

SUMMARY
IFITs are interferon-induced proteins that can bind 5′-triphosphate or ribose-unmethylated capped ends of mRNA to inhibit translation. Although some viruses avoid IFITs by synthesizing RNAs with eukaryotic-like caps, no viral proteins were known to antagonize IFITs. We show that the N- and C-terminal portions of C9, a protein required for vaccinia virus to resist the human type I interferon-induced state, bind IFITs and ubiquitin regulatory complexes, respectively. Together, the two C9 domains target IFITs for proteasomal degradation, thereby providing interferon resistance similar to that also achieved by knockout of IFITs. Furthermore, ectopic expression of C9 rescues the interferon sensitivity of a vaccinia virus mutant with an inactivated cap 1-specific ribose-methyltransferase that is otherwise unable to express early proteins. In contrast, the C9-deletion mutant expresses early proteins but is blocked by IFITs at the subsequent genome uncoating/replication step. Thus, poxviruses use mRNA cap methylation and proteosomal degradation to defeat multiple antiviral activities of IFITs.
In Brief
Liu et al. show that the N- and C-terminal portions of C9, a protein required for vaccinia virus to resist the human type I interferon-induced state, bind IFITs and ubiquitin regulatory complexes, respectively. Together, the two domains target IFITs for proteasomal degradation, thereby enabling viral genome uncoating and replication.
Graphical Abstract
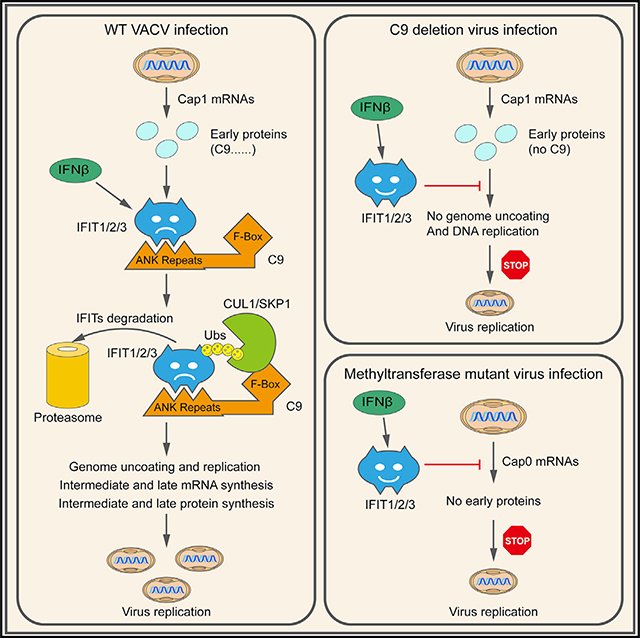
INTRODUCTION
Viruses and their hosts have antagonistic relations in which each entity strives for dominance. Upon recognition of an infection, cells activate programs that increase the synthesis of numerous antiviral proteins. At the same time, viruses synthesize proteins that diminish host defenses. We reported that the vaccinia virus (VACV) C9 protein is required to resist the human interferon (IFN)-induced state, suggesting that it counteracts one or more of the ~300 known interferon-stimulated genes (ISGs) (Liu and Moss, 2018). Here, we set out to answer the following questions: what is the IFN-induced target of C9? How does C9 inactivate the putative target? At what step does the targeted IFN-response factor inhibit virus replication in the absence of C9?
In regard to our first aim, we discovered that human IFN-induced proteins with tetratricopeptide repeats (IFITs) are targets of C9. IFITs can inhibit viruses by mechanisms that include binding to uncapped or partially methylated capped mRNA, which impairs their translation (Diamond and Farzan, 2013). One of the first demonstrations of such antiviral activity was obtained for a VACV mutant with an inactivated ribose methyltransferase (MTase) that is unable to convert IFIT-sensitive cap 0 (m7GpppN-) to IFIT-resistant cap 1 (m7GpppNm-) mRNAs (Daffis et al., 2010). Our finding that the VACV C9 gene, which has no apparent role in mRNA synthesis or modification, is also necessary to counteract IFITs is unexpected.
In regard to our second aim, we show that C9 mediates the proteasomal degradation of IFITs. C9 belongs to a family of poxvirus proteins that contain both ANK repeats and an F-box. The ANK is a 33-residue-repeating motif consisting of two α helices connected by a loop and is commonly associated with protein-protein interactions (Mosavi et al., 2004). Proteins with ANK-repeat motifs are ubiquitous in all of the kingdoms of life and are particularly numerous in Eukaryotes. Nevertheless, ANK-repeat proteins are absent from most viruses, with the notable exception of poxviruses (Herbert et al., 2015). Chordopoxviruses encode multiple ANK-repeat proteins, and phylogenetic studies suggest that the primordial one was acquired by an ancestral poxvirus and has undergone repeated duplication and speciation events that led to the acquisition of new functions. The cellular F-box family of proteins is the substraterecognition components of the Skp1-CUL1-F-box (SCF) ubiquitin ligase E3 complex. The organization of the poxvirus ANKrepeat/F-box proteins suggests that the repeat motifs recognize specific proteins and that the F-box facilitates their polyubiquitination and proteasomal degradation. However, while several poxvirus ANK-repeat/F-box proteins have been shown to associate with Skp1 and CUL1, degradation of biologically important targets recognized by ANK repeats have yet to be demonstrated. Our finding that the C9 protein targets IFITs for proteasomal degradation fulfills the proposed mode of action of ANK-repeat/F-box proteins.
In regard to our third aim, we show that human IFITs prevent a VACV MTase mutant from expressing early proteins, whereas they block a VACV C9 mutant at the later steps of viral genome uncoating and replication, indicating that poxviruses use both mRNA cap methylation and proteosomal degradation to prevent multiple antiviral effects of IFITs.
RESULTS
C9 Binds and Degrades IFIT Proteins
Previously, we showed that the replication of a VACV C9-deletion mutant (vΔC9) was inhibited in A549 cells that had been pretreated for 24 h with 2,000 IU IFN-β and rescued by ectopic expression of an Myc-tagged C9 protein (Liu and Moss, 2018). Components of the SCF and signalosome/neddylation complexes, which regulate protein ubiquitination, were physically associated with Myc-tagged C9, providing a clue to the role of this F-box protein. Nevertheless, bound proteins encoded by ISGs that may be specific targets of C9 were not identified. A plausible explanation for this failure was that C9 induced the proteasomal degradation of the putative target protein, thereby preventing its identification, and that the solution to this impasse would be to avoid degradation by expressing a truncated C9 protein without the F-box (Figure 1A). We constructed a cell line that expresses Myc-tagged C9 lacking the F-box (C9ΔF-box) for comparison with one that expresses Myc-tagged fulllength C9. Following IFN treatment, proteins from the C9- and C9ΔF-box-expressing cell lines were captured on Myc-Trap beads and analyzed by nano-liquid chromatography-tandem mass spectrometry. A complete dataset is shown in Table S1. To identify candidate ISG products, proteins associated with C9ΔF-box that had a mean peak area of ≥106 were filtered via Gene Ontology (GO) term “type I interferon signaling pathway” (GO: 0060337). Twenty-one proteins meeting these criteria from the two cell lines were ranked from a high to a low abundance ratio (C9ΔF-box/full-length C9) in Figure 1B. The ones with the highest ratios were IFIT1, IFIT2, and IFIT3 (Figure 1B), which form a complex. Next was IFIT5; however, its absolute abundance was >1 log lower than the other IFITs in the C9ΔF-box set. The next proteins on the list were ISG20, which is an upregulator of type I IFN response proteins, including IFIT1 (Weiss et al., 2018) and ABCE1, which enhances ribosome recycling (Mancera-Martínez et al., 2017). Based on their preferential binding to C9ΔF-box and known antiviral roles, the IFITs were chosen for follow-up studies.
Figure 1. C9 Binds and Depletes IFIT Proteins.

(A) Left: binding of putative ISG protein to N-terminal ANK repeat domain and CUL1/Skp1 to C-terminal F-box domain of C9 followed by ubiquitination and proteasomal degradation. Right: stable association of ISG protein to C9 with deleted F-box.
(B) Ratios of proteins associated with F-box-deleted (C9ΔF-box) and full-length C9. Cells were transduced to express C9ΔF-box or full-length C9, each with an Myc tag. After IFN-β treatment, proteins bound to Myc-Trap beads were analyzed by mass spectrometry (Table S1). Representative of two biological repeats.
(C) Representative WB. Lysates and Myc-Trap bound proteins (IP) were analyzed from three biological repeats. Mass is expressed in kilodaltons and positions of standard markers are at right.
Western blotting (WB) validated the mass spectrometry results for IFIT1, IFIT2, and IFIT3, but we were unable to detect IFIT5, perhaps because the level was too low or the antibodies were inadequate. IFIT1, IFIT2, and IFIT3 associated with C9ΔF-box from IFN-treated cells, whereas little or no association was seen when full-length C9 was expressed (Figure 1C, immunoprecipitation [IP]). However, full-length C9 but not F-box-deleted C9 bound CUL1, a component of the SCF complex (Figure 1C, IP). A striking finding was that the amounts of IFIT1, IFIT2, and IFIT3 were greatly reduced in IFN-treated cells expressing full-length C9 but not truncated C9, which is consistent with their degradation being dependent on the F-box (Figure 1C, lysate). These data pointed to an IFN-resistance mechanism in which the IFN-inducible IFIT proteins are specifically recognized by the C9 ANK repeats and subjected to F-box-facilitated degradation, as modeled in Figure 1A.
C9-Mediated Depletion of IFITs Determined by Mass Spectrometry
To confirm the specificity of the depletion of IFITs by C9, we performed quantitative mass spectrometry analysis using tandem mass tag (TMT) 6-plex labeling of trypsin-digested total cell extracts from untreated or IFN-pretreated vector control or C9-expressing A549 cells that were mock infected or infected with VACV (Figure 2A). A total of 8,025 proteins, including many encoded by ISGs, were detected (Table S2). To identify IFNinduced proteins that were specifically depleted by C9, abundance ratios for proteins in IFN-treated cells expressing C9 (sample #4)/IFN-treated cells not expressing C9 (sample #2) were determined. The 10 proteins showing the lowest ratios are listed in Figure 2B, alongside the percentile distance of each from the bottom of an ascending distribution of ratios for all detected proteins. IFIT2, IFIT1, and IFIT5 were most severely depleted by C9, and IFIT3 was somewhat less depleted. MX2 was also diminished but to a lesser extent than the IFITs. A STRING database analysis highlighted the enrichment and interaction of the four IFITs and MX2 in the GO categories of “defense response to viruses” (GO: 0051607) and “response to type I interferons” (GO: 0034340) (Figure 2C). MX2 and the other five non-interacting proteins, which were not detectably enriched by association with the C9ΔF-box (Figure 1B), were not further investigated.
Figure 2. C9-Mediated Depletion of IFITs Determined by Mass Spectrometry.

(A) Protocol. Cells transduced with vector or vector expressing full-length C9 were untreated or pretreated with IFN-β and uninfected or infected with VACV WR.
(B) Abundance ratios of proteins from IFN-treated cells expressing C9 (sample #4)/IFN-treated cells not expressing C9 (sample #2), derived from Table S2.
(C) STRING database of protein-protein interactions with significant enrichment for GO “defense response to viruses” (GO: 0051607) in red and “response to type I IFN” (GO: 0034340) in blue.
The effect of VACV strain Western Reserve (WR) infection on cells that had been pretreated with IFN were also examined (Table S2). The host cell proteins listed in Figure 2B were less abundant in the IFN-pretreated VACV-infected A549 cells (sample #6) relative to the IFN-pretreated uninfected cells (sample #2). However, many more proteins showed depletion, presumably due to the expression of other viral proteins in addition to C9. In a temporal proteomic screen of VACV-infected primary human fetal foreskin fibroblasts, Soday et al. (2019) reported the downregulation of 266 host cell proteins, including IFITs.
Degradation of IFIT1 and IFIT2 by C9 Is Proteasome Dependent and Accompanied by Ubiquitination
We investigated the mechanism of IFIT depletion by transfecting expression plasmids encoding individual FLAG-tagged IFITs and fulllength or F-box-deleted Myc-tagged C9 into A549 cells. After 4 h, the cells were treated with PS-341 (bortezomib), which inhibits the chymotryptic activity of the 26S proteasome (Teicher and Tomaszewski, 2015), or with DMSO vehicle control for an additional 18 h. In DMSO-treated cells, IFIT1, IFIT2, and IFIT5 were severely depleted by C9 but not by C9ΔF-box or the vector plasmid, whereas IFIT3 was unaffected (Figure 3A). Notably, PS-341 appreciably restored IFIT1 and IFIT2 levels in cells expressing C9, although IFIT5 levels were rescued only marginally (Figure 3A). Bands lower than full-length IFIT1 may represent PS-341-stabilized partial degradation products. Enhancement of IFIT1 and IFIT2 by PS-341 supported C9-mediated proteasomal degradation of these proteins in the absence of the inhibitor.
Figure 3. Association of C9 with IFITs Mediates Their Ubiquitination and Proteasomal Degradation.

(A) Proteasome inhibitor PS-341 reduced C9-mediated degradation of IFITs. Cells were co-transfected with two plasmids: one encoding a FLAG-tagged IFIT and the other Myc-tagged C9 (C9), or Myc-tagged F-box-deleted C9 (ΔF-box), or the vector. After 4 h, cells were treated with DMSO or PS-341 for an additional 18 h and lysates were analyzed by WB. ns, non-specific.
(B) Ubiquitination of IFITs. Cells co-transfected with a plasmid expressing HA-ubiquitin plus a vector plasmid or plasmid expressing a FLAG-tagged IFIT and with a plasmid expressing Myc-tagged C9 or C9ΔF-box. After 3 h, PS-341 was added for an additional 20 h and lysates and proteins were captured with anti-FLAG antibody and analyzed by WB.
(C) Association of C9 with individual IFITs. Cells were transfected with vector plasmid or a plasmid expressing FLAG-IFITs and Myc-C9. After 20 h, lysates or proteins bound to anti-FLAG antibody (IP) were analyzed by WB.
(D) Association of C9ΔF-box with individual IFITs. Cells expressing Myc-C9ΔF-box were transfected with vector or plasmids expressing FLAG-IFITs. Lysates and proteins captured with anti-FLAG antibody were analyzed by WB.
(A) is representative of three biological repeats and (B)–(D) are representative of two each.
Hemagglutinin (HA)-ubiquitin is a sensitive reagent for detecting transient ubiquitin conjugates (Treier et al., 1994). Cells were transfected with expression plasmids encoding individual FLAG-tagged IFITs along with HA-ubiquitin and Myc-tagged full-length or F-box-deleted C9. After 3 h, the incubation was continued for an additional 20 h in the presence of PS-341 to reduce the degradation of ubiquitin complexes. As expected, total lysates and proteins captured with anti-FLAG antibody revealed that IFIT1 and IFIT2 were reduced by C9 compared to the vector and ΔF-box samples (Figure 3B). Notably, smears representing high-molecular-weight HA-ubiquitin conjugates were detected from all of the lysates, but were selectively captured by FLAG-tagged IFIT1 and IFIT2 from cells in which C9 was expressed. This ubiquitination pattern was consistent with the more extensive C9-dependent proteolysis of IFIT1 and IFIT2 compared to IFIT3.
Additional experiments were carried out to determine whether the degree of ubiquitination correlated with the extent of binding of C9 to specific IFITs. In one scheme, cells were transfected with expression plasmids encoding Myc-tagged C9 and individual FLAG-tagged IFITs and treated with PS-341. The capture of Myc-C9 occurred to a greater extent with IFIT2 than with IFIT1 and the least with IFIT3, even though IFIT3 was the most highly expressed (Figure 3C, IP: FLAG). In another version of this experiment, C9ΔF-box cells were transfected with plasmids encoding individual FLAG-tagged IFITs. Capture of the C9ΔF-box was also the most efficient with IFIT2 (Figure 3D, IP: FLAG). Thus, the relative affinity of individual IFITs for C9 correlated with the extent of ubiquitination (e.g., IFIT2 > IFIT1 > IFIT3).
Depletion of IFITs by VACV Expressing C9
The above experiments demonstrated the degradation of IFITs by cells stably or transiently expressing C9. It was important, however, to investigate C9-dependent degradation during VACV infection. Cells that were untreated or pretreated with IFN for 24 h were infected with wild-type VACV WR expressing C9 or with a C9-deletion mutant (vΔC9) and the lysates were analyzed by WB. As expected, the IFITs were robustly expressed in mock-infected cells and vΔC9-infected cells that had been pretreated with IFN (Figure 4A). However, relative to the mock infection, IFIT1 and IFIT2 were diminished by ~80% at 4 h and were barely detectable at 8 and 16 h following the infection of cells with VACV WR, whereas IFIT3 depletion was delayed and reduced by only 50% at 8 and 16 h after infection (Figure 4A). Depletion of IFITs still occurred in the presence of the DNA replication inhibitor AraC, which allows expression of C9 and other early proteins but secondarily prevents the expression of intermediate and late proteins (Figure 4A).
Figure 4. Degradation of IFITs by VACV and Inhibition of Viral Intermediate and Late Protein Synthesis in IFN-β-Pretreated Cells.

(A) Time course of IFIT degradation following VACV infection. Untreated (−) and IFN-β-pretreated (+) cells were mock infected or infected with 3 plaque-forming units (PFUs) per cell of VACV WR or vΔC9 in the absence or presence of AraC. Lysates after 4, 8, and 16 h were analyzed by WB.
(B) Degradation of individual IFITs by VACV. Cells transfected with vector plasmid or plasmids expressing FLAG-tagged IFITs and mock infected or infected with 0.01 PFU per cell of VACV WR or vΔC9. After 48 h, lysates were analyzed by WB.
(C) Full-length C9 is required for IFN resistance of VACV. Untreated (−) and IFN-β-pretreated (+) cells were infected with 0.01 PFU per cell of VACVWR expressing HA-tagged C9 without F-box (vHA-C9ΔF-box), with F-box (vHA-C9) or without C9 (vΔC9). After 48 h, lysates were analyzed by WB.
(D) Untreated (−) and IFN-β-pretreated (+) cells were incubated with or without AraC and mock infected or infected with 3 PFUs per cell of VACV WR or vΔC9. At 6 h, lysates prepared for WB.
Representative blots from three biological repeats in (A) and (C) and two in (B) and (D).
Strong depletion of IFIT1, IFIT2, and IFIT5 by VACVWR but not vΔC9 was also detected after the transfection of the cells with plasmids that expressed individual IFITs instead of using IFN to induce the expression of the IFIT1/2/3 complex (Figure 4B, upper row). We concluded that the decrease in IFITs was mediated by full-length C9, whether the latter was expressed by transfection or by VACV infection.
The rapid decrease in IFIT proteins following VACV infection was likely also due to proteasomal degradation; however, because proteasome inhibitors interfere with VACV genome uncoating and replication, they could not be effectively tested here. Since VACV infection decreases host mRNAs (Yang et al., 2010), it seemed possible that the depletion of IFIT proteins was partly due to accelerated degradation of mRNA by C9. To investigate this mechanism, we quantified the amounts of IFIT mRNAs in untreated and IFN-treated cells that were mock infected or infected with wild-type VACV or vΔC9 for 6 h. IFN increased the mRNA copy numbers for IFIT1, IFIT2, and IFIT3 in mock-infected cells by 243-, 35-, and 44-fold, respectively (Figure S1). In the IFN-treated cells infected with VACV WR expressing C9, the mRNAs for IFIT1, IFIT2, and IFIT3 were reduced by 56%, 38%, and 50%, respectively, compared to the mock-infected cells (Figure S1), which would not account for the almost complete depletion of the IFIT1 and IFIT2 proteins. In cells infected with vΔC9, the reductions in mRNAs for IFIT1, IFIT2, and IFIT3 of 43%, 21%, and 38%, respectively, were slightly less than in cells infected with wild-type virus, perhaps because the viral decapping enzyme, an intermediate/late protein, that contributes to mRNA degradation was not expressed.
C9 F-Box Is Required for IFN-Resistant Expression of VACV Intermediate and Late Proteins
Although the F-box of C9 was required for the depletion of IFITs, it was possible that IFIT binding of the N-terminal segment containing the ANK repeats would be sufficient to abrogate antiviral activity. A recombinant VACV in which C9 was replaced with an HA-epitope tagged C9 lacking the F-box (vHA-C9ΔF-box) was constructed to compare IFN sensitivity with viruses expressing full-length C9 (vHA-C9) and the C9-deletion mutant (vΔC9). A spread assay was performed by infecting cells with a low multiplicity of virus for 48 h. The expression of HA-C9 and HA-C9ΔF-box was confirmed by WB (Figure 4C). In IFN-untreated cells, VACV proteins were similarly expressed upon infection with each of the three viruses as detected with serum from VACV-infected rabbits and antibodies specific for the I3 early, A3 intermediate/late, and L1 late proteins (Figure 4C). In IFN-treated cells, however, there was a reduction in VACV protein expression in cells infected with vHA-C9ΔF-box or vΔC9 compared to vHA-C9, indicating that the F-box is important for IFN resistance and depletion of IFITs. The presence of two A3 bands in Figure 4C is due to proteolytic processing during a late stage of morphogenesis (Moss and Rosenblum, 1973).
For the experiment in Figure 4D, the cells were synchronously infected at a higher virus multiplicity than in Figure 4C and harvested at 6 h to differentiate the inhibition of early and postreplicative intermediate/late proteins. The early proteins I3 and E3 appeared in similar amounts in cells infected with VACV WR or vΔC9 regardless of IFN or AraC treatment, whereas the intermediate/late protein A3 was barely detected in IFN-treated cells infected with vΔC9 and not at all in AraC-treated cells (Figure 4D). In contrast to the above results obtained by inducing the expression of IFITs with IFN, viral intermediate/late protein synthesis was not inhibited by transfecting individual IFITs (Figure 4B). Whether inhibition requires multiple IFITs or a higher expression of individual IFITs was not ascertained.
The effects of IFN on VACV protein D5 expression was midway between the resistance of the early I3 and E3 and the sensitivity of the intermediate/late A3 protein (Figure 4D). The D5 protein was of particular interest because it has specific roles in core uncoating and DNA replication (Boyle et al., 2007; De Silva et al., 2007; Kilcher et al., 2014), and therefore its depletion may contribute to the IFN-mediated inhibition of a C9-deletion mutant. However, D5 has a TAAAT RNA start site that is characteristic of late promoters, suggesting that it is expressed at late and early times (Yang et al., 2011). This idea was supported by a reduction in D5 expression mediated by AraC in cells infected with VACV WR or vΔC9 (Figure 4D). Therefore, the reduction in D5 expression mediated by IFN in cells infected with vΔC9 (Figure 4D) could be secondary to the inhibition of DNA replication and late gene expression by IFITs, rather than providing a direct target of IFIT activity.
In addition to showing that the expression of representative early proteins by vΔC9 was resistant to IFN, we performed a comprehensive screen of early proteins using TMT 6-plex mass spectrometry. Untreated and IFN-treated cells were infected with wild-type VACV WR or vΔC9 for 4 h in the presence or absence of AraC, and the abundances of the 122 viral proteins detected are provided in Table S3.The median and mean abundance ratios (WR/ΔC9) calculated for all of the proteins from cells treated with IFN without addition of AraC and with AraC were 0.945 and 0.965, respectively, indicating no global reduction in early proteins due to the absence of C9. Further experiments would be needed to evaluate the significance of outlier proteins. We concluded that, under single-step growth conditions, C9-mediated IFN resistance is required to permit viral intermediate/late protein synthesis, but it has no apparent effect on early proteins.
KO of IFIT1 and IFIT3 Reduce IFN Sensitivity of C9-Deficient VACV
To investigate whether each IFIT is required for IFN sensitivity, CRISPR/Cas9 was used to generate A549 cell lines in which IFIT genes were individually inactivated. DNA sequencing demonstrated out-of-frame mutations in the second exon of the IFIT in the corresponding knockout (KO) cell line. The absences of IFIT1, IFIT2, and IFIT3 in the KO cell lines following IFN treatment were demonstrated by WB (Figure 5A). KO of IFIT1 and IFIT2 produced specific depletion of their gene products. However, KO of IFIT3 also reduced IFIT1 and IFIT2, which is consistent with prior studies showing that IFIT3 acts as a scaffold for the stability of the other IFITs (Fleith et al., 2018; Pichlmair et al., 2011). Off-target effects of the IFIT KOs that may affect the expression of ISGs in general were ruled out by demonstrating the IFN induction of protein kinase R (PKR) and ISG15 in control and IFIT KO cells (Figure 5A).
Figure 5. IFN Resistance of C9-Deleted VACV in IFIT KO Cells.

(A) Reduction of IFIT expression in IFIT KO cells. Lysates of A549 or A549 IFIT KO cells untreated (−) or treated (+) with IFN-β were analyzed by WB.
(B) Effects of IFIT KO on IFN resistance. A549 and KO cells were untreated (−) or pretreated (+) with IFN-β and infected with 3 PFUs per cell of VACV WR or vΔC9. After 8 h, lysates were analyzed by WB.
(C) Effect of IFIT KO on RNA levels. A549 and KO cells were infected in triplicate as in (B) for 6 h. RNA was extracted, reverse transcribed, and quantified by ddPCR. Error bars: SEMs.
(D) Antiviral activity of IFIT1 is reduced by the mutation of the RNA-binding motif. Untreated (−) or IFN-β-pretreated (+) A549, IFIT1 KO cells, and IFIT1 KO cells trans-complemented with vector, FLAG-IFIT1, or FLAG-IFIT1R187H by retrovirus transduction were mock infected or infected with 3 PFUs per cell of VACV WR or vΔC9. After 8 h, cell lysates were analyzed by WB.
(E) Antiviral activity of IFIT3 is reduced by mutation of IFIT1-association motif. Untreated (−) or IFN-β-pretreated (+) A549 IFIT3 KO or A549 IFIT3 KO cells transcomplemented with vector, FLAG-IFIT3, or FLAG-IFIT3ΔCTD by retrovirus transduction were mock infected or infected with 3 PFUs per cell of VACV WR or vΔC9. After 8 h, lysates were analyzed by WB.
Representative blots from three biological repeats in (A) and (B) and two in (D) and (E).
Next, we examined the ability of the KO cells to rescue viral protein synthesis in IFN-treated cells infected with vΔC9. In the control cells infected with vΔC9, expression of the VACV intermediate/late A3 protein was barely detected in the presence of IFN, whereas the IFN-mediated downregulation of A3 was substantially mitigated in IFIT1 and IFIT3 KO cells, less strongly so in IFIT2 KO cells, and not at all in IFIT5 KO cells (Figure 5B). Synthesis of the representative viral early E3 protein was undiminished by either IFN treatment or IFIT expression. In view of the specific depletion of IFIT1 in the IFIT1 KO cells (Figure 5A), we conclude that IFIT1 contributes to the IFN-dependent resistance of A549 cells to VACV. Since the absence of IFIT3 also led to a reduced level of IFIT1 (Figure 5A), the role of IFIT3 may be indirect. KO of IFIT2 had a minor effect (Figure 5B), even though binding to C9 was stronger than for the other IFITs (Figure 3C).
The effect of IFN on vΔC9-mediated intermediate/late protein synthesis, as exemplified by the decrease in the A3 protein level and the rescue by KO of IFITs, could occur at either the transcriptional or the translational level. In the absence of IFN, the levels of A3 mRNA quantified by droplet digital PCR (ddPCR) were similar regardless of whether control or KO cells were infected with either VACV WR or vΔC9 (Figure 5C). In control cells infected with VACV WR, IFN reduced A3 mRNA by ~2-fold, whereas a 12-fold reduction occurred with vΔC9 infection (Figure 5C), which could account for the difference in the A3 protein level without invoking an additional translational effect. A3 mRNA levels were rescued in the KO cells in the order IFIT1 > IFIT3 > IFIT2. In contrast to the greater reduction of A3 mRNA in IFNtreated cells infected with vΔC9 than in wild-type VACV, the amounts of the early E3 mRNA were similar, as were the 18S rRNA controls (Figure 5C). We routinely noted increased early mRNAs and proteins in cells treated with IFN, possibly due to the delayed uncoating of cores, which is the site of transcription. We show below that the decrease in intermediate/late mRNAs in IFN-treated vΔC9-infected cells can be explained by the IFITmediated inhibition of genome replication.
Further experiments were carried out, with two aims. The first aim was to determine whether IFN resistance of IFIT1 and IFIT3 KO cells could be reversed by the transfection of expression plasmids encoding IFIT1 or IFIT3. This would rule out off-target CRISPR/Cas9 modifications causing IFN resistance. The second aim was to determine whether known functional motifs within IFIT1 and IFIT3 were involved in their action. Retrovirus vectors expressed FLAG-tagged wild-type IFIT1 or the decreased RNA-binding mutant IFIT1R187H (Abbas et al., 2013) at comparable levels in IFIT1 KO cells, and the expression of both were enhanced by IFN, likely due to stabilization by IFNinduced IFIT3 (Figure 5D). The IFIT1 monoclonal antibody (mAb) failed to react with IFIT1R187H as its epitope is within the mutated RNA-binding site. Notably, the rescue of viral A3 protein expression in IFN-treated IFIT1 KO cells infected with vΔC9 was diminished by the expression of wild-type IFIT1 but to a lesser extent with IFIT1R187H, suggesting the involvement of the RNAbinding motif in the antiviral activity.
Human IFIT3 enhances the stability of IFIT1 and modulates its binding to cap 0 RNA (Johnson et al., 2018). To explore the role of IFIT3 in the present system, IFIT3 KO cells were infected with a retrovirus vector that expressed FLAG-tagged wild-type IFIT3 or IFIT3 with a deletion of the C-terminal domain (CTD) that is required for association with IFIT1 (Fleith et al., 2018). The IFN-dependent reduction in the level of the viral A3 protein in the absence of C9 was greater with the expression of unmutated IFIT3 than IFIT3ΔCTD (Figure 5E). Moreover, IFIT3ΔCTD did not increase IFIT1 levels as did the unmutated IFIT3 (Figure 5E), which is consistent with the importance of IFIT1 stabilization by IFIT3.
VACV MTase and C9-Deletion Mutants Have Different IFIT-Sensitivity Phenotypes
We considered that insights could be gained into the inhibitory role of IFITs by comparing their effects on vΔC9 and the VACV MTase mutant vJ3K175R. The latter virus is unable to convert cap 0 to cap 1 mRNA and was shown to be sensitive to mouse IFIT1, although the stage of virus replication that was inhibited was not investigated (Daffis et al., 2010). Human IFIT1, in contrast to mouse IFIT1, was unable by itself to inhibit vJ3K175R (Daugherty et al., 2016), possibly because human IFIT3 was also needed. We found that human IFN-β inhibited the synthesis of the representative early proteins I3 and E3, as well as the intermediate/late A3 protein in human A549 cells infected with vJ3K175R for 8 h (Figures 6A and 6B), whereas only intermediate/late protein synthesis was inhibited in cells infected with vΔC9 (Figure 6B). The IFN-sensitivity phenotype of the MTase mutant was rescued in IFIT1 KO cells, but not in either IFIT2 or IFIT3 KO cells, indicating an essential role for human IFIT1 (Figure 6A). The rescue of vJ3K175R also occurred in A549 cells expressing Myc-C9 in which the IFITs were degraded, but not in cells expressing Myc-C9ΔF-box in which the IFITs were stable (Figure 6B). These results indicated that human IFIT1 was required for the IFN sensitivity of both mutants but that the inhibition of the MTase mutant occurred at an earlier stage than for the C9-deletion mutant.
Figure 6. Comparison of IFN Resistance of Cap 1 MTase and C9-Deletion Mutants.

(A) IFN resistance of VACV MTase mutant in IFIT1 KO cells. Untreated (−) or IFN-β-pretreated (+) A549 and IFIT KO cells were infected with 3 PFUs per cell of VACV WR or the VACV cap 1 MTase mutant vJ3K175R. After 8 h, lysates were analyzed by WB.
(B) IFN resistance of VACV MTase mutant in cells expressing Myc-C9. Untreated (−) or IFN-β-pretreated (+) A549 cells and A549 cells expressing the vector, Myc-C9, or Myc-C9ΔF-box were mock infected or infected with 3 PFUs per cell of the indicated viruses. After 8 h, lysates were analyzed by WB.
(C) IFN resistance of VACV MTase mutant in cells expressing HA-J3. Untreated (−) or IFN-β-pretreated (+) A549 cells and A549 cells expressing HA-J3 were mock infected or infected with 3 PFUs per cell of the indicated viruses. After 8 h, lysates were analyzed by WB.
Representative blots from three biological repeats in (A) and (B) and two in (C).
The susceptibility of vJ3K175R to IFITs is explained by the ability of IFITs to compete with translation factors for binding to cap 0 mRNAs, thereby preventing viral early protein synthesis. We considered the possibility that even if a virus expresses MTase, IFITs may compete with MTase for binding to nascent mRNAs and prevent cap 1 formation. The expression of viral early proteins by vΔC9 implied that the MTase was active within the core, which may exclude IFITs. However, intermediate and late mRNAs are synthesized by enzymes within the cytoplasm, where competition by IFITs could more readily occur. If that were the case, then increasing the amount of MTase may overcome the competitive inhibition to some degree. To investigate this possibility, a retrovirus vector was used to stably express active epitope-tagged HA-J3 MTase in A549 cells. Expression of the MTase was demonstrated by WB with antibody to the HA tag (Figure 6C). Furthermore, viral early and late protein synthesis in IFN-treated cells infected with vJ3K175R was enhanced, demonstrating that active MTase was made (Figure 6C). Nevertheless, viral protein synthesis was not enhanced in cells infected with vΔC9 (Figure 6C). Since IFIT1 is one of the most abundant proteins following IFN treatment (Pichlmair et al., 2011), it may be difficult to outcompete. To improve the putative competition in favor of J3, we reduced the amount of IFITs by decreasing the time of IFN treatment before infection (Figure S2A). Nevertheless, ectopic expression of HA-J3 enhanced the expression of viral proteins by the MTase mutant but not by vΔC9, even under these conditions (Figure S2B).
Rescue of IFN-Induced Inhibition of Viral DNA Replication in IFIT KO Cell Lines
Thus far, our studies point to a role for IFITs in the inhibition of vΔC9 that is distinct from that due to the absence of ribose methylation of mRNA cap structures. Previous studies (Liu and Moss, 2018) showed that viral DNA replication was severely inhibited in IFN-treated A549 cells infected with vΔC9. In addition, there was evidence of the impaired release of viral DNA from cores. Here, further experiments were undertaken to assess whether these steps would be rescued in IFN-treated IFIT1 and IFIT3 KO cells infected with vΔC9. The basis for these experiments is outlined in Figure 7A. Following entry into the cytoplasm, viral cores synthesize early mRNAs, which encode the factors needed for release and replication of the viral genome. I3, a viral early single-stranded DNA-binding protein, can serve as a localization marker for both released and replicated genomic DNA (Mercer et al., 2012). In the experiment depicted in Figure 7B, fluorescence confocal microscopy detected newly replicated DNA by incorporation of the clickable thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) from 3 to 4 h after infection; I3 was detected with a specific antibody and total DNA in the nucleus and viral factories was stained with DAPI. Where indicated, AraC was used to inhibit viral DNA replication to allow detection of the genome released from the viral core.
Figure 7. Restoration of VACV DNA Replication in IFN-β-Pretreated IFIT1 and IFIT3 KO Cells.

(A) Illustration of virion core synthesizing mRNAs encoding I3 and additional proteins leading to the uncoating and replication of the viral genome. I3 co-localizes with genomes released from the core and newly replicated DNA labeled with EdU.
(B) Fluorescence confocal microscopy images. A549 and IFIT KO cells were mock infected or infected in the absence or presence of IFN and AraC for 3 h and then incubated with 10 μM EdU for 1 h. Individual fluorescence channels are shown in grayscale and the merge in colors (EdU, green; I3, red; DAPI, blue). The images are from two biological repeats. Scale bar: 20 μm.
(C) The percentage of AraC-treated cells positive for I3 punctae relative to the total number of cells expressing I3. Fold increases in the numbers of vΔC9-infected IFIT KO cells relative to A549 cells positive for punctae are shown. Error bars: SEM.
(D) Viral DNA replication. Untreated or IFN-β-pretreated A549 and IFIT KO cells were infected in triplicate with 3 PFUs per cell for 6 h. Viral DNA quantified by ddPCR. Fold increases in DNA copies of IFIT KO cells relative to A549 cells infected with vΔC9 shown. Error bars: SEMs.
In control mock-infected A549 cells, no background I3 staining was detected, and EdU labeled nuclear DNA exclusively (Figure 7B). In A549 cells infected with VACV WR, I3 and EdU colocalized in discrete spots representing DNA factory areas within the cytoplasm with or without IFN pretreatment. Diminution of nuclear labeling following the VACV infection observed here has been previously noted (Jungwirth and Launer, 1968; Pogo and Dales, 1973; Senkevich et al., 2017). In the presence of AraC, I3 dots (much smaller than the factories), representing genomes released from cores over a background of diffuse cytoplasmic I3 staining, were detected in both IFNpretreated and -untreated A549 cells infected with VACV WR. In the absence of IFN, the pattern of I3 and EdU staining was similar in A549 cells infected with vΔC9 and VACV WR. However, in IFNpretreated A549 cells, there was only diffuse I3 staining and no EdU incorporation was detected in the cells infected with vΔC9, even in the absence of AraC; this is consistent with blocks in genome uncoating and replication.
In contrast to the above results obtained with unmodified A549 cells, the I3 and EdU staining pattern of IFIT1 KO cells infected with vΔC9 was not reduced by IFN pretreatment, indicating that IFIT1 was required to inhibit genome replication (Figure 7B). Under the same conditions in IFIT3 KO cells, there was less protection against IFN and little or none in IFIT2 KO cells. The resistance to the IFN-mediated inhibition of uncoating, determined in AraC-treated cells infected with vΔC9, was also greatest in IFIT1 KO cells, followed by IFIT3 and IFIT2 KO cells (Figures 7B and 7C). The effect of IFN on viral DNA replication was quantified using ddPCR. The fold increase in the genome copy numbers in IFN-treated and vΔC9-infected IFIT1, IFIT2, and IFIT3 KO cells compared to A549 cells was approximately 16-, 3-, and 9-fold, respectively (Figure 7D). Thus, the beneficial effects of IFIT KO on genome uncoating and replication in IFN-pretreated cells were IFIT1 > IFIT3 > IFIT2. Furthermore, the inhibition of genome replication can fully explain the reduction in VACV intermediate and late mRNAs and their translation products in IFN-pretreated cells infected with vΔC9.
DISCUSSION
The aims of the present study were to identify the IFN-induced targets of the C9 ANK-repeat/F-box protein, determine how C9 inactivates the putative targets, and investigate the way in which the targeted IFN-response factors inhibit virus replication. Human A549 cells that expressed either the full-length C9 or C9 with the F-box deleted were used to investigate the first two aims. Following IFN-β treatment, full-length C9 was stably associated with the SCF ubiquitin complex, whereas ΔF-box C9 was associated with the IFIT1/2/3 complex. These results suggested that the ANK repeats interacted with the IFITs, while the F-box mediated the degradation of the IFITs. A profound reduction of IFITs in the IFN-treated cells expressing the fulllength C9 was demonstrated both by WB and quantitative mass spectrometry. The latter analysis indicated that IFIT2, IFIT1, and IFIT5 were reduced more than was IFIT3. A similar ranking occurred when IFN treatment was omitted and the IFITs were expressed individually by transfection in cells expressing full-length C9. Stabilization of IFIT1 and IFIT2 by PS-341 supported a proteosomal mechanism for their degradation mediated by C9. Expressed individually, IFIT2 was more highly ubiquitinated than IFIT1, which correlated with their relative binding to C9 and C9ΔF-box. The relative ubiquitination of the IFITs when they are in a complex remains to be determined.
Our next step was to correlate the findings regarding IFITs obtained by the expression of C9 in uninfected cells to a virus infection. A time course experiment in IFN-treated A549 cells revealed that IFIT1 and IFIT2 were greatly reduced at 4 h and virtually undetectable by 8 h after infection with VACV expressing full-length C9, whereas the reduction of IFIT3 was delayed and incomplete. Although the full-length C9 protein was required for IFIT degradation, the C9DΔF-box protein was sufficient for IFIT binding, which in principle may provide IFN resistance. However, the IFN sensitivity of the recombinant virus vC9ΔF-box was similar to vΔC9, in which the entire C9 open reading frame (ORF) was deleted. Thus, both the ANK repeats and F-box were needed for C9-mediated resistance to IFN.
Although the data discussed thus far correlated the ability of a full-length C9 protein to degrade IFITs with the IFN resistance of VACV, they did not prove that IFITs are required for IFN sensitivity. The latter was accomplished using CRISPR/Cas9 to KO expression of individual IFIT genes. The mutations in IFIT1, IFIT2, and IFIT3 prevented the expression of the corresponding proteins; however, KO of IFIT3 also reduced the expression of IFIT1 and IFIT2, consistent with the role of IFIT3 in stabilizing IFIT1 and IFIT2 in a complex (Fleith et al., 2018; Johnson et al., 2018). IFIT1 and IFIT3 KO cell lines each provided greater rescue of the IFN sensitivity of a VACV C9-deletion mutant than an IFIT2 KO, even though IFIT2 exhibited the highest affinity for C9. In addition, no rescue occurred with an IFIT5 KO, even though IFIT5 was degraded in cells that expressed C9. Off-target effects were ruled out by the restoration of IFN sensitivity following transfection of the genes encoding IFIT1 and IFIT3. Restoration was impaired, however, by mutating a site in IFIT1 involved in RNA binding and a site in IFIT3 involved in interaction with IFIT1. We can conclude that IFIT1 is required for IFN resistance and that IFIT3 may have a direct or indirect role due to the stabilization of IFIT1. Recent studies show that human IFIT3 modulates IFIT1 binding to RNA (Johnson et al., 2018).
Another question addressed in this study was how IFITs inhibit the VACV C9-deletion mutant. IFITs may inhibit virus replication at the translational level either by binding to translation factors or to uncapped or incompletely methylated capped RNA (Diamond and Farzan, 2013; Fensterl and Sen, 2015; Habjan et al., 2013). We found that a VACV MTase mutant was inhibited by human IFN at the stage of viral early protein synthesis, which is consistent with the incompletely methylated mRNAs interacting with IFITs and preventing their translation. This model was confirmed by the rescue provided by ectopic expression of the viral MTase. By degrading the IFITs, ectopic expression of C9 also rescued the MTase mutant. The INF-mediated inhibition of the MTase mutant was primarily mediated by human IFIT1 as KO of the corresponding gene rescued viral protein synthesis, whereas KO of IFIT2 or IFIT3 did not. In IFN-pretreated cells infected with the C9-deletion mutant, however, an effect on viral early protein synthesis was not detected by mass spectrometry or analysis of representative early species. We also could not rescue the defect by ectopic expression of the MTase. However, uncoating and replication of the viral genome, which immediately follow early protein synthesis, were inhibited by IFN and rescued by KO of IFIT1, and to a lesser extent by KO of IFIT3. The transcription of intermediate and late genes is dependent on viral DNA replication (Keck et al., 1990). Accordingly, the inhibition of genome replication by IFITs can fully explain the reduction in VACV intermediate and late mRNAs and their translation products in cells infected with vΔC9. In this regard, IFIT1 has been shown to inhibit human papillomavirus genome replication by interacting with the E1 origin binding protein (Saikia et al., 2010; Terenzi et al., 2008). How the IFITs interfere with VACV genome uncoating/replication remains to be determined.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bernard Moss (BMOSS@niaid.nih.gov).
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Cell lines were maintained at 37°C in 5% CO2 humidified incubators. A549 (human, male) was grown in DMEM/F-12 medium containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. BS-C-1 (monkey) cells were grown in EMEM containing 10% FBS, and 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK293T (human embryonic kidney) cells were grown in DMEM containing 10% FBS, and 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
METHOD DETAILS
Plasmids and Transfection
Expression plasmids for pcDNA3.1–3 × FLAG tagged IFIT1, IFIT2, IFIT3, IFIT5 and HA tagged Ub, were from Addgene. Point mutation IFIT1R187H was generated using Q5 Site-Directed Mutagenesis (NEW ENGLAND BioLabs). All constructs were confirmed by DNA sequencing. For transfection of plasmids, Lipofectamine 2000 reagent was used (ThermoFisher).
Retroviral Vectors, Mutant cDNAs
The ORFs corresponding to IFIT1, IFIT1 R187H, full-length IFIT3, and IFIT3 lacking C-terminal residues 403–490 (IFIT3ΔCTD) all with N-terminal 3 × FLAG tags were inserted into the retroviral expression vector pQC-XIP. A549 cells stably expressing the 2 × Myc tagged full length C9 protein were previously made by inserting the eukaryotic codon optimized VACVWR C9 ORF with an N-terminal 2 × Myc tag into pQC-XIP (Liu and Moss, 2018). To generate A549 cells stably expressing 2 × Myc tagged F-box deleted C9, the eukaryotic codon-optimized VACV WR strain C9 ORF lacking the F-box domain (amino acids 552 to 631) with an N-terminal 2 × Myc tag was inserted into pQC-XIP. For A549 cells stably expressing J3, a eukaryotic codon-optimized J3 ORF of VACV WR strain with an N-terminal HA-tag was inserted into pQC-XIP.
VSV-G pseudotyped virus particles were generated by three-plasmid transfection of 293T cells with Lipofetamine 2000 using pMLV-Gag-Pol (murine leukemia viruses gag and pol genes, packaging plasmid), pVSV-G (vesicular stomatitis virus G protein [VSV-G], envelope plasmid), and pQC-XIP encoding cDNAs. A549 cells were infected with the retroviruses in the presence of 5 μg/ml Polybrene (Sigma-Aldrich). The cells were subcultured and passaged several times in selection medium containing 1.5 μg/ml of puromycin (Sigma-Aldrich). Expression of the corresponding protein was determined by WB using anti-epitope antibodies.
Construction of recombinant VACV
The recombinant VACV vΔC9 and vHA-C9 (Liu and Moss, 2018) and the recombinant VACV with a mutationally inactivated MTase site vJ3K175R (Latner et al., 2002) were described previously. The recombinant virus vHA-C9ΔF-box was generated by replacing the P11-GFP ORF in vΔC9 with DNA assembled by overlap-extension PCR with a mixture containing an N-terminal HA tagged F-box domain deleted C9 ORF regulated by the natural promoter flanked by portions of the adjacent genes. Homologous recombination was carried out by infecting BS-C-1 cells with 1 PFU/cell of vΔC9, followed by transfection with assembled PCR products using the Lipofectamine 2000 reagent. After 24 h, cells were harvested and lysed by three freeze-thaw cycles. The lysates were diluted serially and used to infect BS-C-1 cell monolayers. Non-fluorescent recombinant plaques were distinguished from the parental GFP plaques and clonally purified through five rounds of plaque isolation. The purities of the recombinant viruses were confirmed by PCR amplification and sequencing of the modified region. The VACV WR and recombinant viruses were propagated in BS-C-1 cells.
Purification of virus particles
Recombinant viruses grown in BS-C-1 cells were purified by centrifugation through a cushion of 36% (w/v) sucrose in 1 mM Tris-Cl, pH 9.0 for 80 min at 32,900 3 × g at 4°C, followed by centrifugation through a gradient of 24% to 40% sucrose in 1 mM Tris-Cl, pH 9.0 for 50 min at 26,000 × g at 4°C (Cotter et al., 2017). The milky white band of virus was aspirated, diluted with 2 volumes of 1 mM Tris-Cl, pH 9.0 and centrifuged for 60 min at at 32,900 × g at 4°C. After resuspension in 1 mM Tris-Cl, pH 9.0, aliquots of virus were stored at −80°C. Infectivity was determined by plaque assay in BS-C-1 cells as described below (Cotter et al., 2017).
Plaque assay
Virus samples were dispersed in a chilled water bath sonicator with two 30 s periods of vibration, followed by 10-fold serial dilutions in EMEM supplemented with 2.5% FBS. Diluted viruses were distributed onto BS-C-1 cell monolayers. After adsorption for 1.5 h, the medium was aspirated and replaced with medium containing 0.5% methylcellulose. After 48 h, the cells were stained with crystal violet at room temperature for 20 min and the plaques were counted.
Generation of CRISPR-Cas9 genetically modified cell lines
To inactivate human IFIT1, 2, 3, and 5 in A549 cells, sgRNA sequences were designed using the Web-based tool (http://zlab.bio/guide-design-resources/) and are listed in the Key Resources Table. The DNA sequences were synthesized (Eurofins, Luxembourg), annealed and separately introduced into the plasmid vector pSpCas9(BB)-2A-GFP, which drives expression of the Streptococcus pyogenes Cas9, GFP and the chimeric guide RNA in mammalian cells. A549 cells were transfected with 8 μg of individual plasmids with 32 μl Lipofectamine 2000, and after 48 h, GFP-positive cells were sorted by flow cytometry and clonal cell lines were isolated by serial dilution. After 2 weeks, individual colonies were analyzed for IFIT loss-of-expression deletions by WB and mutations were confirmed by Sanger sequencing.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-IFIT1 (clone D2X9Z) | Cell Signaling Technology | Cat # 14769; RRID: N/A |
| Mouse monoclonal anti-IFIT2 (clone F-12) | Santa Cruz Biotechnology | Cat # sc-390724; RRID: N/A |
| Mouse monoclonal anti-IFIT3 (clone B-7) | Santa Cruz Biotechnology | Cat # sc-393512; RRID: N/A |
| Mouse monoclonal anti-CUL-1 (clone D-5) | Santa Cruz Biotechnology | Cat # sc-17775; RRID: AB_627325 |
| Mouse monoclonal anti-ISG15 (clone F-9) | Santa Cruz Biotechnology | Cat # sc-166755; RRID: AB_2126308 |
| Rabbit monoclonal anti-PKR (clone 23H52L96) | Thermo Fisher | Cat # 700286; RRID: AB_2532313 |
| Horseradish peroxidase (HRP)-conjugated c-Myc antibody (clone 9E10) | Santa Cruz Biotechnology | Cat # sc-40 HRP; RRID: AB_627268 |
| Horseradish peroxidase (HRP)-conjugated FLAG antibody (clone M2) | Sigma-Aldrich | Cat # A8592; RRID: AB_439702 |
| Mouse monoclonal anti-HA (clone 16B12) | BioLegend | Cat # 901501; RRID: N/A |
| Rabbit polyclonal anti-VACV WR strain | Moss Laboratory | Davies et al., 2008 |
| Mouse monoclonal anti-E3 (clone 3015B2) | Stuart N. Isaacs | Weaver et al., 2007 |
| Mouse monoclonal anti-I3 (clone 10D11) | David H. Evans | Lin et al., 2008 |
| Rabbit polyclonal anti-D5 | Paula Traktman | Evans and Traktman, 1992 |
| Mouse monoclonal anti-L1 (clone 7D11) | Moss laboratory | Wolffe et al., 1995 |
| Rabbit polyclonal anti-A3 | Moss laboratory | Unpublished |
| Rabbit polyclonal anti-actin | Sigma-Aldrich | Cat # A2066; RRID: AB_476693 |
| Bacterial and Virus Strains | ||
| Vaccinia virus WR strain | ATTC | ATCC VR-1354 |
| Vaccinia virus vΔC9 | Moss laboratory | Liu and Moss, 2018 |
| Vaccinia virus vHA-C9 | Moss laboratory | Liu and Moss, 2018 |
| Vaccinia virus vHA-C9ΔF-box | This paper | N/A |
| Vaccinia virus vJ3K175R | Richard C. Condit | Latner et al., 2002 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human IFN-β1a | Antigenix America | Cat # HC99921B |
| PS-341(Bortezomib) | Sigma-Aldrich | Cat # 5043140001 |
| Cytosine-D-arabinofuranoside (AraC) | Sigma-Aldrich | Cat # C1768 |
| Polybrene | Sigma-Aldrich | Cat # H9268–10G |
| Puromycin | Millipore | Cat # 540411–25MG |
| Protease inhibitor cocktail | Roche | Cat # 5892791001 |
| Phosphatase inhibitor cocktail | Roche | Cat # 04906845001 |
| Lipofectamine 2000 reagent | Thermo Fisher | Cat # 11668–019 |
| Myc-Trap_MA | Chromotek | Cat # ytma-20 |
| Blocked magnetic agarose beads | Chromotek | Cat # bmab-20 |
| Anti-FLAG M2 magnetic beads | Sigma-Aldrich | Cat # M8823 |
| 3X FLAG peptide | Sigma-Aldrich | Cat # F4799 |
| Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher | Cat # 78441 |
| DNase I | Thermo Fisher | Cat # 18068–015 |
| QX200 ddPCR EvaGreen Supermix | BIO-RAD | Cat # 186–4034 |
| Critical Commercial Assays | ||
| Click-iT Plus EdU Alexa Fluor 647 Imaging Kit | Thermo Fisher | Cat # C10640 |
| In-Fusion® HD Cloning Kit | TaKaRa | Cat # 639650 |
| Q5 Site-Directed Mutagenesis Kit | NEW ENGLAND BioLabs | Cat # E0554S |
| DNeasy blood and tissue kit | QIAGEN | Cat # 69504 |
| RNeasy Mini Kit | QIAGEN | Cat # 74104 |
| SuperScript IV First-Strand Synthesis System | Thermo Fisher | Cat # 18091050 |
| Experimental Models: Cell Lines | ||
| Human A549 cells | ATCC | Cat # CCL-185 |
| Human Embryonic kidney 293T cells | ATCC | Cat # CRL-3216 |
| Monkey BS-C-1 cells | ATCC | Cat # CCL-26 |
| Oligonucleotides | ||
| IFIT1 KO sgRNA: ATGACAACCAAGCAAATGTG | Johnson et al., 2018 | N/A |
| IFIT2 KO sgRNA: AATGGCATTTTAGTTGCCGT | This paper | N/A |
| IFIT3 KO sgRNA: ACACCTAGATGGTAACAACG | This paper | N/A |
| IFIT5 KO sgRNA: GGTGTTTCACATAGGCCAAT | This paper | N/A |
| IFIT2 ddPCR F: GGGGAAACTATGCCTGGGTC | This paper | N/A |
| IFIT2 ddPCR R: GTGTCCACCCTTCCTCACAG | This paper | N/A |
| IFIT3 ddPCR F: ACTTGGGGAAACTACGCCTG | This paper | N/A |
| IFIT3 ddPCR R: TCCACCCTTCCTCACAGTCA | This paper | N/A |
| 18 s rRNA F: GGCCCTGTAATTGGAATGAGTC | This paper | N/A |
| 18 s rRNA R: CCAAGATCCAACTACGAGCTT | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3.1(+) | Thermo Fisher | Cat # V790–20 |
| pQC-XIP | TaKaRa | Cat # 631516 |
| pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene #48138 |
| pHA-Ub | Kamitani et al., 1997 | Addgene #18712 |
| pcDNA3.1–3 × FLAG-IFIT1 | Katibah et al., 2013 | Addgene #53554 |
| pcDNA3.1–3 × FLAG-IFIT2 | Katibah et al., 2013 | Addgene #53555 |
| pcDNA3.1–3 × FLAG-IFIT3 | Katibah et al., 2013 | Addgene #53553 |
| pcDNA3.1–3 × FLAG-IFIT5 | Katibah et al., 2013 | Addgene #53556 |
| pQC-XIP-3 × FLAG-IFIT1 | This paper | N/A |
| pQC-XIP-3 × FLAG-IFIT1 R187H | This paper | N/A |
| pQC-XIP-3 × FLAG-IFIT3 | This paper | N/A |
| pQC-XIP-3 × FLAG-IFIT3ΔCTD | This paper | N/A |
| pQC-XIP-2 × Myc-C9 | This paper | N/A |
| pQC-XIP-2 × Myc-C9ΔF-box | This paper | N/A |
| pVSV-G | Stewart et al., 2003 | Addgene #8454 |
| pMLV-Gag-Pol | Chen Liang | McGill University |
| Software and Algorithms | ||
| Geneious R11 | Geneious | https://www.geneious.com |
| Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| iMaris 9.0.1 | Bitplane | https://imaris.oxinst.com |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Mascot 2.6 | Mascot | www.matrixscience |
| SwissProt | UniProt | https://www.uniprot.org |
| String v11.0 | String | https://string-db.org |
| Xcalibur 4.0 | ThermoFisher | https://www.thermofisher.com/us/en/home.html |
| Proteome Discoverer 2.2 | ThermoFisher | https://www.thermofisher.com/us/en/home.html |
Affinity purification and mass spectrometry
Cells were washed twice with cold phosphate buffered saline (PBS) on ice, harvested by scraping, and lysed in lysis buffer (20 mM Tris [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) containing protease inhibitor and phosphatase inhibitor (Roche) on wet ice for 30 min with frequent agitation. Lysates were centrifuged for 10 min at 20,000 × g at 4C, and the supernatant was incubated with anti-FLAG M2 beads (Sigma Aldrich) or Myc-Trap agarose beads (ChromoTek) at 4°C for 3 h. After extensive washing with lysis buffer, the bound proteins were eluted with 3 × FLAG peptide or 1 × reducing sample buffer and resolved by SDS-PAGE. To determine the association of IFITs with C9 and ubiquitination of IFITs, cell lysates and captured proteins were analyzed by WB with antibodies to specific proteins.
For mass spectrometry, the affinity purified proteins were reduced and alkylated and size-fractionated on SDS-polyacrylamide gels. Gel slices were treated with trypsin and the supernatant and two washes (5% formic acid in 50% acetonitrile) of the gel digests were pooled and concentrated with a Speed Vac (Labconco, Kansas, MO) to dryness directly in 200 μl polypropylene auto-sampler vials (Sun Sri, Rockwood, TN). The recovered peptides were dissolved in 10 μl of Solvent A (0.1% formic acid).
Mass spectrometry analysis was performed on an Orbitrap Fusion Tribrid with in-line chromatography. Nano LC-MS (LC-MS/MS) was performed with a ProXeon Easy-nLC 1000 multi-dimensional liquid chromatograph and temperature controlled Nanospray Flex Ion Source (ThermoFisher). Peptides were separated at 200 nl/min at 60°C using a PepMap100 C18 reverse phase media trap column (3 μm particle size, 75 μm ID, 2 cc(2 μm particle size, 75 μm ID, 50 cm length) (ThermoFisher). The mobile phase comprised a linear gradient from solvent A to 40% solvent B (0.1% formic acid, and 99.9% acetonitrile) over 100 minutes, followed by a rapid 2-minute increase to 80% solvent B where it was held for 8 minutes before a 5-minute return to 100% solvent A. Computer controlled data dependent automated switching to MS/MS by Xcalibur software was used for the generation of fragmentation spectra. Data processing and database searching were performed with Proteome Discoverer v2.2 (ThermoFisher). The data were searched against human and VACV WR proteins deposited in the Uniprot KB (6/2017) and the common Repository of Adventitious Proteins (theGPM.org) with oxidation of methionine as a dynamic modification and carbamindomethylation of cysteine as a fixed modification. Peptides were filtered at a 1% FDR (False Discovery Rate) calculated using a target-decoy approach and a 2 peptide per protein minimum. Label-free area quantification based on unique and razor peptides after retention time alignment was performed using nodes within Proteome Discoverer with normalization to total peptide load.
Mass spectrometry of TMT 6-Plex-labeled proteins
Untreated or IFN-treated A549, A549-C9 and VACV-infected A549 cells were harvested and solubilized in 8 M urea, 0.1 M triethylammonium bicarbonate (TEAB), pH 8.0, before addition of 5 volumes of cold acetone, incubation at −20°C for 60 min then centrifugation in a benchtop centrifuge at 13,000 rpm at 4°C for 60 min. The resulting pellets were dissolved in 8 M urea, 0.1 M TEAB followed by Bicinchonic acid (BCA) protein assay. Aliquots corresponding to 0.120 mg protein from each of the six samples were supplemented with Tris(2-carboxyethyl)phosphine (TCEP) to 10 mM (final) then incubated at 37°C for 30 min. After dilution to 6 M urea with 0.1 M TEAB and addition of Lys-C (Promega; 1:100 Lys-C:substrate mass ratio) samples were incubated at 37°C overnight then supplemented with an equivalent aliquot of Lys-C for a subsequent overnight incubation at 37°C. After dilution to 1 M urea with 0.1 M TEAB, samples were supplemented with trypsin (1:100 trypsin:substrate mass ratio) and incubated at 37°C overnight then supplemented with an equivalent aliquot of trypsin and incubated again overnight at 37°C. nanoLC-MS/MS of a small aliquot of each sample showed the presence of missed trypsin sites in < 12% of identified peptides. The remaining peptides in each sample were subjected to solid-phase extraction (Sep-Pak C18; Waters Inc.), eluting with 80% CH3CN/0.1% formic acid (FA) followed by solvent evaporation under vacuum. After re-solubilizing in 0.1 M TEAB, 0.10 mg of peptides from each of the six samples were incubated with, respectively, 0.8 mg of TMT6-#1, TMT6-#2, TMT6-#3, TMT6-4, TMT6-#5 and TMT6-#6 reagent (ThermoFisher) according to manufacturer’s instructions, and reactions quenched with hydroxylamine. The six individually labeled samples were combined and the pool was acidified with FA then subjected to C18 solid-phase extraction using Sep-Pak C18 as above. Tryptic peptides were re-dissolved in strong cation exchange (SCX) solvent mixture A (30% CH3CN; H3PO4 to pH 2.7) then loaded on a Polysulfethyl A (200 × 3.0 mm, 5-μm particle size, 200 Å pore size) column (PolyLC Inc.) that had been thoroughly equilibrated with solvent mixture A using a Waters 600E multisolvent delivery system/486 detector and monitoring OD214 via Clarity chromatography software (DataApex Inc.). After washing with SCX solvent A until the OD214 approached zero, the column was eluted with a concave gradient of 0 – 24% SCX solvent B (solvent A plus 0.5 M KCl) at a flow rate of 0.5 mL/min over 198 min, collecting 1 mL fractions. The volume of each fraction was reduced under vacuum almost to dryness, then diluted in 0.1% FA in water for C18 stage-tipping (Rappsilber et al., 2007). StageTip eluates were dried under vacuum then redissolved in 0.1% FA in water for injection, via an Easy-nLC 1000 (ThermoFisher) to a 250 × 0.075 mm (ID) nanospray tip packed in house with ReproSil-Pur C18-AQ (1.9 μm diameter; Dr. Maisch GmbH) and pre-equilibrated with 0.1% FA in water.
For NanoLC-MS/MS, spectra were acquired using an LTQ Orbitrap Velos Pro (ThermoFisher) while running a bipartite linear gradient of 5%–23% C18 solvent B (CN3CN in 0.1% FA/water) over 205 min followed by 23 – 35% C18 solvent B over 30 min at a flow rate of 250 nl/min. In each 380 – 1600 m/z precursor spectrum (centroid; resolution = 60000) the 15 most intense ions above a threshold of 5000 counts with a charge of +2 to +4 were subjected to HCD fragmentation (30% NCE) followed by product spectrum acquisition (centroid; resolution = 7500). Ions otherwise eligible for fragmentation a second time within a period of 40 s were added to a 500 member (maximum) exclusion list for a period of 30 s unless expiring from the list earlier on the basis of either priority or increased signal:noise (S:N) by a factor of 2.0.
Analysis was performed using Mascot 2.6. Target/decoy searches of instrument raw data file data were against SwissProt (taxonomy: Human, Vaccinia) plus a database of common contaminants, with trypsin specificity allowing up to 1 missed site, charge states of +2 to +4 and Oxidation (M), deamidated (NQ) as variable modifications, with precursor and product mass tolerances of ± 20 ppm each, and the TMT 6plex quantitation method. Search results from all fractions of a single SCX gradient (above) were combined then thresholded to < 2% false discovery rate (FDR) for data export.
Exports of peptide identification and absolute TMT reporter ion intensities from Mascot Server were collated and analyzed: Pairwise ratios of TMT reporter ion intensity for individual peptides were annealed to protein accessions taking, as the protein abundance ratio, the weighted geometric mean of all peptide ratios for that protein. Protein abundance ratios based on the quantitation of multiple peptide species were then ordered into a continuous distribution used for simultaneous graphical display of protein abundance ratios overlaying all constituent peptide quant ratios, as follows: The topmost 5% of the continuously ascending distribution of protein abundance ratios for mock-infected A549-Vector cells in the presence/absence of IFN (the most highly IFN-induced proteins, represented by TMT6 #2/TMT6 #1) was examined and every protein for which all contributing peptide quants fell above the 90th percentile for the distribution as a whole was written to a list. Simultaneously written was the ranking of each of the proteins (as a percentile) within the continuously ascending protein abundance ratio distribution of mock-infected, IFN-treated A549-C9 (#4)/A549-Vector (#2). This second distribution represents the degree of protein under-abundance as a result of C9 expression, all other factors being equal.
Western blotting
Cells were washed once with PBS, lysed in sample buffer (1 × LDS loading buffer, 1 × reducing agent, protease inhibitor (ThermoFisher)). The total cell lysates were sonicated for two 30 s periods; the proteins were resolved on 4 to 12% NuPAGE Bis-Tris gels (ThermoFisher) and transferred to a nitrocellulose membrane with an iBlot2 system (ThermoFisher). The membrane was blocked with 5% nonfat milk in Tris-buffered saline (TBS) for 1 h, washed with TBS with 0.1% Tween 20 (TBST), and then incubated with the primary antibody in 5% nonfat milk in TBST overnight at 4°C. The membrane was incubated with the secondary antibody conjugated with horseradish peroxidase (Jackson ImmunoResearch) or secondary antibody conjugated with IRDye 800CW (Li-Cor Biosciences) for 1 h. After washing the membrane, the bound proteins were detected with SuperSignal West Dura substrates (ThermoFisher) or a Li-Cor Odyssey infrared imager (Li-Cor Biosciences).
Confocal microscopy
A549 and IFIT KO cells grown on glass coverslips were treated with IFNβ for 24 h and then mock infected or infected with 3 PFU/cell of purified virus in the presence or absence of AraC (44 μg/ml). At 3 h after infection, the cells were incubated with 10 μM 5-ethynyl-2′deoxyuridine (EdU; ThermoFisher) for 1 h, after which the cells were fixed with 4% formaldehyde and permeabilized with 0.5% Triton X-100. EdU was detected by click reaction with Alexa Fluor 647 azide according to the manufacturer’s protocol (ThermoFisher). Samples were then blocked with 3% bovine serum albumin and stained with antibody to the I3 protein followed by Alexa Fluor 568 secondary antibody (ThermoFisher). Nuclei and virus factories were stained with 4’,6-diamidino-2-phenylindole (DAPI). Coverslips were mounted on slides using ProLong Diamond Antifade reagent (ThermoFisher). Images were collected on a Leica SP5 confocal microscope with a 63 × oil immersion objective and processed using ImageJ software to adjust the brightness. For quantification, images were collected using an automated tiling method to obtain an unbiased data pool from two independent experiments. Acquired images were further analyzed using Imaris image-processing software (Bitplane AG) to count the number of cells with I3 punctae and the number of cells expressing the I3 protein.
Quantification of viral genome and specific mRNA copies by ddPCR
A549 cells and IFIT KO cells were untreated or treated with IFNβ for 24 h and then infected with 3 PFU/cell of purified viruses. After 6 h, cells were harvested for extraction of DNA and RNA. DNA was extracted using a DNeasy blood and tissue kit (QIAGEN) according to the manufacturer’s protocols. Total RNA was extracted using the RNeasy Mini Kit (QIAGEN), treated with DNase I (Thermo Fisher), and then reverse transcribed with a SuperScript IV First-Strand Synthesis System (Thermo Fisher). The DNA was serially diluted and analyzed with gene-specific primers and QX200 ddPCR EvaGreen Supermix (Bio-Rad) by droplet digital PCR following the protocol described previously (Americo et al., 2017). After 40 reaction cycles, the droplets were digitally analyzed with a droplet reader (Bio-Rad), and absolute DNA copy numbers were determined. Viral genome copies were measured by primers targeting E11 gene (Liu and Moss, 2018). For analysis of viral mRNAs, the primer sequences for viral early gene E3 and intermediate/ late gene A3 were described previously (Liu and Moss, 2018; Hyun et al., 2017). For analysis of IFITs mRNAs, the primer sequences for IFIT1 was from Li and coworkers (Li and Swaminathan, 2019). The 18 s rRNA served as the endogenous control for the mRNA experiment.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with Graphpad Prism 7. All data represent mean ± standard error of measurement (SEM).
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
Highlights.
Viral C9 protein is required for vaccinia virus to resist interferon
C9 N-terminal ankyrin repeats bind IFITs and C-terminal F-box binds Cullin1/SKP1
Full-length C9 mediates ubiquitination and proteasomal degradation of IFITs
IFITs prevent genome uncoating and replication in the absence of C9
ACKNOWLEDGMENTS
We thank Catherine Cotter for cell culture; Juraj Kabat for help with confocal microscopy; Stuart N. Isaacs, David H. Evans, and Paula Traktman for antibodies; and Richard C. Condit for vJ3K175R. The research was supported by the NIAID Division of Intramural Research. Mass spectrometry at the University of California was performed on an instrument that had been acquired with NIH grant 1S10OD016328-01.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.039.
REFERENCES
- Abbas YM, Pichlmair A, Górna MW, Superti-Furga G, and Nagar B (2013). Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 494, 60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Americo JL, Earl PL, and Moss B (2017). Droplet digital PCR for rapid enumeration of viral genomes and particles from cells and animals infected with orthopoxviruses. Virology, 19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle KA, Arps L, and Traktman P (2007). Biochemical and genetic analysis of the vaccinia virus d5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J. Virol 81, 844–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter CA, Earl PL, Wyatt LS, and Moss B (2017). Preparation of cell cultures and vaccinia virus stocks. Curr. Protoc. Mol. Biol. 117, 5.12.1–5.12.18. [DOI] [PubMed] [Google Scholar]
- Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, et al. (2010). 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Schaller AM, Geballe AP, and Malik HS (2016). Evolution-guided functional analyses reveal diverse antiviral specificities encoded by IFIT1 genes in mammals. eLife 5, e14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DH, Wyatt LS, Newman FK, Earl PL, Chun S, Hernandez JE, Molina DM, Hirst S, Moss B, Frey SE, and Felgner PL (2008). Antibody profiling by proteome microarray reveals the immunogenicity of the attenuated smallpox vaccine modified vaccinia virus ankara is comparable to that of Dryvax. J. Virol 82, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva FS, Lewis W, Berglund P, Koonin EV, and Moss B (2007). Poxvirus DNA primase. Proc. Natl. Acad. Sci. USA 104, 18724–18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, and Farzan M (2013). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol 13, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, and Traktman P (1992). Characterization of vaccinia virus DNA replication mutants with lesions in the D5 gene. Chromosoma 102 (1 Suppl), S72–S82. [DOI] [PubMed] [Google Scholar]
- Fensterl V, and Sen GC (2015). Interferon-induced Ifit proteins: their role in viral pathogenesis. J. Virol 89, 2462–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleith RC, Mears HV, Leong XY, Sanford TJ, Emmott E, Graham SC, Mansur DS, and Sweeney TR (2018). IFIT3 and IFIT2/3 promote IFIT1-mediated translation inhibition by enhancing binding to non-self RNA. Nucleic Acids Res. 46, 5269–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, et al. (2013). Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 9, e1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert MH, Squire CJ, and Mercer AA (2015). Poxviral ankyrin proteins. Viruses 7, 709–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun SI, Weisberg A, and Moss B (2017). Deletion of the Vaccinia Virus I2 Protein Interrupts Virion Morphogenesis, Leading to Retention of the Scaffold Protein and Mislocalization of Membrane-Associated Entry Proteins. J. Virol 91, e00558–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, VanBlargan LA, Xu W, White JP, Shan C, Shi PY, Zhang R, Adhikari J, Gross ML, Leung DW, et al. (2018). Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity 48, 487–499.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungwirth C, and Launer J (1968). Effect of poxvirus infection on host cell deoxyribonucleic acid synthesis. J. Virol 2, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, and Yeh ET (1997). Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem 272, 28557–28562. [DOI] [PubMed] [Google Scholar]
- Katibah GE, Lee HJ, Huizar JP, Vogan JM, Alber T, and Collins K (2013). tRNA binding, structure, and localization of the human interferoninduced protein IFIT5. Mol. Cell 49, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck JG, Baldick CJ Jr., and Moss B (1990). Role of DNA replication in vaccinia virus gene expression: a naked template is required for transcription of three late trans-activator genes. Cell 61, 801–809. [DOI] [PubMed] [Google Scholar]
- Kilcher S, Schmidt FI, Schneider C, Kopf M, Helenius A, and Mercer J (2014). siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 15, 103–112. [DOI] [PubMed] [Google Scholar]
- Latner DR, Thompson JM, Gershon PD, Storrs C, and Condit RC (2002). The positive transcription elongation factor activity of the vaccinia virus J3 protein is independent from its (nucleoside-2′-O-) methyltransferase and poly(A) polymerase stimulatory functions. Virology 301, 64–80. [DOI] [PubMed] [Google Scholar]
- Li D, and Swaminathan S (2019). Human IFIT proteins inhibit lytic replication of KSHV: a new feed-forward loop in the innate immune system. PLoS Pathog. 15, e1007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Li J, Irwin CR, Jenkins H, DeLange L, and Evans DH (2008). Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J. Virol 82, 5922–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, and Moss B (2018). Vaccinia Virus C9 Ankyrin Repeat/F-Box Protein Is a Newly Identified Antagonist of the Type I Interferon-Induced Antiviral State. J. Virol 92, e00053–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera-Martínez E, Brito Querido J, Valasek LS, Simonetti A, and Hashem Y (2017). ABCE1: a special factor that orchestrates translation at the crossroad between recycling and initiation. RNA Biol. 14, 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Snijder B, Sacher R, Burkard C, Bleck CK, Stahlberg H, Pelkmans L, and Helenius A (2012). RNAi screening reveals proteasomeand Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2, 1036–1047. [DOI] [PubMed] [Google Scholar]
- Mosavi LK, Cammett TJ, Desrosiers DC, and Peng ZY (2004). The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 13, 1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B, and Rosenblum EN (1973). Letter: protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol 81, 267–269. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Lassnig C, Eberle CA, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, et al. (2011). IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol 12, 624–630. [DOI] [PubMed] [Google Scholar]
- Pogo BGT, and Dales S (1973). Biogenesis of poxviruses: inactivation of host DNA polymerase by a component of the invading inoculum particle. Proc. Natl. Acad. Sci. USA 70, 1726–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Saikia P, Fensterl V, and Sen GC (2010). The inhibitory action of P56 on select functions of E1 mediates interferon’s effect on human papillomavirus DNA replication. J. Virol 84, 13036–13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Katsafanas G, Weisberg A, Olano LR, and Moss B (2017). Identification of vaccinia virus replisome and transcriptome proteins by iPOND coupled with mass spectrometry. J. Virol 91, e01015–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soday L, Lu Y, Albarnaz JD, Davies CTR, Antrobus R, Smith GL, and Weekes MP (2019). Quantitative Temporal Proteomic Analysis of Vaccinia Virus Infection Reveals Regulation of Histone Deacetylases by an Interferon Antagonist. Cell Rep. 27, 1920–1933.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher BA, and Tomaszewski JE (2015). Proteasome inhibitors. Biochem. Pharmacol 96, 1–9. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Saikia P, and Sen GC (2008). Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 27, 3311–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, and Bohmann D (1994). Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78, 787–798. [DOI] [PubMed] [Google Scholar]
- Weaver JR, Shamim M, Alexander E, Davies DH, Felgner PL, and Isaacs SN (2007). The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res. 130, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss CM, Trobaugh DW, Sun C, Lucas TM, Diamond MS, Ryman KD, and Klimstra WB (2018). The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. MSphere 3, e00209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe EJ, Vijaya S, and Moss B (1995). A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211, 53–63. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bruno DP, Martens CA, Porcella SF, and Moss B (2010). Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. USA 107, 11513–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Bruno DP, Martens CA, Porcella SF, and Moss B (2011). Genome-wide analysis of the 5′ and 3′ ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J. Virol 85, 5897–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.