

Immunogenicity is a major challenge in drug development and patient care. Currently, most efforts are dedicated to the elimination of the unwanted immune responses through T‐cell epitope prediction and protein engineering. However, because it is unlikely that this approach will lead to complete eradication of immunogenicity, we propose that quantitative systems pharmacology models should be developed to predict and manage immunogenicity. The potential impact of such a mechanistic model‐based approach is precedented by applications of physiologically‐based pharmacokinetics.
Immunogenicity (IG) is defined as the propensity of the therapeutic protein to generate immune responses to itself and to related proteins or to induce certain immunologically related adverse clinical events.1, 2 In a recent US Food and Drug Administration review of 121 approved biological products, 89% of the products had reported IG, and in 49% of the cases this impacted its efficacy.3
Currently, IG is mostly tackled pre‐emptively by bioinformatics and in vitro analysis of protein sequence to prioritize compounds with a low risk of generating an immune response or alter compound sequence by protein engineering before it is tested in the clinic. The most frequently used strategy is to predict peptides that bind strongly to major histocompatibility (MHC) II receptors and subsequently select or engineer protein sequences, particularly those of nonhuman origin, in such a way as to avoid peptide motifs that will bind strongly to MHC II. In our view, this strategy is unlikely to completely eradicate antidrug antibody (ADA) responses. For example, for monoclonal antibodies, the target binding sites are one likely source of T‐cell epitopes. However, any engineering within this region could affect target binding or other aspects of developability and a strategy based on “engineering out” all potential epitopes would frequently lead to the rejection of potentially valuable compounds. In addition, the consideration of T‐cell epitope content alone does not take into account a number of other important factors related to the drug product, the patients, or the route of administration. For example, in combination therapies, the mechanism of action of one drug could influence the immune system or the population variability of immune system components in a way that influences the immune response to a second drug. Another scenario could be that a particular T‐cell epitope might not be strong enough to initiate a T‐cell‐mediated immune response in a healthy volunteer, but could be sufficient for a response to be initiated in a subject with immune dysfunction disease. Also, as the immune status of a patient or comedications change, a drug that had not appeared immunogenic for many years of treatment could begin to induce an immune response. Moreover, a marketed drug may exhibit IG for the first time in a new and sensitive target population, such as patients with an autoimmune disease or children. We believe that it is very unlikely that IG can be completely eradicated by targeting just one process (MHC II binding) in the complex cascade of events that culminates in an unwanted immune response.
Numerous approved drugs on the market benefit patients despite inducing the development of ADAs in a significant number of individuals3. In these cases, IG is usually managed in an empirical manner either by changes of dosing regimens or cotherapy with immune‐suppressive drugs.
A major limitation of current bioinformatic strategies is that these only calculate a static risk score rather than a time‐dependent profile that could provide insights into whether and to what extent IG impacts pharmacokinetics (PK), pharmacodynamics, or both. They do not take into account concurrent medications, disease state, or other patient characteristics, such as age, gender, body weight, and other physiological parameters. Therefore, bioinformatic approaches provide a good basis for screening and optimizing compounds, but they cannot be used to manage IG once a protein therapeutic has entered human trials. We argue that to more effectively address the major challenges posed by IG, quantitative systems pharmacology (QSP) models need to be developed to complement the bioinformatics toolbox. A QSP approach may provide the basis for a quantitative framework to manage and predict IG at all stages of drug development and clinical care. It could be argued that this concept bears many similarities to the way physiologically‐based PK (PBPK) modeling has impacted the issue of drug–drug interactions (DDIs) in small‐molecule development.
PBPK modeling is a bottom‐up, mechanistic modeling approach routinely used in drug discovery, development, and regulatory submissions.4 Detailed mechanistic models of drug absorption, distribution, metabolism, and excretion are built based on physiological knowledge on tissue volumes, blood and lymph flows, and metabolic enzyme and transport kinetics. Parameters are adopted from literature or in vitro assays rather than inferred empirically from data. In a typical scenario, a PBPK model is used to simulate a clinical trial, where virtual subjects are randomly generated using distributions of physiological parameters. Genetic background is taken into account through allele frequencies of gene‐encoding enzymes and transporters. Mechanistic models capturing fundamental processes underpinning PK are capable of substantial extrapolation outside of a particular clinical data set. The most frequent application of PBPK is the prediction of DDIs and the confidence in this approach is such that regulators accept simulations as a substitute for clinical trials and as the basis for label statements.4 Thus, although DDIs still cannot be “engineered out” completely, they can be predicted and managed effectively through virtual trial simulation using models with sufficient mechanistic detail.
We propose that a QSP model integrating biologics PBPK and mechanistic models of immune response can be used to inform management of IG in a similar way as PBPK of small molecules informs DDI management and regulatory interaction. Mechanistic modeling of the immune system dates back to the 1970s, and there is a legacy of models reflecting the growing understanding of biology. Chen et al.5, 6 published the first QSP model of IG, integrating a mechanistic model of immune response, with two‐compartment PK. In 2017, we established a QSP IG Consortium to create a comprehensive mechanistic model of IG integrated in a PBPK context and implemented in software of regulatory submission quality.
The IG Simulator7 (Figure 1) integrates literature knowledge with consortium‐member data. The mechanistic model is composed of two parts representing immune response and biologics PBPK. The PBPK is simulated by the Simcyp simulator (Certara UK, Sheffield, UK), whereas the mechanistic model of the immune system is implemented in a bespoke biological process map interface. Both models are integrated through common variables, which are compound concentrations in physiological compartments. The QSP model is compiled to one ordinary differential equation (ODE) system, which includes all feedback between PBPK and immune response and is submitted to a virtual trial simulation with the Simcyp‐correlated Monte Carlo algorithm.8 Distributions of physiological parameters in the population of interest are used to instantiate the model with randomly generated virtual subject parameters and subsequently run the simulation with the dosing regimen specified in the trial protocol. A population of virtual subjects is simulated, allowing the prediction of between‐subject variability in a clinical trial. In addition to population parameters already included in Simcyp, the IG Simulator uses human leukocyte antigen (HLA) allele frequencies and variability in the immune system as a function of age or disease will soon be included. Once the virtual trial simulation is completed, the time profiles of all model variables in all individual subjects are available for analysis.
Figure 1.
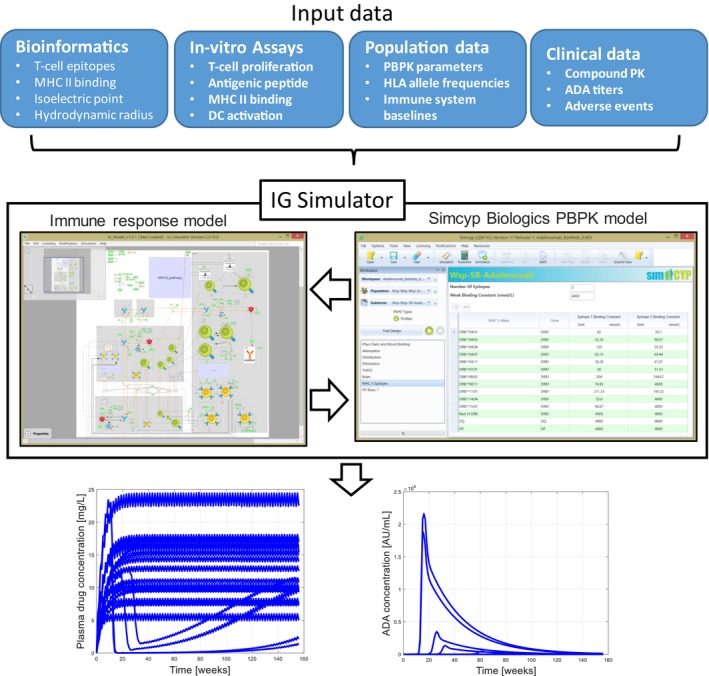
Overview of IG Simulator. The quantitative systems pharmacology model includes a mechanistic model of immune response and Simcyp Biologics PBPK. The model has sufficient mechanistic granularity to use MHC II binding constants predicted by bioinformatics as inputs. The model can use binding constants determined in vitro as well as the results of other in vitro assays, such as T‐cell proliferation. Population data for virtual clinical trial simulation include frequencies of HLA genes, immune system baselines, and physiological parameters in target population. When the model is applied to extrapolation from first‐in‐human data or extrapolation between different clinical populations, clinical data on PK and ADA titers are used. The mechanistic model integrates diverse inputs and simulates a virtual trial—a population of individuals subject to a specific dosing regimen. The figure shows the example individual time profiles for drug and ADA, which can be used to determine ADA‐positive subjects using the same criteria as used in clinical trials and regulatory submissions. Individual time profiles of other model variables are available as well, facilitating the investigation of biomarkers. The quantitative systems pharmacology model can be expanded in the future to incorporate additional assays. The model capable of integrating data from a wide array of experimental approaches will motivate the inclusion of additional biomarkers into clinical trials and enable extrapolation from these biomarkers to clinical outcomes. ADA, antidrug antibody; DC, dendritic cell; HLA, human leukocyte antigen; IG, immunogenicity; MHC, major histocompatibility; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetics.
The QSP model used in the IG Simulator has sufficient mechanistic detail to integrate diverse inputs, including bioinformatics predictions of MHC II binding to antigenic peptides, in vitro cell‐based assays and clinical measurements of compound concentrations, and ADA titers. Moreover, a detailed simulation of complex immune system interactions allows for the incorporation of important sources of patient variability. This can be done through the generation of virtual patients with cell numbers following population distributions of immune‐system baselines corresponding to different age or disease groups. Thus, the mechanistic modeling approach expands rather than replaces T‐cell epitope prediction. Predicted binding constants and HLA allele frequencies are used as parameters of the model, and thus MHC II binding is considered as an important factor in the multifaceted biological process rather than the sole determinant of IG.
An example of a virtual trial simulation with the IG Simulator7 is shown in Figure 2. We simulated a clinical trial of adalimumab to replicate a study described by Bartelds et al.9 Because the time profiles of both the compound and ADA are simulated, the virtual subjects can be classified into those who do and do not exhibit IG using the same criteria that were applied in the clinic. Figure 2 shows PK profiles for three groups of subjects and ADA incidence at each time point determined by the criteria used in the clinical protocol.9 The simulation describes the clinical data well, when HLA allele frequencies for a European population are used in accordance with subjects participating in the trial. When HLA data for a North American population are used, the results are in lesser concordance with the clinical study (data not shown). We also note that the simulation outputs all other model variables, such as the dynamics of immune‐cell populations, which are not routinely measured in clinical trials but may be highlighted for consideration as valuable biomarkers. This example virtual trial simulation (Figure 2) demonstrates that the IG Simulator is capable of simulating clinical population of interest, with accuracy sufficient to inform IG management. In a typical IG management scenario, the simulation would be used to prioritize compound doses and dosing frequencies, where the impact of ADAs on PK is minimal. Although ADA synthesis would not be eliminated, the exposure would remain within the therapeutic window. For example, Bartelds et al.9 reported that six patients lost antibodies to adalimumab after dose escalation. Studies where dosing regimen changes were used to overcome immunogenicity of anti‐tumor necrosis factor therapeutics are reviewed elsewhere.10
Figure 2.
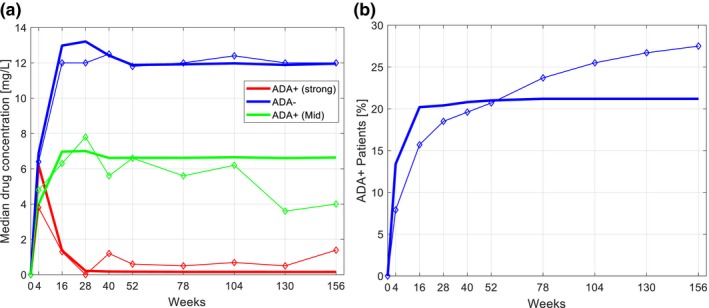
Virtual trial simulation of adalimumab. The IG Simulator was used to simulate clinical trial protocol of Bartelds et al. 9 applied to a European population. The 500 virtual subjects were dosed 40 mg of adalimumab every 2 weeks. Both adalimumab and ADA concentration time profiles were simulated and analyzed using the same criteria as in a clinical trial. The subject was considered to be ADA+ if ADA concentration was > 12 AU/mL (AU = 12 ng) on at least one occasion in combination with plasma adalimumab levels of < 5 mg/L. The ADA+ subjects were further split into low‐titer and high‐titer groups, with the high‐titer group defined by ADA concentration > 100 AU/mL: (a) compound pharmacokinetics; (b) ADA incidence. Simulated time profiles (lines) were analyzed and plotted only at the time points where clinical data (diamonds) were collected. The model reproduces clinical data with sufficient accuracy to inform IG management through the in silico testing of different dosing regimes in different populations. ADA, antidrug antibody; IG, immunogenicity; PK, pharmacokinetics.
In conclusion, we propose that an integrated QSP approach can provide the basis for the model‐informed management of IG. Different target populations and patient cohorts can be considered to guide treatment optimization in a rational manner. “Virtual‐twin” subjects can be created using HLA genotype, ex vivo assays, and peripheral blood flow cytometry data for actual individual patients, thus enabling a personalized‐medicine approach to IG. We believe that the mechanistic, QSP model‐informed management of IG is a natural extension of the well‐established PBPK approach and has the potential to also become a prominent feature of drug development and regulatory science.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
References
- 1. Biopharma Dive . The Future of Biologics Development: Insights to Help Biopharma Companies Win the Race to Market (Industry Dive, Washington, DC, 2018). [Google Scholar]
- 2. US Department of Health and Human Services, Food and Drug Administration . Guidance for Industry. Immunogenicity Assessment for Therapeutic Protein Products (US Department of Health and Human Services, Food and Drug Administration, Silver Spring, MD, 2014). [Google Scholar]
- 3. Wang, Y.M. , Wang, J. , Hon, Y.Y. , Zhou, L. , Fang, L. & Ahn, H.Y. Evaluating and reporting the immunogenicity impacts for biological products—a clinical pharmacology perspective. AAPS J. 18, 395–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shebley, M. et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. 104, 88–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen, X. , Hickling, T.P. & Vicini, P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 1‐theoretical model. CPT Pharmacometrics Syst. Pharmacol. 3, e133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen, X. , Hickling, T.P. & Vicini, P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 2‐model applications. CPT Pharmacometrics Syst. Pharmacol. 3, e134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Graaf, P.H. & Kierzek, A.M. The development of an Immunogenicity Simulator through a quantitative systems pharmacology consortium approach. Workshop on Predictive Immunogenicity for Better Clinical Outcomes, Silver Spring, MD, October 3, 2018. [Google Scholar]
- 8. Jamei, M. , Dickinson, G.L. & Rostami‐Hodjegan, A. A framework for assessing interindividual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: A tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab. Pharmacokinet. 24, 53–75 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Bartelds, G.M. et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long‐term follow‐up. JAMA. 305, 1460–1468 (2011). [DOI] [PubMed] [Google Scholar]
- 10. Kothari, M.M. , Nguyen, D.L. & Nimisha, K.P. Strategies for overcoming anti‐tumor necrosis factor drug antibodies in inflammatory bowel disease: Case series and review of literature. World J. Gastrointest. Pharmacol. Ther. 8, 155–161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]