

Abstract
An 80-year-old woman with rheumatoid arthritis presented with chest pain. Clinical examination revealed new-onset paroxysmal atrial fibrillation with symptomatic sinus pauses and worsening mitral regurgitation, which were both resistant to conventional therapies. Based on her skin lesions, an increase in pleural and pericardial effusion, possible myocardial involvement, and a positive finding for immune complex testing, rheumatoid vasculitis was diagnosed. Subsequent glucocorticoid therapy suppressed systemic inflammation, resulting in structural, functional, and electrical reverse remodeling of the left atrium with complete remission of atrial arrhythmias and also an improvement of mitral regurgitation. This case highlights the importance of evaluating the underlying disease activity in a case of de novo paroxysmal atrial fibrillation associated with systemic autoimmune disease.
Keywords: mitral regurgitation, paroxysmal atrial fibrillation, prednisolone, rheumatoid vasculitis, sick sinus syndrome
Introduction
A systemic inflammatory disease, such as rheumatoid arthritis can be associated with the development and persistence of atrial fibrillation (1). Inflammatory cytokines play a significant role in creating an arrhythmogenic substrate and trigger (2). We herein report a case of rheumatoid vasculitis associated with paroxysmal atrial fibrillation, sick sinus syndrome, and a transient exacerbation of mitral regurgitation. Glucocorticoid therapy alone successfully suppressed all of these pathological conditions.
Case Report
An 80-year-old woman with rheumatoid arthritis, which had been diagnosed at 57 years of age, presented with chest pain. She had been treated with bucillamine and tocilizumab for the last 3 years. Her past history included angina pectoris, hypertension, dyslipidemia, hyperuricemia, and bronchial asthma. On admission, she was afebrile, and her blood pressure was 106/42 mmHg, with a regular pulse rate of 72 beats/min. A prominent Levine grade II/VI systolic regurgitant murmur was audible at the cardiac apex along with a third heart sound. She showed a z-deformity, buttonhole deformity, bilateral ulnar deviation of the fingers, rheumatoid nodules on the lower arms, skin ulcers on the small toes, and pitting edema of the lower limbs. Both inflammatory markers and cardiac enzymes were elevated (Table 1). The serum brain natriuretic peptide level was elevated at 660 pg/dL. Chest radiography revealed cardiomegaly and congestion (Fig. 1). The electrocardiogram indicated normal sinus rhythm with ST-segment depression in leads V4-V6 (Fig. 2). Transthoracic echocardiography showed severe mitral regurgitation (Fig. 3), which had been mild on a previous reading 5 months before admission. No echocardiographic findings indicating mitral valvar degeneration, prolapse, ruptured chordae tendineae, tethering, and papillary muscle dysfunction were observed. Left atrial dilation, augmentation of E-wave velocity, and the appearance of an L-wave were also detected (Fig. 4). The left ventricular ejection fraction (65%) remained normal without asynergy. Emergent coronary angiography revealed no significant stenosis. With the tentative diagnosis of congestive heart failure secondary to mitral regurgitation, she was transferred to the intensive care unit. Because the left ventricular afterload had not increased, but instead had decreased, and the patient’s fluid status did not indicate volume retention as revealed by transthoracic echocardiography (the inferior caval vein was not distended, but instead had somewhat collapsed), acute left heart failure due to mitral regurgitation could not be controlled with conventional regimens, including several diuretics. The serum brain natriuretic peptide level was further elevated at 994 pg/dL on day 9 (Fig. 5). Furthermore, de novo paroxysmal atrial fibrillation was frequently observed, combined with symptomatic sinus pauses (>5 seconds) at the termination of paroxysmal atrial fibrillation. Several antiarrhythmic drugs, including amiodarone, disopyramide, carvedilol, and bisoprolol, were unable to reduce or eliminate the episodes of atrial fibrillation and repeated sinus pauses.
Table 1.
Laboratory Data on Admission.
| Peripheral blood | Total protein | 6.3 | g/dL | |||||
| White blood cells | 10,000 | /μL | Serum albumin | 2.8 | g/dL | |||
| Red blood cells | 358×104 | /μL | Total bilirubin | 0.9 | mg/dL | |||
| Hemoglobin | 11.3 | g/dL | Aspartate aminotransferase | 33 | IU/L | |||
| Platelets | 13.0×104 | /μL | Alanine aminotransferase | 22 | IU/L | |||
| Lactate dehydrogenase | 205 | IU/L | ||||||
| Biochemistry | Creatine kinase | 193 | IU/L | |||||
| Sodium | 125 | mEq/L | Creatine kinase MB | 19 | IU/L | |||
| Potassium | 4.5 | mEq/L | Troponin I | 1.18 | ng/mL | |||
| Chloride | 96 | mEq/L | Brain natriuretic peptide | 660 | pg/mL | |||
| Blood urea nitrogen | 35.9 | mg/dL | C-reactive protein | 11.8 | mg/dL | |||
| Serum creatinine | 1.40 | mg/dL | ||||||
| Estimated glomerular filtration rate | 28.2 | mL/min/1.73m2 |
Figure 1.

Chest radiography and computed tomography. After prednisolone (PSL) therapy, cardio-thoracic ratio, congestion, and both pleural and pericardial effusion improved.
Figure 2.
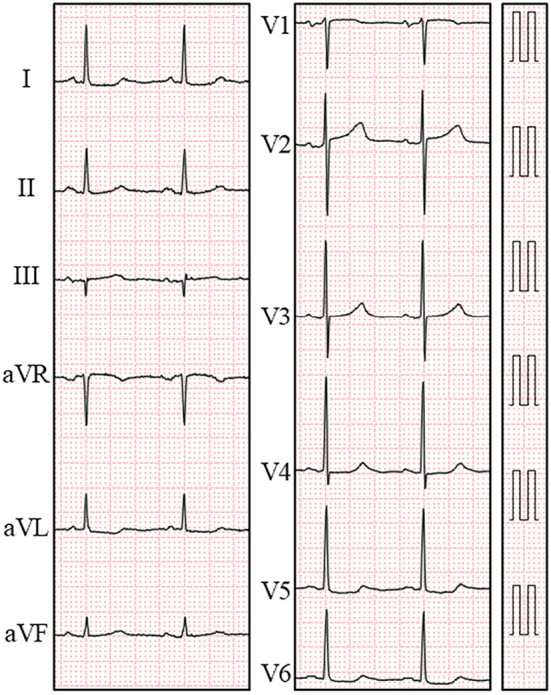
Electrocardiogram on admission. An electrocardiogram showed a normal sinus rhythm with ST-segment depression in leads V4-V6.
Figure 3.

Transthoracic echocardiographic findings (1): Left atrial dimension and mitral regurgitation. The left atrial dimension decreased after prednisolone (PSL) therapy along with the elimination of paroxysmal atrial fibrillation. Mitral regurgitation improved from severe to mild.
Figure 4.

Transthoracic echocardiographic findings (2): Transmitral flow pattern. A significant improvement in the transmitral flow pattern was observed following the administration of prednisolone (PSL).
Figure 5.

Clinical course during admission. The levels of troponin I and C-reactive protein improved promptly following the administration of glucocorticoid therapy. The episodes of paroxysmal atrial fibrillation disappeared after day 5 of glucocorticoid therapy.
During the patient’s stay in intensive care, her chest pain gradually improved after admission. However, she had worsening articular pain, relative hypotension suggesting a decreased systemic vascular resistance, and increased pleural and pericardial effusions (Fig. 1). Accordingly, pleuritis and pericarditis was considered as the cause of her initial chest pain on admission. Furthermore, rheumatoid nodules, skin ulcers, pleural and pericardial effusions, possible myocardial involvement suggested by elevated cardiac enzymes, and additional laboratory findings (Table 2), including a positive finding for immune complex testing, led us to diagnose rheumatoid vasculitis (3). On day 9, oral prednisolone (30 mg/day) was administered. Her arthritis rapidly improved after the administration of prednisolone. The patient’s inflammatory markers and elevated cardiac enzyme levels decreased over time (Fig. 5). An electrocardiogram on day 20 showed an improvement of ST-segment depression (Fig. 6), and a shortening of P-wave dispersion and the QT-interval were confirmed (Fig. 6). Echocardiography demonstrated an improvement of mitral regurgitation (Fig. 3) and the transmitral flow pattern (Fig. 4), combined with a reduction in the left atrial size (Fig. 3). Although all antiarrhythmic drugs had been discontinued before the administration of prednisolone, paroxysmal atrial fibrillation and sinus arrest disappeared after day 14 (Fig. 5). Accordingly, the patient’s congestive heart failure improved (Fig. 1) even though all diuretics had been discontinued as well. Computed tomography on day 15 revealed the disappearance of pleural and pericardial effusions (Fig. 1). She was discharged on day 31 with the prednisolone dose reduced to 25 mg/day.
Table 2.
Additional Laboratory Data.
| Complement C3 | 98 | mg/dL (73-138) | |
| Complement C4 | 31.4 | mg/dL (11-31) | |
| Erythrocyte sedimentation rate | 126 | mm (0-15) | |
| Anti-nuclear antibody | 1:1,280 | (Centromere pattern) (0-39.99) | |
| Rheumatoid factor | 24 | IU/mL (0-14.99) | |
| Anti-cyclic citrullinated peptide antibodies | 1,412 | U/mL (0-4.499) | |
| Monoclonal rheumatoid factor binding immune complex | 4.4 | ug/mL (0-4.19) | |
| Soluble interleukin-2 receptor | 4,515 | U/mL (121-613) | |
| Proteinase 3 antineutrophil cytoplasmic autoantibody | 0.2 | U/mL (0-3.49) | |
| Myeloperoxidase antineutrophil cytoplasmic autoantibody | <0.1 | U/mL (0-3.49) |
Numbers in parentheses indicate normal range.
Figure 6.

Electrocardiographic changes in P-wave dispersion and the QT-interval. P-wave dispersion improved from 54 ms to 30 ms, and the corrected QT-interval decreased from 440 ms to 418 ms after prednisolone (PSL) therapy.
Discussion
We herein described a case demonstrating the transient exacerbation of antiarrhythmic drug-resistant paroxysmal atrial fibrillation, sick sinus syndrome, and mitral regurgitation, associated with acute systemic inflammation secondary to rheumatoid vasculitis.
Systematic autoimmune disease, including rheumatoid arthritis, is a risk factor for atrial fibrillation. Inflammatory cytokines, particularly tumor necrosis factor-α, interleukin-2, interleukin-6, and interleukin-8, play a key pathophysiological role in atrial fibrillation by promoting both structural and electrical atrial remodeling. Several mechanisms, including atrial fibroblast activation, gap junction impairment via changes in connexins, and intracellular calcium-handling abnormalities, have been demonstrated (1). In addition to humoral factors, infiltration of inflammatory cells, including CD45+ cells, CD3+ T lymphocytes, and CD68+ macrophages, into the endomyocardium has been observed in patients with atrial fibrillation. These cellular factors may play a role in the pathogenesis of atrial fibrillation (4). The resultant fibrosis in the atrial myocardium and conduction delay can act as an arrhythmogenic substrate, and premature atrial contractions induced by delayed afterdepolarization can serve as an arrhythmogenic trigger (1). Because of elevated cardiac enzyme levels in the present case, it seemed rational to hypothesize the existence of atrial inflammation. However, whether atrial fibrillation in the general population is the cause or consequence of inflammation remains unclear based on the available evidence. It is likely that both statements are true (2). Although the effect of anti-inflammatory therapy on prevention and treatment of atrial fibrillation has not been confirmed, the suppression of inflammation led to the complete remission of paroxysmal atrial fibrillation in the present case. Furthermore, we observed structural (left atrial dimension), functional (transmitral flow), and electrical (P-wave dispersion) reverse remodeling of the atrium (1) in response to prednisolone therapy. All of these findings and the clinical course indicated that inflammation was the primary cause of atrial fibrillation in the present case. In the present case, however, we could not perform any imaging modalities to directly prove inflammation, including 18F isotope-labeled positron emission tomography, because her general status was poor and therefore she was deemed to not be stable enough to undergo such diagnostic procedures.
Systemic autoimmune disease may also cause mitral valve deterioration leading to mitral regurgitation (5). Although no structural deterioration of the mitral valve was observed in this case, mitral regurgitation improved after the administration of prednisolone, accompanied by the remission of paroxysmal atrial fibrillation. Based on the findings of a serial echocardiographic evaluation, the primary cause of exacerbation of mitral regurgitation in the present case was considered to be insufficient leaflet coaptation due to an enlargement of the mitral annulus in association with frequent episodes of paroxysmal atrial fibrillation. This led to a malignant cycle in which a worsening of mitral regurgitation increased the left atrial load and provoked atrial fibrillation. The frequent episodes of atrial fibrillation induced an enlargement of the left atrium, leading to a worsening of mitral regurgitation. This cycle was successfully treated with a single administration of prednisolone, presumably by suppressing atrial inflammation, leading to a reduction of the left atrial size and mitral annular diameter. A drastic improvement in the transmitral flow (Fig. 4), furthermore, indicated possible improvement in left ventricular diastolic properties in addition to left atrial function, thereby suggesting ventricular involvement during active inflammation. The possibility of ventricular involvement was supported by an improvement in the QT-interval (1) during the clinical course (Fig. 6).
We experienced a case of antiarrhythmic agent-resistant, but glucocorticoid-sensitive paroxysmal atrial fibrillation, sick sinus syndrome, and mitral regurgitation due to rheumatoid vasculitis. The standard treatment for paroxysmal atrial fibrillation includes antiarrhythmic drugs or radiofrequency catheter ablation (6). In the present case, radiofrequency catheter ablation for drug-resistant paroxysmal atrial fibrillation with congestive heart failure, as well as pacemaker implantation for symptomatic sick sinus syndrome, was avoided owing to the administration of glucocorticoid therapy. This case highlights the need to focus on the underlying disease in cases of atrial fibrillation associated with systemic autoimmune disease.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Lazzerini PE, Capecchi PL, Laghi-Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J 38: 1717-1727, 2017. [DOI] [PubMed] [Google Scholar]
- 2. Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol 60: 2263-2270, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Japan Intractable Disease Information Center. [Malignant Rheumatoid Vasculitis (designated intractable disease 46)] [Internet]. [cited 2018 May 15]. Available from: http://www.nanbyou.or.jp/entry/205(in Japanese)
- 4. Liu L, Lee J, Fu G, et al. Activation of peripheral blood CD 3(+) T-lymphocytes in patients with atrial fibrillation. Int Heart J 53: 221-224, 2012. [DOI] [PubMed] [Google Scholar]
- 5. Sugiura A, Funabashi N, Ozawa K, Kobayashi Y. Immunological and inflammatory processes in systemic autoimmune disease may not only cause pericardium inflammation, but may also cause mitral valve deterioration and left ventricular wall thickening. Int J Cardiol 215: 466-471, 2016. [DOI] [PubMed] [Google Scholar]
- 6. Kirchhof P, Benussi S, Kotecha D, et al. ESC Scientific Document Group. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J 37: 2893-2962, 2016. [DOI] [PubMed] [Google Scholar]