

Abstract
Protein disulfide isomerase (PDI) is a redox-dependent protein with oxidoreductase and chaperone activities. It is a U-shaped protein with an abb’xa’ structural organization where the a and a' domains have CGHC active sites, the b and b' domains are involved with substrate binding, and x is a flexible linker. PDI exhibits substantial flexibility and undergoes cycles of unfolding and refolding in its interaction with cholera toxin, suggesting PDI can regain a folded, functional conformation after exposure to stress conditions. To determine whether this unfolding-refolding cycle is a substrate-induced process or an intrinsic physical property of PDI, we used circular dichroism to examine the structural properties of PDI subjected to thermal denaturation. PDI exhibited remarkable conformational resilience that is linked to its redox status. In the reduced state, PDI exhibited a 54°C unfolding transition temperature (Tm) and regained 85% of its native structure after nearly complete thermal denaturation. Oxidized PDI had a lower Tm of 48-50°C and regained 70% of its native conformation after 75% denaturation. Both reduced and oxidized PDI were functional after refolding from these denatured states. Additional studies documented increased stability of a PDI construct lacking the a’ domain and decreased thermal stability of a construct lacking the a domain. Furthermore, oxidation of the a domain limited the ability of PDI to refold. The stability and conformational resilience of PDI are thus linked to both redox-dependent and domain-specific effects. These findings document previously unrecognized properties of PDI and provide insight into the physical foundation of its biological function.
Graphical Abstract
INTRODUCTION
Protein disulfide isomerase (PDI) is a 57 kDa protein that is predominantly localized to the endoplasmic reticulum (ER) and is required for cell survival.1, 2 It assists protein folding by acting as both a chaperone and oxidoreductase.3, 4 These functions are linked to the “U” shape and modular abb'xa'c organization of PDI.5, 6 The a and a' domains at either end of the U are functional thioredoxin-like domains with conserved -CGHC- regions. The b and b' domains at the base of the U are inactive thioredoxin-like domains that appear to act as a single unit7, 8 and are involved with substrate binding, mainly through the b' domain.9-11 The x linker is a dynamic, flexible region that facilitates a conformational shift between the oxidized and reduced states of PDI. A short, acidic c region at the C-terminus of PDI contains an ER-targeting KDEL motif.
The structure of PDI is affected by its redox state.12, 13 For the reduced form of PDI (rPDI), the U is a flat structure with all domains in the same plane. This is considered a “closed” conformation because it limits substrate access to the binding domains at the internal base of the U in both human and fungal PDI.14, 15 For the oxidized form of PDI (oPDI), disulfide bonds connect the cysteine residues within each active site. This is considered an “open” conformation because the a' arm is positioned at a ~45° angle away from the plane of the abb'x domains. The redox status of PDI thus influences its binding capacity, with most unfolded substrate interactions involving oPDI due to the accessibility of its substrate-binding domain.14-18
In addition to its redox-sensitive structural change, PDI also exhibits substantial conformational flexibility. Structural studies and molecular dynamics simulations have identified a range of inter-domain movements within PDI.18-20 The bb' structure is rigid, with limited hinge-like motion and no rotation at the bb' interface. The ab structure is more flexible, with some hinge-like motion and some rotation at the ab interface. The b'xa' structure is the most dynamic region, with extensive motional and rotational flexibility. The a' domain of rPDI is also more susceptible to proteolysis than the rest of the protein, consistent with its high flexibility.21 The inter-domain flexibility of PDI, along with intra-domain flexibility in the b' domain,18 are key factors that allow PDI to accommodate a wide variety of substrates within its binding pocket at the inner base of the U. Structural analysis of full-length PDI, as well as PDI with whole domain deletions, may therefore shed light on the physical basis of the protein’s chaperone function.
The dynamic, redox-dependent nature of PDI influences its interaction with cholera toxin (CT). This AB-type protein toxin consists of a catalytic A1 subunit, an A2 linker, and a cell-binding B pentamer.22 PDI only recognizes the CTA1 subunit, and only rPDI binds to CTA1.23, 24 Binding occurs after CT travels from the cell surface to the ER by retrograde transport.25 PDI then facilitates the release of CTA1 from its holotoxin, which is a prerequisite for CTA1 to enter the cytosol where it elicits a cAMP response through ADP-ribosylation of the heterotrimeric G protein stimulatory alpha subunit (Gsα). PDI can reduce the disulfide bond that anchors CTA1 to CTA2, but this can also occur at the resident redox state of the ER and does not itself cause holotoxin disassembly.26-28 The essential role of PDI in CT dissassembly instead involves the physical displacement of CTA1 from its reduced holotoxin.29 We have shown that rPDI unfolds when it binds to CTA1 and have proposed the expanded hydrodynamic size of partially unfolded PDI acts as a wedge to dislodge CTA1 from CTA2/CTB5. In support of this model, the inhibition of rPDI unfolding blocks its disassembly of the reduced CT holotoxin without affecting its binding to CTA1. rPDI returns to a folded conformation after it is released from CTA1, thereby allowing it to repeat the unfolding process during additional interactions with CT. These observations indicate PDI is a resilient protein that can regain a folded, functional conformation after shifting to a disordered state.
To further examine the stability and conformational resilience of PDI, we subjected both rPDI and oPDI to thermal denaturation. We define conformational resilience as the ability of PDI to refold from a disordered state and consider this to be a linked but distinct phenomenon from its inter-domain conformational flexibility. Our collective data have documented the remarkable redox-dependent conformational resilience of PDI and have identified the contributions of individual PDI domains to both its stability and resilience. These observations provide new insight into the chaperone function of PDI and its role in stress response.
MATERIALS AND METHODS
Structural Analysis by Circular Dichroism (CD).
Lyophilized proteins (30 μg) were dissolved in 220 μL of 20 mM sodium borate buffer (pH 7.0) containing 100 mM NaCl and, with reduced protein samples, 1 mM dithiothreitol (DTT). GSH was not used as a reductant for these studies because the tripeptide contains two chiral centers that generate substantial noise in the CD signal. Furthermore, as we found in preliminary experiments, that noise becomes more pronounced with increasing temperature and overwhelms the PDI spectrum. Samples were placed in a 0.1 mm path-length quartz cuvette and read in a Jasco J-1100 CD Spectrophotometer (Jasco Corp., Tokyo, Japan) at various temperatures. Thermal melts were conducted from 20°C to 90°C, with readings taken every 2°C during both heating and cooling back to 20°C. Samples were incubated at each measured temperature for 4 min before measurement. Transition temperatures (Tm) and melt curves were calculated as previously described.30 Briefly, a value of specific heat capacity ΔC = 0.39 kcal mol−1 K−1 was used, while the enthalpy of transition, ΔH, was varied to fit the transition cooperativity. For experiments involving transient thermal treatment, readings were first taken at 25°C and then 10 min after incubation at the indicated elevated temperature. A third reading was taken after cooling the sample to 25°C. A baseline reading of buffer alone or buffer with DTT was background subtracted from all protein samples before data analysis. For each sample, CD spectra were measured 5 times in the 195–260 nm range and averaged to produce the final spectrum. Data were compiled in Igor Pro software (Wavemetrics, Portland, OR ) to produce a final figure. A line of best fit was calculated as previously described.31 All experiments were performed at least twice.
The fraction of unfolded protein at a given temperatute T, fT, was deduced from the CD spectra using the following formula:
where [θ] is the ellipticity at 222 nm and the subscripts T, F, and U indicate the test temperature, a low temperature where the protein is fully folded, and 90°C where the protein is maximally unfolded, respectively. All calculations were done on individual traces and averaged from duplicate measurements. For refolding experiments, the native structure was defined based on the ellipticity at 222 nm for the initial measurement at 20°C. Thus, the native state of each construct was not calculated but was instead established from the initial 20°C measurement and then normalized to a standard ellipticity value. The percentage of refolding was calculated from the difference in 222 nm ellipticity between a refolded PDI construct and its normalized starting value, as described in the equation above.
CT Disassembly Assay.
The wells of a 96-well ELISA plate were coated with 6 μg of the CT receptor (ganglioside GM1, EMD Millipore, Burlington, MA) in 100% EtOH and left to evaporate overnight. All subsequent additions to the plate were made in phosphate-buffered saline (pH 7.0) with 0.1% Tween 20 (PBS-T) containing 2.5% bovine serum albumin, and all washes were done 4 times with 400 μL of PBS-T. CT or CTB (both from Sigma-Aldrich, St. Louis, MO) were added in 100 ng quanities to each well and incubated at 4°C for 1 h. The plate was washed, and 2 μg of PDI was added to each well for 1 h at 37°C. When indicated, rPDI or oPDI was heated to the designated temperature for 10 min in a Proflex PCR System (Thermo Fisher Scientific, Waltham, MA) and then allowed to cool for 10 min before addition to the toxin-coated wells. For heated oPDI samples, 1 mM DTT was added to the sample after cooling to place PDI in the proper state for interaction with CT. After washing, primary antibody (1:1,000 polyclonal rabbit anti-CTA, Advanced Targeting Systems, San Diego, CA) was added to the plate for 1 h at 4°C and washed before adding the secondary antibody (1:1,000 HRP-conjugated goat anti-rabbit IgG, Jackson ImmunoResearch, West Grove, PA) for 30 min at 4°C. Preliminary experiments confirmed the anti-CTA antibody recognizes CTA1 but not the CTA2 subunit that remains with CTB after toxin disassembly. After incubation with the secondary antibody, the plate was washed again and blotted dry. TMB substrate (Thermo Fisher Scientific, Waltham, MA) was then added for 5 min before stopping the reaction with 4 N sulfuric acid. Absorbance at 412 nm was read in a BioTek Synergy 2 plate reader (BioTek, Winooski, VT). Values obtained from the CTB negative control were subtracted from all results before calculating the percentage of toxin disassembly as (1.00 - [PDI-treated CT signal / untreated CT control signal]) x 100. The untreated CT control for oPDI was incubated in buffer alone. The untreated CT control for rPDI and heated oPDI samples was incubated in buffer containing 1 mM DTT. As determined from preliminary experiments, treatment with DTT alone released a minor quantity of GM1 from the ELISA plate and accordingly reduced the CT holotoxin signal. Using holotoxin incubated in DTT-containing buffer as the untreated CT control signal compensated for this loss, which represented an 11 ± 5% loss of signal (n = 13, standard error of the mean) in comparison to the CT control incubated in buffer alone.
Cloning and Purification of PDI Deletion Constructs.
The pOLR130 plasmid encoding mature human PDI with an N-terminal His6 tag32 was generously provided by Dr. Lloyd Ruddock (University of Oulu, Finland). The bb'x construct was cloned from pOLR130 using the primers listed in Table 1. A new vector, pET-His-TEV-LIC encoding mature human PDI, was used as a PCR template to clone the additional PDI deletion constructs with the primers listed in Table 3. The pET-His-TEV-LIC vector was cloned from pcDNA3 LIC cloning vector (6A), which was a gift from Scott Gradia (Addgene plasmid # 30124; http://n2t.net/addgene:30124; RRID:Addgene_30124). Insertion of the PCR products into pET-His-TEV-LIC was performed using the LIC-PCR strategy.33 The empty plasmid was linearized using Ssp1 and treated, along with the PCR products, with T4 DNA polymerase. The linearized plasmid was then mixed with an individual PCR product to form a closed, complete vector. Escherichia coli strain DH5α was transformed with recombinant plasmids and plated overnight. Colonies were selected for plasmid minipreps that were used to confirm the proper coding sequences for our PDI deletion constructs with N-terminal hexahistidine tags. The plasmids were then used to transform E. coli strain BL21(DE3)pLysS .
Table 1.
Primers (5' to 3' orientations) for cloning of the PDI deletion constructs.
| Construct | Primer | Sequence |
|---|---|---|
| abb'x | Forward | TACTTCCAATCCAATGCAGACGCCCCCGAGGAG |
| Reverse | TTATCCACTTCCAATGTTATTAAGGCTGCTTGTCCCAGTC | |
| bb'x | Forward | TTGGATCCATGCATCACCATCACCATCACATGGCTGCCACCACC |
| Reverse | TTGAATTCTTACAGTTCATCTTTCACAGCTTTCTGATCATCGTCTTCCTCCATG TCTGGCTCC | |
| bb'xa' | Forward | TACTTCCAATCCAATGCAGCTGCCACCACCCTG |
| Reverse | TTATCCACTTCCAATGTTATTACAGTTCATCTTTCACAGCTTTCTG |
Table 3.
Conformational resilience of PDI and PDI deletion constructs under reducing (r) or oxidizing (o) conditions. Because redox and the individual domains of PDI influence its thermal stability, we could not heat all constructs to a single temperature in order to reach a common state of denaturation. The listed temperatures represent the minimal thermal treatment required for near-complete denaturation. The extent of denaturation and renaturation for each construct (± range) was calculated from two independent experiments represented in Figures 2 and 8.
| Construct | Denaturation Temperature |
% Folded Structure after Denaturation |
% Folded Structure after Renaturation |
|---|---|---|---|
| rPDI | 65°C | 2 ± 1 | 85 ± 2 |
| oPDI | 70°C | 12 ± 4 | 62 ± 5 |
| rbb'xa'c | 55°C | 12 ± 8 | 90 ± 0 |
| obb'xa’c | 55°C | 11 ± 8 | 90 ± 0 |
| rabb'x | 65°C | 6 ± 6 | 96 ± 4 |
| oabb'x | 60°C | 14 ± 5 | 72 ± 8 |
| rbb'x | 95°C | 8 ± 8 | 74 ± 5 |
| obb'x | 70°C | 0 ± 0 | 88 ± 3 |
For protein purification, cultures of transformed E. coli strain BL21(DE3)pLysS were induced at an O.D.600 of 0.4 with 1 mM IPTG for an overnight incubation at 20°C. For the production of uniformly 13C-labeled PDI, 13C-D-glucose (Cambridge Isotope Laboratories, Cambridge, MA) was used as the sole carbon source in M9 minimal medium. Cultures were pelleted, and protein was purified by Talon affinity chromatography as previously described.29 To confirm the purity of each construct, SDS-PAGE with Coomassie blue stain was used to visualize a 2 μg sample of each protein. Aliquots (30 μg) of each protein were then frozen overnight at −80°C before lyophilization.
Fourier Transform Infrared (FTIR) Spectroscopy.
CTA1 and/or uniformly 13C-labeled PDI (50 μg each) were dissolved in a D2O-based buffer containing 20 mM sodium borate and 1 mM GSH, for a total volume of 100 μL. CTA1 with a C-terminal hexahistidine tag was purified as previously described.34 All samples were read on a Jasco 4200 FTIR spectrometer (Jasco, Easton, MD) with a Peltier temperature controller (Pike Technologies, Madison, WI). Samples were incubated at either 10°C or 48°C for 5 min before measurements were taken. All traces were baseline corrected and, when necessary, smoothed. Spectral subtraction was used to reveal the gain or loss of amide I components (i.e., respective secondary structures) of PDI resulting from either elevated temperature or interaction with CTA1. Spectral subtractions were preceded by normalization, to obtain similar total amide I (1700–1600 cm−1) intensities of the operand spectra. The spectral ranges for assignment of each secondary structure are as follows: 1690–1660 cm−1 for turns, 1659–1646 cm−1 for α-helix, 1645–1638 cm−1 for irregular structure, and 1637–1620 cm−1 for β sheet.35 The intensity at lower wavenumbers was assigned to side chains. The respective spectral ranges for a 13C-labeled protein were lower by 45 cm−1,36 which accounted for the isotope effect on infrared vibrational frequencies. Final figures showing each individual trace, along with the subtraction of the two comparative traces, were compiled in Igor Pro software.
Insulin Reduction Assay.
The reductase activity of PDI was monitored by observing the aggregation of insulin.37, 38 rPDI and oPDI were either kept at 25°C or heated to 90°C for 10 min prior to mixing at 3 μM concentrations with 104 μM insulin (Sigma-Aldrich) in the absence or presence of 450 μM of the PDI inhibitor quercetin-3-rutinoside (Q3R, Sigma-Aldrich).39 The total volume was 135 μL in a buffer of 100 mM potassium phosphate, 2 mM EDTA, and 1 mM DTT. The 650 nm optical density of samples in a 96-well plate was read every minute for 90 min using a BioTek Synergy 2 plate reader. For clarity, only end-point measurements are shown.
RESULTS
Thermal Stability of PDI.
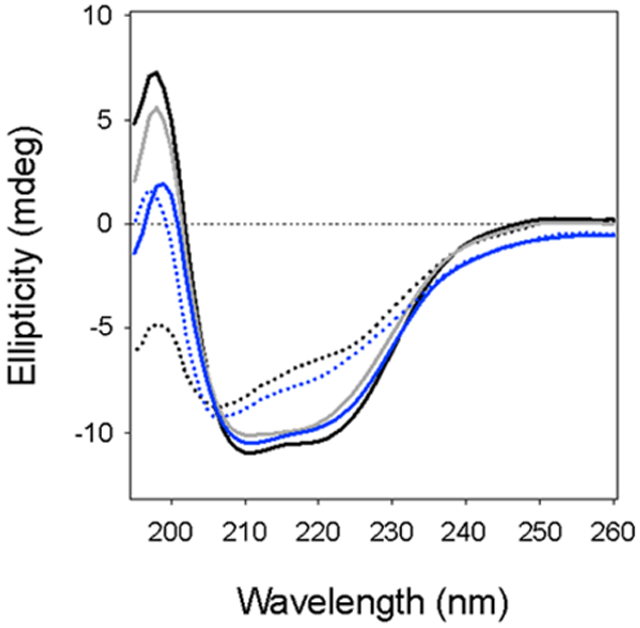
To establish the thermal stability of PDI, the protein was heated in step-wise fashion from 20°C to 90°C under reducing (1 mM DTT) or oxidizing conditions. A CD spectrum was recorded after every 2°C increase in temperature, and thermal unfolding profiles were then generated by plotting the ellipticity at 222 nm, [θ]222, versus temperature. When the data were plotted as sigmoidal unfolding profiles, rPDI (Fig. 1A, blue line) exhibited a higher transition temperature (Tm) of 54°C than the 48–50°C Tm of oPDI (Fig. 1B, Table 2). A nearly identical Tm of 56°C for rPDI was obtained for a PDI sample pre-treated with 10 mM DTT (not shown). rPDI also displayed a reproducible intermediate state, occuring at the transition midpoint of 54°C. When plotted as a two-step transition (Fig. 1A right panel, black line), rPDI exhibited a first stage Tm of 40–45°C and a second stage Tm of 61–63°C (n = 2). Neither rPDI nor oPDI could fully return to their native conformation after heating over the course of 4 h to 90°C, where the effect of thermal unfolding was saturated. rPDI did regain some of its native structure upon cooling to 20°C, but oPDI did not regain any structure after cooling. Thus, rPDI was more resistant to thermal denaturation than oPDI.
Figure 1.

Thermal stability of PDI. Reduced (A) or oxidized (B) PDI was heated stepwise from 20°C to 90°C, with CD measurements taken every 2°C. The proteins were then cooled from 90°C to 20°C in the same manner. Representative spectra are shown in the left panels: the initial 20°C measurement (black line), the Tm measurement (dashed line), the 90°C measurement (dotted line), and the final 20°C measurement after cooling (grey line). The right panels plot ellipticities at 222 nm as a function of temperature. Closed circles represent measurements taken during heating; open circles represent measurements taken during cooling. For rPDI, the blue and black lines represent thermal unfolding profiles modeled from one-step and two-step transitions, respectively.
Table 2.
Thermal stability of PDI and PDI deletion constructs under reducing or oxidizing conditions. Values of the transition temperature and enthalpy representing the ranges of two independent experiments per condition were derived from one-step unfolding profiles as shown in the experiments of Figures 1 and 7.
| Tm | ΔH | |||
|---|---|---|---|---|
| Construct | Reduced | Oxidized | Reduced | Oxidized |
| PDI | 54°C | 48-50°C | 31-35 | 30-32 |
| bb'xa'c | 50-51°C | 42°C | 30-46 | 20-26 |
| abb'x | 62-63°C | 51-52°C | 60-66 | 40 |
| bb'x | 58-60°C | 56°C | 50-66 | 50-66 |
PDI Resilience, Function in Toxin Disassembly, and Reductase Activity After Thermal Denaturation.
To further examine the ability of PDI to refold after thermal denaturation, we rapidly heated rPDI (Fig. 2A) or oPDI (Fig. 2B) to a set temperature for 10 min before cooling back to 25°C. CD spectra were recorded before heating, after 10 min at the elevated temperature, and after cooling to 25°C. The protocol for thermal denaturation in this experiment was therefore distinct from the prolonged thermal stress used in the experiments of Figure 1. For these studies, we wanted to compare the propensity for refolding from a common state of denaturation (≤ 10% native structure). We therefore ran preliminary experiments at several temperatures and chose the minimal temperature that resulted in nearly complete protein denaturation for each condition. rPDI only retained 2% of its native α-helical structure after heating to 65°C, yet it regained 85% of its structure after cooling to 25°C (Table 3). This demonstrated rPDI could refold from a state of nearly complete thermal denaturation. oPDI exhibited a substantially weaker ability to recover its structure after thermal denaturation, regaining only 62% of its structure after cooling from a 70°C temperature where it maintained even more (12%) of its original structure than the heated rPDI sample (Table 3). rPDI thus exhibited a greater propensity for renaturation than oPDI.
Figure 2.

Structure and function of refolded PDI. (A-B) CD spectra for rPDI (A) or oPDI (B) were measured at 25°C (black line) before heating rPDI to 65°C and oPDI to 70°C. Spectra were measured again after 10 min at elevated temperature (dotted line) and after cooling back to 25°C (grey line). (C) oPDI and rPDI samples were left at room temperature or heated to the indicated temperatures for 10 min. After cooling to room temperature, each sample was added to a 96-well plate coated with CT. Loss of the CTA1 subunit due to PDI-driven CT disassembly was subsequently detected by ELISA. Final values were calculated based on the maximum CTA1 signal from CT holotoxin incubated in the absence of PDI, as detailed in the Materials and Methods. Error bars report standard error of the means from at least five independent experiments per condition, each with four replicate samples. All values represent statistically significant differences from the rPDI control (Student's t test, p < 0.05).
Heating rPDI to 90°C for 5 min inhibits its ability to disassemble reduced CT at room temperature.40 However, it is not known if this loss-of-function is an all-or-none process or if PDI gradually loses function with increasing temperature. To answer this question, an ELISA-based CT disassembly assay was performed after exposing PDI to the thermal denaturation conditions used in Figure 2A-B. PDI was heated and cooled as previously described, and oPDI was then reduced with 1 mM DTT to allow an interaction with CT since only rPDI can bind to CTA1.23, 24 All PDI samples were then added to a 96-well plate coated with CT and incubated for 1 h at 37°C to initiate CT disassembly. The release of CTA1 from plate-anchored CTA2/CTB5 was detected by the loss of signal detected with a primary anti-CTA1 antibody and a secondary antibody conjugated to horse radish peroxidase (HRP) (Fig. 2C). Consistent with previous reports,23, 24, 40 CT disassembly did not occur when oPDI was added in the absence of reductant. CT disassembly by rPDI, which is an inefficient process,24, 40 resulted in a 19% loss of the CTA1 signal. Remarkably, the heat-treated rPDI sample was able to displace CTA1 from CTA2/CTB5 with greater efficiency than the rPDI control. The gain-of-structure in refolded rPDI thus corresponded to a gain-of-function for its interaction with CT. The oPDI sample that was heated to 70°C and then reduced after cooling could also disassemble CT, but with lower efficiency than the untreated rPDI control. To determine if CT disassembly by oPDI was more efficient after moderate thermal treatment, we repeated the disassembly assay with an oPDI sample that was heated to 60°C before cooling, reduction, and addition to CT. This sample of oPDI regained 70 ± 4% of its native structure after 75 ± 5% denaturation (n = 2). Like the heat-treated rPDI sample, this sample of heat-treated oPDI was more effective at CT disassembly than the rPDI control. These data showed that heat-treated PDI is not only functional, but it is able to regain a more active conformation after denaturation from thermal treatment.
PDI performs linked but independent functions as a chaperone and oxidoreductase.13 The physical displacement of CTA1 from its holotoxin represents a unique, chaperone-related function of PDI29 that is inactivated by a 90°C heat treatment.40 To examine the effect of thermal denaturation on the reductase activity of PDI, we employed a standard assay that monitors the aggregation of insulin resulting from reduction of its disulfide-linked homodimer (Fig. 3). We began this experiment with samples of rPDI and oPDI that had been heated to 90°C for 10 min, which was expected to inactive PDI and produce a negative control signal. As with the CT disassembly assay, the oPDI sample was reduced with 1 mM DTT after cooling in order to facilitate its interaction with insulin. Surprisingly, both rPDI and oPDI retained some enzymatic activity after complete thermal denaturation (Fig. 3A). After a 90 min incubation, both PDI samples exhibited 40–50% of the activity recorded for the rPDI control that had not undergone thermal denaturation. In addition, the onset of insulin aggregation occurred about 10 min sooner with the rPDI control than with the heated PDI samples (not shown). When DTT was not included in the assay bufer, an oPDI sample that had not undergone thermal denaturation did not reduce insulin. Consistent with previous reports,37, 38 there was little to no reduction of insulin in the presence of DTT alone. The latter two results demonstrated the specificity of insulin reduction by heat-treated PDI samples.
Figure 3.

Reductase activity of heat-treated PDI. rPDI and oPDI were incubated at 25°C or 90°C for 10 min. After cooling, PDI samples were incubated with insulin in DTT-containing buffer for 90 min. The reduction and resulting aggregation of insulin was detected by a change in optical density at 650 nm. Data are reported as the averages ± standard deviations of triplicate samples. (A) The 25°C oPDI sample was incubated with insulin in buffer lacking DTT. Insulin incubated in DTT-containing buffer without PDI was used as an additional control. One of three representative experiments is shown. (B) PDI samples were incubated in the absence or presence of the PDI inhibitor Q3R, as indicated.
A second insulin reduction assay was run in order to further confirm the activity of heat-treated PDI (Fig. 3B). For this assay, a subset of PDI samples were incubated with Q3R after thermal denaturation. Q3R is a polyphenolic compound that specifically binds to the b'x region of PDI and inhibits its reductase activity.38, 39 Both rPDI and oPDI again retained enzymatic activity after thermal denaturation, but none of the Q3R-treated samples could reduce insulin. The chaperone40 and enzymatic (Fig. 3) functions of PDI thus appear to exhibit different levels of recovery from thermal denaturation, with PDI reductase activity exhibiting greater resilience.
PDI Refolding After Repeated Thermal Denaturation.
Additional CD experiments documented the response of PDI to repeated transient thermal denaturation (Fig. 4). PDI was subjected to four sequential rounds of heating and cooling. rPDI generated the same spectrum and returned to the same level of folded conformation (~83% native structure) after each cycle of heating and cooling (Fig. 4A). This experiment used the same temperature for rPDI denaturation (65°C) that was used in Figure 2. We used 60°C for the repeated thermal treatment of oPDI because it could return to a fully active state after a single 10 min incubation at 60°C (Fig. 2C) but not at the 70°C temperature that left it with 62% native structure upon cooling (Fig. 2B-C). A single round of heating to 60°C allowed oPDI to regain more of its native structure upon cooling (Fig. 4B, grey line), which explains its high level of activity in the CT disassembly assay. Yet oPDI exhibited a substantially altered structure at the end of the fourth heating/cooling cycle, with a prominent minimum around 208 nm that was not seen in the original spectrum before heating (Fig. 4B, solid blue line). Thus, a major difference between rPDI and oPDI is the unique capability of rPDI to regain its initial folded structure after multiple cycles of thermal unfolding and refolding.
Figure 4.

Resilience of PDI to repeated thermal denaturation. CD spectra for rPDI (A) or oPDI (B) were measured at 25°C (black line) before heating rPDI to 65°C and oPDI to 60°C. Spectra were measured again after 10 min at elevated temperature (dotted black line) and after cooling back to 25°C (grey line). The heating/cooling cycle was repeated three times, with the dotted blue lines representing spectra measured at the elevated temperatures of the final (fourth) cycle and solid blue lines representing spectra measured after the final cooling to 25°C.
Similarities Between the Substrate-Induced Unfolding and Thermal Unfolding of PDI.
rPDI can refold after substrate-induced unfolding29 or thermal denaturation (Figs. 2, 4). However, it was unclear whether substrate-induced conformational changes are analogous to those caused by thermal stress. To answer this question, stable isotope-edited FTIR spectroscopy was used to examine substrate- and temperature-induced changes in the secondary structure of PDI. As shown in Figure 5A, the secondary structure of 13C-rPDI was analyzed at 10°C either alone (dashed line) or in the presence of unlabeled CTA1 at 1:1 molar ratio (black line). rPDI does not recognize the disordered conformation of free CTA1 that is present at 37°C,23 which necessitated the use of low temperature for the measurement of CTA1-bound PDI. For reference, Figure 5A also presents the FTIR spectrum of unlabeled CTA1 alone (grey line). These data highlight the expected FTIR spectral shift between the amide I bands of unlabeled and 13C-labeled proteins, which permits evaluation of the structures of both proteins combined in one sample.
Figure 5.

Substrate-induced and thermal unfolding of PDI. FTIR spectroscopy was used to analyze the secondary structure of rPDI during its interaction with CTA1 or during thermal stress. (A) Spectra of 13C-labeled rPDI (dashed line), unlabeled CTA1 (grey line), and both 13C-labeled rPDI and CTA1 combined at an equimolar ratio (black line) at 10°C. (B) Spectra of 13C-labeled rPDI alone (black line) and 13C-labeled rPDI in the presence of CTA1, with the CTA1 spectrum at 10°C removed (dashed line). (C) Spectra of 13C-labeled rPDI at 10°C (black line) and 48°C (dashed line). (D) Using the data presented in panels B-C, the spectrum for CTA1-treated PDI (black line) was subtracted from the spectrum of heat-treated PDI (dashed line) to generate the difference spectrum (grey line).
To compare the structure of PDI alone with that in the presence of CTA1, the CTA1 spectrum was subtracted from the spectrum of the combined 13C-rPDI / CTA1 sample. This allowed us to resolve the structure of 13C-rPDI during its interaction with CTA1. The resulting spectrum for toxin-treated 13C-rPDI (Fig. 5B, dashed line) was broader compared to the spectrum of 13C-rPDI alone (Fig. 5B, black line), which was indicative of a more disordered secondary structure of PDI resulting from its interaction with CTA1.
In addition to our previous CD studies, FTIR experiments were conducted to document the change in rPDI secondary structure resulting from its thermal unfolding. The FTIR spectrum of 13C-rPDI was recorded at 48°C (dashed line) and compared to the 10°C measurement (black line) (Fig. 5C). 48°C was chosen for the temperature of thermal unfolding because, based on calculations from the CD thermal melt (Fig. 1), the loss of PDI α-helical structure at this elevated temperature matched the previously reported loss of PDI α-helical structure resulting from its interaction with CTA1.29 The spectrum measured at 48°C was broader and shifted to higher wavenumbers compared to the spectrum at 10°C, again indicating a more disordered structure.
We next compared the thermal unfolding of PDI to its CTA1-induced unfolding (Fig. 5D). The spectrum of heat-treated 13C-rPDI (Fig. 5D, dashed line) was shifted toward higher wavenumbers as compared to CTA1-treated 13C-rPDI (Fig. 5D, black line). The difference spectrum (Fig. 5D, grey line) indicated a higher fraction of irregular (1604–1595 cm−1) and β-sheet structures (1594–1570 cm−1) in rPDI resulting from its interaction with CTA1 as compared to its partially unfolded structure resulting from thermal treatment. This indicated that PDI unfolding caused by its interaction with CTA1 involves a greater loss of α-helical structure than its thermal unfolding, which involves the loss of more β-sheet structure. Duplicate FTIR experiments using uniformly 13C-labeled CTA1 and unlabeled rPDI confirmed this result (not shown). PDI thus unfolds in distinct ways during thermal denaturation and upon its interaction with CTA1, yet it can still refold from either disordered state.
Contribution of Individual Domains to the Stability and Conformational Resilience of PDI.
We generated PDI deletion constructs that lack either the a domain, the a' domain, or both a and a' domains in order to examine how the individual domains of PDI contribute to its overall structural stability. SDS-PAGE was used to confirm the purity of our constructs (Fig. 6).
Figure 6.

Purified PDI constructs. Proteins purified from E. coli using Talon affinity chromatrography were visualized on SDS-PAGE with Coomassie stain. Lanes 1-4 contain 2 μg of full-length PDI, bb'xa', abb'x, and bb'x, respectively. The mobility of molecular mass standards is noted on the left.
Thermal melts were performed in order to determine the stability of each reduced and oxidized construct (Fig. 7). We found a bb'xa'c deletion construct lacking the a domain was less stable than full-length PDI, with a Tm of 50–51°C in the reduced state and 42°C in the oxidized state (Fig. 7A, Table 2). The oxidized form of bb'xa'c also exhibited a distinct thermal melt, with unfolding beginning immediately upon heating from 20°C and an apparent two-step unfolding transition with a first stage Tm of 30–33°C and a second stage Tm of 54–56°C (n = 2). In contrast to these results, an abb'x deletion construct lacking the a'c region was more stable than full-length PDI (Fig. 7B, Table 2). The reduced form of abb'x exhibited a Tm of 62–63°C, with a sharp transition from its native conformation to its final disordered state. The oxidized form of abb'x was substantially less stable than its reduced form, with a Tm of 51–52°C (Fig. 7B, Table 2). The oxidized forms of bb'xa'c and abb'x did not regain any structure after cooling from 90°C, while the reduced constructs gained some amount of α-helical structure after cooling. These collective results again demonstrated that the physical properties of PDI are linked to its redox state. Our observations also suggested a stabilizing role for the a domain and a destabilizing role for the a' domain in the PDI response to thermal stress, which has also been suggested by other investigators.21
Figure 7.

Thermal stability of PDI deletion constructs. Reduced or oxidized forms of bb'xa'c (A), abb'x (B), and bb'x (C) were heated stepwise from 20°C to 90°C, with CD measurements taken every 2°C. The proteins were then cooled from 90°C to 20°C in the same manner. Representative spectra are shown in the left panels of each column: the initial 20°C measurement (black line) the Tm measurement (dashed line), the 90°C measurement (dotted line), and the final 20°C measurement after cooling (grey line). The right panels of each column plot ellipticities at 222 nm as a function of temperature. Closed circles represent measurements taken during heating; open circles represent measurements taken during cooling. For oxidized bb'xa'c, the blue and black lines represent thermal unfolding profiles modeled from one-step and two-step transitions, respectively.
Like abb'x, a bb'x deletion construct lacking both a and a' domains was more stable than full-length PDI (Fig. 7C, Table 2). However, in comparison to abb'x, the oxidized and reduced forms of bb'x displayed less dramatic differences in stability: reduced bb'x exhibited a Tm of 58–60°C, and oxidized bb'x exhibited a 56°C Tm. Given the absence of a disulfide bridge in the b' domain of native PDI,14, 41 our results with the reduced form of bb'x likely reflect the physiological properties of this PDI region.
To examine how the individual domains in PDI contribute to its overall conformational resilience, each PDI deletion construct was subjected to transient thermal stress under reducing or oxidizing conditions. As previously performed with full-length PDI, we monitored the refolding of each deletion construct after 10 min at the lowest temperature that left the protein in a nearly complete state of denaturation. Thus, all constructs began at a common state of denaturation that allowed a direct comparison of the propensity for PDI refolding under various conditions. Heating bb'xa'c in either reduced or oxidized forms to 55°C left the deletion construct with 11–12% of its native structure. Upon cooling to 25°C, both reduced and oxidized bb'xa'c regained 90% of its native structure (Fig. 8A, Table 3). This demonstrated that the a domain is not essential for the conformational resilience (i.e., refolding) of PDI. Likewise, reduced abb'x had only 6% of its native α-helical structure remaining after 10 min at 65°C but regained 96% of its structure upon cooling to 25°C. Oxidized abb'x was less resilient than its reduced form, as it only regained 72% of its native structure after heating to 60°C (Fig. 8B, Table 3). These results indicated both the a and a' domains can refold after thermal denaturation, with a specific redox-dependent effect on the extent of a domain refolding.
Figure 8.

Conformational resilience of the PDI deletion constructs. CD spectra for bb'xa'c (A), abb'x (B), and bb'x (C) were measured at 25°C (black line) before heating to temperatures, listed in Table 3, that resulted in nearly complete denaturation. Spectra were measured again after 10 min at elevated temperature (dotted line) and after cooling to 25°C (grey line).
The bb'x construct was less resilient than bb'xa'c and reduced abb'x, yet it was extremely stable: temperatures of 95°C and 70°C were required for nearly complete denaturation of reduced and oxidized bb'x, respectively. These properties were consistent with the rigid nature of bb'x.18 Reduced bb'x was able to refold 74% after retaining only 8% of its original structure, while oxidized bb'x could refold 88% after retaining none of its native conformation (Fig. 8C, Table 3). Although the two cysteine residues in the b' domain of full-length human PDI do not form a disulfide bond,14, 41 the oxidation of unconserved, non-catalytic cysteine residues in the Humicola insolens PDI has been shown to enhance its refolding and stability.42 It is possible that removal of the a and a' domains in our bb'x construct allowed a disulfide bond to form between the b' cysteine residues, which would explain the redox-dependent changes seen for bb'x stability and resilience. Regardless, our collective data suggest bb'x confers a high degree of stability to PDI, while both a and a' domains further contribute to the conformational resilience of PDI.
DISCUSSION
The redox-dependent conformational changes to PDI and its conformational flexibility are well-established, but the conformational resilience of PDI has not been examined. Our previous work on PDI-CTA1 interactions documented the ability of PDI to refold after undergoing substrate-induced unfolding.29 Here, we report that PDI can also return to a functional conformation after thermal denaturation. The conformational resilience of PDI thus appears to derive from a general physical property rather than from a specific, toxin-induced effect. We further document how the functionally important conformational properties of PDI are modulated by its redox state and pinpoint the roles of specific domains in this relationship.
Our data demonstrate that rPDI is substantially more stable and resilient than oPDI. rPDI had a higher Tm than oPDI and, unlike oPDI, could regain some of its native conformation after a thermal melt that exposed it to extreme temperatures for over 3 hours. rPDI was also more resilient to transient thermal stress than oPDI, regaining 85% of its α-helical structure after a 10 min incubation at 65°C left it with just 2% of its native structure. Previous work has shown that the isolated a' domain of human PDI is more stable in the reduced state.43 Likewise, PDI from the thermophilic fungus H. insolens is more stable when its active sites are reduced.42 Here, we demonstrated that the redox state of full-length human PDI greatly affects both its stability and propensity for renaturation.
We hypothesize the thermal stability of PDI is affected by its flexibility and the dramatic conformational change that occurs between its oxidized and reduced states. rPDI adopts a “closed” conformation in which the x linker positions the a' domain in line with the rest of the protein.12, 21 This could stabilize its overall structure, as the a and a' domains of rPDI are only 27.6 Å apart in the crystal structure, and possibly even closer in soluble PDI.12, 18, 20 Conversely, the “open” conformation of oxidized PDI positions the a' domain at a 45° angle away from the protein core. This separates the a and a' domains by 40.3 Å in the crystal structure of PDI. The expanded structure of oPDI would likely have fewer inter-domain contacts than rPDI, which is consistent with fungal PDI,44 and would therefore be less stable than rPDI.
With our model, the redox-dependent positioning of the a' domain influences the extent of inter-domain contacts and, thus, the overall stability of PDI. However, the intrinsic properties of distinct regions within PDI could also influence its overall stability. This issue was addressed with a panel of deletion constructs that provided insight regarding which PDI domains contribute to its stability and resilience. The bb'x core of PDI confers a high degree of stability to PDI, which is consistent with the rigid nature of this region.18 The a and a' domains of PDI appear to play opposing roles in its thermal stability, with a stabilizing role for the a domain and a destablizing role for the a' domain. Full-length PDI is less stable than the abb'x deletion construct, which indicates the a domain cannot completely compensate for the destablizing effect of the a' domain. The a and a' domains also appear to exhibit redox-dependent properties, as both abb'x and bb'xa'c deletion constructs were less stable in the oxidized state than the reduced state. This is consistent with the observed destabilization of the isolated a' domain upon formation of a disulfide bond between its active site cysteines.43 Our collective studies thus indicate the overall stability full-length PDI is influenced by several factors, including redox-dependent changes to the three-dimensional shape of PDI; the stability of the bb'x region; a redox-linked stabilizing role for the a domain; and a redox-linked destabilizing role for the a' domain.
In contrast to their differing roles in stability, both the a and a' domains appear to facilitate the conformational resilience of PDI as demonstrated by the extensive refolding of both abb'x and bb'xa'c deletion constructs after their nearly complete denaturation. However, bb'xa'c exhibited a redox-independent response to transient thermal stress: the minimal temperature required for denaturation and the extent of refolding after denaturation were identical for both oxidized and reduced bb'xa'c. This redox-independent response was unique to bb'xa'c, which suggests the conformational resilience of PDI is regulated by oxidation of the a domain. The mechanism for this process is currently unknown, but a precedent for redox-dependent regulation of an individual PDI domain has already been established with the redox-dependent stability of the isolated a' domain.43
The stability of the bb'x region may influence the two-step thermal transitions of rPDI and obb'xa'c as well as the functions of full-length PDI. Although clearly manifested intermediate states in thermal melts have been observed for the full-length rPDI and the obb’xa’c construct (Figs. 1A, 7A), “hidden,” unresolved sequential transitions may be present in other PDI constructs.45 Such multi-step thermal transitions have been reported for other proteins and described in terms of sequential unfolding of protein domains45, 46 that may be displayed to different degrees depending on subtle changes in experimental conditions.47 The second, higher temperature transition of rPDI has a Tm of 61–63°C, which may reflect an individual transition of rbb’x with Tm = 58–60°C, and that of obb’a’c (Tm = 54–56°C) may be due to obb’x (Tm = 56°C). The values of thermal melting enthalpies, ΔH, should be considered as apparent, rather than absolute, values, as they have been obtained as best fit parameters for a constant value of ΔC = 0.39 kcal mol−1 K−1, consistent with earlier reports.30, 48 However, there is a general correlation between these apparent ΔH values and Tm, as expected.
The results of our insulin reduction assay suggest the substrate-binding bb'x region of PDI retains and/or regains sufficient structure after a transient 90°C heat treatment for continued binding to both Q3R and insulin. Our structural analysis of the PDI deletion constructs is consistent with this interpretation: the reduced bb'x region was highly resistant to transient thermal denaturation, with moderate resilience, while the oxidized bb'x region was moderately resistant to transient thermal denaturation but exhibited a high level of resilience/refolding (Table 3). Both conditions would allow PDI to bind insulin or Q3R after cooling from 90°C. It is possible that the active site cysteines in the a and a' domains of PDI can reduce insulin without a highly ordered secondary structure. It remains to be determined if these catalytic domains, which are less resistant but generally more resilient to thermal treatment than bb'x (Table 3), regain the thioredoxin-like fold after cooling from 90°C.
We have previously shown that a transient 90°C heat treatment of rPDI does not disrupt its binding to CTA1 but does prevent its disassembly of the CT holotoxin.40 The continued binding of heat-treated PDI to CTA1 is consistent with the stability/resilience of its bb'x region (Table 3) and the interaction of heat-treated PDI with insulin (Fig. 3). However, PDI heated to 90°C could still reduce insulin to some extent (Fig. 3) but could not disassemble CT.40 PDI could only disassemble CT after lower levels of thermal denaturation (Fig. 2C). The linked but independent enzymatic and chaperone functions of PDI thus appear to exhibit different levels of recovery from thermal denaturation, with PDI reductase activity exhibiting greater resilience. These collective observations also suggest the PDI-driven disassembly of CT requires a greater level of structure in its a and/or a' domains than is necessary for insulin reduction, which may only require a structured bb'x region.
The ER-localized pool of PDI exists in an equilibrium between its oxidized and reduced states, and these levels are in dynamic equilibrium due to the oxidoreductase activity of PDI.49 oPDI assists oxidative protein folding by creating and rearranging disulfide bonds in exchange for reducing its own. rPDI can then interact with additional substrates such as CTA1, or it can regenerate its active site disulfide bonds through an interaction with either Ero1p or oxidized glutathione.50, 51 Previous studies have determined about 15% of the ER-localized pool of PDI is oxidized,52 which indicates a large pool of stable, rPDI is consistently available to withstand stress conditions. This pool of PDI would likely remain functional after ER stress and could thus help rapidly restore homeostasis to the ER.
The conformational resilience of PDI may also play a direct role in its chaperone activity. We have suggested the unfolding/refolding cycle PDI exhibits in its interaction with CT represents a normal property of PDI that could be used to disrupt protein aggregates in the same way its substrate-induced unfolding leads to the physical displacement of CTA1 from the CT holotoxin.29 In support of this hypothesis, we found a transient thermal treatment that resulted in the nearly complete denaturation of PDI did not prevent it from returning to a functional conformaton that could still disassemble CT (Fig. 2). In fact, our data suggest the unfolding/refolding cycle may actually enchance PDI-driven CT disassembly. Repeated substrate interactions that lead to the unfolding and refolding of PDI could thus prime its chaperone-linked ability to block or possibly reverse protein aggregation. It should be noted, however, that PDI is not currently known to act as a disaggregase for any substrate. Further examination of this hypothesis will require the identification of an appropriate aggregation-prone substrate for the chaperone activity of PDI.
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the Florida Department of Health, Ed and Ethel Moore Alzheimer’s Disease Research Program Grant 8AZ12 and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI137056, both to KT. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Florida Department of Health or National Institutes of Health.
REFERENCES
- (1).Soares Moretti AI, and Martins Laurindo FR (2017) Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys 617, 106–119. [DOI] [PubMed] [Google Scholar]
- (2).Turano C, Coppari S, Altieri F, and Ferraro A (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J. Cell. Physiol 193, 154–163. [DOI] [PubMed] [Google Scholar]
- (3).Ali Khan H, and Mutus B (2014) Protein disulfide isomerase: a multifunctional protein with multiple physiological roles. Front. Chem 2, doi: 10.3389/fchem.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bulleid NJ, and Ellgaard L (2011) Multiple ways to make disulfides. Trends Biochem. Sci 36, 485–492. [DOI] [PubMed] [Google Scholar]
- (5).Kozlov G, Maattanen P, Thomas DY, and Gehring K (2010) A structural overview of the PDI family of proteins. FEBS J. 277, 3924–3936. [DOI] [PubMed] [Google Scholar]
- (6).Appenzeller-Herzog C, and Ellgaard L (2008) The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta 1783, 535–548. [DOI] [PubMed] [Google Scholar]
- (7).Karamzadeh R, Karimi-Jafari MH, Sharifi-Zarchi A, Chitsaz H, Salekdeh GH, and Moosavi-Movahedi AA (2017) Machine learning and network analysis of molecular dynamics trajectories reveal two chains of red/ox-specific residue interactions in human protein disulfide isomerase. Sci. Rep 7, 3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Denisov AY, Maattanen P, Dabrowski C, Kozlov G, Thomas DY, and Gehring K (2009) Solution structure of the bb' domains of human protein disulfide isomerase. FEBS J. 276, 1440–1449. [DOI] [PubMed] [Google Scholar]
- (9).Hatahet F, and Ruddock LW (2007) Substrate recognition by the protein disulfide isomerases. FEBS J. 274, 5223–5234. [DOI] [PubMed] [Google Scholar]
- (10).Klappa P, Ruddock LW, Darby NJ, and Freedman RB (1998) The b' domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cheung PY, and Churchich JE (1999) Recognition of protein substrates by protein-disulfide isomerase. A sequence of the b' domain responds to substrate binding. J. Biol. Chem 274, 32757–32761. [DOI] [PubMed] [Google Scholar]
- (12).Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, and Wang CC (2013) Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal 19, 36–45. [DOI] [PubMed] [Google Scholar]
- (13).Wang L, Wang X, and Wang CC (2015) Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med 83, 305–313. [DOI] [PubMed] [Google Scholar]
- (14).Wang C, Yu J, Huo L, Wang L, Feng W, and Wang CC (2012) Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a'. J. Biol. Chem 287, 1139–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yagi-Utsumi M, Satoh T, and Kato K (2015) Structural basis of redox-dependent substrate binding of protein disulfide isomerase. Sci. Rep 5, 13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Serve O, Kamiya Y, Maeno A, Nakano M, Murakami C, Sasakawa H, Yamaguchi Y, Harada T, Kurimoto E, Yagi-Utsumi M, Iguchi T, Inaba K, Kikuchi J, Asami O, Kajino T, Oka T, Nakasako M, and Kato K (2010) Redox-dependent domain rearrangement of protein disulfide isomerase coupled with exposure of its substrate-binding hydrophobic surface. J. Mol. Biol 396, 361–374. [DOI] [PubMed] [Google Scholar]
- (17).Hatahet F, and Ruddock LW (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal 11, 2807–2850. [DOI] [PubMed] [Google Scholar]
- (18).Freedman RB, Desmond JL, Byrne LJ, Heal JW, Howard MJ, Sanghera N, Walker KL, Wallis AK, Wells SA, Williamson RA, and Römer RA (2017) ‘Something in the way she moves': The functional significance of flexibility in the multiple roles of protein disulfide isomerase (PDI). Biochim. Biophys. Acta Proteins Proteom 1865, 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Römer RA, Wells SA, Emilio Jimenez-Roldan J, Bhattacharyya M, Vishweshwara S, and Freedman RB (2016) The flexibility and dynamics of protein disulfide isomerase. Proteins 4, 1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang S, Wang X, Cui L, Ding X, Niu L, Yang F, Wang C, Wang CC, and Lou J (2014) Compact conformations of human protein disulfide isomerase. PLoS ONE 9, e103472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang C, Chen S, Wang X, Wang L, Wallis AK, Freedman RB, and Wang CC (2010) Plasticity of human protein disulfide isomerase: evidence for mobility around the X-linker region and its functional significance. J. Biol. Chem. 285, 26788–26797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Heggelund JE, Bjornestad VA, and Krengel U (2015) Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond, in The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th edition (Alouf JE, Ladant D, and Popoff MR, Eds.) pp 195–229, Elsevier, Waltham, MA. [Google Scholar]
- (23).Taylor M, Banerjee T, Ray S, Tatulian SA, and Teter K (2011) Protein disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem 286, 22090–22100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tsai B, Rodighiero C, Lencer WI, and Rapoport TA (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948. [DOI] [PubMed] [Google Scholar]
- (25).Wernick NLB, Chinnapen DJ-F, Cho JA, and Lencer WI (2010) Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2, 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Majoul I, Ferrari D, and Soling HD (1997) Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 401, 104–108. [DOI] [PubMed] [Google Scholar]
- (27).Mekalanos JJ, Collier RJ, and Romig WR (1979) Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J. Biol. Chem 254, 5855–5861. [PubMed] [Google Scholar]
- (28).Orlandi PA (1997) Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem 272, 4591–4599. [PubMed] [Google Scholar]
- (29).Taylor M, Burress H, Banerjee T, Ray S, Curtis D, Tatulian SA, and Teter K (2014) Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathogens 10, e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pande AH, Scaglione P, Taylor M, Nemec KN, Tuthill S, Moe D, Holmes RK, Tatulian SA, and Teter K (2007) Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol 374, 1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Banerjee T, Pande A, Jobling MG, Taylor M, Massey S, Holmes RK, Tatulian SA, and Teter K (2010) Contribution of subdomain structure to the thermal stability of the cholera toxin A1 subunit. Biochemistry 49, 8839–8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nguyen VD, Saaranen MJ, Karala AR, Lappi AK, Wang L, Raykhel IB, Alanen HI, Salo KE, Wang CC, and Ruddock LW (2011) Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol 406, 503–515. [DOI] [PubMed] [Google Scholar]
- (33).Aslanidis C, and de Jong PJ (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Massey S, Banerjee T, Pande AH, Taylor M, Tatulian SA, and Teter K (2009) Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J. Mol. Biol 393, 1083–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tatulian SA (2013) Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy, in Lipid-Protein Interactions: Methods and Protocols (Kleinschmidt JH, Ed.) pp Chapter 9, pp. 177–218, Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- (36).Tatulian SA (2010) Structural analysis of proteins by isotope-edited FTIR spectroscopy. Spectroscopy Int. J. 24, 37–43. [Google Scholar]
- (37).Dupuy A, and Passam F (2019) Functional assays of thiol isomerase ERp5. Methods Mol. Biol 1967, 149–163. [DOI] [PubMed] [Google Scholar]
- (38).Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, Xue G, Yuan C, Furie BC, Flaumenhaft R, Huang M, and Furie B (2015) Quercetin-3-rutinoside inhibits protein disulfide isomerase by binding to Its b'x domain. J. Biol. Chem 290, 23543–23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, and Flaumenhaft R (2012) Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Invest 122, 2104–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cherubin P, Guyette J, Taylor M, O'Donnell M, Herndon L, Burress H, Riad A, Tatulian SA, and Teter K (2018) Protein disulfide isomerase does not act as an unfoldase in the disassembly of cholera toxin. Biosci. Rep 38, pii: BSR20181320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nguyen VD, Wallis K, Howard MJ, Haapalainen AM, Salo KE, Saaranen MJ, Sidhu A, Wierenga RK, Freedman RB, Ruddock LW, and Williamson RA (2008) Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b' domain. J. Mol. Biol 383, 1144–1155. [DOI] [PubMed] [Google Scholar]
- (42).Harada T, Kurimoto E, Tokuhiro K, Asami O, Sakai T, Nohara D, and Kato K (2001) Disulfide bond formation in refolding of thermophilic fungal protein disulfide isomerase. J. Biosci. Bioeng 91, 596–598. [DOI] [PubMed] [Google Scholar]
- (43).Darby NJ, and Creighton TE (1995) Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry 34, 11725–11735. [DOI] [PubMed] [Google Scholar]
- (44).Nakasako M, Maeno A, Kurimoto E, Harada T, Yamaguchi Y, Oka T, Takayama Y, Iwata A, and Kato K (2010) Redox-dependent domain rearrangement of protein disulfide isomerase from a thermophilic fungus. Biochemistry 49, 6953–6962. [DOI] [PubMed] [Google Scholar]
- (45).Varhač R, Sedláková D, Stupák M, and Sedlák E (2015) Non-two-state thermal denaturation of ferricytochrome c at neutral and slightly acidic pH values. Biophys. Chem 203–204, 41–50. [DOI] [PubMed] [Google Scholar]
- (46).Sherrer SM, Maxwell BA, Pack LR, Fiala KA, Fowler JD, Zhang J, and Suo Z (2012) Identification of an unfolding intermediate for a DNA lesion bypass polymerase. Chem. Res. Toxicol 25, 1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Malhotra P, and Udgaonkar JB (2016) How cooperative are protein folding and unfolding transitions? Protein Sci. 25, 1924–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Pande AH, Moe D, Jamnadas M, Tatulian SA, and Teter K (2006) The pertussis toxin S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. Biochemistry 45, 13734 –13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Margittai É, Enyedi B, Csala M, Geiszt M, and Bánhegyi G (2015) Composition of the redox environment of the endoplasmic reticulum and sources of hydrogen peroxide. Free Radic. Biol. Med 83, 331–340. [DOI] [PubMed] [Google Scholar]
- (50).Kulp MS, Frickel EM, Ellgaard L, and Weissman JS (2006) Domain architecture of protein-disulfide isomerase facilitates its dual role as an oxidase and an isomerase in Ero1p-mediated disulfide formation. J. Biol. Chem 281, 876–884. [DOI] [PubMed] [Google Scholar]
- (51).Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB, and Wang CC (2009) Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a' domains of protein-disulfide isomerase. J. Biol. Chem 284, 199–206. [DOI] [PubMed] [Google Scholar]
- (52).Appenzeller-Herzog C, and Ellgaard L (2008) In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid. Redox Signal 10, 55–64. [DOI] [PubMed] [Google Scholar]