

Abstract
The comparative performances of physiologically‐based pharmacokinetic (PBPK) modeling and allometric scaling for predicting the pharmacokinetics (PKs) of large molecules in pediatrics are unknown. Therefore, both methods were evaluated for accuracy in translating knowledge of infliximab PKs from adults to children. PBPK modeling was performed using the base model for large molecules in PK‐Sim version 7.4 with modifications in Mobi. Eight population PK models from literature were reconstructed and scaled by allometry to pediatrics. Evaluation data included seven pediatric studies (~4–18 years). Both methods performed comparably with 66.7% and 68.6% of model‐predicted concentrations falling within twofold of the observed concentrations for PBPK modeling and allometry, respectively. Considerable variability was noted among the allometric models. Therefore, pediatric clinical trial planning would benefit from using approaches that require predictions depending on the specific question i.e., PBPK modeling and allometry.
Physiologically‐based pharmacokinetic (PBPK) modeling and allometric scaling are the two most common methods for translating knowledge of adult pharmacokinetics (PKs) to the pediatric space for the planning of pediatric clinical trials.1 Although drug developers and regulatory agencies alike maintain high confidence in both methods for small molecule drugs, little is known about the performance and utility of either method for large molecule drugs.2 Here, we evaluate the two approaches for the prediction of large molecule PKs in pediatric patients with infliximab as a working example, because plenty of adult and pediatric data are available in published literature for the exercise.
The practice of allometry applies an empiric and inherently simple method to perform body‐weight‐based scaling of adult PK parameters that have been derived from population pharmacokinetic (PopPK) studies (i.e., clearance and volume of distribution).3 The accuracies of allometric models have been demonstrated with numerous small molecule drugs for children older than 2 years of age,4, 5 when distribution and clearance processes have achieved adult performance. Below this threshold, empirical adjustments for maturation and ontogeny are required and the resulting functions are not translatable between molecules.6, 7 In keeping with this observation, our evaluation of allometric scaling in the large molecule space first includes studies with children older than 4 years of age, when the mechanisms governing PKs are thought to have reached full maturity.8
PBPK models have long been recognized for offering translational utility by maintaining mechanistic approaches to distribution and clearance. They enable PK predictions in unique populations or unknown exposure scenarios based on known physiological and anatomic characteristics but are considerably more complex to develop. PBPK models must first be calibrated, informed, and evaluated with PK data in adults before their predictions can become reliable in the pediatric space. The practice of PBPK modeling to support pediatric clinical trial planning in the context of small molecule drugs has achieved critical mass with regulatory support from both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA).9 In contrast to allometric models, PBPK models can use physiologic knowledge of growth and maturation to account for the ontogeny of key distribution and clearance processes in young children, even neonates and preterm infants.10
The same confidence must yet be earned for large molecule drugs. The mechanisms governing the PKs of large molecules are not at all similar to those of small molecules.11 Platform PBPK models for large molecules in adults have emerged in the last 7 years,12, 13 and, to date, only one minimal PBPK modeling effort has been made to characterize PKs in children.14 We continue the exploration with a whole‐body approach to the PKs of infliximab in children because a large amount of pediatric PK data are available for this purpose in published literature. The assessment of pediatric PBPK modeling in older children and adolescents must be completed prior to considering children <4 years of age for proper evaluation of size‐dependent scaling alone without additionally considering age‐dependent factors, namely the maturation and ontogeny of key distribution and clearance mechanisms.8
Infliximab is a chimeric monoclonal antibody directed against tumor necrosis factor alpha “(TNF)” that is used to treat inflammation associated with many autoimmune conditions, such as inflammatory bowel disease, rheumatoid arthritis, psoriasis, ankylosing spondylitis, and Kawasaki disease. It is most often dosed at 5 mg/kg by a 2‐hour intravenous infusion at 0, 2, and 6 weeks for induction of remission and then every 8 weeks thereafter for maintenance of remission, although standard regimens may vary slightly among disease states and clinical treatment centers.
The drug exhibits complex but generally linear PKs due to its large size, antibody‐based design, and low target burden. Two‐compartment models are often used to describe its time course in the adult body. The drug possesses an extensive plasma residence time due to poor permeability across blood vessel walls, with a mean plasma half‐life of 9.5 days. High affinity interactions with the neonatal Fc receptor (FcRn) in endothelial, epithelial, and hematopoietic cells further protect the drug from intracellular degradation. Infliximab neutralizes soluble TNF in most extracellular fluid domains, but most importantly in the interstitial spaces of inflamed organs. From the interstitial space, the lymphatic system recirculates infliximab via the thoracic duct to the venous blood, where the distribution cycle can begin again. Over time, and in part due to its makeup with portions of murine protein, the body may mount an immune response against infliximab. At sufficient titers, antidrug antibodies (ADAs) can render infliximab undetectable in plasma.
Monoclonal antibodies are a rapidly expanding drug class, and pediatric investigations are well underway. The FDA has recently highlighted the gaps in modeling and simulation efforts to support pediatric drug approvals.2 By completing this work, we aim to assess the confidence with which PBPK modeling and/or allometric scaling can be applied in the context of pediatric drug development and clinical trial planning.
Methods
Software
PBPK models were built and evaluated using the base model for large molecule drugs in PK‐Sim and Mobi version 7.4 (www.open‐systems‐pharmacology.org)13 with minor modifications. PK data in scientific literature were digitized using Plot Digitizer version 2.6.8 by Joseph Huwaldt. MATLAB R2018b and the Intiquian toolbox (IQMTools version 1.2.2.2) by Henning Schmidt were used for reconstruction of PopPK models, allometric scaling, and graphical presentation of results.
PK data for evaluation of PBPK modeling and allometric scaling
After a comprehensive literature search, seven pediatric PK trials were located and the data were digitized for evaluation of the pediatric PK predictions between PBPK modeling and allometric scaling15, 16, 17, 18, 19, 20, 21, 22 (Table 1). The data sets encompass children with inflammatory bowel disease or juvenile idiopathic arthritis almost exclusively between 4 and 18 years of age with infliximab doses ranging from 1 to 10 mg/kg i.v.
Table 1.
Data sets used for evaluation of pediatric PK predictions
| Study | Dose, mg/kg i.v. | N | Age, years | Weight, kg |
|---|---|---|---|---|
| Inflammatory bowel disease | ||||
| Candon et al.15 | Induction with 5 | 20 | 10.5 [0.5–15] | NA |
| Hyams et al.16, 17 | Induction with 5 then 5 q8w and q12w with dose escalation to 10 | 112 | 13.3 ± 2.5 | 43.8 ± 14.6 |
| Hamalainen et al.18 | Induction with 5 then 5 q8w | 37 | 14 [5.6–18] | 43.5 [19.6–67.1] |
| Baldassano et al.19 | 1, 5, and 10 single dose | 21 | 15.1 [8–17] | 49.1 |
| Singh et al.20 | Induction with 5 then 5 q8w | 58 | 11.4 [6.6–18.4] | NA |
| Adedokun et al.21 | Induction with 5 then 5 q8w and q12w with dose escalation | 60 | 14.5 [6–17] | 50.8 (36.3–59.4) |
| Juvenile idiopathic arthritis | ||||
| Ruperto et al.22 | Induction with 3 or 6 then 3 or 6 q8w | 122 | 11.2 [4–18] | NA |
Data presented as mean or median and [range], (interquartile range), or ± SD. “Induction” refers to intensive dosing at weeks 0, 2, and 6 before regular maintenance dosing.
NA, not applicable; PK, pharmacokinetic.
PBPK modeling
Virtual individuals
PBPK modeling enables a mechanistic representation of drug disposition in virtual individuals with known anatomic and physiological properties. Virtual adults and children for the analysis were generated using PK‐Sim version 7.4. Algorithms for the generation of virtual individuals in PK‐Sim are reported elsewhere.23 In the PBPK modeling process, one virtual individual (whether adult or child) was created to represent the mean individual in each PK study. Each virtual individual had the same age, height, weight, sex, and disease state as the mean individual in the corresponding real‐world study along with all associated anatomic and physiological characteristics. Maturation was assumed not to have an impact on these characteristics in children older than 4 years of age.8
Adult PBPK model development
In agreement with pediatric PBPK model development workflows for small molecules, an adult PBPK model was constructed for infliximab first to gain confidence in the global parameterization before scaling the model to pediatric individuals.
Model structure
All PBPK modeling was carried out in PK‐Sim and Mobi version 7.4. The base model for large molecule drugs in the software package has been described previously.13 Fifteen organs were included in the model structure to represent a virtual human, and each organ compartment was further divided into plasma, endosomal, interstitial, and cellular subcompartments. The base model featured two‐pore extravasation, endothelial uptake, salvage by FcRn, and lymphatic recycling. The model structure was adapted in Mobi version 7.4 to include target‐mediated interactions with TNF and ADAs. Model parameters as inputted into PK‐Sim and Mobi are presented in Table 2.
Table 2.
Model parameters
| Parameter | Final value [Ref.] | |
|---|---|---|
| Infliximab | ||
| Molecular weight | 149,100 kDa | |
| Hydrodynamic radius | 5.34 nm [Ref. 13] | |
| Rate of uptake into endosomal space () | 0.35 min −1 | |
| Dissociation constant for FcRn binding | 727 nM [Ref. 45] | |
| Dissociation constant for TNF binding | 30 pM [Ref. 46] | |
| Dissociation constant for ADA binding | 500 pM [Ref. 47] | |
| TNF | ||
| TNF maximum plasma concentrationa | 0.5 pM [Ref. 25, 26, 37] | |
| TNF factor for autoimmune disease | 2 [Ref. 25, 26, 27, 28] | |
| TNF degradation rate | 0.0231 min−1 [Ref. 48, 49] | |
| mAb‐TNF complex degradation rate | 0.0231 min−1 | |
| ADA | ||
| Duration of IgM production | 30.7 days [Ref. 36] | |
| Lag time prior to IgG production | 38.8 days [Ref. 36] | |
| IgM maximum plasma concentrationa |
|
|
|
IgG maximum plasma concentration
a
|
136 nM | |
| ADA (IgM) degradation rate | 0.1 day−1 [Ref. 36] | |
| ADA (IgG) degradation rate in healthy | 0.014 day−1 [Ref. 36] | |
| ADA (IgG) degradation rate in autoimmune | 0.008 day−1 [Ref. 36] | |
| mAb‐ADA complex degradation rate | 0.48 day−1 [Ref. 50] | |
| Inflamed organ | ||
| Inflammation factor for inflamed pores | 1.5 [Ref. 31, 32, 33] | |
| TNF factor for inflamed organ | 3 [Ref. 29, 30] | |
Italicized values were mathematically optimized to their final value in the model building process.
ADA, antidrug antibody; FcRn, neonatal Fc receptor; mAb, monoclonal antibody; TNF, tumor necrosis factor.
Zero order synthesis rates were calculated by multiplying the degradation rate by the maximum concentration of the molecule.
Tumor necrosis factor alpha
Infliximab binds to TNF in circulation with high affinity, and the complex is later eliminated by the immune system.24 In the PBPK model, TNF was present in all plasma and interstitial spaces with a relative expression of 1 across all organs. The natural synthesis () and turnover () of TNF were represented with zero order and first order rate constants, respectively. TNF molecules were noncirculating and stationary within each subcompartment. Binding of infliximab to TNF caused the formation of a complex, which was eliminated from the system by a first order degradation rate ().
Inflammation
In PopPK studies, infliximab is often noted to exhibit faster clearance in individuals with severe inflammation, possibly mediated through higher TNF concentrations, vascular hyperpermeability, or cachexia. In the PBPK model, the presence of autoimmune disease was represented with a global twofold increase in TNF concentrations.25, 26, 27, 28 In inflamed organs, local TNF concentrations were further increased by a factor of 3.29, 30
Large molecule drugs abide by permeability‐rate‐limited distribution and are subject to vascular hyperpermeability in inflamed organs. To represent this effect, the pore sizes in inflamed organs were increased proportional to an inflammation factor in agreement with previous minimal PBPK models of inflammatory conditions by other studies31, 32, 33 that observed the requirement for lower vascular reflection coefficients in inflamed organs. The large intestine was inflamed in patients with inflammatory bowel disease, the bone was inflamed in patients with rheumatoid arthritis and ankylosing spondylitis, the skin was inflamed in patients with psoriasis, and the lungs were inflamed in patients with non‐small cell lung cancer.
Immunogenicity
Immunogenicity is a key determinant of infliximab elimination. Results are conflicting due to inconsistency in ADA assays, although up to 60% of patients may express an immunogenic response against infliximab over the course of 1 year of therapy.34 ADA molecules (IgM and IgG) bind to infliximab to form a complex, neutralizing its therapeutic potential and eventually resulting in degradation. In the PBPK model, ADA molecules (IgM and IgG) were synthesized, released, and contained within the plasma of venous and arterial blood, similar to the assumptions made by Chen et al.35 Mean ADA synthesis and degradation rates in the adult population were parameterized according to Ren et al.,36 who quantified the typical immune response after first‐dose exposure to four therapeutic proteins with a PopPK modeling approach. In the absence of additional data, the zero order molar synthesis rates of IgM and IgG were assumed equal (). The typical ADA profile featured an initial period of IgM synthesis, a lag phase, and finally a sustained IgG response.36 The group noted differences in IgG degradation rates between healthy and autoimmune individuals, which were implemented in the model.36 Mechanistically, binding of infliximab to IgM or IgG caused the formation of a complex, which was eliminated from the system by a first order degradation rate ().
Optimization
Two parameters in the adult PBPK model were uncertain and required optimization: the rate of uptake into endosomal space () and the zero order molar synthesis rate of ADA molecules against infliximab (), which was mediated via the maximum concentration of IgG parameter (). Optimization was carried out in Mobi using a Monte‐Carlo approach for exploring the parameter space. The parameters were optimized separately in two steps. First, was optimized to ADA‐negative PK data available in four studies (Table S1 ). Following this step, was optimized to PK data available from 20 studies in healthy and diseased individuals (Table S1 ). Therefore, represents the mean immunogenic response across the general adult population, including both ADA‐positive and ADA‐negative individuals.
Model accuracy following optimization was assessed statistically by the squared Pearson correlation coefficient (Pearson R 2), the absolute average fold error (AAFE) across all data points, the root mean squared error (RMSE), and the percent of model‐predicted concentrations falling within twofold of the corresponding observed concentrations. An external evaluation of the final adult model was not performed because the purpose of the model was to extrapolate knowledge of adult PKs to pediatrics, and this workflow maximizes learning from adult data sets.
Pediatric extrapolation
After establishing confidence in the performance of the adult PBPK model, it was then translated to the pediatric space. In simple terms, the virtual adult individual was replaced with a pediatric individual with all associated anatomic and physiological parameters to explore the a priori PK predictions of the model in children. Physiological parameters related to disease pathology and immunogenicity were kept constant between adults and children, with some literature to inform this decision.15, 37, 38 Observed PK data from seven pediatric clinical trials were available for evaluation of the predictions15, 16, 17, 18, 19, 20, 21, 22 (Table 1). As above, prediction accuracy was assessed by four metrics: Pearson R 2, AAFE, RMSE, and the percent of model‐predicted concentrations falling within twofold of the corresponding observed concentrations in children.
Allometric scaling
Allometric scaling offers a simple and empirical approach to translating the statistical knowledge gained from an adult PopPK study to a pediatric population by acknowledging differences in body weight. Many adult PopPK models for infliximab exist in published literature. In a typical drug development case, only one model would exist, built after collation of PK data from two or three small studies. For this exercise, a total of eight adult PopPK models were reconstructed from literature for application of allometric scaling (Table 3).
Table 3.
Adult infliximab PopPK studies used for allometric scaling
| Study | N | Age, years | Weight, kg | CL, mL/h | Q, mL/h | V1, L | V2, L |
|---|---|---|---|---|---|---|---|
| Inflammatory bowel disease | |||||||
| Fasanmade et al.51 | 580 | 37.5 ± 11.9 | 71.1 ± 18.3 | 15.27 | 6.09 | 3.58 | 1.29 |
| Fasanmade et al.52 | 482 | 41.2 ± 13.9 | 78.8 ± 18.4 | 16.96 | 297.5 | 3.29 | 4.13 |
| Ternant et al.53 | 33 | 33 [19–53] | 67 [44–110] | 12 | 5.4 | 2.9 | 1.9 |
| Dotan et al.54 | 54 | 35.6 [20–70] | NA | 15.8 | 5.08 | 2.37 | 1.37 |
| Aubourg et al.55 | 133 | NA | 60 [41–120] | 14 | 83 | 2.6 | 4.5 |
| Buurman et al.56 | 42 | 44 [19–80] | 75 [51–145] | 8.29 | 2.58 | 4.94 | 3.13 |
| Brandse et al.39 | 332 | 38.6 ± 13.9 | 72.3 ± 16.3 | 14.96 | 2.9 | 4.72 | 2.4 |
| Rheumatoid arthritis | |||||||
| Ternant et al.57 | 84 | 58 [27–84] | 65 | 19 | 180 | 2.3 | 3.6 |
Data presented as mean or median and [range], (interquartile range), or ± SD.
CL, clearance; NA, not applicable; PopPK, population pharmacokinetic; Q, intercompartmental clearance. V1, volume in the central compartment; V2, volume in the peripheral compartment.
Each PopPK model was constructed, scaled, and evaluated independently. First, values for clearance (), intercompartmental clearance (), volume of distribution in the central compartment (), and volume of distribution in the peripheral compartment () were extracted for the typical adult individual in each study (Table 3). The typical adult had mean or median covariate status for age, height, weight, sex, disease status, immunomodulator use, and inflammatory biomarker concentration where applicable. The four PK parameters were then scaled by allometry from the typical adult to the typical child in each of the pediatric PK studies designated for evaluation (pediatric body weights presented in Table 1). Allometric scaling was implemented as described by Tod et al.3 Mathematically,
Pediatric PK predictions were only made in the context of the same or similar disease states. Seven PopPK models developed in adults with inflammatory bowel disease were used to make pediatric PK predictions for children with inflammatory bowel disease, and one PopPK model developed in adults with rheumatoid arthritis was used to make pediatric PK predictions for children with juvenile rheumatoid arthritis. Pediatric PK profiles were simulated with the updated parameters and compared with the mean pediatric observed concentrations from literature (Table 1). Once again, the accuracy of PK profile prediction through allometric scaling was assessed by the same four statistical metrics for direct comparison to the PBPK model‐driven predictions (Pearson R 2, AAFE, RMSE, and percent of model‐predicted concentrations falling within twofold of the corresponding observed concentrations).
Results
A comprehensive PBPK model was developed using the base model for large molecule drugs in PK‐Sim and Mobi version 7.4 with modifications to feature the effects of inflammation and target‐mediated interactions with TNF and ADA molecules. The adult model was then scaled to represent children by updating all anatomic and physiological parameters accordingly. Additionally, eight two‐compartment PopPK models from literature were reconstructed and scaled by allometry to the pediatric space, according to the methods proposed by Tod et al.3 Equipped with these tools, the performances of PBPK modeling and allometric scaling for the prediction of infliximab PKs in children and adolescents were systematically evaluated.
PBPK modeling
In the first optimization step, the final value for was 0.35 min−1 (Figure S1 ). In the second optimization step, the final value for was 136 nM. The adult PBPK model well characterized the observed PK profiles from all data sets used in both optimization steps achieving a Pearson R 2 of 0.90, an AAFE of 1.16, and an RMSE of 30.9. Notably, 88.4% of the model‐predicted infliximab concentrations were within twofold of the corresponding observed concentrations. Figure 1 shows sample PK profiles in healthy subjects, ankylosing spondylitis, and rheumatoid arthritis, and a comparison of the model‐predicted vs. observed concentrations across all studies. The possible effect of inflammation on clearance was modestly underestimated for patients with inflammatory bowel disease and psoriasis, although a significant portion of this observed data was obtained from nonresponder subsets. Nonresponders have lower trough concentrations than responders, possibly due to higher TNF concentrations, ADA positivity, or severely inflamed, damaged, or leaky organs.
Figure 1.
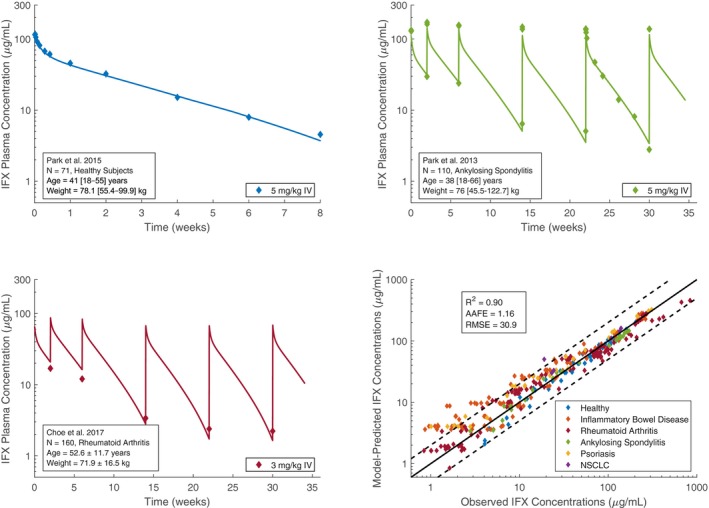
Sample infliximab pharmacokinetic profiles in healthy subjects, ankylosing spondylitis, and rheumatoid arthritis, and a comparison of model‐predicted vs. observed infliximab concentrations. References to observed data provided in Table S1 . AAFE, absolute average fold error; IFX, infliximab; NSCLC, non‐small cell lung cancer; R 2, squared Pearson correlation coefficient; RMSE, root mean squared error.
Evaluation of PBPK modeling and allometric scaling
Both methods performed similarly for predicting the mean PKs in children obtained from studies that included individuals almost exclusively between the ages of 4 and 18 years. Figure 2 displays a plot‐by‐plot comparison of allometrically derived PK profiles, PBPK profiles, and the mean pediatric observed concentrations from literature. Considerable differences were noted among the profiles derived from allometric scaling that stemmed from variable PK parameter values from the eight adult PopPK models. Visually, the pediatric PBPK model was more proficient for predicting peak concentrations after infusion and volume of distribution in the central compartment, while the allometrically‐derived models performed better for predicting trough concentrations.
Figure 2.
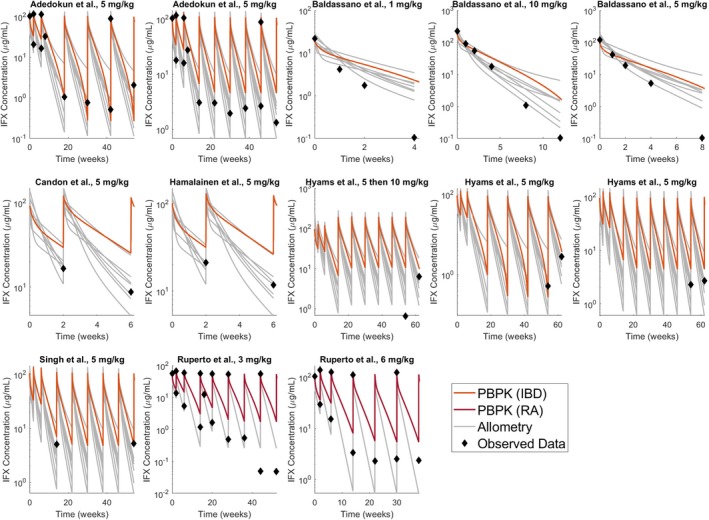
Comparison of pediatric physiologically‐based pharmacokinetic (PBPK) modeling and allometric scaling for the prediction of infliximab (IFX) pharmacokinetics in children with inflammatory bowel disease (IBD) or juvenile idiopathic arthritis; data digitized from seven pediatric studies (Table 1). RA, rheumatoid arthritis.15, 16, 17, 18, 19, 20, 21, 22
Across all allometric models, model‐predicted concentrations were within twofold of the observed concentrations 68.6% of the time, while the best allometric model39 enabled predictions within twofold of the observed concentrations 84.3% of the time (Table 4). The PBPK model performed similarly overall, achieving predictions within twofold of the observed concentrations 66.7% of the time (Table 4). No discernable correlations were detected between the sample size of a PopPK study (N) and the accuracy of its pediatric predictions. The Pearson R 2 and RMSE values identified trends in model performance but no conclusions could be made about overall model performance from these metrics.
Table 4.
Statistical evaluation of pediatric PK model performance
| Model | N | AAFE | R 2 | RMSE | Twofold error |
|---|---|---|---|---|---|
| Inflammatory bowel disease | |||||
| Fasanmade et al. 51 | 580 | 0.77 | 0.977 | 8.1 | 60.8% |
| Fasanmade et al. 52 | 482 | 1.09 | 0.964 | 9.7 | 76.5% |
| Ternant et al. 53 | 33 | 1.35 | 0.980 | 10.1 | 80.4% |
| Dotan et al. 54 | 54 | 0.60 | 0.957 | 21.4 | 51.0% |
| Aubourg et al. 55 | 133 | 1.36 | 0.973 | 8.5 | 82.4% |
| Buurman et al. 56 | 42 | 2.82 | 0.913 | 17.0 | 47.0% |
| Brandse et al. 39 | 332 | 1.18 | 0.970 | 14.0 | 84.3% |
| Rheumatoid arthritis | |||||
| Ternant et al. 57 | 84 | 0.83 | 0.987 | 20.7 | 66.7% |
| All allometric models | |||||
| 1.25 | 13.7 | 68.6% | |||
| PBPK modeling | |||||
| This study | 1.79 | 0.96 | 7.0 | 66.7% | |
AAFE, absolute average fold error; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetic; R 2, squared Pearson correlation coefficient; RMSE, root mean squared error.
Finally, Figure 3 compares the model‐predicted and observed infliximab concentrations across all pediatric studies. Low concentrations were overpredicted by both methods, although there is uncertainty in these data values with regard to the lower limits of quantification for the assays in the studies. Overall, the PBPK model predictions fell near the median of the allometric predictions for children.
Figure 3.
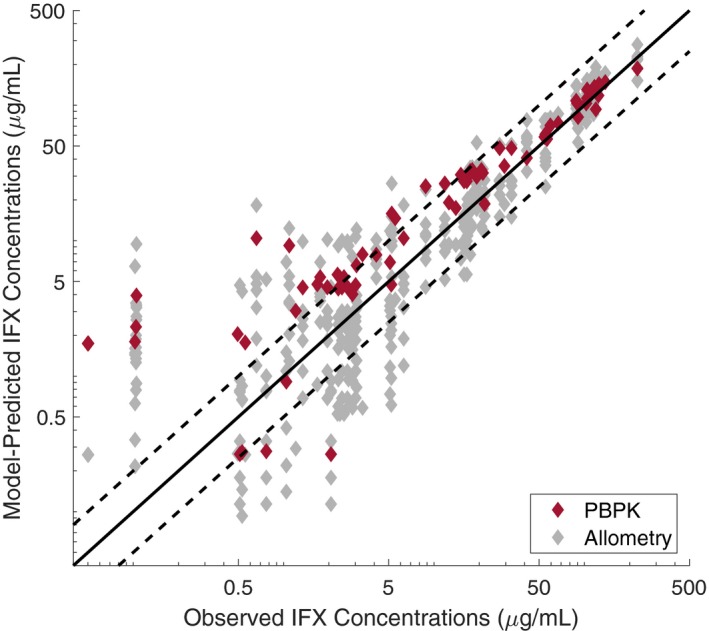
Model‐predicted vs. observed infliximab (IFX) concentrations in pediatrics. PBPK, physiologically‐based pharmacokinetic.
Discussion
The evaluations of PBPK modeling and allometric scaling for the prediction of small molecule PKs in children have been extensive.4, 5 Both methods are often reported to achieve predictions within twofold of the observed values up to 90% of the time for children older than 2 years of age. However, this evaluation has not yet been performed in the context of large molecule drugs. With infliximab as a case example, we demonstrate that PBPK modeling and allometric scaling in their current states provide similar pediatric PK predictions for children older than 4 years of age in known exposure scenarios. There is significant room for improvement in the large molecule space, as neither method in this example achieved the accuracies reported in small molecule evaluations (66.7% and 68.6% of model‐predicted concentrations falling within twofold of the observed concentrations for PBPK modeling and allometric scaling, respectively). Refinement of allometric exponents for large molecule drugs and investigations into the relevance of pediatric physiology for predicting PKs in young children8 are required to advance the predictive accuracy of these methods.
A closer inspection of model performances may offer guidance about the utility of each method for specific purposes (Figure 2). The pediatric PBPK model performed stronger than the allometrically derived models for the prediction of peak drug concentrations after infusion (Figure 3), suggesting that volume of distribution in the central compartment for children is most reliably estimated by physiological methods. Clearance in children older than 4 years of age was modestly underestimated by PBPK modeling, suggesting that there may be mild effects of maturation on anatomical and physiological parameters that govern elimination. Allometric scaling may more reliably estimate trough concentrations and clearance in known exposure scenarios. Last, allometric scaling was performed for eight different PopPK models in a small fraction of the time that was required to develop the pediatric PBPK model (~100 hours) and would be preferred when time is a constraint.
The findings from this study can be generalized to the pediatric PBPK modeling and allometric scaling of monoclonal antibodies with similar FcRn affinity and a low target burden to guide drug development and selection of first‐in‐pediatric doses. Antibodies with a high target burden often exhibit nonlinear kinetics (e.g., trastuzumab40), and there is no reliable method for scaling the target‐mediated drug disposition component of a nonlinear PopPK model by allometry, especially when disease pathology may vary between adults and children. PBPK modeling would be highly desired in this case.
Wide variability was noted in the predictions among the eight allometric models, and the accuracy among the models was independent of adult study sample size (Figure 2 and Table 3). This observation draws attention to the need for optimal sampling in the design of adult PopPK studies to increase confidence when applying allometric scaling. Without dense sampling and variable exposure scenarios, fundamental model structures cannot be determined. In the case of infliximab, estimates for PK parameters are highly variable among the eight models and are often poorly defined due to inadequate mid‐profile sampling (Table 3). Limited sampling analyses for large molecule drugs are beginning to be used to guide the sampling times for appropriate derivation of pivotal PK parameters, some of which govern curve shape.41
PBPK modeling for large molecules is gaining traction in academia, industry, and regulatory agencies. Here, we present the first evaluation of a whole‐body PBPK model for a large molecule drug in pediatrics and the first PBPK model to incorporate immunogenicity. Infliximab is one of the oldest monoclonal antibodies, and there is an abundance of PK data available for model parameterization, yet the drug has often been avoided due to uncertainty around modeling immunogenicity, which is a key driver of its elimination. The immunogenic response is a modeling challenge because ADA assays often do not quantify concentrations, only titers of ADA molecules.42 As a first step toward quantifying the response, the ADA‐negative PK data for infliximab available in literature combined with the parameterization of the time course of ADA formation in humans by Ren et al.36 have enabled the first mechanistic representation of the immunogenic response to a therapeutic drug product. Figure S2 displays the simulated profile of mean ADA formation following first dose administration in a typical population of adults with inflammatory bowel disease. Not yet addressed mechanistically is the impact of concurrent drug therapy with immunomodulators on ADA formation and clearance.
The adult PBPK model showed a modest underprediction of clearance in inflammatory bowel disease, although a significant portion of this observed data was extracted from subsets including only nonresponders to treatment. As previously mentioned, this result is expected because nonresponders often have higher ADA concentrations, higher TNF levels, and more severely inflamed tissues. Loss of infliximab into the feces is an alternate explanation for the error.43
For therapeutic proteins, the observation that children receive lower exposures than adults with the same weight‐based doses has been reported before.8 Drug developers are beginning to anticipate this phenomenon and derive pediatric‐specific doses according to age or weight using PBPK modeling or allometric scaling.2, 44 The results of this study highlight the degree of error that can be expected when only adult information is used to predict pediatric PKs in maturation‐independent age groups for the purposes of deriving age or weight‐tiered doses with therapeutic proteins.44
To summarize, the methods of PBPK modeling and allometric scaling were comprehensively evaluated in this work for their accuracy in translating infliximab PK knowledge from adults to children. Both methods performed comparably, yet neither method achieved the prediction accuracy that is recorded in studies with small molecule drugs. Considerable variability was noted among the predictions made by the eight allometric models and accuracy was not driven by the sample sizes from the parent popPK studies. Therefore, a comprehensive pediatric clinical trial planning approach would benefit from both PBPK modeling and allometric scaling, being cognizant that PBPK modeling may be more appropriate for predicting peak concentrations and allometric scaling may be more appropriate for predicting trough concentrations in children.
Funding
This study received funding from the Canadian Institutes of Health Research (CIHR) through the Frederick Banting and Charles Best Canada Graduate Scholarship (CGS‐D).
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
P.M. wrote the manuscript. P.M. and A.E. designed the research. P.M. performed the research. P.M. analyzed the data. P.M. and A.E. contributed new reagents/analytical tools.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ At present, pediatric drug development for large molecules is poorly supported by modeling and simulation. The comparative performances of physiologically‐based pharmacokinetic (PBPK) modeling and allometric scaling for the prediction of large molecule pharmacokinetics in children are unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Which method(s) for pediatric PK prediction can be used reliably in pediatric drug development and clinical trial planning for large molecules?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ With infliximab as an example for the evaluation, PBPK modeling and allometric scaling had comparable accuracy in known exposure scenarios. Two‐thirds of the model‐predicted concentrations fell within twofold of the observed concentrations. PBPK modeling was more accurate for predicting peak drug concentrations, while allometry was more accurate for predicting trough drug concentrations.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ No pediatric PBPK modeling has been completed to support regulatory submissions to the US Food and Drug Administration (FDA) for a monoclonal antibody.2 We highlight the opportunity for PBPK modeling to be used in tandem with allometry to contribute to this growing field of drug development.
Supporting information
Figure S1. Comparison of model‐fitted profiles to observed data after the first optimization step.
Figure S2. Simulated profile of mean ADA formation following first dose administration in a typical population of adults with inflammatory bowel disease.
Table S1. Datasets used for adult PBPK model building.
Project Files. Contains the adult Mobi file, pediatric Mobi file and the Intiquan toolbox code for allometric simulations.
Project Files. Contains the adult Mobi file, pediatric Mobi file and the Intiquan toolbox code for allometric simulations.
Acknowledgments
P.M. acknowledges Stephan Schaller and Pierre Chelle for assistance in modeling.
ASCPT Communities: Pharmacometrics & Pharmacokinetics, Biologics
References
- 1. Edginton, A.N. Knowledge‐driven approaches for the guidance of first‐in‐children dosing. Pediatric Anesthesia 21, 206–213 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Liu, X.I. et al Monoclonal antibodies and Fc‐fusion proteins for pediatric use: dosing, immunogenicity, and modeling and simulation in data submitted to the US Food and Drug Administration. J. Clin. Pharmacol. 59, 1130–1143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tod, M. , Jullien, V. & Pons, G. Facilitation of drug evaluation in children by population methods and modelling. Clin. Pharmacokinet. 47, 231–243 (2008). [DOI] [PubMed] [Google Scholar]
- 4. Wu, Q. & Peters, S.A. A retrospective evaluation of allometry, population pharmacokinetics, and physiologically‐based pharmacokinetics for pediatric dosing using clearance as a surrogate. CPT Pharmacometrics Syst. Pharmacol. 8, 220–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahmood, I. & Tegenge, M.A. A comparative study between allometric scaling and physiologically based pharmacokinetic modeling for the prediction of drug clearance from neonates to adolescents. J. Clin. Pharmacol. 59, 189–197 (2019). [DOI] [PubMed] [Google Scholar]
- 6. Wang, J. , Edginton, A.N. , Avant, D. & Burckart, G.J. Predicting neonatal pharmacokinetics from prior data using population pharmacokinetic modeling. J. Clin. Pharmacol. 55, 1175–1183 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Mansoor, N. , Ahmad, T. , Alam Khan, R. , Sharib, S.M. & Mahmood, I. Prediction of clearance and dose of midazolam in preterm and term neonates: a comparative study between allometric scaling and physiologically based pharmacokinetic modeling. Am. J. Ther. 26, e32–e37 (2019). [DOI] [PubMed] [Google Scholar]
- 8. Malik, P. & Edginton, A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin. Drug Metab. Toxicol. 14, 585–599 (2018). [DOI] [PubMed] [Google Scholar]
- 9. Yellepeddi, V. , Rower, J. , Liu, X. , Kumar, S. , Rashid, J. & Sherwin, C.M. State‐of‐the‐art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin. Pharmacokinet. 58, 1–13 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Claassen, K. et al Development of a physiologically‐based pharmacokinetic model for preterm neonates: evaluation with in vivo data. Curr. Pharm. Des. 21, 5688–5698 (2015). [DOI] [PubMed] [Google Scholar]
- 11. Ferl, G.Z. , Theil, F.‐P. & Wong, H. Physiologically based pharmacokinetic models of small molecules and therapeutic antibodies: a mini‐review on fundamental concepts and applications. Biopharm. Drug Dispos. 37, 75–92 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Shah, D.K. & Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67–86 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Niederalt, C. et al A generic whole body physiologically based pharmacokinetic model for therapeutic proteins in PK‐Sim. J. Pharmacokinet. Pharmacodyn. 45, 235–257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hardiansyah, D. & Ng, C.M. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically‐based pharmacokinetic modelling. MAbs 10, 1144–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Candon, S. , Mosca, A. , Ruemmele, F. , Goulet, O. , Chatenoud, L. & Cézard, J.‐P. Clinical and biological consequences of immunization to infliximab in pediatric Crohn's disease. Clin. Immunol. 118, 11–19 (2006). [DOI] [PubMed] [Google Scholar]
- 16. Hyams, J. et al Safety and efficacy of maintenance infliximab therapy for moderate‐to‐severe Crohn's disease in children: REACH open‐label extension. Curr. Med. Res. Opin. 27, 651–662 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Hyams, J. et al Induction and maintenance infliximab therapy for the treatment of moderate‐to‐severe Crohn's disease in children. Gastroenterology 132, 863–873 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Hämäläinen, A. , Sipponen, T. & Kolho, K.‐L. Serum infliximab concentrations in pediatric inflammatory bowel disease. Scand. J. Gastroenterol. 48, 35–41 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Baldassano, R. et al Infliximab (REMICADE) therapy in the treatment of pediatric Crohn's disease. Am. J. Gastroenterol. 98, 833–838 (2003). [DOI] [PubMed] [Google Scholar]
- 20. Singh, N. et al Early infliximab trough levels are associated with persistent remission in pediatric patients with inflammatory bowel disease. Inflamm. Bowel Dis. 20, 1708–1713 (2014). [DOI] [PubMed] [Google Scholar]
- 21. Adedokun, O.J. et al Pharmacokinetics of infliximab in children with moderate‐to‐severe ulcerative colitis: results from a randomized, multicenter, open‐label, phase 3 study. Inflamm. Bowel Dis. 19, 2753–2762 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Ruperto, N. et al A randomized, placebo‐controlled trial of infliximab plus methotrexate for the treatment of polyarticular‐course juvenile rheumatoid arthritis. Arthritis Rheum. 56, 3096–3106 (2007). [DOI] [PubMed] [Google Scholar]
- 23. Willmann, S. et al Development of a physiology‐based whole‐body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J. Pharmacokinet. Pharmacodyn. 34, 401–431 (2007). [DOI] [PubMed] [Google Scholar]
- 24. Chen, X. , DuBois, D.C. , Almon, R.R. & Jusko, W.J. Interrelationships between infliximab and recombinant tumour necrosis factor‐alpha in plasma using minimal physiologically‐based pharmacokinetic models. Drug Metab. Dispos. 45, 790–797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manicourt, D.H. , Triki, R. , Fukuda, K. , Devogelaer, J.P. , Nagant de Deuxchaisnes, C. & Thonar, E.J. Levels of circulating tumor necrosis factor alpha and interleukin‐6 in patients with rheumatoid arthritis. Relationship to serum levels of hyaluronan and antigenic keratan sulfate. Arthritis Rheum. 36, 490–499 (1993). [DOI] [PubMed] [Google Scholar]
- 26. Bal, A. , Unlu, E. , Bahar, G. , Aydog, E. , Eksioglu, E. & Yorgancioglu, R. Comparison of serum IL‐1 beta, sIL‐2R, IL‐6, and TNF‐alpha levels with disease activity parameters in ankylosing spondylitis. Clin. Rheumatol. 26, 211–215 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Korolkova, O.Y. , Myers, J.N. , Pellom, S.T. , Wang, L. & M'Koma, A.E. Characterization of serum cytokine profile in predominantly colonic inflammatory bowel disease to delineate ulcerative and Crohn's colitides. Clin. Med. Insights Gastroenterol. 8, 29–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arican, O. , Aral, M. , Sasmaz, S. & Ciragil, P. Serum levels of TNF‐alpha, IFN‐gamma, IL‐6, IL‐8, IL‐12, IL‐17, and IL‐18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005, 273–279 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olsen, T. et al Tissue levels of tumor necrosis factor‐alpha correlates with grade of inflammation in untreated ulcerative colitis. Scand. J. Gastroenterol. 42, 1312–1320 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Olszewski, W.L. , Pazdur, J. , Kubasiewicz, E. , Zaleska, M. , Cooke, C.J. & Miller, N.E. Lymph draining from foot joints in rheumatoid arthritis provides insight into local cytokine and chemokine production and transport to lymph nodes. Arthritis Rheum. 44, 541–549 (2001). [DOI] [PubMed] [Google Scholar]
- 31. Li, X. , Jusko, W.J. & Cao, Y. Role of interstitial fluid turnover on target suppression by therapeutic biologics using a minimal physiologically based pharmacokinetic model. J. Pharmacol. Exp. Ther. 367, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen, X. , Jiang, X. , Jusko, W.J. , Zhou, H. & Wang, W.J. Minimal physiologically‐based pharmacokinetic (mPBPK) model for a monoclonal antibody against interleukin‐6 in mice with collagen‐induced arthritis. J. Pharmacokinet. Pharmacodyn. 43, 291–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen, X. , DuBois, D.C. , Almon, R.R. & Jusko, W.J. Biodistribution of etanercept to tissues and sites of inflammation in arthritic rats. Drug Metab. Dispos. 43, 898–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baert, F. et al Influence of immunogenicity on the long‐term efficacy of infliximab in Crohn's disease. N. Engl. J. Med. 348, 601–608 (2003). [DOI] [PubMed] [Google Scholar]
- 35. Chen, X. , Hickling, T.P. & Vicini, P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 1‐theoretical model. CPT Pharmacometrics Syst. Pharmacol. 3, e133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ren, Y. , Li, L. , Kirshner, S. , Wang, Y. , Sahajwalla, C. & Ji, P. A model‐based approach to quantify the time‐course of anti‐drug antibodies for therapeutic proteins. Clin. Pharmacol. Ther. 105, 970–978 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Kleiner, G. , Marcuzzi, A. , Zanin, V. , Monasta, L. & Zauli, G. Cytokine levels in the serum of healthy subjects. Mediators Inflamm. 2013, 6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kosmač, M. , Avčin, T. , Toplak, N. , Simonini, G. , Cimaz, R. & Šerbec, V.Č. Exploring the binding sites of anti‐infliximab antibodies in pediatric patients with rheumatic diseases treated with infliximab. Pediatr. Res. 69, 243–248 (2011). [DOI] [PubMed] [Google Scholar]
- 39. Brandse, J.F. et al A real‐life population pharmacokinetic study reveals factors associated with clearance and immunogenicity of infliximab in inflammatory bowel disease. Inflamm. Bowel Dis. 23, 650–660 (2017). [DOI] [PubMed] [Google Scholar]
- 40. Malik, P.R.V. , Hamadeh, A. , Phipps, C. & Edginton, A.N. Population PBPK modelling of trastuzumab: a framework for quantifying and predicting inter‐individual variability. J. Pharmacokinet. Pharmacodyn. 44, 277–290 (2017). [DOI] [PubMed] [Google Scholar]
- 41. Brekkan, A. , Berntorp, E. , Jensen, K. , Nielsen, E.I. & Jönsson, S. Population pharmacokinetics of plasma‐derived factor IX: procedures for dose individualization. J. Thromb. Haemost. 14, 724–732 (2016). [DOI] [PubMed] [Google Scholar]
- 42. Gunn, G.R. 3rd , Sealey, D.C.F. , Jamali, F. , Meibohm, B. , Ghosh, S. & Shankar, G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin. Exp. Immunol. 184, 137–146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brandse, J.F. et al Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology 149, 350–355.e2 (2015). [DOI] [PubMed] [Google Scholar]
- 44. Hanke, N. , Kunz, C. , Thiemann, M. , Fricke, H. & Lehr, T. Translational PBPK modeling of the protein therapeutic and CD95L inhibitor asunercept to develop dose recommendations for its first use in pediatric glioblastoma patients. Pharmaceutics 11, 152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki, T. et al Importance of neonatal FcR in regulating the serum half‐life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc‐fusion proteins to human neonatal FcR. J. Immunol. 184, 1968–1976 (2010). [DOI] [PubMed] [Google Scholar]
- 46. Kaymakcalan, Z. et al Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 131, 308–316 (2009). [DOI] [PubMed] [Google Scholar]
- 47. van Schie, K.A. et al Restricted immune activation and internalisation of anti‐idiotype complexes between drug and antidrug antibodies. Ann. Rheum. Dis. 77, 1471–1479 (2018). [DOI] [PubMed] [Google Scholar]
- 48. Stepensky, D. Local versus systemic anti‐tumour necrosis factor‐alpha effects of adalimumab in rheumatoid arthritis. Clin. Pharmacokinet. 51, 443–455 (2012). [DOI] [PubMed] [Google Scholar]
- 49. Chen, X. , DuBois, D.C. , Almon, R.R. & Jusko, W.J. Characterization and interspecies scaling of rhTNF‐alpha pharmacokinetics with minimal physiologically based pharmacokinetic models. Drug Metab. Dispos. 45, 798–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rojas, J.R. et al Formation, distribution, and elimination of infliximab and anti‐infliximab immune complexes in cynomolgus monkeys. J. Pharmacol. Exp. Ther. 313, 578–585 (2005). [DOI] [PubMed] [Google Scholar]
- 51. Fasanmade, A.A. , Adedokun, O.J. , Blank, M. , Zhou, H. & Davis, H.M. Pharmacokinetic properties of infliximab in children and adults with Crohn's disease: a retrospective analysis of data from 2 phase III clinical trials. Clin. Ther. 33, 946–964 (2011). [DOI] [PubMed] [Google Scholar]
- 52. Fasanmade, A.A. et al Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur. J. Clin. Pharmacol. 65, 1211–1228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ternant, D. et al Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther. Drug Monit. 30, 523–529 (2008). [DOI] [PubMed] [Google Scholar]
- 54. Dotan, I. et al Patient factors that increase infliximab clearance and shorten half‐life in inflammatory bowel disease: a population pharmacokinetic study. Inflamm. Bowel Dis. 20, 2247–2259 (2014). [DOI] [PubMed] [Google Scholar]
- 55. Aubourg, A. , Picon, L. , Lecomte, T. , Bejan‐Angoulvant, T. , Paintaud, G. & Ternant, D. A robust estimation of infliximab pharmacokinetic parameters in Crohn's disease. Eur. J. Clin. Pharmacol. 71, 1541–1542 (2015). [DOI] [PubMed] [Google Scholar]
- 56. Buurman, D.J. , Maurer, J.M. , Keizer, R.J. , Kosterink, J.G. & Dijkstra, G. Population pharmacokinetics of infliximab in patients with inflammatory bowel disease: potential implications for dosing in clinical practice. Aliment. Pharmacol. Ther. 42, 529–539 (2015). [DOI] [PubMed] [Google Scholar]
- 57. Ternant, D. et al Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br. J. Clin. Pharmacol. 78, 118–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of model‐fitted profiles to observed data after the first optimization step.
Figure S2. Simulated profile of mean ADA formation following first dose administration in a typical population of adults with inflammatory bowel disease.
Table S1. Datasets used for adult PBPK model building.
Project Files. Contains the adult Mobi file, pediatric Mobi file and the Intiquan toolbox code for allometric simulations.
Project Files. Contains the adult Mobi file, pediatric Mobi file and the Intiquan toolbox code for allometric simulations.