signaling cascade. Journal of Cellular Biochemistry. 2007;100(5):1217–1231. doi: 10.1002/jcb.21110. [DOI] [PubMed] [Google Scholar]
- 119.Camandola S., Poli G., Mattson M. P. The lipid peroxidation product 4-hydroxy-2, 3-nonenal increases AP-1-binding activity through caspase activation in neurons. Journal of Neurochemistry. 2000;74(1):159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- 120.Ruef J., Moser M., Bode C., Kübler W., Runge M. S. 4-Hydroxynonenal induces apoptosis, NF-κB-activation and formation of 8-isoprostane in vascular smooth nmuscle cells. Basic Research in Cardiology. 2001;96(2):143–150. doi: 10.1007/s003950170064. [DOI] [PubMed] [Google Scholar]
- 121.Lee S. J., Seo K. W., Yun M. R., et al. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-κB signaling pathways. Free Radical Biology & Medicine. 2008;45(10):1487–1492. doi: 10.1016/j.freeradbiomed.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 122.Lee S. J., Kim C. E., Seo K. W., Kim C. D. HNE-induced 5-LO expression is regulated by NF-κB/ERK and Sp1/p38 MAPK pathways via EGF receptor in murine macrophages. Cardiovascular Research. 2010;88(2):352–359. doi: 10.1093/cvr/cvq194. [DOI] [PubMed] [Google Scholar]
- 123.Ramana K. V., Reddy A. B. M., Tammali R., Srivastava S. K. Aldose reductase mediates endotoxin-induced production of nitric oxide and cytotoxicity in murine macrophages. Free Radical Biology and Medicine. 2007;42(8):1290–1302. doi: 10.1016/j.freeradbiomed.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yin G., Wang Y., Cen X. M., Yang M., Liang Y., Xie Q. B. Lipid Peroxidation-Mediated Inflammation Promotes Cell Apoptosis through Activation of NF-κB Pathway in Rheumatoid Arthritis Synovial Cells. Mediators of Inflammation. 2015;2015 doi: 10.1155/2015/460310.460310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Herbst U., Toborek M., Kaiser S., Mattson M. P., Hennig B. 4-Hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. Journal of Cellular Physiology. 1999;181(2):295–303. doi: 10.1002/(SICI)1097-4652(199911)181:2<295::AID-JCP11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 126.Vladykovskaya E., Sithu S. D., Haberzettl P., et al. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. The Journal of Biological Chemistry. 2012;287(14):11398–11409. doi: 10.1074/jbc.M111.320416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hammond C. L., Lee T. K., Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. Journal of Hepatology. 2001;34(6):946–954. doi: 10.1016/S0168-8278(01)00037-X. [DOI] [PubMed] [Google Scholar]
- 128.Yun M. R., Park H. M., Seo K. W., Lee S. J., Im D. S., Kim C. D. 5-lipoxygenase plays an essential role in 4-HNE-enhanced ROS production in murine macrophages via activation of NADPH oxidase. Free Radical Research. 2010;44(7):742–750. doi: 10.3109/10715761003758122. [DOI] [PubMed] [Google Scholar]
- 129.Lee J. Y., Jung G. Y., Heo H. J., et al. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicology Letters. 2006;166(3):212–221. doi: 10.1016/j.toxlet.2006.07.305. [DOI] [PubMed] [Google Scholar]
- 130.Usatyuk P. V., Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. The Journal of Biological Chemistry. 2004;279(12):11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- 131.Usatyuk P. V., Parinandi N. L., Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. The Journal of Biological Chemistry. 2006;281(46):35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 132.Uchida K., Shiraishi M., Naito Y., Torii Y., Nakamura Y., Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. The Journal of Biological Chemistry. 1999;274(4):2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 133.Harry R. S., Hiatt L. A., Kimmel D. W., et al. Metabolic impact of 4-Hydroxynonenal on macrophage-like RAW 264.7 function and activation. Chemical Research in Toxicology. 2012;25(8):1643–1651. doi: 10.1021/tx3001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ramana K. V., Bhatnagar A., Srivastava S., et al. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. The Journal of Biological Chemistry. 2006;281(26):17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 135.Laurora S., Tamagno E., Briatore F., et al. 4-Hydroxynonenal modulation of p53 family gene expression in the SK-N-BE neuroblastoma cell line. Free Radical Biology and Medicine. 2005;38(2):215–225. doi: 10.1016/j.freeradbiomed.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 136.Sharma A., Sharma R., Chaudhary P., et al. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Archives of Biochemistry and Biophysics. 2008;480(2):85–94. doi: 10.1016/j.abb.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haynes R. L., Brune B., Townsend A. J. Apoptosis in RAW 264.7 cells exposed to 4-hydroxy-2-nonenal: dependence on cytochrome C release but not p53 accumulation. Free Radical Biology & Medicine. 2001;30(8):884–894. doi: 10.1016/S0891-5849(01)00476-2. [DOI] [PubMed] [Google Scholar]
- 138.Jang E. J., Jeong H. O., Park D., et al. Src tyrosine kinase activation by 4-Hydroxynonenal upregulates p38, ERK/AP-1 signaling and COX-2 expression in YPEN-1 cells. PLoS One. 2015;10(10) doi: 10.1371/journal.pone.0129244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jang E. J., Kim D. H., Lee B., et al. Activation of proinflammatory signaling by 4-hydroxynonenal-Src adducts in aged kidneys. Oncotarget. 2016;7(32):50864–50874. doi: 10.18632/oncotarget.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang X. M., Guo L., Huang X., Li Q. M., Chi M. H. 4-Hydroxynonenal Regulates TNF-α Gene Transcription Indirectly via ETS1 and microRNA-29b in Human Adipocytes Induced From Adipose Tissue-Derived Stromal Cells. The Anatomical Record. 2016;299(8):1145–1152. doi: 10.1002/ar.23371. [DOI] [PubMed] [Google Scholar]
- 141.Chapple S. J., Cheng X., Mann G. E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biology. 2013;1:319–331. doi: 10.1016/j.redox.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Uchida K. HNE as an inducer of COX-2. Free Radical Biology and Medicine. 2017;111:169–172. doi: 10.1016/j.freeradbiomed.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 143.Wang Y., Wang W., Yang H., Shao D., Zhao X., Zhang G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of toll-like receptor 4 signaling. Free Radical Biology and Medicine. 2019;131:237–242. doi: 10.1016/j.freeradbiomed.2018.11.037. [DOI] [PubMed] [Google Scholar]
- 144.West J. D., Marnett L. J. Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chemical Research in Toxicology. 2005;18(11):1642–1653. doi: 10.1021/tx050211n. [DOI] [PubMed] [Google Scholar]
- 145.Jacobs A. T., Marnett L. J. Heat shock factor 1 attenuates 4-Hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. The Journal of Biological Chemistry. 2007;282(46):33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- 146.Rahman I., van Schadewijk A. A. M., Crowther A. J. L., et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2002;166(4):490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 147.Vigh L., Smith R. G., Soós J., Engelhardt J. I., Appel S. H., Siklós L. Sublethal dose of 4-hydroxynonenal reduces intracellular calcium in surviving motor neurons in vivo. Acta Neuropathologica. 2005;109(6):567–575. doi: 10.1007/s00401-004-0977-1. [DOI] [PubMed] [Google Scholar]
- 148.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Molecular Aspects of Medicine. 2003;24(4-5):293–303. doi: 10.1016/S0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 149.Shichiri M. The role of lipid peroxidation in neurological disorders. Journal of Clinical Biochemistry and Nutrition. 2014;54(3):151–160. doi: 10.3164/jcbn.14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen S.-H., Fahmi H., Shi Q., Benderdour M. Regulation of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase-activating protein/5-lipoxygenase by 4-hydroxynonenal in human osteoarthritic chondrocytes. Arthritis Research & Therapy. 2010;12(1):R21–R21. doi: 10.1186/ar2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shi Q., Vaillancourt F., Côté V., et al. Alterations of metabolic activity in human osteoarthritic osteoblasts by lipid peroxidation end product 4-hydroxynonenal. Arthritis Research & Therapy. 2006;8(6):R159–R159. doi: 10.1186/ar2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shoeb M., Ansari N. H., Srivastava S. K., Ramana K. V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Current Medicinal Chemistry. 2014;21(2):230–237. doi: 10.2174/09298673113209990181. [DOI] [PMC free article] [PubMed] [Google Scholar]