

Abstract
The thrombospondin type-1 domain containing 7A (THSD7A) protein is known to be one of the antigens responsible for the autoimmune disorder idiopathic membranous nephropathy. The structure of this antigen is currently unsolved experimentally. Here we present a homology model of the extracellular portion of the THSD7A antigen. The structure was evaluated for folding patterns, epitope site prediction, and function was predicted. Results show that this protein contains 21 extracellular domains and with the exception of the first two domains, has a regular repeating pattern of TSP-1-like followed by F-spondin-like domains. Our results indicate the presence of a novel Trp-ladder sequence of WxxxxW in the TSP-1-like domains. Of the 21 domains, 18 were shown to have epitope binding sites as predicted by epitopia. Several of the F-spondin-like domains have insertions in the canonical TSP fold, most notably the coiled coil region in domain 4, which may be utilized in protein-protein binding interactions, suggesting that this protein functions as a heparan sulfate binding site.
Keywords: Homology modeling, THSD7A, IMN, epitope site prediction, thrombospondin
Introduction
Primary (also known as idiopathic) membranous nephropathy (PMN) is a kidney specific autoimmune disorder characterized by the accumulation of immune complexes within the glomerular basement membrane and the leakage of massive amounts of protein into the urine. Patients suffering from this condition experience signs and symptoms of the nephrotic syndrome as a result of this damage to the glomerular filtration barrier. Ten to twenty percent of patients suffering from PMN will eventually progress to end stage renal disease whether treatment is undertaken or not.1 Two antigens that contribute to this disorder have been discovered in the last decade; the phospholipase A2 receptor (PLA2R) antigen2 and thrombospondin type-1 domain containing 7A (THSD7A).3 While some electron microscopy studies have been reported for PLA2R providing some structural information about this antigen,4,5 no work has been reported detailing structural information about THSD7A.
THSD7A is an extracellularly located type-1 transmembrane protein known to contain multiple thrombospondin repeat (TSR) domains.6 This 250 kDa glycoprotein has been discovered to contain multiple epitope-containing domains that are targeted by the immune system.7 This protein serves as an autoantigen in approximately 3–5% of PMN patients, and both human and rabbit anti-THSD7A have been shown to induce proteinuria and features of membranous nephropathy when passively transferred to mice.8,9 Specific residues on the THSD7A antigen which are being targeted by the human autoantibodies are still unknown.
THSD7A is a member of the thrombospondin (TSP) family of glycoproteins. Crystal structures of other members in this superfamily (Thrombospondin related anonymous protein (TRAP),10,11 F-spondin,12 ADAMTS13,14 have shown a characteristic thrombospondin repeat (TSP-1) containing a three stranded motif (Figure 1a). The three strands (A, B, and C) alternate in direction and have a unique conserved residue composition. The A strand typically contains two or three Trp residues and two Arg residues on the B strand.15 The Trp located on the A strand intercalates between the Arg residues on the B strand. The conserved A and B strand motifs, WSxWS and R*R, respectively, together create a Trp-ladder that typically consist of six rungs or more. These rungs are stabilized by cation-pi interactions between the indole portion of the Trp and the guanidinium portion of the Arg residues on the A and B strands, respectively.16 The ladder is held into place by a pair of disulfide bonds which flank the top and bottom of the Trp-ladder. A third disulfide bond is often present at the bottom of each domain to further stabilize this portion of the TSR. Two major groups of TSR domains, distinguished by the location of the Cys residues forming the disulfide bonds, have been described by Tan and colleagues.15 The crystal structure of human TSP-1 having the second and third tandem repeats showed the presence of jar handles, bulges stemming from the BC loop.17
FIGURE 1. Structural features of the second thrombospondin repeat in Thrombospondin-1, and THSD7A.
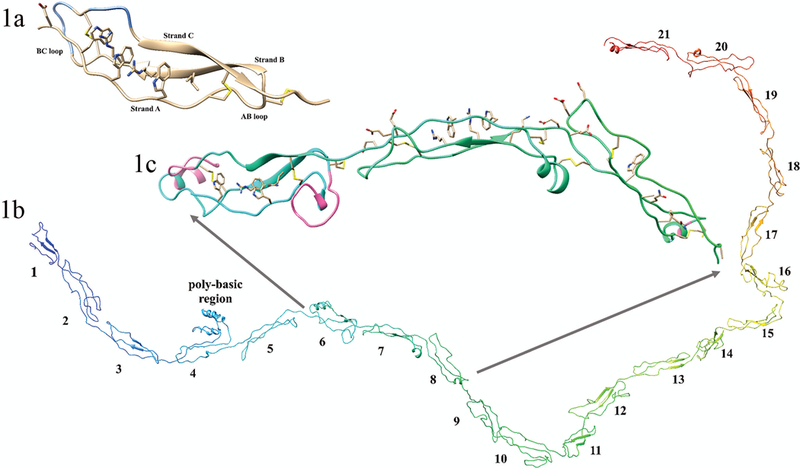
(1a) Crystal structure image of the second TSR repeat in thrombospondin (created using pdb ID 1LSL). This TSP-1-like domain has 3 strands; strand A, demonstrating the characteristic ripple, and strands B and C showing the characteristic short beta sheets, and the AB and BC loops are indicated. The regions of the BC loop ribbon colored blue are the jar handles. Shown is the Trp-ladder with three Trp residues and two Arg residues and an N-terminal hydrophobic substitution. The disulfides bonds show the C1-C5, C2-C6, and C3-C4 bonding pattern of the TSP-1-like domain. (1b) complete homology of the 21 extracellular TSR domains of THSD7A; N-terminus (blue) and C-terminus (red). Domain numbers are shown for each individual TSR domain. (1c) Expanded image of domains 6 through 8 of THSD7A. Shown are the disulfide bonds of consecutive TSP-1-like (domains 6 and 8) interrupted by a F-spondin-like domain (domain 7) in THSD7A. In domain 6 important structural features of note are the AB loop insertion, the jar handle insertion (insertions shown in pink), and the WxxxxW Trp-ladder. In domain 7 the Trp-ladder comprising three positively charged residues and two Trp residues is demonstrated. Both domains 7 and 8 have negatively charged residues in the BC loop. Domain 8 has a relatively small BC loop insertion (shown in pink) and no Trp-ladder forming cation-pi interactions.
The function of THSD7A in the podocyte is currently unknown. Other proteins containing TSR domains for which functions have been determined are involved in angiogenesis, cell migration, axon guidance, and cell-cell and cell-extracellular matrix interactions.17,18 The WSxWS motif in TSP-1 grooves have been observed to interact with heparan sulfate glycosaminoglycans (HSG).15,16
Knowledge of the structure and function of THSD7A antigen would be both clinically useful in terms of understanding the autoimmune pathogenesis of PMN, as well as instructive about potential biological mechanisms of this protein in the podocyte and other cells. Further knowledge of the specific residues involved in the epitope sites would assist in development of therapeutic interventions for PMN, such as epitope-specific therapies. Therefore, we developed a homology model of the extracellular portion of the THSD7A antigen in silico. Homology models have been utilized to develop novel therapeutics and explore structure of proteins.19,20 Our model was used to predict which residues might specifically be targeted by the autoantibodies in PMN.
Materials and Methods
Computational approaches
Homology modeling was performed using Protein Homology/analogy Recognition Engine V 2.0 (Phyre2).21 To create the THSD7A homology model smaller sections of the extracellularly located portion of the protein were submitted to the Phyre2 server. Residues 59–1475 of THSD7A were successfully modeled. Residues 1476 to 1606 which make up the rest of the extracellular portion of THSD7A were omitted from the model. In total 20 smaller sections containing pairs of consecutive domains (i.e., D1-D2; D2-D3; D3-D4… D20-D21) based on our sequence alignment were submitted to Phyre2. Overlapping regions in the 20 models were aligned and linked together in UCSF Chimera22 to create the full structure representing the majority of the THSD7A extracellular domain. Individual insertions (regions of primary peptide sequence interrupting the consensus TSP-1-like domain homology) were modeled independently then inserted back into the overall domain structure. Template selection was performed in Phyre2. Refinement of the THSD7A model was performed in UCSF Chimera. Minimization of the THSD7A homology model was performed using steepest descent followed by conjugate gradient to relax the structure and ensure good bond angles. The final model was submitted to RAMPAGE to determine quality assessment.23
Alignment of THSD7A domains
A Basic Local Alignment Search Tool (BLAST) search was performed to identify sequences which had high homology to THSD7A. Individual domains were identified using the pattern of six Cys residues and other landmark residues such as Trp common to the two major groups of TSRs, as suggested by Tan et al.15 Domain 4 could be best aligned as a TSP-1-like domain with a large, poly-basic insert after the first Cys residue and the omission of the usual disulfide bond between C2 and C6. Using sequence alignment, similar smaller insertions within other TSR domain structures were identified. The sequences comprising the identified domains were modeled to determine their individual structures.
Normal mode analysis
Conformational changes and protein flexibility of individual domains were investigated using normal mode analysis. PDB files were created for individual THSD7A domains, pairs of consecutive domains (i.e., D1-D2, D2-D3, D3-D4…), and segments containing three consecutive domains (i.e., D1-D3, D2-D4, D3-D5…) and submitted to ElNémo, the Elastic Network server24 for analysis. Default parameters were selected and low frequency normal modes were calculated. Modes with the lowest frequency and highest degree of collectivity were analyzed.
Conservation Analysis
An evaluation of the conservation of residues in THSD7A was performed to determine which regions were highly conserved. The THSD7A homology model was submitted to the ConSurf server.25,26 Results were evaluated in Chimera. The ConSurf coloring scheme was used to denote degrees of conservation.
Prediction of B-cell epitope sites
B-cell epitope prediction was performed using the Epitopia server.27,28 The Epitopia server uses physicochemical (residue composition) and structural aspects (frequency of helices, surface accessibility to solvent and antibody CDR loops, and curvature) of the submitted antigen to determine which residues are the most immunogenic. 27 File formatting for the Epitopia server was completed using the in house script Epitopia_prep3.1.py. The completed THSD7A homology model was submitted to the Epitopia server. Results were visualized with the Epitopia coloring scheme in Chimera.
Results and Discussion
THSD7A model validation
The Ramachandran plot produced using RAMPAGE showed that 75.3% of the phi and psi angles were in the favored region, and 18.6% of residues were found in the allowed region. Only 87 residues out of 1417 (6.1%) were shown to be in the outlier region. Thus 94% of the residues in the model were shown to have phi and psi angles in allowed regions. Taken together this model represents an excellent example of a large system homology model.
Overall structure of THSD7A extracellular portion
1417 residues (amino acids 59–1475) of the extracellular portion of THSD7A were successfully modeled. Our model of this region demonstrates 21 tandem extracellular domains (Figure 1b) with the potential for a 22nd domain that begins similarly to the other domains but degenerates after strand A (see Figure 2). This model, which was developed before the recent publication by Seifert and colleagues,7,29 shows that the first 21 domains are indeed a mixture of TSP-1-like (Figures 1c, 3a) and F-spondin-like (Figures 1c, 3b) domains (or “C6-like,” as in Seifert).
FIGURE 2. Sequence alignment of THSD7A.

FASTA sequence alignments for the TSP-1-like (group 1) and F-spondin-like (group 2) domains of THSD7A is shown. The sequence alignment for a hypothetical domain 22 is added to show similarities in the A and B strands, but a degenerate strand C. shown with the shortened strand C. Conserved residues that define the structural features of the two groups of TSRs are shown at the top. Basic residues are shown in blue and acidic residues are in red. Note the variability and length of the insertions within the AB loop (especially in Domain 4) and the BC loop (Domain 6) in the TSP-1-like domains.
Tan and colleagues present two major groups of TSR domains: TSP-1 is the prototype for the group 1 TSRs, while F-spondin serves as the prototype for the group 2 TSRs.15 We will refer to these group 2 TSRs as “F-spondin-like” (instead of C6-like) following this initial classification and due to the fact that the third strand of the F-spondin-like TSRs are more similar to their counterparts in THSD7A than are the analogous domains in C6 (Supplementary Figure 1). Similar to Seifert et al., we found that domains 1 and 2 of THSD7A were the only tandem repeating TSP-1-like domains. Our work here shows that domains 4, 6, 8, 10, 12, 14, 16, 18, and 20 are TSP-1-like domains, whereas domains 3, 5, 7, 9, 11, 13, 15, 17, 19, and 21 are F-spondin-like domains. Like Seifert et al. we show that the structure of THSD7A contains alternating TSP-1-like and F-spondin-like domains. This change from TSP-1-like to F-spondin-like is the result of a disulfide bond pattern shift from a late to early cysteine (Figure 3c).
FIGURE 3. Homology models of (a) TSP-1-like domains and (b) F-spondin-like domains.
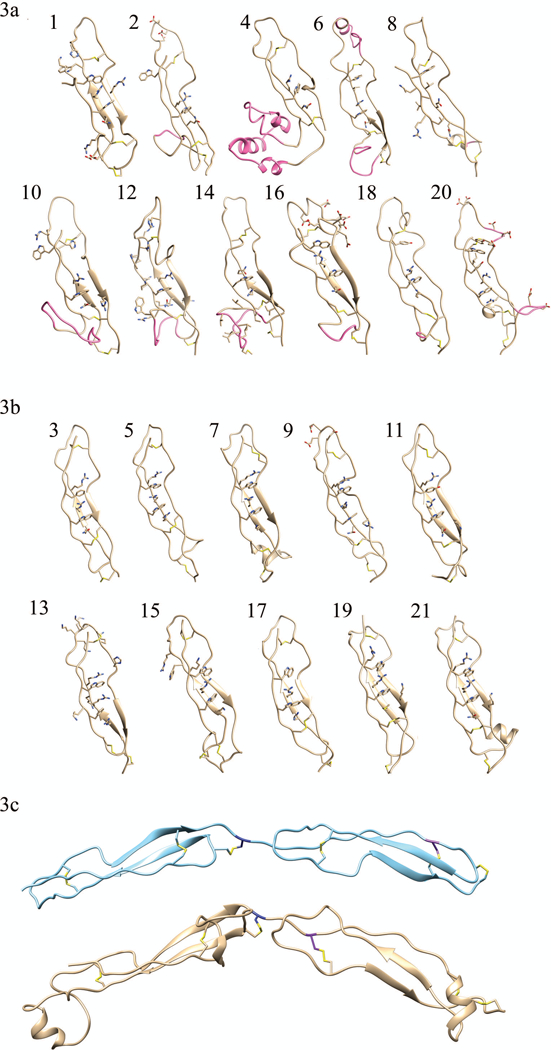
Individual domain homology models of the eleven TSP-1-like domains (1, 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20) and the ten F-spondin-like domains (3, 5, 7, 9, 11, 13, 15, 17, 19, 21). In each domain the residues participating in disulfide bonds and those forming the Trp-ladder are shown. Insertions into the canonical fold (AB loop insertion, the jar handle insertion) of the TSP-1-like domains are shown in pink. Domain 8 has a relatively small BC loop insertion (shown in pink) and no Trp-ladder forming cation-pi. (3c) Disulfide bonding pattern switch is shown. The consecutive second and third TSP-1 domains of thrombospondin are shown in light blue ribbon structure, (pdb ID: 1LSL). TSP-1-like domain (domain 6) followed by a F-spondin-like domain (domain 7) in THSD7A are shown beneath the thrombospondin structure in gold. The C6 Cys is colored in navy blue of the first TSR repeat. It can be seen that the C1 Cys residue (purple) of the following tandem repeat shifts from a late position in thrombospondin to an early position in THSD7A. This alternating late then early C1 Cys results in the TSP-1 like followed by F-spondin like pattern of THSD7A.
Based on our sequence analysis shown in Figure 2, we determined that each of the A strands of the TSP-1-like domains in THSD7A follows a YxWxxxxWxxC consensus pattern. We were only able to observe this unusual TSP-1-like sequence in the 8th domain of ADAMTS13. While all TSP-1-like domains begin with a Tyr residue, the F-spondin-like domains begin with a charged residue with the exception of domain 19.
Trp-ladders in TSP-1-like versus F-spondin-like domains
Trp-ladders have been suggested to be sulphate/glycosaminoglycan-binding motifs.16 Twenty of the 21 modeled TSR domains of THSD7A were shown to have a recognizable Trp-ladder of some sort (Figure 3a, 3b). The TSP-1-like domains were found to have truncated ladders usually containing only one Trp and one Arg residue with the exception of domain 8. It is the latter WS in the typical WSxWS ladder motif which is maintained. This structural feature is similar to the 8th TSR domain of ADAMTS13 and in the CCN TSR family models30 which were also shown to have only one Trp and one Arg residue present in the TSR domains. In our model THSD7A domain 20 was found to be the only TSP-1-like domain containing a Tyr residue that participates in the WSxWS Trp-ladder sequence (YGQWS).
Further evaluation of these TSP-1-like domains demonstrated that, while the WSxWS sequence was not present in many of these TSP-1-like domains, a YxWxxxxW consensus sequence was consistently present instead (Figure 2). Domains 1, 2, 6, 10, 12, 16, 18, 20, and 22 all have this same basic sequence homology. In domain 18, a Tyr residue replaces the first Trp and domain 20 has an additional Tyr residue present. We evaluated the models to determine if any ladder structure was possible. We found that structurally a Trp-ladder was still able to form having a novel Trp-ladder sequence of WxxxxW and, in one case, utilizing the entire YxWxxxxW sequence. Domains 1, 6, 16, 18, and 20 were shown to form similar Trp-ladder structures using this WxxxxW sequence. These domains are the only TSP-1-like domains having more than one aromatic residue present in the Trp-ladder. This extended Trp-ladder is formed by the A strand creating a small bulge to allow for the more distant Trp residue to participate as a rung. In domain 20 there is an extended ladder having a sequence of YxWxYxxW, which also contains the WxxxxW sequence. This could constitute a new Trp-ladder consensus sequence. It is interesting to note that group 1 TSRs typically have all three Trp residues participating in the ladder. In THSD7A six of the eleven TSP-1-like domains only include one Trp residue. This disparity may be due to a divergence in function.
Domain 8 does not seem to form a true Trp-ladder but does utilize the first Trp residue of the WxxxxW consensus sequence as a rung in the incomplete ladder. This ladder does not produce any cation-pi interactions at all, but does create hydrophobic interactions between the Trp and the Leu residue located on the B strand. This is interesting since it has been previously thought that the cation-pi interactions in the Trp-ladder were important for maintaining the fold of the TSR domain.10,15 This suggests that the disulfide bonding pattern may actually be more important for maintaining the TSR fold than the stabilization through cation-pi interactions.
In each TSP-1-like domain except 18 and 20 a Gln residue replaces one of the Arg residues in the ladder. This substitution of a Gln residue in the Trp-ladder scaffold is sometimes seen in TSR domains, although this is primarily seen in the group 2 (F-spondin-like) not group 1 (TSP-1-like) domains.15 However the truncation of the ladder in addition to the substitution of the polar uncharged Gln residues in place of a positively charged Arg residue would produce a much less positively charged binding grove. It is the positively charged groove that is thought to be the glycosaminoglycan binding site in the TSR domain, thus these TSP-1-like domains may have weaker binding to glycosaminoglycans or serve as binding partners for other types of macromolecules.
The F-spondin-like domains were shown to have more rungs in the ladder. Each F-spondin-like domain contained two aromatic rungs, usually Trp residues, and typically three basic residues intercalated in between them. Only four of the ten F-spondin like domains (9, 11, 19, and 21) have a Gln or Met substitution. Even though these domains have a Gln or Met residue they all have a positively charged residue on the C strand which interacts with the base of the Trp-ladder. The only TSP-1-like domain having this additional positively charged residue from the C strand interacting at the base of the ladder is domain 10. This positive charge from the neighboring strand may serve to maintain a net charge in the groove. This type of interaction is seen in the modeled structure of TSP-1-like domains of the CCN family of proteins.30 We developed a homology model for the 8th domain of ADAMTS13 and determined that this interaction was also present in this protein. It is notable that no F-spondin-like domains have insertions into the fold (see below). These domains having a much fuller Trp-ladder in addition to fewer Gln substitutions would contain a much stronger and larger positively charged groove. Taken together this suggests that THSD7A creates an alternating weaker positive, stronger positive electrostatic gradient within the grooves.
Insertions in THSD7A are located in TSP-1-like domains
A typical TSP-1-like domain is consists of approximately 50–55 residues. We noted that domains 2, 4, 6, 10, 12, 14, 16 and 20 contained at least 10 more residues than the expected amount of residues in a typical TSP-1-like structure. Domains 4 and 6 contained up to 30+ residues more than are usually present. We determined that each of these domains contained one or two insertions in the canonical TSP-1-like fold (Figure 3a). Each domain, except for domain 20, contained an insertion that was found to extend from the base of the A strand leading into the B strand (AB-loop insertion). The AB-loop insertions are structurally contained between the C1 and C2 cysteines in each domain, with the exception of the poly-basic insert in domain 4, which does not have a C2 residue. Our model suggests that domain 6 contained 2 insertions: an AB-loop insertion and a second insertion extending from the first jar handle on the BC-loop. These insertions in domain 6 are located on opposite sides of the TSP-1-like fold. Domain 20 was shown to have 2 insertions as well. The first insertion is located in the BC loop similar to the second insertion in domain 6, while the second insertion in domain 20 protrudes out from the end of the C-strand.
We found that many of the insertions contained charged or polar uncharged residues. Domains 8 and 14 both have a small cluster of hydrophobic residues within the insertions. These insertions may be structurally relevant for the function of this protein as post-translational modification sites or alternatively may be binding sites for other proteins molecules.
Since we could not find other evidence detailing distinct insertions into a TSP-1-like fold, we looked for other TSR domains possessing approximately 10 or more additional residues and modeled them to determine the possibility that they also contained insertions. Relatively few TSP-1-like domains were found that had an additional 10 or more residues present in the sequence. However in SCO-spondin, the 11th, and 16th TSP-1-like domains were found to have 10 or more residues in excess of the 55 residues typical of a TSR domain. The 11th TSP-1-like domain has 68 residues while the 16th TSP-1-like domain was listed as having 123 residues. After modeling the 11th TSP-1-like domain we determined that it also had an insertion in the jar handle region (Supplementary Figure 1). The 16th TSP-1-like domain of SCO-spondin (D3812-T3934) was not found to have an insertion but was actually determined to be two tandem TSP-1-like domains with a longer linker region between the domains (Figure 4b). This indicates that SCO-spondin has 25 TSP-1-like domains instead of 24, which is consistent with work from Meiniel et al.31 Thus we suggest that sequence nomenclature for this protein should actually be listed as residues D3812-G3869 as the 16th TSP-1-like domain and E3876-T3934 is 17th TSP-1-like domain. In addition the currently listed 17th through 24th TSP-1-like domains should be updated as TSP-1-like domains 18 through 25 to represent the additional TSP-1-like domain in the SCO-spondin protein. While structural determination of SCO-spondin was not the focus of this work, this determination of the location of the missing TSP-1-like domain is something worth noting.
FIGURE 4. Electrostatic potential maps of TSR domains in THSD7A.
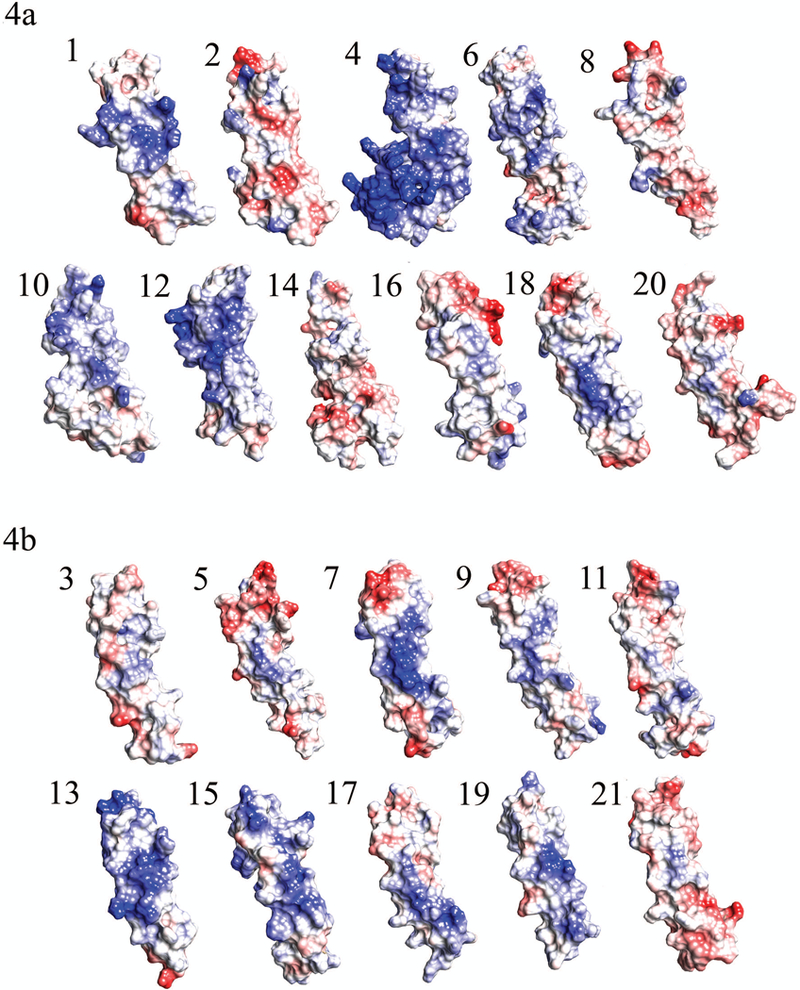
(5a) Charge density maps of the eleven TSP-1-like domains of THSD7A. (5b) Charge density maps of the ten F-spondin-like domains in THSD7A. Blue color indicates region of positive charge, red color indicates regions of negative charge, and white color indicates regions of neutral charge.
Electrostatic and hydrophobicity considerations of domains
It is known that the jar handles characteristically contain charged residues, thus we analyzed the electrostatic composition of individual domains in THSD7A (Figure 4). Several domains were shown to have negatively charged residues near the jar handles, namely domains 2, 5, 7, 8, 9, 11, 16, 17, 18, 20, and 21. Five domains (domains 2, 5, 16, 20, 21) seemed to have the greatest degree of negatively charged regions in the jar handle. Only five domains (domains 4, 10, 12, 13, and 15) were shown to have positively charged residues in the jar handle. Domains 4, 12, and 13 had the largest degree of positive charge in the jar handle. Domain 4 which contains the poly-basic region has the greatest surface area and number of positively charged residues. These localized regions of positive charge would likely attract very negatively charged molecules.
Domains 8, 11, 14, and 21 had hydrophobic patches present (Figure 5). The hydrophobic patches in these domains were located on the TSR side opposite the Trp-ladder. In domain 14 the hydrophobic patch was part of the AB-loop insertion. Domain 21 has the largest hydrophobic patch opposite the Trp-ladder side.
FIGURE 5. Hydrophobicity plot of domains containing hydrophobic patches in THSD7A.
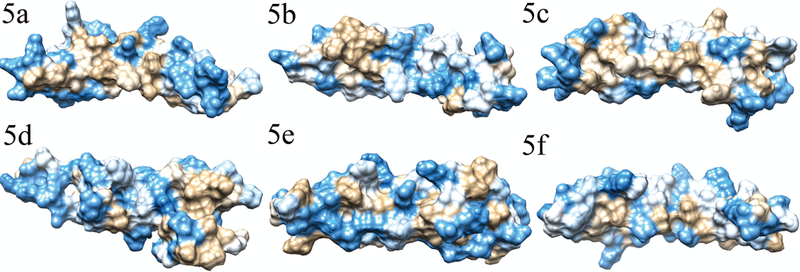
Hydrophobicity plots of domains found to have hydrophobic patches. Blue color indicates hydrophilic regions, tan color indicates hydrophobic regions, and white color indicates regions with average hydrophobicity. Panels 6a (domain 8), 6b (domain 11), and 6c (domain 21) show the hydrophobic patches observed on the side opposite the Trp-ladder, while 6d shows the hydrophobic patch found in domain 14 within the insertion. For purposes of comparison, the sides opposite the Trp-ladder for domains 2 and 15 are shown in panels 6e and 6f.
Domain 4 is a TSP-1-like domain with a poly-basic insert.
The UNIPROT website currently lists the region from residues 267–315 as a coiled coil domain. Seifert et al. describe the region from A248 to T358 as a coiled coil region. When we submitted the Y253 to V355 to Phyre2 we originally did not get a TSP-1-like domain structure, however after submitting the Y253-V533 omitting R269-E313 (polybasic region) the sequence was shown to assume a perfect TSP-1-like domain architecture (Figure 3a). Thus this work demonstrates that this region is not a complete coiled coil as a distinct entity, but rather a coiled-coil insertion within the overall TSR fold which juts out from the base of the A strand leading into the B strand. While this poly-basic insertion is significantly larger than the other inserts in the TSP-1-like domains, it is consistent with the other AB-loop insertions identified in this study. This larger domain, which contains only 2 disulfide bonds, C1-C5 and C3-C4, was also shown to form a truncated TRP ladder produced with Trp-260 and Arg-326 residues.
Glycosaminoglycan binding sites are highly positively charged. It has been demonstrated that the Trp-ladder groove of TSR domains can be utilized as a HSG binding site. We believe that domain 4 of THSD7A, which contains this large poly-basic insert, may also be a glycosaminoglycan binding site for heparan sulfate or similar proteoglycans.
The extracellular, juxtamembrane portion of THSD7A
The structure of the THSD7A region C-terminal to domain 21 was not shown to have a complete TSR domain and instead has a largely uncharacterized structure. Sequence alignment suggests the potential presence of a 22nd domain that has A and B strands similar to those of the TSP-1-like domains, with C2, C3, and potentially C4 residues but lacking the first (C1) Cys residue. Seifert et al. reported that this domain was a TSP-1-like domain. When we attempted to model this domain we were unable to produce a model that adopted a TSR structure of any kind. We believe that, while this domain does have several Cys residues that can be forcibly aligned to match a consensus TSR sequence, it does not actually fold in this pattern. The C4, C5, and C6 residues, which are all utilized in creating the disulfide bonding pattern are positioned in the sequence in areas which disallow them to fold into a TSR domain properly. In a typical TSR domain, strands A, B and C all contain a similar number of residues (between 8 and11). In the juxtamembrane region of THSD7A following domain 21, the A and B strands would contain 11 residues while the appropriate segment of the sequence corresponding to the C strand would contain only 3 residues. This discrepancy in length would prevent a full TSR structure from folding.
Transmembrane and cytoplasmic region
The transmembrane region spans from W1607 to M1657. Modeling of the transmembrane and cytoplasmic region resulted in a single alpha helix (transmembrane portion), followed by a smaller coil with some alpha helix character (cytoplasmic region) (Supplementary Figure 3). There is a short cytoplasmic region that contains many positively charged residues immediately adjacent to the inner leaflet of the plasma membrane. While this may merely serve to anchor the protein to the negatively charged phospholipid residues of the lipid bilayer, sequence homology suggests that it may contain a calmodulin binding site (Figure 6). L-selectin undergoes regulated cleavage of its ectodomain in a calcium/calmodulin mediated fashion.32 The sequence similarity between THSD7A, L-selectin, and several other transmembrane molecules known to undergo calmodulin-mediated shedding of the ectodomain and the finding of a large extracellular domain of THSD7A that can be shed into culture medium of human umbilical vein endothelial cells33 lends credence to a similar modus operandi in podocytes. For example, if THSD7A serves as a strong electrostatic mechanism of cell adhesion of the podocyte to negatively charged heparan-sulfate proteoglycans in the glomerular basement membrane (GBM), a calcium/calmodulin cleavage event might allow rapid disengagement of the podocyte foot processes from the GBM.
FIGURE 6. Sequence alignment of regions C-terminal to domain 21.

The transmembrane (gray box) and cytoplasmic region of THSD7A bears a striking resemblance to the corresponding domains of other proteins, such as L-selectin, known to undergo calcium-dependent shedding of the ectodomain. The residues in bold, which extend into the transmembrane region and include a series of basic amino acids in the cytoplasmic juxtamembrane region, are sites of known or proposed (for THSD7A) calmodulin binding. Modified from Gifford et al 2012.
Normal Mode Analysis
In THSD7A most of the junctions between the individual domains in were not shown to have more than one linker residue. Only the linker regions between domains 1 and 2, 3 and 4, and 4 and 5 were shown to have multiple residues (6, 6, and 4 residues respectively). Overall protein flexibility can be impacted by residues sandwiched between domains. Normal mode analysis can be used to determine the flexibility of proteins. In this work we studied the movement of THSD7A domains. When the full THSD7A structure was submitted for analysis relatively little motion was observed. We believe this is due to the large nature of the system, which reveals an inherent limitation of normal mode analysis. In light of this, each individual domain was then submitted to evaluate if there was any potential for movement. All domains were shown to have a correlated motion between the AB-loop and the BC-loop of the TSR folds (Figure 7). This folding motion further exposes the Trp-ladder which opens due to the correlated motion of both loops forcing the Trp-ladder in the opposite direction. This suggests that the Trp-ladders become more accessible through fluctuations in the THSD7A protein.
FIGURE 7. Normal mode fluctuations in THSD7A TSR domains and tandem segments.
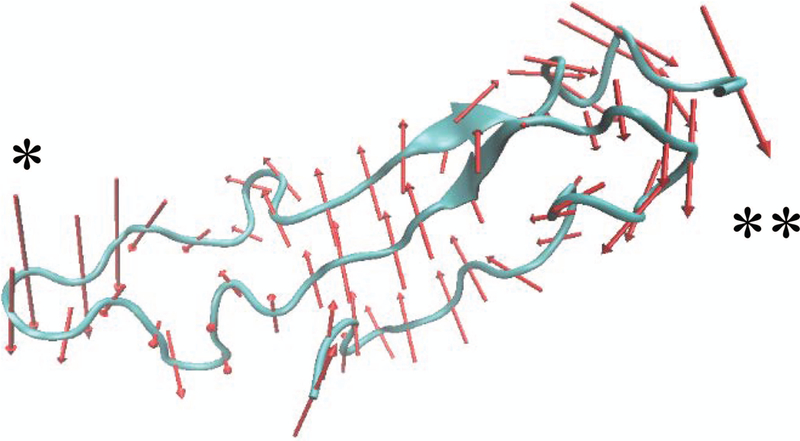
The lowest frequency and highest collectivity mode (mode 7) of THSD7A domain 1 is shown as a representative example of individual domain protein motions. Red arrows are vectors representing the direction and magnitude of individual motions in the protein structure. It can be seen that the AB loop (*) and the BC loop (**) move in a correlated fashion both regions of the protein moving together and in an anti-correlated motion with respect to the Trp-ladder.
When pairs of consecutive domains were analyzed, a correlated motion was still observed; however the BC loop of the first domain moved in correlation to the AB loop of the second domain. The linker region in between the two individual TSR domains was shown to be anti-correlated with the BC loop of the first domain and the AB loop of the second domain. Thus the linker region moved in the same fashion as the Trp-ladder moved in with respect to the AB-loop and BC-loop in a single domain NMA simulation. When three consecutive domains were evaluated, a correlated motion was observed between the two TSR domains on the ends of the protein segment. As the length of the protein segment was increased a correlated motion between the ends of the structure was noted in addition to an anti-correlated motion to the middle of the protein structure, whether that was the Trp-ladder, the linker region, or an entire domain. These motions are just as likely to occur as the motions of the individual domains. It is also possible that the insertions in THSD7A could contribute to additional motions between domains. A protein NMR study would be ideal to determine which of these motions is occurring to the largest degree, and how protein flexibility of TSR domains might change as successive TSR domains are added onto the protein structure.
While there are limitations to the normal mode analysis, as the entire THSD7A system is quite large, we do believe that these results taken together this data suggest that THSD7A is a very flexible protein. This protein flexibility might dictate exposure of regions of THSD7A, such as Trp-ladder to promote binding to other macromolecules. We believe that the decrease in motion in the individual domains as larger portions of the structure were evaluated could be due to a limitation of the normal mode analysis.
Domain conservation
Conservation in protein sequences can suggest essential functions of proteins, or individual domains in proteins. We analyzed the degree of conservation in THSD7A across other species to evaluate if any critical functions might be present. Our results show that there is a high degree of conservation in domains 1, 3, 7, 9, 13, 14, 15, and 18 (Supplementary Figure 4). The F-spondin-like domains were much more likely to have a larger number of conserved residues, as five out of the ten F-spondin-like domains were shown to have greater than 40 residues with more than average conservation (approximately 70–75% conservation): compared to only three out of eleven TSP-1-like domains having greater than 40 residues. Domain 15 has the largest degree of conservation thus suggesting a critical functional role for this domain. Notably the insertions in the TSP-1-like domains were shown to be part of the highly variable regions, further supporting that they are indeed insertions into the canonical TSP-1-like fold in THSD7A. It is interesting that the less conserved TSP-1-like domains contain insertions while the more conserved F-spondin-like domains do not. An open question is whether the conserved domains are critical for biological function and structure, or if the more variable insertions in the TSP-1-like domains represent evolutionary adaptations to modulate THSD7A function. The overall domain structure of THSD7A is an equal and patterned mixture of both insertions (notably within the TSP-1-like domains) and conservation (represented by the F-spondin-like domains). Intriguingly, the related protein THSD7B is remarkably similar to THSD7A in terms of domain structure; modeling of this protein is underway.
Epitope prediction in the THSD7A extracellular domain
Epitope predication has been utilized for a variety of receptors to garner useful information facilitating therapeutic design.34–36 We combined epitope prediction algorithms with our structural model to investigate favorable epitopes in the extracellular domain of THSD7A. Eighteen domains (domains 1–17 and 19) were predicted to contain epitope sites, and three domains (18, 20, and 21) were not shown to have epitope sites (Figure 8). Experimental work by Seifert et al. has used serum from patients with THSD7A-associated PMN to investigate domains targeted by human anti-THSD7A autoantibodies.7 It should be noted that Seifert’s work utilizes differing domain numbers than ours due to the coiled-coil region being identified as a separate domains, and not as an insert within a larger TSP-1-like domain. Supplementary Table 1 demonstrates the corresponding domains between the two articles. All predicted epitope sites corresponded well to the regions identified by Seifert except for domains 4 and 5. Additionally domains 20 and 21 which were not predicted to have epitope sites also correspond well to experimental work performed by Seifert. Accurate prediction of epitope sites is quite challenging, however using our model we accurately predicted all domains which were shown to have an epitope sites by Seifert and two of the domains not shown to have epitope sites. The predicted residues comprising these epitopes are listed in Supplementary Table 2.
FIGURE 8. Predicted epitope sites on THSD7A.

Homology model of THSD7A with residues predicted to have higher than average probability of immunogenicity shown in sphere conformation. Magenta residues = 5, Pink residues = 4 in immunogenicity on 5-tier scale.
Epitopia uses a 5-tier scale to rank immunogenicity probability. A value of four and five is suggested to be immunogenic, a value of three is average, and values of two or one are unlikely to be immunogenic. The d15–16 construct in Seifert et al. was recognized by approximately 60% of patient sera, yet corresponds to domains 16–17 in our model, which is the region predicted to have the lowest degree of overall immunogenicity. These two domains also have much smaller clusters of immunogenic residues. This suggests the size of the epitope cluster may not necessarily determine degree of immunogenicity in patients. We also noted that hydrophobic and polar uncharged residues were most likely to be involved in epitope sites (Supplementary Table 3). Eleven of the epitope containing domains were shown to have mostly hydrophobic residues in the epitope sites, five domains contained mostly polar uncharged residues, and in the remaining two domains there was an equal mixture of both hydrophobic and polar uncharged residues. Residue composition of all domain is listed in Supplementary Table 4).
Seifert et al. also utilized two domain truncations of the THSD7A antigen to determine epitope locations. Seifert’s work shows that the d17–18 construct was immunogenic, which would correspond to our domains 18–19. Our results show that only domain 19 was predicted to have any immunogenic residue. This suggests that only d18 of Seifert is immunogenic. Taken together this work demonstrates an accurate prediction of epitope sites through the use of an in silico generated homology model of a very large antigen system. Homology models are quite often generated for much smaller systems.20,37–39 Typically when homology models are developed for large system it is in combination with cryo-electron microscopy, and individual pieces of the model are fit into the larger overall surface map.4,40 In this work, even though we developed a very large scale homology model independently of cyro-electron microscopy data, the model proves to be useful for accurate prediction of epitope regions in THSD7A, as it is corroborated by the work of Seifert. Thus, we can suggest residues that are part of the epitope sites (Supplementary Table 2). Knowledge of the specific immunogenic residue in THSD7A antigen will be critical in advancing the quest for antigen specific therapies for IMN.
Conclusions
In conclusion we have shown that the THSD7A antigen associated with PMN contains 21 TSR domains and an uncharacterized region between the last TSR and the transmembrane portion. The uncharacterized region does show features of a TSP-1 domain, but seems to devolve quickly. It is unclear whether this regions folds into a TSP-1 domain or not. Characterization of our model helps to further define the TSP-1-like fold that seems to be able to tolerate small to large insertions of residues jutting from the end of the AB and BC loops. These insertions we believe correspond to functional aspects of THSD7A. The most notable insertion, the poly-basic region located in domain 4, most likely acts as a HSG binding site. The structural aspects revealed in this in silico work should help to further enable essential experimental functional studies. In silico models do have some limitations when predicting the side chain rotamers of amino acids. However the accurate prediction of the epitope regions and most non-epitope regions on THSD7A support that the utility of the model presented in the work here. More structural aspects of THDS7A can be further evaluated using cryo EM, X-ray crystallography, or NMR. In addition to this work we determined that domain 16 of SCO-spondin as listed in the UNIPROT website is actually two TSP-1 domains thus, SCO-spondin has 25 TSP-1-like domains. While SCO-spondin was not the focus of the paper this is significant enough to note.
Antigen specific therapies can be considered the holy grail for the definitive treatment of autoimmune disorders. Knowledge of the antigen structure is essential for development of therapeutics in this class. Herein we present an in silico 3D structure of the THSD7A antigen. Homology modeling of the extracellular portion of this protein produced a very reliable model which can be utilized to further rational design of drug therapies for PMN, and characterization of THSD7A function in vivo. This work identifies the most probable residues eliciting the immune response associated with PMN. Knowledge of these individual residues will greatly aid in the sought after gold standard of antigen specific therapies for PMN.
Supplementary Material
SUPPLEMENTARY FIGURE 1: Alignment of F-spondin-like THSD7A domains six TSRs from F-spondin and three TSRs C6.
SUPPLEMENTARY FIGURE 2: Homology model of 6th TSP-1-like domains in SCO-spondin.
(a) Eleventh TSP-1-like domain of SCO-spondin. The insertion, located in the BC loop and similar to those found in THSD7A, is shown in pink. (b) Homology model of what was found to be the sixteenth and seventeenth domain of SCO-spondin, instead of a single domain.
SUPPLEMENTARY FIGURE 3: Transmembrane and intracellular portion of THSD7A protein.
The transmembrane domain of THSD7A was determined to be a single alpha helix. The intracellular portion is a coil with some alpha helix character. Shown is a representation of the transmembrane and intracellular portion aligned in a representative lipid layer from pdb ID 1NEN. Residues on the intracellular portion of THSD7A are shown.
SUPPLEMENTARY FIGURE 4: Conservations plots for each TSR domain in THSD7A.
Conservation plots demonstrating the variation in amino acid probability in individual TSR domains of THSD7A. Individual domain numbers are indicated. Shown are domains which represent the variety of variation in TSR domains of THSD7A. Key domains to note are domain 15 (shown to be highly conserved, domain 14 (demonstrating the variability in loop the insertion regions, domain 18 demonstrating the variability in the C-strand, and domain 8 (the only domain to be primarily variable). Scale indicates degree of conservation (modified from consurf website).
SUPPLEMENTARY TABLE 1: Corresponding Domain Architecture for THSD7A between UNIPROT, Seifert, and Stoddard
SUPPLEMENTARY TABLE 2: THSD7A Amino Acid Characterization of Epitope by Domain
SUPPLEMENTARY TABLE 3: Characterization of residue composition in epitope sites by domain.
Acknowledgements:
The authors would like to acknowledge the William Randolph Hearst Foundation, and the Rhodes College Faculty Development Endowment grant for financial support of the work. L.H.B. Jr. is supported by NIH/NIDDK grant R01 097053.
Footnotes
Conflicts of Interest: The authors declare that they have no conflict of interest
References
- 1.Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol 2017;12:983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck LH Jr., Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-Type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, Helmchen U, Dabert-Gay AS, Debayle D, Merchant M, Klein J, Salant DJ, Stahl RAK, Lambeau G. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371:2277–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fresquet M, Jowitt TA, Gummadova J, Collins R, O’Cualain R, McKenzie EA, Lennon R, Brenchley PE. Identification of a Major Epitope recognized by PLA2R Autoantibodies in Primary Membranous Nephropathy. J Am Soc Nephrol. 2015;26:302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Y, Cao L, Tang H, Shi X, He Y, Structure of Human M-type Phospholipase A2 Receptor Revealed by Cryo-Electron Microscopy. J Mol Biol. 2017;429:3825–3835. [DOI] [PubMed] [Google Scholar]
- 6.Wang CH, Su PT, Du XY, Kuo MW, Lin CY, Yang CC, Chan HS, Chang SJ, Kuo C, Seo K, Leung LL, Chuang YJ. Thrombospondin type I domain containing 7A (THSD7A) mediates endothelial cell migration and tube formation. J Cell Physiol. 2010;222:685–694. [DOI] [PubMed] [Google Scholar]
- 7.Seifert L, Hoxha E, Eichhoff AM, Zahner G, Dehde S, Reinhard L, Zoch-Nolte F, Stahl RAK, Tomas NM. The most N-terminal region of THSD7A is the predominant target for autoimmunity in THSD7A-associated membranous nephropathy. J Am Soc Nephrol. 2018;29:1536–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, Bachmann F, Budde K, Koch-Nolte F, Zahner G, Rune G, Lambeau G, Meyer-Schwesinger C, Stahl RAK. Autoantibodies against type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest. 2016;126:2519–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomas NM, Meyer-Schwesinger C, von Spiegel H, Kotb AM, Zahner G, Hoxha E, Helmchen U, Endlich N, Koch-Nolte F, Stahl RAK. A Heterologous Model of Thrombospondin Type 1 Domain-Containing 7A-Associated Membranous Nephropathy. J Am Soc Nephrol. 2017;28:3262–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tassavainen H, Pihlajamaa T, Huttunen TK, Raulo E, Rauvala H, Permi P, Kilpelainen I. The layerd fold of the TSR domain of P. falciparum TRAP contains a heparin binding site. Protein Sci. 2006;15:1760–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song G, Koksal AC, Lu C, Springer TA. Shape change in the receptor for gliding motility in Plasmodium sporozites. Proc Natl Acad Sci USA. 2012;109:21420–21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pääkkönen K, Tossavainen H, Permi P, Rakkolainen H, Rauvala H, Raulo E, Kilpeläinen I, Güntert P. Solution structures of the first and fourth TSR domains of F-spondin. Proteins. 2006;64:665–672. [DOI] [PubMed] [Google Scholar]
- 13.Akiyamma M, Takeda S, Kokame K, Takagi J, Miyata T. Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc Natl Acad Sci USA. 2009;106:19274–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng XL. Structure-function and regulation of ADAMTS-13 protease. J Thromb Haemost. 2013;11 Suppl 1:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan K, Duquette M, Liu J-H, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang, J-H. Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication. J Cell Biol. 2002;159:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsen JG, Kragelund BB. Who climbs the tryptophan ladder? On the structure and function of the WSXWS motif in cytokine receptors and thrombospondin repeats. Cytokine Growth Factor Rev. 2014;25:337–341. [DOI] [PubMed] [Google Scholar]
- 17.Tucker RP. The thrombospondin type 1 repeat superfamily. Int J Biochem Cell Biol. 2004;36:969–974. [DOI] [PubMed] [Google Scholar]
- 18.Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011;3:a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo J, Medina-Franco JL. Homology modeling, docking and structure-based pharmacophore of inhibitors of DNA methyltransferase. J Comput Aided Mol Des. 2011;25:555–567. [DOI] [PubMed] [Google Scholar]
- 20.Nandy A, Saenz-Mendez P, Gorman AM, Samali A, Eriksson LA. Homology model of the human tRNA splicing ligase RtcB. Proteins. 2017;85:1983–1993. [DOI] [PubMed] [Google Scholar]
- 21.Kelly LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera a visualization system for exploratory research and analysis. Journal of Computational Chemistry 2004;13:1605–1612 [DOI] [PubMed] [Google Scholar]
- 23.Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. [DOI] [PubMed] [Google Scholar]
- 24.Suhre K, Sanejouand YH. ElNémo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. [DOI] [PubMed] [Google Scholar]
- 26.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubinstein ND, Mayrose I, Pupko T. A machine-learning approach for predicting B-cell epitopes. Mol Immunol. 2009;46:840–847. [DOI] [PubMed] [Google Scholar]
- 28.Rubinstein ND, Mayrose I, Martz E, Pupko T. Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinformatics 2009;10:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck LH Jr. PLA2R and THSD7A: Disparate paths to the same disease? J Am Soc Nephrol. 2017;28:2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holbourn KP, Acharya KR, Perbal B. The CCN family of proteins: structure-function relationships Trends Biochem Sci. 2008;33:461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meiniel O, Meiniel A. The complex organization of SCO-spondin protein is highly conserved in mammals. Brain Research Reviews. 2007;53:321–327 [DOI] [PubMed] [Google Scholar]
- 32.Gifford JL1, Ishida H, Vogel HJ. Structural insights into calmodulin-regulated L-selectin ectodomain shedding. J Biol Chem. 2012;287:26513–26527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuo MW, Wang CH, Wu HC, Chang SJ, Chuang YJ. Soluble THSD7A is an N-glycoprotein that promotes endothelial cell migration and tube formation in angiogenesis. PLoS One. 2011;6:e29000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huerta-Saquero A, Evangelista-Martinez Z, Moreno-Enriquez A, Perez-Rueda E. Rhizobium etli asparaginase II: an alternative for acute lymphoblastic leukemia (ALL) treatment. Bioengineered. 2013;4:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandra S, Singh TR, Linear B cell epitope prediction for epitope vaccine design against meningococcal disease and their computational validations through physiochemical properties. Network Modeling Analysis in Health Informatics and Bioinformatics. 2012;1:153–159. [Google Scholar]
- 36.Kirk K, Poh CL, Fecondo J, Pourianfar H, Shaw J, Grollo L. Cross-reactive neutralizing antibody epitopes against Enterovirus 71 identified by an in silico approach. Vaccine. 2012;30:7105–7110. [DOI] [PubMed] [Google Scholar]
- 37.Gomez Barroso JA, Aguilar CF. Chagas disease: a homology model for the three-dimensional structure of the Trypanosoma cruzi ribosomal P0 antigenic protein. Eur Biophys J..2014;43:361–366. [DOI] [PubMed] [Google Scholar]
- 38.Bora L Homology modeling and docking to potential novel inhibitor for chikungunya (37997) protein nsP2 protease. J. Proteomics Bioinform, 2012;5:054–059. [Google Scholar]
- 39.Hasan MdA, Alauddin SM, Amin MA, Nur SM, Mannan A In silico Moledular Characterization of cysteine protease YopT from Yersinia pestis by homology modeling and binding site identification. Drug Target Insights 2014;8:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature, 2014;505: 515–521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY FIGURE 1: Alignment of F-spondin-like THSD7A domains six TSRs from F-spondin and three TSRs C6.
SUPPLEMENTARY FIGURE 2: Homology model of 6th TSP-1-like domains in SCO-spondin.
(a) Eleventh TSP-1-like domain of SCO-spondin. The insertion, located in the BC loop and similar to those found in THSD7A, is shown in pink. (b) Homology model of what was found to be the sixteenth and seventeenth domain of SCO-spondin, instead of a single domain.
SUPPLEMENTARY FIGURE 3: Transmembrane and intracellular portion of THSD7A protein.
The transmembrane domain of THSD7A was determined to be a single alpha helix. The intracellular portion is a coil with some alpha helix character. Shown is a representation of the transmembrane and intracellular portion aligned in a representative lipid layer from pdb ID 1NEN. Residues on the intracellular portion of THSD7A are shown.
SUPPLEMENTARY FIGURE 4: Conservations plots for each TSR domain in THSD7A.
Conservation plots demonstrating the variation in amino acid probability in individual TSR domains of THSD7A. Individual domain numbers are indicated. Shown are domains which represent the variety of variation in TSR domains of THSD7A. Key domains to note are domain 15 (shown to be highly conserved, domain 14 (demonstrating the variability in loop the insertion regions, domain 18 demonstrating the variability in the C-strand, and domain 8 (the only domain to be primarily variable). Scale indicates degree of conservation (modified from consurf website).
SUPPLEMENTARY TABLE 1: Corresponding Domain Architecture for THSD7A between UNIPROT, Seifert, and Stoddard
SUPPLEMENTARY TABLE 2: THSD7A Amino Acid Characterization of Epitope by Domain
SUPPLEMENTARY TABLE 3: Characterization of residue composition in epitope sites by domain.