

Summary
Pyridoxal 5′-phosphate (PLP) is a versatile cofactor that catalyzes a plethora of chemical transformations within a cell. Although many human PLP-dependent enzymes (PLP-DEs) with crucial physiological and pathological roles are known, a global method enabling their cellular profiling is lacking. Here, we demonstrate the utility of a cofactor probe for the identification of human PLP-binding proteins in living cells. Striking selectivity of human pyridoxal kinase led to a customized labeling strategy covering a large fraction of known PLP-binding proteins across various cancer-derived cell lines. Labeling intensities of some PLP-DEs varied depending on the cell type while the overall protein expression levels of these proteins remained constant. In addition, we applied the methodology for in situ screening of PLP-antagonists and unraveled known binders as well as unknown off-targets. Taken together, our proteome-wide method to study PLP-DEs in human cancer-derived cells enables global understanding of the interactome of this important cofactor.
Keywords: chemical proteomics, pyridoxal 5′-phosphate, cofactor, enzyme, mass spectrometry
Graphical Abstract
Highlights
-
•
Enrichment of human vitamin B6-binding proteins with cofactor-derived probes
-
•
In situ target screening of vitamin B6 antagonists
-
•
Comparison of human cell lines suggests cell-type-dependent cofactor loading states
Fux et al. applied cofactor mimics to study human pyridoxal 5′-phosphate (PLP)-dependent proteins in situ, covering a large fraction of known vitamin B6-binders. Moreover, cellular targets of anti-vitamin B6 compounds were unraveled. Comparison of different human cell lines resulted in signature labeling profiles, suggesting different cofactor loading states of PLP-binding proteins.
Introduction
Vitamin B6 refers to six interconvertible pyridine compounds: pyridoxine (PN), pyridoxamine (PM), pyridoxal (PL), and their 5′-phosphorylated versions (PNP, PMP, and PLP). The biologically active form is PLP, one of the most ubiquitous cofactors found in nature, that catalyzes around 238 different types of biochemical transformations, corresponding to 4% of all classified activities (Percudani and Peracchi, 2003). Although all living organisms rely on vitamin B6, only plants and microorganisms are able to synthesize the cofactor de novo. All other organisms, including humans, acquire vitamin B6 from nutrients and interconvert the vitamers to the active PLP form by a salvage pathway. Pyridoxal kinase (PLK), a ubiquitously expressed enzyme, phosphorylates PL, PM, and PN to PLP, PMP, and PNP. The latter two are subsequently oxidized to PLP by pyridoxine/pyridoxamine 5′-phosphate oxidase (PNPO) (Di Salvo et al., 2011). Finally, the cofactor is loaded onto PLP-dependent enzymes (PLP-DEs) via formation of a Schiff base (the internal aldimine) between the PLP aldehyde group and an ϵ-amino group of the active-site lysine (Toney, 2005).
PLP-DEs catalyze a diverse set of chemical reactions mainly with amino acid substrates, including decarboxylation, transamination, racemization, α, β, γ-substitution or α, β, γ-elimination (Percudani and Peracchi, 2003). Although sequence similarity is low among PLP-DEs, they are categorized into five distinct fold types guided by structural similarity (El-Sayed and Shindia, 2012). Due to the large number of chemical reactions catalyzed by PLP-DEs, they play a crucial role in cellular metabolism, ranging from amino acid biosynthesis and turnover to synthesis of neurotransmitters or tetrapyrroles (Parra et al., 2018). Therefore, dysregulated expression levels of PLP-DEs correlate with several diseases, which have sparked the development of inhibitors for these enzymes. For example, drugs targeting γ-aminobutyric acid (GABA) aminotransferase are used for the treatment of severe epilepsy (Amadasi et al., 2007). In addition, several human PLP-DEs, such as serine hydroxymethyltransferase (SHMT) (Renwick et al., 1998) or ornithine aminotransferase (OAT) (Zigmond et al., 2015), represent attractive targets for drug development due to their role in cell proliferation and tumor development. However, as all PLP-DEs share the same cofactor, the design of selective inhibitors is challenging.
We recently introduced a library of B6 cofactor mimics composed of alkyne- and azide-functionalized PL-probes to study the Staphylococcus aureus PLP-binding proteome (PLP-ome) (Hoegl et al., 2018). With this strategy, we were able to detect 73% of all annotated PLP-DEs in S. aureus and assign functions to previously uncharacterized proteins. Here, we customize our methodology for eukaryotic cells and thereby cover a large fraction of the human PLP-ome. We validated our results exemplarily for two PLP-DEs. As an example of application, we demonstrated the utility of our labeling strategy to screen for in situ targets of vitamin B6 antagonists, revealing insights into the selectivity of the examined compounds. Finally, we uncovered differences in labeling profiles among certain cell types that have the potential to inspire downstream applications for therapeutic purposes.
Results
Bioactivation of PL-Probes by Human PLK
As PL phosphorylation is the limiting step for generating the biologically active form of vitamin B6 (Figure 1A), we first monitored the turnover of PL-probes (Figure 1B) by human PLK (hPLK). PL-probes were synthesized as described previously (Hoegl et al., 2018) with slight modifications in the synthetic route. We purified hPLK as a dimer (Cao et al., 2006) (Figure S1A) and subsequently utilized the recombinant enzyme to record PL turnover at 392 nm, corresponding to the absorbance maxima of PLP (Figures 1C and S1B). The calculated Km value of about 2.16 μM for natural PL is in good agreement with previous observations (Figures 1D and S1C) (Kimura et al., 2014). The absorbance maxima of the probes were slightly redshifted, with PL1P (λmax = 416 nm) exhibiting the most pronounced change compared with PLP. Although all PL-probes were phosphorylated by hPLK, the corresponding catalytic efficiencies varied significantly. For example, the Km value of PL1, bearing a short substitution, increased about 10-fold, while its catalytic efficiency was only about 3-fold reduced compared with the natural vitamer (Figures 1D and S1D). In contrast, the longer 2′-substitution of PL2 and PL3 prevented efficient turnover by hPLK (Figures 1D, S1E, and S1F). Interestingly, S. aureus PLK (SaPLK) was able to phosphorylate PL2 and PL3 at levels comparable with PL (Hoegl et al., 2018), suggesting that distinct catalytic mechanisms and active-site geometries account for these differences. This is supported by previous work showing that in SaPLK, PL forms a hemithioacetal with a cysteine located in a flexible lid and remains covalently linked during the catalytic cycle (Nodwell et al., 2014). In contrast, in hPLK, an aspartate positions PL exclusively through hydrogen bonds and hydrophobic interactions (Cao et al., 2006, Gandhi et al., 2009). Thus, these distinct evolutionary paths of substrate interaction have tailored a more flexible accommodation of larger 2′ substitutions in SaPLK and more restrictive binding in hPLK. As PL1 is effectively phosphorylated by hPLK, we proceeded with our in situ profiling studies in human cell lines using this probe.
Figure 1.

Bioactivation of PL and PL-probes by hPLK
(A) To generate biologically active vitamin B6, PL is phosphorylated to PLP by hPLK inside the cell. PLP is then bound as an internal aldimine to PLP-DEs.
(B) Chemical structures of PL and PL1–PL3.
(C) UV-vis spectra of PLP and PLP-probes. Curves were recorded for equal vitamer and probe concentrations.
(D) Kinetic parameters (n = 3 biological replicates; data presented as means ± SEM) for PL and PL-probe turnover.
See also Figure S1.
Metabolic Labeling in HEK293 with PL1 Provides Access to a Large Fraction of the Human PLP-ome
As previous experiments in bacteria demonstrated a superior labeling efficiency upon supplementation of growth media with PL-probes (Hoegl et al., 2018), we cultivated human adenovirus-transfected embryonic kidney cells (HEK293) in the presence of PL1. We maintained cells for seven passages on PL1-containing media lacking natural B6 vitamers to allow for complete turnover of the proteome (Ong, 2002) and therefore accumulation of PL1P in PLP-DEs. We included HEK293 cells passaged in defined complete media with natural PL as control. To account for the lower concentrations (5 μM) of natural PL in standard cell culture media and the increased Km value of the probe, we cultured cells in the presence of 10 μM PL1. Notably, the doubling rate and morphology of HEK293 was not affected by incubation with PL1 compared with the PL control, indicating that the probe supports essential functions of natural vitamin B6. To exclude that remaining intracellular vitamin B6 levels may impair the labeling results, we relatively quantified PL and PLP levels over the passaging time. Satisfyingly, concentrations of endogenous vitamers decreased markedly after three passages with no PL and PLP left at the time point of the proteomic experiments (seven passages in PL1-containing media; Figure 2A).
Figure 2.

Mining Human PLP-DEs with PL1 in HEK293
(A) Targeted metabolomics analysis of PL and PLP levels in HEK293 cells. Concentrations of both vitamers decreased over time in media with PL1 (n = 4 biological replicates). No endogenous B6 vitamers were present at the time point of labeling after seven passages.
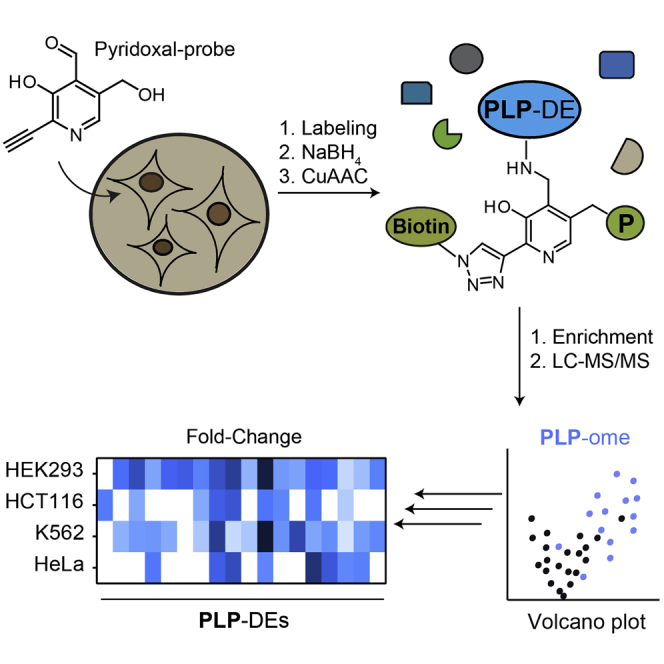
(B) Labeling strategy using PL1 and PL as a control. Cells were passaged in media containing either PL1 or PL. Following cell lysis, the internal aldimine of PLP-DEs was reduced with sodium borohydride (NaBH4) and proteins were subjected to CuAAC to attach biotin. After avidin enrichment, proteins were digested with trypsin for subsequent liquid chromatography-tandem mass spectrometry analysis (P = OPO32−).
(C) Volcano plot representing the t test results of PL1 labeling in HEK293 compared with the PL control revealed significant enrichment of many PLP-DEs (blue) (n = 6 biological replicates). Cut-off values were defined as an enrichment factor of log2 = 1 (2-fold enrichment) and −log10 (p value) of 1.3 (solid lines).
See also Table S1.
Cells were lysed in the presence of sodium borohydride (NaBH4) to attach the probe irreversibly to target proteins by reductive amination. Subsequent bioorthogonal ligation of the alkyne tag to biotin-azide using copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC, click chemistry) allowed for enrichment of probe-bound proteins on avidin beads (Rostovtsev et al., 2002, Speers and Cravatt, 2004, Tornøe et al., 2002). After tryptic digest, peptides were analyzed using liquid chromatography combined with tandem mass spectrometry (LC-MS/MS) and compared with the PL-cultured control using label-free quantification (LFQ) (Figure 2B) (Cox et al., 2014). We detected 15 PLP-DEs (GO term 0030170: PLP binding; Ashburner et al., 2000) significantly enriched upon PL1 labeling in HEK293 compared with the control (Figure 2C and Table S1), which accounts for around 30% of the complete human pool of PLP-binding enzymes (53 reviewed proteins annotated with GO term 0030170: PLP binding within the human proteome [UniProt: 9606]; Ashburner et al., 2000), including members of fold types I, II, and IV (El-Sayed and Shindia, 2012). Several PLP-DEs related to cancer proliferation and tumor development (e.g., SHMT1; Renwick et al., 1998 or cytosolic aspartate aminotransferase, GOT1; Thornburg et al., 2008) or to Parkinson disease (Bras et al., 2008) and atherosclerosis (Park et al., 2008) (serine palmitoyltransferase 2, SPTLC2, and sphingosine-1-phosphate lyase 1, SGPL1) were identified. These therapeutically relevant proteins are currently being targeted through the development of selective inhibitors (Ducker et al., 2017, Lowther et al., 2010). Interestingly, pyridoxal 5′-phosphate binding protein (PLPBP), a PLP-DE with an unknown cellular function, displayed the highest fold change by comparing LFQ intensities of PL1 labeling with the PL control.
PL1P Is the Species Bound to PLP-DE Active Sites
In order to validate binding of PL1P to PLP-DEs, we exemplarily selected the well-studied anti-cancer target SHMT1 as well as PLPBP, the strongest enriched protein upon PL1 labeling. Both enzymes are highly conserved throughout evolution and expressed in all tissues (Ikegawa et al., 1999, Renwick et al., 1998). SHMT1 catalyzes the conversion of serine and tetrahydrofolate (THF) into glycine and 5,10-methylene-THF (Ducker et al., 2017), which donate carbon for the de novo synthesis of purine and pyrimidine nucleotide bases for DNA replication (Snell et al., 1988). PLPBP was shown to bind PLP, but no enzymatic activity toward any of the 20 proteinogenic amino acids or their corresponding d-enantiomers was detected (Ito et al., 2013).
Inspection of the active-site pocket of SHMT1 with bound PLP (PDB: 1BJ4; Renwick et al., 1998) indicates sufficient space for the additional alkyne group of PL1 (Figure 3A). To test PL1P binding, recombinant SHMT1 was purified as the holoenzyme and transformed to the apo-form by nucleophilic displacement of bound PLP using hydroxylamine (Demoss, 1962). Internal aldimine absorbance re-appeared upon reconstitution of apo-SHMT1 with PL1P and PLP as a control (SHMT1-PL1P and SHMT-PLP) (Figure 3B). Notably, the absorbance maximum of SHMT1-PL1P was redshifted compared with holo-SHMT1 and SHMT1-PLP, which is in line with changes observed for unbound PL1P. Thermal denaturation studies revealed a selective increase in melting temperature of apo-SHMT1 upon incubation with increasing PLP amounts, whereas the un-phosphorylated vitamer did not show these characteristics (Figure 3C, left). PL1P also resulted in significant stabilization of the apo-enzyme, suggesting that probe and natural vitamin B6 behave similarly (Figure 3C, right). Finally, apo-SHMT1 was incubated with PL1 or PL1P, the corresponding internal aldimines were reduced with NaBH4, and the protein-bound alkynes conjugated to a Rhodamine-azide tag via CuAAC. SDS-PAGE with fluorescence readout revealed specific and concentration-dependent labeling of SHMT1 with PL1P and only weak background labeling when incubated with PL1 (Figure 3D). This demonstrates that only the phosphorylated version of the probe can bind the enzyme.
Figure 3.

PL1P Is Bound to Recombinant Human SHMT1
(A) Active-site pocket of SHMT1 with corresponding distances to residues surrounding the 2′-methyl group of PLP (PDB: 1BJ4; Renwick et al., 1998).
(B) UV/Vis spectra of holo- and apo-SHMT1 reconstituted with PLP or PL1P (SHMT1-PLP, SHMT1-PL1P) showing internal aldimine absorbance (P = OPO32−).
(C) Thermal denaturation studies of apo-SHMT1 revealed a selective stabilization upon PLP (left) and PL1P (right) incubation (n = 3, data presented as means ± SEM).
(D) In vitro labeling of recombinant apo-SHMT1 displayed selective and concentration-dependent labeling by PL1P.
See also Figure S2.
Next, we expressed and purified poorly characterized recombinant human PLPBP. The protein is predominantly dimeric even under reductive conditions, indicating that dimerization is not caused by a disulfide bridge (Figure S2A). To validate binding of PL1P to PLPBP, we also performed thermal stability assays and in vitro labeling. We observed a melting temperature shift of the dimer from 57°C to around 66°C upon incubation with increasing amounts of PLP, whereas no changes were observed upon incubation with PL (Figure S2B, left). As for SHMT1, similar results were obtained with PL1 and PL1P, where only PL1P led to stabilization (Figure S2B, right). In line with thermal stability assays, we detected a concentration-dependent fluorescence increase exclusively for PL1P, whereas only weak background labeling was observed for PL1 (Figure S2C). PLPBP monomer behaved similarly (Figures S2D and S2E). These validation results are in line with previous experiments using recombinant S. aureus alanine racemase (Hoegl et al., 2018).
Finally, to monitor whether the probe binds to the active-site lysine residues of PLP-DEs, we exemplarily incubated PL1P-bound PLPBP with increasing amounts of the natural cofactor (Figure S2F). Satisfyingly, labeling was outcompeted by a 10-fold molar excess of PLP over probe, validating its binding into the active-site pocket.
In Situ Target Screening of Vitamin B6 Antagonists
As inhibitor development for PLP-DEs is often challenged by conserved cofactor binding sites, in situ target screening of vitamin B6 antagonists is an important step to characterize their cellular selectivity. We previously applied our cofactor mimics to uncover new off-targets of d-cycloserine (DCS) in S. aureus (Hoegl et al., 2018). To adapt this selectivity screen for human cells, we exemplarily selected the two enantiomers of the PLP-DE inhibitor penicillamine (Pen), which display poorly characterized cellular specificities and thus represent excellent candidates for competition experiments in PL1-labeled HEK293.
Pen is a degradation product of penicillins (Weigert et al., 1975), which is therapeutically applied in the treatment of rheumatoid arthritis (Jaffe, 1964) and Wilsons' disease (Walshe, 1956). Although the d-enantiomer is the natural occurring isomer, both configurations of Pen display an anti-vitamin B6 effect (Kuchinskas et al., 1957, Rumsby and Shepherd, 1981, Takahashi and Matsuda, 1976). Several PLP-DEs, including cystathionine γ-lyase (CTH) (Brancaleone et al., 2016), SHMT1 (Sukanya et al., 2008), and GOT1 (du Vigneaud et al., 1957), were shown to be inhibited by Pen in vitro. However, our competitive proteomics studies in HEK293 using l- and d-Pen revealed only a limited number of PLP-DE targets (Figures 4A and 4B, Table S2), suggesting a higher selectivity in situ. The cofactor is displaced by Pen via formation of a thiazolidine adduct (Figure 4C) (Heyl et al., 1948). Both enantiomers show a similar target scope, including PLPBP and SGPL1. Contrary to previous postulations, the primary target within the H2S pathway is cystathionine β–synthase (CBS) instead of CTH (Brancaleone et al., 2016). Interestingly, mitochondrial cysteine desulfurase (NFS1) competed exclusively with l-Pen, indicating an absolute configuration-dependent selectivity. To validate our competition results, we incubated recombinant PLPBP with increasing concentrations of l- and d-Pen. UV-vis spectra displayed a decrease in absorbance of the internal aldimine (Figures 4D and S2G), confirming that Pen indeed binds to PLPBP.
Figure 4.

In Situ Target Screening of Penicillamine
(A and B) PL1-labeled HEK293 cells were incubated with 10 mM of l-Pen (A) and d-Pen (B) for 2 h, respectively. Volcano plots representing t test results of the competition compared with the PL1-labeled control revealed significant depletion of PLP-DEs (blue) (n = 3 biological replicates). Cut-off values were defined as the depletion factor of log2 = −1 (2-fold depletion) and −log10 (p value) of 1.3 (solid lines).
(C) Mechanism of thiazolidine adduct formation of Pen with PLP (P = OPO32−).
(D) UV-vis spectra of PLPBP dimer incubated with increasing concentrations of l-Pen.
Expanding Metabolic Labeling to Study Cell-type-Dependent Targeting Profiles
With a validated PLP-DE profiling tool for human cells in hand, we determined if labeling of this enzyme class across various cell lines would reveal individual signature profiles. For this, we cultivated human cancer-derived cell lines K562 (chronic myelogenous leukemia), HeLa (cervix epithelioid carcinoma), as well as HCT116 (colorectal carcinoma) in the presence of PL1. Satisfyingly, we obtained significant enrichment of several PLP-DEs in all cell lines examined (Figure S3, Table S1). However, throughout all experiments, we observed a certain amount of non-PLP-DE background binders, which are not annotated as putative PLP-DEs. We selected glucosylceramidase (GBA) as common non-vitamin B6-dependent protein detected in all cell lines to evaluate putative PLP-dependency (Figure S4A). Neither a characteristic internal aldimine peak at around 430 nm (Figure S4B) nor substantial stabilization of GBA upon incubation with PLP and PL1P, which is a characteristic feature of PLP-DEs, was observed (Figure S4C), indicating that GBA is most likely bound to the probe in an unspecific manner. Indeed, PLP exhibits a highly reactive aldehyde, which can in principle bind to any ϵ-amino group on reactive lysine residues (Phillips, 2015). In fact, several of the significantly enriched non-PLP-binders carry nucleophilic lysine residues as identified by an amine-reactive probe (Hacker et al., 2017).
Taken together, we were able to enrich 18 PLP-DEs, which account for 34% of the complete human PLP-ome. Interestingly, differences in the number and fold changes of significantly enriched PLP-DEs were observed dependent on the type of cell (Figure 5A, Table S1). Enzymes such as molybdenum cofactor sulfurase (MOCOS), sphingosine-1-phosphate lyase 1 (SGPL1), or PLPBP were enriched throughout all cell lines studied. Remarkably, other PLP-DEs were exclusively detected by our method in particular cell lines, such as mitochondrial 5-aminolevulinate synthase (ALAS1) solely in HCT116 or mitochondrial 2-amino-3-ketobutyrate coenzyme A ligase (GCAT) in HEK293. This is an intriguing finding as expression levels of PLP-DEs as well as vitamin B6 salvage enzymes PLK and PNPO were comparable throughout all cell lines (Figures 5B, 5C, and S5) (Schmidt et al., 2018), indicating that the level of cofactor binding is cell-type specific and does not necessarily correlate with protein abundance. This was corroborated by a PLP-ome analysis with an anti-PL antibody (Whittaker et al., 2015). First, we monitored the specificity of the antibody toward reduced PLP-DEs. Satisfyingly, NaBH4-treated PLPBP and SHMT1 were recognized with only a slight signal resulting from the non-reduced PLP-DEs, whereas hPLK, which does not covalently bind the cofactor, was not detected at all (Figures 5D, left and S5G). We proceeded with the analysis of NaBH4-treated and non-treated proteomes (Figures 5D, right and S5H). In line with the chemoproteomic data, we observed different cofactor loading states dependent on the type of cell for the reduced samples, whereas only slight labeling was observed for the non-reduced control, further verifying antibody specificity. Notably, for HeLa cells, we observed the lowest number of PLP-DEs in both the proteomic and the western blot analysis. This result further supports cell-type specific alterations in PLP-DEs cofactor loading sates independent of labeling with the probe, which might indicate a so far unrecognized means of enzyme regulation.
Figure 5.

Comparison of PL1-Labeling Profiles in Various Human Cell Lines
(A–C) (A) Heatmap illustrating the corresponding fold changes (log2PL1/PL) of PLP-DEs in the different cell lines. The map depicts only PLP-DEs that are significantly enriched in at least one cell line. In the case that a PLP-DE was not detected in samples of a certain cell line, the corresponding rectangle is colorless. Western blots against the PLP-DEs ALAS1, PLPBP, SHMT1, and SHMT2 (B), as well as vitamin B6 salvage enzymes PNPO and hPLK (C) revealed a similar expression of corresponding proteins in all cell lines studied.
(D) PLP-ome analysis using an anti-PL antibody raised against reduced cofactor in conjugation with BSA. Antibody specificity was validated upon incubation with hPLK (1), which does not covalently bind PLP, NaBH4-reduced forms of PLPBP (2), and SHMT1 (3), as well as the non-reduced PLPBP (4) and SHMT1 (5) counterparts (left). Western blot analysis of cofactor loading states of PLP-DEs in NaBH4-treated as well as not reduced proteomes (right).
See also Figures S3–S5 and Table S1.
Discussion
PLP-DEs play crucial physiological and pathological roles in human cells. Here, we customize the application of cofactor mimics to the study of the human PLP-ome in living cells. We observed altered metabolic conversion of the probes by hPLK compared with previous studies with SaPLK, and only probe PL1, with a minimal alkyne substituent, could be efficiently phosphorylated. Despite this restricted scope and the intrinsic electrophilicity of the alkyne linker in PL1, which is prone to nucleophilic attack by cellular cysteines (Lehmann et al., 2016), the probe significantly enriched a large fraction of the human PLP-ome in HEK293, including several clinically relevant PLP-DEs such as SHMT1 and little characterized proteins such as PLPBP. Both proteins were subjected to in-depth studies validating that PL1P is the species bound to recombinant PLP-DEs. The methodology was further utilized for in situ target screening of both enantiomers of the PLP-DE inhibitor Pen, demonstrating an unexpected high cellular PLP-DE selectivity contrary to the results of previous in vitro studies and opening new perspectives for therapeutic applications. Finally, we compared PLP-DE labeling in cancer-derived cell lines K562, HCT116, and HeLa with labeling in HEK293. Interestingly, we observed signature profiles depending on the cell type although corresponding PLP-DEs were ubiquitously expressed. Such changes in cofactor loading are largely unknown and highlight the utility of the probe for a direct readout. This was also supported by an antibody-based PLP-ome analysis of unmodified cells, revealing differences in vitamer loading supporting chemoproteomic data, which might display a mechanism of cell-type-specific enzyme regulation.
Significance
Many essential cellular processes such as glucose, lipid, and amino acid metabolism are driven by PLP-DEs. Thus, mining the human PLP-ome represents an attractive strategy for accessing a large and important enzyme class associated with multiple physiological and pathological functions. We show that probe design, including the chemical positioning and length of the bioorthogonal tag, is a crucial determinant for the success of labeling, which, contrary to bacteria, does not work efficiently for hPLK if the tag is too large. We figured out that a probe with a small modification is able to utilize native PL uptake systems to be incorporated across a significant portion of cellular PLP-DEs. Trapping the transient binding state within PLP-DE active sites permitted labeling and downstream identification of cancer-associated PLP-DEs. Interestingly, individual enzymes largely varied in the cofactor loading status across the panel of cells studied. Furthermore, application of the probe in a competitive mode revealed targets and off-targets of human PLP-DE inhibitors, which highlight this methodology as a general approach to test for inhibitor selectivity in drug development. Thus, our studies not only provided unprecedented insights into the human PLP-ome but also allowed direct monitoring of cofactor loading as well as inhibitor off-targets.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PLPBP antibody (clone 1G2) | OriGene | Cat# TA505162; RRID: AB_2622886 |
| Rabbit polyclonal anti-SHMT2 antibody | abbexxa | Cat# abx128462 |
| Rabbit polyclonal anti-PNPO antibody | Sigma | Cat# HPA023204; RRID: AB_1855506 |
| Rabbit polyclonal anti-ALAS1 antibody | Thermo Fisher Scientific | Cat# PA5-57434; RRID: AB_2637832 |
| Rabbit polyclonal anti-SHMT1 antibody | abcam | Cat# ab55736; RRID: AB_2285970 |
| Goat anti-rabbit antibody conjugated to horseradish peroxidase | invitrogen | Cat# 32260 |
| Goat anti-mouse antibody conjugated to horseradish peroxidase | invitrogen | Cat# 32230 |
| Mouse monoclonal anti-PLK antibody | Santa Cruz Biotechnology, Inc. | Cat# sc-365173, RRID: AB_10708566 |
| Rabbit polyclonal anti-PL antibody | GeneTex | Cat# GTX12625 |
| Bacterial and Virus Strains | ||
| Escherichia coli Rosetta 2 (DE3) | Merck | Cat# 71400 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PL1 | Hoegl et al., 2018 | N/A |
| PL2 | Hoegl et al., 2018 | N/A |
| PL3 | Hoegl et al., 2018 | N/A |
| Glycosylceramidase (GBA) | ProSpec | Cat# enz-908 |
| Lysyl Endopeptidase | Wako | Cat# 125-05061 |
| Trypsin, Sequencing grade | Promega | Cat# V5111 |
| l-Penicillamine | TCI | Cat# P1370 |
| d-Penicillamine | Acros Organics via Fisher Scientific | Cat# 10224750 |
| BTTAA ligand | Jena Bioscience | Cat# CLK-067-25 |
| Critical Commercial Assays | ||
| Roti Quant Universal | Roth | Cat# 0120.1 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| ECI western blotting substrate solution | Pierce | Cat# PIER80196 |
| Ponceau S | Sigma | Cat# P3504 |
| Deposited Data | ||
| raw files, Fasta files, and MaxQuant analysis to PRIDE | This manuscript | https://www.ebi.ac.uk/pride/archive/; PXD014771 |
| Experimental Models: Cell Lines | ||
| Human HeLa cell line | ECACC via Sigma Aldrich | Cat# 93021013; RRID: CVCL_0030 |
| Human K562 cell line | ECACC via Sigma Aldrich | Cat# 89121407; RRID: CVCL_0004 |
| Human HCT116 cell line | ECACC via Sigma Aldrich | Cat# 91091005; RRID: CVCL_0291 |
| Human HEK293 cell line | ECACC via Sigma Aldrich | Cat# 85120602; RRID: CVCL_0045 |
| Oligonucleotides | ||
| hPLK forward primer (5’->3’): ggggacaagtttgtacaaaaaa gcaggctttgagaatctttattttcagggcgaggaggagtgccgggtg |
eurofins | custom made |
| hPLK reverse primer (5’->3’): ggggaccactttgtacaagaaa gctgggtgtcacagcaccgtggcctgg |
eurofins | custom made |
| SHMT1 forward primer (5’->3’): ggggacaagtttgtacaaaaa agcaggCtttgagaatctttattttcagggcacgatgccagtcaacgggg |
eurofins | custom made |
| SHMT1 reverse primer (5’->3’): ggggaccactttgtacaagaaa gctgggtgtcagaagtcaggcaggccag |
eurofins | custom made |
| PLPBP forward primer (5’->3’): ggggacaagtttgtacaaaaaa gcagGctttgagaatctttattttcagggctggagagctggcagcatgtcg |
eurofins | custom made |
| PLPBP reverse primer (5’->3’): ggggaccactttgtacaagaaa gctgggtgtcagtgctcctgtgccacctc |
eurofins | custom made |
| Recombinant DNA | ||
| Human cDNA library from HeLa | BioAcademia | Cat# 02-723 |
| SHMT1 ORF clone, NM_004169 | GeneScript | Cat# OHu21374 |
| PLPBP ORF clone, NM_007198.3 | GeneScript | Cat# OHu09065 |
| SHMT1_pDest007 | This manuscript | Addgene ID: 131229 (http://www.addgene.org) |
| hPLK_pDest007 | This manuscript | Addgene ID: 131230 (http://www.addgene.org) |
| PLPBP_pDest007 | This manuscript | Addgene ID: 131231 (http://www.addgene.org) |
| Software and Algorithms | ||
| MaxQuant software | MPI Biochemistry Martinsried | http://www.biochem.mpg.de/5111795/maxquant |
| Perseus software | MPI Biochemistry Martinsried | http://www.biochem.mpg.de/5111810/perseus |
| Gene Ontology annotation file | Ashburner et al., 2000 | http://geneontology.org/page/download-go-annotations |
| Prism 6 software | GraphPad | RRID: SCR_002798 |
| Protein Deconvolution Software | Thermo Fisher Scientific | Cat# IQLAAEGAB SFANOMBAQ |
| Other | ||
| InfiniteM200 PRO reader | TECAN | Cat# IN-MNANO |
| StrepTrap HP column | GE Healthcare | Cat# 28-9075 |
| Superdex 200 10/300 GL | GE Healthcare | Cat# 17517501 |
| Superdex 75 10/300 GL | GE Healthcare via Sigma Aldrich | Cat# GE17-5174-01 |
| HiTrap Desalting column | GE Healthcare | Cat# 17-1408-01 |
| SYPRO orange protein gel stain | Thermo Fisher Scientific | Cat# S6650 |
| CFX96 Real-Time System | Bio-Rad | Cat# 20421 |
| Roti-PVDF, 0.2 μm | Roth | Cat# 8989.1 |
| Trans-Bot SD Semi-Dry Transfer Cell | Bio-Rad | Cat# 1703940 |
| Luminescent LAS 4000 image analyzer | Fujifilm, ordered via GE Healthcare | Cat# 28955810 |
| Sep-Pak C18 columns | Waters | Cat# WAT054960 |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephan A. Sieber (stephan.sieber@tum.de). Plasmids generated in this study have been deposited to Addgene (http://www.addgene.org, Deposit No. 77114; IDs 131231, 131230, 131229).
Experimental Model and Subject Details
Cell Lines
All cell lines were obtained from ECACC via Sigma Aldrich. K562 (female, Cat# 89121407; RRID: CVCL_0004), HEK293 (female, Cat# 85120602; RRID: CVCL_0045), and HCT116 (male, Cat# 91091005; RRID: CVCL_0291) were cultivated in RPMI-1640, HeLa (female, Cat# 93021013; RRID: CVCL_0030) in DMEM media supplemented with 10% l-glutamine and 10% FCS at 37°C and humidified 5% CO2 atmosphere. The cells were routinely tested for mycoplasma contamination.
Microbe Strains
For protein expression, Escherichia coli Rosetta2 (DE3) (Merck, Cat# 71400) were grown in lysogeny broth (LB)-media containing ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) at 37°C and with shaking at 200 rpm until reaching the exponential phase (OD600nm 0.6-0.8).
Method Details
Chemical Synthesis
Synthesis of PL2 and PL3 was performed as described previously (Hoegl et al., 2018). Synthesis of PL1 was performed as described with slight modifications. PN hydrochloride was protected with TBS-Cl. Meta-chloro perbenzoic acid (m-CPBA) (77%, 3.86 g, 17.2 mmol, 1.2 eq) was added to a solution of TBS-protected PN (8.00 g, 15.7 mmol, 1.0 eq) in anhydrous dichloromethane (DCM, 150 mL) at 0°C in five portions. After addition, the reaction was stirred vigorously at r.t for 2 h. Then, the reaction mixture was washed with 20% sodium sulfite solution (1×80 mL) and saturated sodium bicarbonate solution (1×80 mL), dried over sodium sulfate and concentrated to yield crude TBS-protected N-oxide quantitatively as a white solid, which was used directly in the next step.
Trifluoroacetic anhydride (TFAA, 10.3 mL, 15.5 g, 73.9 mmol, 5.0 eq) was slowly added to a solution of crude TBS-protected N-oxide (7.80 g, 14.8 mmol, 1.0 eq) in anhydrous DCM (120 mL) via syringe at 0°C. The ice bath was removed and the reaction was stirred overnight. Then, anhydrous MeOH (30 mL) was slowly added at 0°C and the reaction was stirred for 20 min and subsequently allowed to warm to r.t. while stirring for another 20 min. The mixture was diluted with DCM (200 mL) and neutralized with saturated sodium bicarbonate solution (1×200 mL). After separation, the organic phase was dried over sodium sulfate and concentrated under reduced pressure. The residue obtained was purified by flash chromatography (EtOAc/hexanes, 10-50%) to yield TBS-protected diol (3.64 g, 8.80 mmol, 60%) as a pale yellow solid. All further steps were carried out as described (Hoegl et al., 2018).
Biological Methods
Cloning and Overexpression of Proteins
GBA was purchased from ProSpec (Cat# enz-908). N-terminally Strep-II tagged hPLK, SHMT1, and PLPBP (Addgene IDs No.: 131231, 131230, 131229) were cloned and expressed using the primers and conditions listed in Table S3. Cloning was performed using the Invitrogen Gateway cloning system with pDONR201Kan as the donor vector and pDest007Amp as the destination vector. E. coli Rosetta2 (DE3) (Merck, Cat# 71400) carrying the expression plasmids were cultured as described and expression was induced through the addition of 0.2 μg/mL anhydrotetracycline (ATET). Bacteria were harvested and washed with PBS (6,000 x g, 4°C) prior to cell lysis and protein purification.
Purification and Analytics of Proteins
Buffer compositions, as well as columns used for purification are listed in Table S4. The bacterial overexpression culture was resuspended in strep binding buffer and lysed by sonication. The lysate was clarified by centrifugation (36,000 x g, 30 min, 4°C). Supernatant was loaded onto a StrepTrap HP column (5 mL, GE Healthcare, Cat# 28-9075) equilibrated with binding buffer using an Äkta purification system (GE Healthcare). After extensive washing, proteins were eluted in binding buffer containing 2.5 mM desthiobiotin. For preparation of apo-SHMT1, the column was washed with 20 mL binding buffer containing 25 mM hydroxylamine prior to elution. SEC was applied for further purification. Proteins were loaded onto columns equilibrated with SEC buffer. Oligomeric state of the proteins was determined according to calibration curves. Concentration of hPLK was calculated according to Lambert-Beer’s law from the absorbance at 280 nm recorded in an InfiniteM200 PRO reader (TECAN, Cat# IN-MNANO) and an extinction coefficient of 40,000 M-1⋅cm-1 at 280 nm (calculated with ProtParam). Concentrations of holo- and apo-SHMT1 were determined with the Bradford assay (Roti Quant Universal, Roth, Cat# 0120.1) and of PLPBP with the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Cat# 23225). Molecular weights of the proteins were confirmed by intact-protein MS (Table S4). Proteins were stored at -80°C in small aliquots. UV/Vis spectra of protein samples (100 μM) were recorded in corresponding SEC buffers or in phosphate buffered saline (PBS) in the case of GBA at 37°C on an InfiniteM200 PRO reader (TECAN, Cat# IN-MNANO) (300-600 nm, 2 nm increments).
hPLK Kinetic Assay
PL-probes were phosphorylated using SaPLK as described previously (Hoegl et al., 2018). UV/Vis-spectra of PL and PL-probes, as well as their phosphorylated counterparts were recorded in assay buffer (20 mM HEPES pH 7.0, 100 mM KCl, 1 mM MgCl2) at 37°C using an InfiniteM200 PRO reader (TECAN, Cat# IN-MNANO). The initial rate of product formation was measured by following the increase in absorbance at the maxima of PLP (392 nm) and PLP-probes (PL1P: 416 nm, PL2P: 396 nm, PL3P: 400 nm). A typical assay contained 0.25 μM hPLK in assay buffer and activity was monitored at 37°C using an InfiniteM200 PRO reader (TECAN, Cat# IN-MNANO) in the presence of 1 mM ATP. PL and PL-probes were added from 100x stocks in dimethylsulfoxide (DMSO) to varying concentrations including no enzyme controls. Three biological replicates with three technical replicates each were performed.
Reconstitution of apo-SHMT1 with PLP and PL1P
For reconstitution with PLP and PL1P, 50 μM apo-SHMT1 were incubated with 5-fold molar excess of cofactor in 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 100 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM ethylenediaminetetraacetic acid (EDTA), and 10% glycerol overnight at 4°C under rotation. Residual PLP or PL1P was removed by desalting the proteins using a HiTrap Desalting column (5 mL, GE Healthcare, Cat# 17-1408-01) and an Äkta purification system (GE Healthcare) simultaneously placing them into 10 mM HEPES pH 7.5, 100 mM NaCl, 1 mM DTT, and 0.5 mM EDTA.
Sample Preparation for Intact-Protein MS
10 μM protein samples (25 μL, SHMT1 and PLPBP in corresponding SEC buffer) were treated with 10 mM NaBH4 (2 μL of 250 mM stock prepared fresh in 0.1 M NaOH) at r. t. for 30 min. Residual NaBH4 was quenched by acidification to pH 5-6 with HCl (5-10 μL of 0.5% FA) and neutralized to pH 7 with NaOH (5-10 μL of 0.1 M NaOH). Samples were diluted to 50 μL with PBS (5 μM final enzyme concentration). 2 μM hPLK was prepared in SEC-buffer for measurement by intact-protein MS.
Intact-Protein MS
Full-length protein measurements were performed as described previously (Hoegl et al., 2018). Proteins were measured on a MassPREP On-Line Desalting Cartridge (Waters) on an Ultimate 3000 HPLC system (Dionex) coupled to a Finnigan LTQ-FT Ultra mass spectrometer (Thermo Fisher Scientific) with electrospray ionization (spray voltage 4.0 kV, tube lens 110 V, capillary voltage 48 V, sheath gas 60 arb, aux gas 10 arb, sweep gas 0.2 arb). Protein Deconvolution Software (Thermo Fisher Scientific, Cat# IQLAAEGABSFANOMBAQ) was used for data analysis and deconvolution.
Gel-Based Labeling of Recombinant Proteins
Gel-based labeling of recombinant proteins was performed as described previously (Hoegl et al., 2018). In brief, proteins were labeled with PL1 or PL1P and reduced with NaBH4 previous to CuAAC to attach rhodamine azide. For competitive labeling, PLPBP dimer at 10 μM was incubated with five equivalents of PL1P (added from a 2 mM aqueous stock) in SEC buffer for 30 min at 37°C. Probe-labeled protein was further incubated with indicated molar excess of PLP (added from 2 and 100 mM stocks, respectively) for 30 min at 37°C previous to reduction and CuAAC. Samples were analyzed by SDS-PAGE and fluorescence scanning.
Thermal Stability Assay
2 μM of protein in SEC buffer or GBA in PBS were incubated with varying excess of PL(P) and PL1(P) (2 μL of corresponding stocks) and SYPRO orange protein gel stain (Thermo Fisher Scientific, Cat# S6650) was added to a final concentration of 1 x from a 5,000 x stock. Temperature was increased from 20 to 89.6°C with a heating rate of 0.3°C per min, monitoring fluorescence at 569 nm in a CFX96 Real-Time System (Bio-Rad, Cat# 20421). Three independent replicates were performed for each condition.
Protein Expression Analysis
Cell lines were grown as described and incubated for 15 min at 4°C in lysis buffer (1% (w/v) sodium deoxycholate, 1% (v/v) NP-40 in PBS pH 7.4). Supernatant was clarified at 21,000 x g for 20 min at 4°C. Protein concentration was adjusted with a BCA-assay. Samples were separated on a 12.5% SDS-gel (80 μg of lysate per cell line) and plotted on a PVDF membrane (Roti-PVDF, 0.2 μm, Roth, Cat# 8989.1) using a semi-dry blotting station (Trans-Bot SD Semi-Dry Transfer Cell, Bio-Rad, Cat# 1703940). For PLPBP, SHMT2, PNPO, ALAS1, and SHMT1 blocking was performed using 5% milk-powder (w/v) in PBS-T (PBS supplemented with 0.5% Tween-20) at r.t. for 1 h. Antibodies (mouse monoclonal anti-PLPBP antibody (clone 1G2, OriGene, Cat# TA505162, RRID: AB_2622886); rabbit polyclonal anti-SHMT2 antibody (abbexa, Cat# abx128462); rabbit polyclonal anti-PNPO antibody (Sigma, Cat# HPA023204, RRID: AB_1855506); rabbit polyclonal anti-ALAS1 antibody (Thermo Fisher Scientific, Cat# PA5-57434, RRID:AB_2637832); rabbit polyclonal anti-SHMT1 antibody (abcam, Cat# ab55736, RRID: AB_2285970)) were diluted according to manufactures protocol in 5% milk-powder in PBS-T and added to the membranes. Immunobinding carried out overnight at 4°C. After extensive washing the membranes were incubated with secondary antibodies (goat anti-rabbit antibody conjugated to horseradish peroxidase (0.5 mg/mL, Invitrogen, Cat# 32260) or goat anti-mouse antibody conjugated to horseradish peroxidase (0.5 mg/mL, Invitrogen, Cat# 32230)) diluted 1:10,000 in 5% milk-powder in PBS-T for 1 h at r.t. For PLK, blocking was performed using 3% BSA (w/v) in PBS-T at r.t. for 1 h. Mouse monoclonal anti-PLK antibody (Santa Cruz Biotechnology, Inc., Cat# sc-365173, RRID: AB_10708566) was diluted 1:100 in 3% BSA (w/v) in PBS-T and added to the membrane. Immunobinding carried out overnight at 4°C. After extensive washing the membranes were incubated with the secondary antibody (goat anti-mouse antibody conjugated to horseradish peroxidase (0.5 mg/mL, invitrogen)) diluted 1:2,500 in 3% BSA in PBS-T for 1 h at r.t. The membranes were washed and chemo-luminescence was detected after incubation with freshly prepared ECI western blotting substrate solution (Pierce, Cat# PIER80196) in a Luminescent LAS 4000 image analyzer (Fujifilm, ordered via GE Healthcare, Cat# 28955810). Membranes were stained with ponceau S (Sigma, Cat# P3504) to inspect for equal protein loading amounts.
PLP-ome Analysis with the anti-PL Antibody
Cell lines were grown as described and incubated for 15 min at 4°C in lysis buffer (1% (w/v) sodium deoxycholate, 1% (v/v) NP-40 in PBS pH 7.4) either without or with 20 mM NaBH4 added from a 500 mM stock prepared fresh in cold 0.1 M NaOH. Supernatant was clarified at 21,000 x g for 20 min at 4°C. Protein concentration was adjusted with a BCA-assay. For analysis of recombinant proteins, hPLK was diluted to 30 μM in SEC buffer and mixed with 2-fold gel loading buffer. PLPBP and SHMT1 at 30 μM were saturated with 10-fold molar excess of PLP for 30 min at 37°C. Non reduced samples were diluted with 2-fold loading buffer. Reduced samples were incubated with 10 mM NaBH4 for 30 min at r.t. (added from a 250 mM stock prepared fresh in cold 0.1 M NaOH) previous to quenching with 5 μL 1% FA in H2O and subsequent neutralization with 5 μL 0.2 M NaOH. Samples were diluted by addition of 2-fold loading buffer, separated on a 12.5% SDS-gel (70 μg of lysate per cell line and 8 μL of the 15 μM protein samples) and plotted on a PVDF membrane (Roti-PVDF 2.0, 0.2 μm, Roth, Cat# 8989.1) using a semi-dry blotting station (Trans-Bot SD, Bio-Rad, Cat# 1703940). Blocking was performed using 5% milk-powder (w/v) in PBS-T (PBS supplemented with 0.5% Tween-20) at r.t. for 1 h. The rabbit polyclonal anti-PL antibody (reduced vitamer, conjugated to BSA; GeneTex, Cat# GTX12625) was diluted 1:500 in 5% milk-powder in PBS-T and added to the membranes. Immunobinding carried out overnight at 4°C. After extensive washing the membranes were incubated with secondary antibody (goat anti-rabbit antibody conjugated to horseradish peroxidase (0.5 mg/mL, Invitrogen, Cat# 32260) diluted 1:5,000 in 5% milk-powder in PBS-T for 2 h at r.t. The membranes were washed and chemo-luminescence was detected after incubation with freshly prepared ECI western blotting substrate solution (Pierce, Cat# PIER80196) in a Luminescent LAS 4000 image analyzer (Fujifilm, ordered via GE Healthcare, Cat# 28955810). Membranes were stained with ponceau S (Sigma, Cat# P3504) to inspect for equal protein loading amounts.
PLPBP Incubation with Penicillamine (Pen)
130 μM of PLPBP were incubated with 10, 25, or 50 mM l-Pen (TCI, Cat# P1370) and d-Pen (Acros Organics via Fisher Scientific, Cat# 10224750) (added from 10x stocks in water, pH adjusted to neutral), respectively, for 20 min at 25°C in a total volume of 10 μL. Afterwards, UV/Vis-spectra were recorded as described.
Proteomics
Metabolic Labeling with PL1 and Competition
All cell lines were cultivated as described. HeLa were transferred into in-house prepared DMEM media (Table S5) lacking pyridoxine, supplemented with 10 μM PL1 (added fresh from a 100 mM stock in DMSO) and 10% dialyzed FCS (Sigma, Cat# F0392). K562, HEK293, and HCT116 were transferred into in-house prepared RPMI media (Table S5) lacking pyridoxine, supplemented with 10 μM PL1 (added fresh from a 100 mM stock in DMSO) and 10% dialyzed FCS. All cell lines were additionally transferred into corresponding in-house prepared media containing 5 μM PL added fresh from a 50 mM stock in DMSO to ensure for equal amounts of organic solvent.
Cells were passaged seven times in media with PL1 or PL, respectively, and afterwards seeded onto plates (148 cm2) and grown until 90% confluence. One separate plate was seeded per biological replicate (HEK293, HCT116, K562: n = 6; HeLa: n = 4). For competition experiments l-, and d-Pen were diluted in in-house prepared media lacking dialyzed FCS to a final concentration of 10 mM (l- and d-Pen) and pH values were adjusted. PL1-labeled HEK293 cells were incubated with B6-antagonists for 2 h at 37°C. Cells were washed once with cold PBS (4°C) and then detached in cold PBS. After harvesting at 600 x g at r.t., cells were resuspended in 1 mL lysis buffer (1% (w/v) sodium deoxycholate, 1% (v/v) NP-40 in PBS pH 7.4) containing 20 mM NaBH4 (added from a 500 mM stock prepared fresh in 0.1 M NaOH) and incubated for 30 min at 4°C under rotation. Cell debris was removed at 4°C for 20 min (21,000 xg). Soluble proteins were precipitated by adding 4 x volume of cold acetone (-80°C) overnight at -20°C. Precipitated proteins were pelleted by centrifugation (21,000 x g, 15 min, 4°C) and washed with ice-cold methanol (MeOH, 2 × 1 mL), using sonication to resuspend the pellets between washes. Proteins were solubilized in 0.5 mL PBS containing 0.4% (w/v) SDS and protein concentrations were adjusted to 2 mg/mL (= 1 mg total per sample). Samples were subjected to CuAAC by adding 0.1 mM biotin-azide (5 μL of 10 mM stock in DMSO), 0.5 mM 2-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (BTTAA, Jena Bioscience, Cat# CLK-067-25) ligand (25 μL of 10 mM stock in ddH2O), 1 mM CuSO4 (10 μL of 50 mM stock in ddH2O) and 2 mM sodium ascorbate (10 μL of 100 mM prepared fresh in ddH2O) to each sample and incubating for 1 h at r. t. in the dark. Upon precipitation and washing (as described above), the protein pellets were re-solubilized in 0.5 mL PBS containing 0.4% SDS (w/v) and centrifuged (18,000 x g, 5 min, r.t.) prior to avidin bead enrichment. Enrichment, reduction, digestion, desalting, and sample preparation for MS was performed as described previously (Hoegl et al., 2018) with the exception that enrichment was performed for 1.5 h. In brief, proteins were added to pre-equilibrated beads, enriched under rotation and washed extensively to remove background binders. Samples were reduced and alkylated previous to pre-digest with Lys-C (Wako, Cat# 125-05061) for 2 h at 37°C. Trypsin (Promega, Cat# V5111) digestion took place overnight at 37°C and samples were desalted on Sep-Pak C18 columns (Waters, Cat# WAT054960) afterwards. Peptides were evaporated in vacuo and re-dissolved in water containing 1% FA previous to LC-MS/MS analysis.
LC-MS/MS Analysis of Proteomics Samples
Samples were analyzed via LC-MS/MS using a UltiMate 3000 nano HPLC system (Thermo Fisher Scientific) equipped with an Acclaim C18 PepMap100 75 μm ID x 2 cm trap (Thermo Fisher Scientific, Cat# ES803A) and an Acclaim PepMap RSLC C18 separation column (75 μm ID x 50 cm, Thermo Fisher Scientific, Cat# 164535) coupled to an EASY-source equipped Thermo Fisher LTQ Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). Samples were loaded onto the trap column at a flow rate of 5 μL/min with aqueous 0.1% trifluoracetic acid (TFA) and then transferred onto the separation column at 0.3 μL/min. Buffers for the nano-chromatography pump were aqueous 0.1% FA (buffer A) and 0.1% FA in acetonitrile (ACN, buffer B). Samples were separated using a gradient raising buffer B from 5 to 22% in 112 min, followed by a buffer B increase to 32% within 10 min. Buffer B content was further raised to 90% within the next 10 min and held another 10 min at 90%. Subsequently buffer B was decreased to 5% and held until end of the run (total: 152 min). During sample separation MS full scans were performed at 120,000 resolution in the orbitrap with quadrupole isolation. The MS instrument was operated in a 3 s top speed data dependent mode. The scan range was set from 300 to 1,500 m/z with 60% RF lens amplitude. The automatic gain control (AGC) target was set to 2.0e5, the maximum ion injection time was 50 ms and internal calibration was performed using the lock mass option. Peptides with intensity higher than 5.0e3 and charge state 2-7 were fragmented with higher-energy collisional dissociation (HCD) (30%). MS2 scans were recorded in the ion trap operating in rapid mode. The isolation window was set to 1.6 m/z and the AGC target to 1.0e4 with maximum injection time of 100 ms. Ions were injected for all available parallelizable time. Dynamic exclusion time was 60 s with 10 ppm low and high mass tolerance. Samples for HCT116 PL1 labeling were analyzed on a UltiMate 3000 nano HPLC system (Thermo Fisher Scientific) equipped with an Acclaim C18 PepMap100 75 μm ID x 2 cm trap (Thermo Fisher Scientific, Cat# ES803A ) and an Acclaim PepMap RSLC C18 separation column (75 μm ID x 50 cm, Thermo Fisher Scientific, Cat# 164535) coupled to an EASY-source equipped Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). Sample loading and separation was performed as for measurements on the Fusion instrument. Full scans were performed at 140,000 resolution over a scan range of 300-1,500 m/z using an AGC target of 3e6 and a maximum ion injection time of 80 ms. For data-dependent MS2 scans the AGC target was set to 1e5 at 17,500 resolution and 1.6 m/z isolation window. Fragmentation was performed at a normalized collision energy of 30%. The maximum injection time was set to 100 ms. Q Exactive Plus was operating in a TopN = 10 mode. Dynamic exclusion time was set to 60 s.
Metabolomics
Metabolomics Sample Preparation
HEK293 cells were cultured as described. For time-point zero measurements, cells were seeded into 148 cm2 dishes in chemically defined media complemented with 5 μM PL (added fresh from a 50 mM stock in DMSO). For later time points, cells were transferred into chemically defined media complemented with 10 μM PL1 (added fresh from a 100 mM stock in DMSO) and cultured till the desired passaging numbers previous to seeding them onto 148 cm2 dishes. Cells were grown to 90% confluence. After that, media was removed and cells were scraped to detach in 10 mL cold (4°C) 0.9% NaCl (m/v) in water per plate. Cells were harvested at 600 x g for 2 min. Supernatant was removed and cells were lysed in 1 mL ice-cold MeOH (-80°C, containing 1 μM trimethoprim as internal standard) overnight at -20°C. Samples were centrifuged for 20 min at 21,000 x g and 4°C. Supernatant was dried in vacuo and metabolites were re-dissolved in 90 μL 50/50 Vol-% MeOH/H2O containing 1% FA for LC-MS/MS measurement.
LC-MS/MS Analysis of Metabolomics Samples
Metabolic profiling and MS/MS analysis was carried out on an UltiMate 3000 RSLC system (Thermo Fisher Scientific) coupled to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Chromatographic separation was performed using an Accucore HILIC column (150 x 2.1 mm, 2.6 μm, 80 Å) (Thermo Fisher Scientific) at 40°C and 80 mM NH4OAc in 100% H2O pH = 6.8 (A) and 100% ACN (B). After 5 min pre-equilibration with 95% B, samples were eluted with a linear gradient from 95% to 0% B over 30 min at a flow rate of 250 μL/min followed by 8 min re-equilibration with 95% B. Mass spectrometric measurements were accomplished in positive ion mode (HESI-II source, Thermo Fisher Scientific) with following source parameters: capillary voltage 4.5 kV, tube lens 40 V, vaporizer temperature 43°C, sheath gas 50 L/h, aux gas 10 L/h, capillary voltage 12 V and capillary temperature 320°C. Full scan measurements were recorded between 50 – 1,000 m/z in profile mode at 30,000 resolution in the orbitrap. Parent ions of interest (PL m/z 168.06 and PLP m/z 248.03) were isolated (Isolation width 1.0 m/z) subjected to collision-induced dissociation (normalized collision energy 35 V) in the MRM mode and most intense daughter ions (PL/PLP m/z 150.05) were used for quantification (SIM width 10 m/z) at a resolution of 7,500 in the orbitrap. Prior to measurement 8 pooled QC samples were injected to equilibrate the column. In order to take metabolic degradation over time into account, sample order was randomized. Overall instrument performance was monitored using trimethoprim.
Quantification and Statistical Analysis
Statistical Analysis of hPLK Kinetic Data
Data were processed using Prism 6 (GraphPad, RRID: SCR_002798). Absorbance values were transformed into concentrations using molar extinction coefficients of PLP and PLP-probes at corresponding absorbance maxima derived from the recorded UV/Vis-spectra (ϵ392nm (PLP) = 6,054.42 M-1cm-1, ϵ416nm (PL1P) = 9,657.49 M-1cm-1, ϵ396nm (PL2P) = 9,374.44 M-1cm-1, ϵ400nm (PL3P) = 6,240.76 M-1cm-1). Initial slopes from linear regions were determined from three technical replicates (with corresponding error) and plotted against concentrations of PL or PL-probe, respectively. Kinetic values (kcat, Km, vmax) and corresponding errors (SEM) were derived from non-linear regression analysis (kcat and Michaelis-Menten) using default parameters. Means of kinetic values and corresponding errors from the three biological replicates were calculated using error propagation. The number of replicates together with mean values and corresponding SEM are reported in the figure and legend, as well as Supplemental figure and legend for clarity.
Statistical Analysis of Thermal Stability Data
Denaturation curves were fitted according to a sigmoidal trace. Melting temperatures were calculated as mean value with corresponding error (SEM). The number of replicates together with mean values and corresponding SEM are reported in the figure and legend, as well as Supplemental figure and legend for clarity.
Statistical Analysis of Proteomics Data
MS raw data were searched with MaxQuant software (version 1.6.0.1) (Cox and Mann, 2008) and default settings (with the exceptions that label-free quantification and match between runs were activated). All replicates from one condition (e.g. PL1 treated samples) were defined as one fraction. Searches were performed against a UniProt database of Homo sapiens proteome (taxon identifier: 9606, reference reviewed and unreviewed, downloaded on 25.12.2018). Resulting data were further analyzed using Perseus software version 1.6.0.0 (Tyanova et al., 2016). The rows were filtered (only identified by site, potential contaminant, reverse) and log2 transformed. Biological replicates were grouped, filtered for 100% valid values in at least one group, and missing values were imputed for total matrix using default settings. A both sided, two-sample Student’s t-test was performed and derived p-values were corrected for multiple testing by the method of Benjamini and Hochberg with a significance level of p = 0.05. Volcano plots were generated by plotting log2 (fold change of different conditions) against –log10 (p-value). PLP-dependent proteins (gene ontology (GO)-term: 0030170; PLP-binding) were identified with the help of a GO annotation file for H. sapiens submitted on 26.06.2018 (Ashburner et al., 2000). The number of replicates, fold-change and p-value cut-offs are reported in the figures and legends, Supplemental figures and legends, as well as Supplemental tables for clarity.
Statistical Analysis of Metabolomics Data
Raw data were processed with Xcalibur Quan Browser (Thermo Fisher Scientific) using Genesis algorithm and manual integration mode. Intensities of the corresponding daughter ions were extracted and further processed using Excel. Finally, daughter ion intensities were plotted for PL and PLP for respective time points using Prism 6 (GraphPad, RRID: SCR_002798). The number of replicates is reported in the figure legend.
Data and Code Availability
All mass-spectrometric data and corresponding analysis generated during this study have been deposited at the ProteomeXchange Consortium (https://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository (Vizcaíno et al., 2016) with the dataset identifier PXD014771. Plasmids generated in this study have been deposited to Addgene (Deposit No. 77114; IDs 131231, 131230, 131229). The authors declare that all other data supporting the findings of this study are available within the article and its Supplemental Information or from the corresponding author upon request.
Acknowledgments
This project has received funding from the European Research Council (ERC) and the European Union's Horizon 2020 research and innovation program (grant agreement no. 725085, CHEMMINE, ERC consolidator grant). Further financial support was provided by the Deutsche Forschungsgemeinschaft (DFG) SFB 749. M.P. acknowledges a PhD fellowship from the Studienstiftung des deutschen Volkes. We thank Mona Wolff, Katja Gliesche, and Katja Bäuml for technical assistance. We thank Stephan M. Hacker for critical evaluation of the manuscript.
Author Contributions
A.F. designed, planned, and conducted all experiments, including kinetic studies, proteomics, and metabolomics sample preparation, as well as protein expression and analysis. A.F. performed proteomic and intact protein mass spectrometry measurements and statistical analysis of the data. V.C.K. contributed expertise to metabolomics sample preparation, measured metabolomics samples, and analyzed metabolomics data. M.P. and A.H. synthesized PL-probes. S.A.S. supervised experiments. A.F. and S.A.S. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chembiol.2019.08.003.
Supplemental Information
References
- Amadasi A., Bertoldi M., Contestabile R., Bettati S., Cellini B., di Salvo M.L., Borri-Voltattorni C., Bossa F., Mozzarelli A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007;14:1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaleone V., Esposito I., Gargiulo A., Vellecco V., Asimakopoulou A., Citi V., Calderone V., Gobbetti T., Perretti M., Papapetropoulos A. D-Penicillamine modulates hydrogen sulfide (H2S) pathway through selective inhibition of cystathionine-γ-lyase. Br. J. Pharmacol. 2016;173:1556–1565. doi: 10.1111/bph.13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras J., Singleton A., Cookson M.R., Hardy J. Emerging pathways in genetic Parkinson’s disease: potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P., Gong Y., Tang L., Leung Y.C., Jiang T. Crystal structure of human pyridoxal kinase. J. Struct. Biol. 2006;154:327–332. doi: 10.1016/j.jsb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoss J.A. Studies on the mechanism of the tryptophan synthetase reaction. Biochim. Biophys. Acta. 1962;62:279–293. doi: 10.1016/0006-3002(62)90041-0. [DOI] [PubMed] [Google Scholar]
- Ducker G.S., Ghergurovich J.M., Mainolfi N., Suri V., Jeong S.K., Hsin-Jung Li S., Friedman A., Manfredi M.G., Gitai Z., Kim H. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U S A. 2017;114:11404–11409. doi: 10.1073/pnas.1706617114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed A.S., Shindia A.A. PLP-dependent enzymes: a potent therapeutic approach for cancer and cardiovascular diseases. In: You Y., editor. Targets in Gene Therapy. IntechOpen; 2012. [Google Scholar]
- Gandhi A.K., Ghatge M.S., Musayev F.N., Sease A., Aboagye S.O., di Salvo M.L., Schirch V., Safo M.K. Kinetic and structural studies of the role of the active site residue Asp235 of human pyridoxal kinase. Biochem. Biophys. Res. Commun. 2009;381:12–15. doi: 10.1016/j.bbrc.2009.01.170. [DOI] [PubMed] [Google Scholar]
- Hacker S.M., Backus K.M., Lazear M.R., Forli S., Correia B.E., Cravatt B.F. Global profiling of lysine reactivity and ligand ability in the human proteome. Nat. Chem. 2017;9:1181–1190. doi: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyl D., Harris S.A., Folkers K. The chemistry of vitamin B6. VI. pyridoxylamino acids. J. Am. Chem. Soc. 1948;70:3429–3431. doi: 10.1021/ja01190a061. [DOI] [PubMed] [Google Scholar]
- Hoegl A., Nodwell M.B., Kirsch V.C., Bach N.C., Pfanzelt M., Stahl M., Schneider S., Sieber S.A. Mining the cellular inventory of pyridoxal phosphate-dependent enzymes with functionalized cofactor mimics. Nat. Chem. 2018;10:1234–1245. doi: 10.1038/s41557-018-0144-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegawa S., Isomura M., Koshizuka Y., Nakamura Y. Cloning and characterization of human and mouse PROSC (proline synthetase co-transcribed) genes. J. Hum. Genet. 1999;44:337–342. doi: 10.1007/s100380050172. [DOI] [PubMed] [Google Scholar]
- Ito T., Iimori J., Takayama S., Moriyama A., Yamauchi A., Hemmi H., Yoshimura T. Conserved pyridoxal protein that regulates Ile and Val metabolism. J. Bacteriol. 2013;195:5439–5449. doi: 10.1128/JB.00593-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe I.A. Rheumatoid arthritis with arteritis. Ann. Intern. Med. 1964;61:556. doi: 10.7326/0003-4819-61-3-556. [DOI] [PubMed] [Google Scholar]
- Kimura T., Shirakawa R., Yaoita N., Hayashi T., Nagano K., Horiuchi H. The antimalarial drugs chloroquine and primaquine inhibit pyridoxal kinase, an essential enzyme for vitamin B6 production. FEBS Lett. 2014;588:3673–3676. doi: 10.1016/j.febslet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Kuchinskas E.J., Horvath A., du Vigneaud V. An anti-vitamin B6 action of l-penicillamine. Arch. Biochem. Biophys. 1957;68:69–75. doi: 10.1016/0003-9861(57)90327-2. [DOI] [PubMed] [Google Scholar]
- Lehmann J., Wright M.H., Sieber S.A. Making a long journey short: alkyne functionalization of natural product scaffolds. Chemistry. 2016;22:4666–4678. doi: 10.1002/chem.201504419. [DOI] [PubMed] [Google Scholar]
- Lowther J., Yard B.A., Johnson K.A., Carter L.G., Bhat V.T., Raman M.C.C., Clarke D.J., Ramakers B., McMahon S.A., Naismith J.H. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol. Biosyst. 2010;6:1682–1693. doi: 10.1039/c003743e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodwell M.B., Koch M.F., Alte F., Schneider S., Sieber S.A. A subfamily of bacterial ribokinases utilizes a hemithioacetal for pyridoxal phosphate salvage. J. Am. Chem. Soc. 2014;136:4992–4999. doi: 10.1021/ja411785r. [DOI] [PubMed] [Google Scholar]
- Ong S.-E. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Park T.S., Rosebury W., Kindt E.K., Kowala M.C., Panek R.L. Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol. Res. 2008;58:45–51. doi: 10.1016/j.phrs.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Parra M., Stahl S., Hellmann H. Vitamin B6 and its role in cell metabolism and physiology. Cells. 2018;7:84. doi: 10.3390/cells7070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R., Peracchi A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003;4:850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.S. Chemistry and diversity of pyridoxal-5′-phosphate dependent enzymes. Biochim. Biophys. Acta. 2015;1854:1167–1174. doi: 10.1016/j.bbapap.2014.12.028. [DOI] [PubMed] [Google Scholar]
- Renwick S.B., Snell K., Baumann U. The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure. 1998;6:1105–1116. doi: 10.1016/s0969-2126(98)00112-9. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Rumsby P.C., Shepherd D.M. The effect of penicillamine on vitamin B6 function in man. Biochem. Pharmacol. 1981;30:3051–3053. doi: 10.1016/0006-2952(81)90492-5. [DOI] [PubMed] [Google Scholar]
- Di Salvo M.L., Contestabile R., Safo M.K. Vitamin B 6 salvage enzymes: mechanism, structure and regulation. Biochim. Biophys. Acta. 2011;1814:1597–1608. doi: 10.1016/j.bbapap.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Schmidt T., Samaras P., Frejno M., Gessulat S., Barnert M., Kienegger H., Krcmar H., Schlegl J., Ehrlich H.C., Aiche S. ProteomicsDB. Nucleic Acids Res. 2018;46:D1271–D1281. doi: 10.1093/nar/gkx1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell K., Natsumeda Y., Eble J.N., Glover J.L., Weber G. Enzymic imbalance in serine metabolism in human colon carcinoma and rat sarcoma. Br. J. Cancer. 1988;57:87–90. doi: 10.1038/bjc.1988.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A.E., Cravatt B.F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Sukanya N., Vijaya M., Radhakrishnan A.N., Rao N.A., Savithri H.S. Serine hydroxymethyltransferase from mung bean (Vigna radiata) is not a pyridoxal-5′-phosphate-dependent enzyme. Plant Physiol. 2008;95:351–357. doi: 10.1104/pp.95.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Matsuda M. Effects of penicillamine on the contents of B6 vitamers of the mouse brain. J. Nutr. Sci. Vitaminol. (Tokyo) 1976;22:375–379. doi: 10.3177/jnsv.22.375. [DOI] [PubMed] [Google Scholar]
- Thornburg J.M., Nelson K.K., Clem B.F., Lane A.N., Arumugam S., Simmons A., Eaton J.W., Telang S., Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney M.D. Reaction specificity in pyridoxal phosphate enzymes. Arch. Biochem. Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Tornøe C.W., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- du Vigneaud V., Kuchinskas E.J., Horvath A. l-Penicillamine and rat liver transaminase activity. Arch. Biochem. Biophys. 1957;69:130–137. doi: 10.1016/0003-9861(57)90480-0. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J.A., Csordas A., Del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshe J.M. Penicillamine, a new oral therapy for Wilson’s disease. Am. J. Med. 1956;21:487–495. doi: 10.1016/0002-9343(56)90066-3. [DOI] [PubMed] [Google Scholar]
- Weigert W.M., Offermanns H., Degussa P.S. D-penicillamine—production and properties. Angew. Chem. Int. Ed. 1975;14:330–336. doi: 10.1002/anie.197503301. [DOI] [PubMed] [Google Scholar]
- Whittaker M.M., Penmatsa A., Whittaker J.W. The Mtm1p carrier and pyridoxal 5′-phosphate cofactor trafficking in yeast mitochondria. Arch. Biochem. Biophys. 2015;568:64–70. doi: 10.1016/j.abb.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E., Ben Ya’acov A., Lee H., Lichtenstein Y., Shalev Z., Smith Y., Zolotarov L., Ziv E., Kalman R., Le H.V. Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med. Chem. Lett. 2015;6:840–844. doi: 10.1021/acsmedchemlett.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass-spectrometric data and corresponding analysis generated during this study have been deposited at the ProteomeXchange Consortium (https://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository (Vizcaíno et al., 2016) with the dataset identifier PXD014771. Plasmids generated in this study have been deposited to Addgene (Deposit No. 77114; IDs 131231, 131230, 131229). The authors declare that all other data supporting the findings of this study are available within the article and its Supplemental Information or from the corresponding author upon request.