

Abstract
Canonical WNT/β-catenin signaling is involved in most of the mechanisms that lead to the formation and development of cancer cells. It plays a central role in three cyclic processes, which are the cell division cycle, the immune cycle, and circadian rhythms. When the canonical WNT pathway is upregulated as in cancers, the increase in β-catenin in the nucleus leads to activation of the expression of numerous genes, in particular CYCLIN D1 and cMYC, where the former influences the G1 phase of the cell division cycle, and the latter, the S phase. Every stage of the immune cycle is disrupted by the canonical WNT signaling. In numerous cancers, the dysfunction of the canonical WNT pathway is accompanied by alterations of the circadian genes (CLOCK, BMAL1, PER). Induction of these cyclic phenomena leads to the genesis of thermodynamic mechanisms that operate far from equilibrium, and that have been called “dissipative structures.” Moreover, upregulation of the canonical WNT/β-catenin signaling is important in the myofibroblasts of the cancer stroma. Their differentiation is controlled by the canonical WNT /TGF-β1 signaling. Myofibroblasts present ultraslow contractile properties due to the presence of the non-muscle myosin IIA. Myofibroblats also play a role in the inflammatory processes, often found in cancers and fibrosis processes. Finally, upregulated canonical WNT deviates mitochondrial oxidative phosphorylation toward the Warburg glycolysis metabolism, which is characteristic of cancers. Among all these cancer-generating mechanisms, the upregulated canonical WNT pathway would appear to offer the best hope as a therapeutic target, particularly in the field of immunotherapy.
Keywords: canonical WNT/β-catenin signaling, immune cycle, cell division cycle, myofibroblast, circadian rhythms, Warburg glycolysis, TGF-β1, immunotherapy
Introduction
Since the discovery in 1982 of Int1 (WNT1a) as an oncogene reported in murine breast cancers, the canonical WNT/β-catenin pathway has often been found to be associated with cancer (1). Indeed, the canonical WNT pathway represents a key element in the genesis and development of a large number of human cancers. This makes the pathway a prime target in the search for new treatments for cancers, with high mortality and morbidity. In this review, we summarize the main findings regarding the dysfunctions of the canonical WNT signaling encountered in many cancers (Figure 1). It has been reported that three major cyclic processes namely the cell division cycle (2–4), the immune cycle (5), and circadian rhythms (CRs) (6) become highly disturbed due to upregulation of the canonical WNT signaling. In the division cell cycle, canonical WNT signaling was initially described as a trigger for the G1 phase advancement through CYCLIN D1 and c-MYC transcription (7–11). This signaling is also involved at every stage of the immune cycle (12) and has led to the development of new modalities of immunotherapy. In addition, the dysfunction of the canonical WNT signaling is involved in the generation and development of the cancer stroma (13, 14). In particular, aerobic glycolysis in tumor cells, which is partly responsible for shunting the mitochondrial oxidative phosphorylations [Warburg effect (15)], is related to the upregulation of the canonical WNT pathway. Finally, cancers fit into the thermodynamic framework of dissipative structures, in a manner similar to CRs or by generating oscillating phenomena in which canonical WNT pathway plays a central role (16). Since β-catenin regulates the expression of a considerable number of genes, such as CYCLIN D1 and cMYC, it is not surprising that this signaling interferes in numerous cancer-generating processes. Other pathways such as TGF-β1, activate the canonical WNT signaling, and will therefore potentially play a role upstream.
Figure 1.
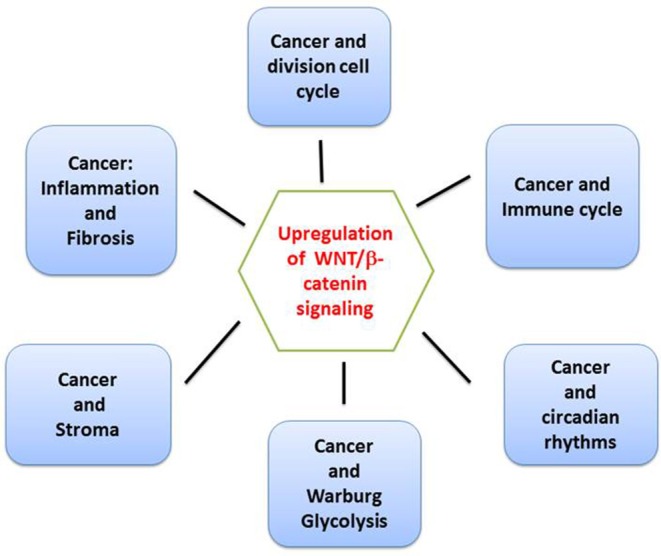
Overview of checkpoints of the canonical WNT/β-catenin signaling. The canonical WNT signaling is involved in at least six key areas of cancer pathophysiology, i.e., cell division cycle, immune cycle, circadian rhythms, cancer stroma, Warburg glycolysis, inflammation, and fibrosis.
General Remarks About the Canonical WNT/β-Catenin Signaling
The canonical WNT signaling is involved in numerous physiological processes such as embryonic development, metabolism, cell fate and epithelial-mesenchymal transition (EMT). Upregulation of the canonical WNT pathway increases the level of β-catenin in both the cytoplasm and nucleus. The dysfunction of this pathway is involved in numerous pathological processes, particularly in cancers and fibrosis (17–20). The T-cell /lymphoid enhancer (TCF/LEF) transcription factors represent the final effectors of the canonical WNT signaling (Figure 2). The destruction complex is composed of AXIN, tumor suppressor adenomatous polyposis coli (APC), and glycogen synthase kinase-3 (GSK-3β). GSK-3β exerts a tight control on the canonical WNT pathway. When WNT ligands are absent (“off state”), the cytosolic β-catenin is continuously ubiquitinylated leading to degradation via sequential phosphorylations by GSK-3β and caseine kinase 1 (CK1). The destruction complex phosphorylates β-catenin. Then β-catenin is degraded within the proteasome. When WNT ligands are present (“on state”), the WNT receptor interacts with Frizzled (FZL) and LDL receptor-related protein 5/6 (LRP5/6). WNT receptor is linked with Disheveled (DSH). This favors the disruption of the destruction complex and hinders degradation of β-catenin in the proteasome. β-catenin translocates to the nucleus and interferes with the TCF- LEF co-transcription factors. This leads to the stimulation of several β-catenin target genes (CYCLIN D1, c-MYC, PDK, MTC-1, MMP7, fibronectin, COX-2, AXIN-2) (7, 8, 21, 22) (Figure 2). Mutations in the β-catenin gene (CTNNB1), mutations or loss-of-function in APC, AXIN-1 and AXIN-2 genes, all induce a continuous stimulation of the β-catenin signaling and have been reported in a large number of human cancers.
Figure 2.

The canonical WNT/β-catenin pathway: “on” and “off” states. The canonical WNT pathway can be in either “on-state” or “off-state.” In the “off state”, i.e., in the absence of WNT ligand or in the presence of the active form of the glycogen synthase kinase-3β (GSK-3β) (i.e., the unphosphorylated form GSK-3β), cytosolic β-catenin is phosphorylated by GSK-3β and undergoes the destruction process into the proteasome. In the “on-state,” a WNT ligand binds both FZD and LRP5/6 receptors. GSK-3β is then under the inactive form (i.e., the phosphorylated form pGSK-3β). This leads to activation of the phosphoprotein Disheveled (DSH). DSH recruits the destruction complex (pGSK-3β + AXIN +APC) to the plasma membrane, where AXIN directly binds the cytoplasmic tail of LRP5/6. APC is the adenomatous polyposis coli. Activation of DSH leads to the inhibition of both phosphorylation and degradation of β-catenin. β-catenin accumulates into the cytosol and then translocates to the nucleus to bind the LEF-TCF co-transcription factors. This induces the WNT-response gene transcription of numerous genes such as CYCLIN D1, cMYC, MCT1, PDK, fibronectin. DKK1 inhibits the canonical WNT signaling.
There is an opposed link between the canonical WNT pathway and PPARγ (23) (Figure 3). This has been established in several cancers, neuro-degenerative diseases, cardiovascular diseases, fibrosis, and numerous other pathological processes (24–32). Transforming growth factor1 (TGF-β1) acts as a driver of these two major pathways (33). TGF-β1 and canonical WNT signaling are closely related, whereby the former activates the later. TGF-β1 receptors are the transmembrane proteins Type I (TGFβRI) and Type II (TGFβRII) (Figure 3). Moreover, TGF-β1 activates both SMAD and non-SMAD pathways such as PI3K-AKT, MAPK and RHO, and downregulates PPARγ expression through the SMAD pathway (34). TGF-β1 and consequently the canonical WNT pathway play a central role in both cancers and fibrosis. In alveolar epithelial cells (AEC), TGF-β1 induces EMT and leads to lung fibrosis. In idiopathic pulmonary fibrosis, myofibroblasts induce lung fibrosis (35–37). In fibrotic lungs, TGF-β1-induced EMT in type II AEC results in differentiation of fibroblasts into myofibroblasts (38–42).
Figure 3.

WNT/β-catenin, TGF-β1, PI3K-AKT, SMAD, and PPARγ pathways. “On state” canonical WNT: In the presence of WNT ligands, the WNT receptor binds LRP5/6 and FZD receptors and initiates LRP phosphorylation and DSH-mediated Frizzled internalization. Then, the pGSK-3β/AXIN/APC destruction complex is dissociated. Phosphorylation of β-catenin is inhibited and β-catenin increases in the cytosol and then translocates to the nucleus. β-catenin binds the TCF-LEF transcription factors and induces the WNT-response gene transcription (CYCLIN D1, cMYC, PDK, MCT-1, AXIN2, fibronectin). DKK1 is activated by PPARγ and inhibits WNT. TGF-β1 binds the TGF-βR2 receptor, which recruits the TGF-βR1 receptor. This leads to the formation of a heterotetramer that phosphorylates SMAD. The SMAD complex then translocates to the nucleus and regulates the transcription of Smad target genes (CTGF, COL1A). PI3K-AKT pathway is a non-SMAD signaling and inactivates GSK-3β. PTEN inhibits PI3K-AKT and PPARγ inhibits AKT. PPARγ inhibits the β-catenin/TCF-LEF-induced activation of WNT target genes. TGF-β1 enhances the canonical WNT pathway through the inhibition of DKK1, itself an inhibitor of the canonical WNT signaling.
Canonical WNT/β-Catenin Signaling, Cell Division Cycle and Cancer
The Cell Division Cycle
The canonical WNT signaling plays a central role in the cell division cycle, particularly because the two nuclear β-catenin targets CYCLIN D1 and cMYC intervene at two key checkpoints of the cell cycle. CYCLIN D1 and cMYC act on the G1 phase and cMYC on the S phase (Figure 4). An oscillator rotates the cell cycle, which consists of replicating the DNA. This then leads to a division into two cells, each containing a set of chromosomes (mitosis). In cancer cells, the regulation of the cell division cycle is impaired. Nurse, Hunt, and Hartwell have identified two factors that play an important role in mitosis, namely cyclins and cyclin-dependent kinases (Cdk) that control the transitions between the M, G1, S, and G2 phases of the cell division cycle (4). The key process is a feedback mechanism in which cyclins play a driving role in the cell division cycle (2, 3). Each phase is controlled by a given CYCLIN/Cdk complex. The transition to a later phase of the cycle can only be made if the previous phase has been correctly completed. The cell can remain in the quiescent phase (Go) from which it exits when the level of growth factors is high enough and then allows the onset of the G1 phase. CYCLIN D1 is a key regulator of the checkpoint allowing the G1-S transition, via phosphorylation of the retinoblastoma protein (pRB) complex, thus increasing the cyclin E level. The transcription factor EF2 promotes the cell division cycle and the tumor suppressor pRB slows it down. Cdk kinases are activated by dephosphorylation (cdc25 phosphatase) and inhibited by phosphorylation (Wee1 kinase). The activation of each phase of the cell cycle is sequential and leads to inactivation of the previous phases.
Figure 4.

Cell division cycle. Four CYCLIN/Cdk kinase complexes control transitions between the 4 phases of the cell cycle, namely, M, G1, S (DNA replication), and G2 (separating the S phase from the M phase of mitosis). Each phase is controlled by a given CYCLIN/Cdk complex. The transition to a later phase of the cycle can develop only if the previous phase has been totally completed. The cell remains in the quiescent phase (Go) from which it exits when the level of growth factors becomes sufficient and then enters the G1 phase. The G1 phase is controlled by the CYCLIN D1/Cdk4 complex. Phase S is controlled by the CYCLIN E/Cdk2 complex. The G2 phase is controlled by the CYCLIN A/Cdk2 complex. The M phase is controlled by the cyclin B/Cdk1 complex. The canonical WNT signaling increases the expression of CYCLIN D1 and cMYC. CYCLIN D1 induces the G1 phase whereas cMYC induces the S phase.
WNT Transcriptional and Non-transcriptional Effects During the G1 Phase
Several pathways that regulate the cell proliferation act through the G1 phase (43). CYCLIN D1 is inhibited by p21 and p27, allowing the entry of the cell into the quiescence phase Go. The canonical WNT signaling triggers the G1 phase advancement through CYCLIN D1 and c-MYC transcriptional processes (Figures 2, 4) (7–11). The canonical WNT pathway operates through the G1 phase by inducing the transcription of CYCLIN D1 and cMYC which are involved in cell proliferation (10, 44–46). CYCLIN D1 has been one of the first transcriptional target genes of β-catenin to be reported in the colorectal cancer. The transcription factor c-MYC has a dual role in G1 phase by promoting CYCLIN D1 (47) and repressing p21 and p27 (45). pGSK-3β phosphorylates CYCLIN D1, CYCLIN E1, and c-MYC which are direct regulators of G1 progression (48–50).The phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway (PI3K-AKT) allows the G1-S transition phase of the cell cycle through the phosphorylation of GSK-3β (Figure 4). GSK-3β phosphorylation (i.e., pGSK-3β is the inactivated form) decreases the degradation of β-catenin within the proteasome. Thus, TCF/LEF transcription factors are activated, which in turn leads to transcription of the target gene CYCLIN D1 (51). The phosphatase and tensin homolog (PTEM) also decreases cancer cell proliferation due the arrest of the cell cycle in G1 phase (Figures 2, 6).
Canonical WNT/β-Catenin Pathway, CYCLIN D1, and Cancer
CYCLIN D1 is considered as key target of the canonical WNT pathway. In endothelial cells, inorganic polyphosphate induces CYCLIN D1 synthesis via activation of the mTOR/WNT/β-catenin pathway (52). CYCLIN D1 is critical for intestinal adenoma development (53). The expression of both CYCLIN D1 and β-catenin, and the inactive pGSK-3β are significantly increased in papillary thyroid carcinoma primary tumors. Expressions of β-catenin and CYCLN D1 are correlated suggesting that β-catenin is a factor driving high CYCLIN D1 levels (54).
WNT/β-Catenin Signaling, Thermodynamics, Oscillations, and Dissipative Structures
A complete oscillating model of cyclins and cyclin-dependent kinases has been proposed by Gérard and Goldbeter (55). In the presence of a sufficient level of growth factors, the CYCLIN/Cdk complexes are activated sequentially. This complex system is capable of self-organization over time (16, 55, 56). The canonical WNT pathway modulates the cell cycle and conversely the cell cycle influences it. During the cell cycle, the expression of β-catenin oscillates and the maximum activation of β-catenin occurs at the G2/M transition (Figure 4) (57, 58). Moreover, expression of the β-catenin target AXIN2 also peaks at the G2/M transition (59). This requires the LRP6 phosphorylation by CYCLIN Y which mediates phosphorylation of LRP6. The CYCLIN D1/Cdk4 complex peaks at G1. The CYCLIN E/Cdk2 complex peaks at S (Figure 4). The CYCLIN A/Cdk2 complex peaks at the end of S. The peaks of CYCLINS D1, E, and A follow each other, leading to the peak of CYCLIN B. The CYCLIN B/Cdk1 complex peaks at M and triggers the cell division (Figure 4). The sustained oscillations of the various CYCLIN/Cdk complexes reflect the spontaneous self-organization of the cell division cycle over time. Thermodynamically, the cell division cycle represents a dissipative structure which behaves far from equilibrium (60, 61). When a CYCLIN/Cdk complex is graphically represented in an X-Y manner as a function of another complex or as a function of the transcription factor E2F that induces the CYCLIN synthesis, there is an evolution toward a “limit cycle” regardless of initial conditions (55). Below a critical value of growth factor amount corresponding to a bifurcation point, the cell remains in a stable stationary state (quiescent phase Go). Beyond this point, the stationary system becomes unstable and the network of CYCLIN/Cdk complexes begins to oscillate autonomously. Cell proliferation persists as long as the concentration levels of growth factors allow. When the balance between the transcription factor E2F and the suppressor pRB tilts toward a certain level, sustained oscillations appear and the cell division cycle operates. However, even in the absence of growth factor, and under particular conditions, the cell cycle can start beyond a critical point in G1 (“Pardee restriction point”) (62). It must be mentioned that the canonical WNT pathway is involved in another important physiological process. Due to a periodic signal emitted by the segmentation clock, the WNT, NOTCH, and FGF oscillator synchronizes the activation of segmentation genes in successive steps. This leads to the formation of somites during the vertebrate segmentation (63, 64). This represents a supplementary oscillatory dissipative structure controlled by the WNT pathway.
Canonical WNT/β-Catenin Signaling, Immune Cycle and Cancer
The immune cycle (Figure 5) is defined by natural homeostatic oscillations of the immune system when chronic inflammation is occurring such as in cancers, and typically repeated every 7 days. This is due to the synchronous division of T cells over time. T-effector cells boost the immune activity. This is followed by T-regulatory cells that suppress the immune response (65). The cancer-immune cycle has been described by Chen and Mellman (5). The effects of the canonical WNT signaling on the immune cycle have been recently reported in an extensive review by Wang et al. (12). The canonical WNT signaling impacts all the steps of the immune cycle (Figure 4), and we summarize here some of the numerous works concerning this subject. The canonical WNT pathway is a well-recognized cancer promoter as it regulates the cancer immune cycle at most of the checkpoints, composed of dendritic cells, T cells (especially effector T cells, T helper cells, and regulatory T cells) and tumor cells. The treatment of cancers by immunotherapy is being revolutionized by activating or reactivating the tumor-immune cycle (5).
Figure 5.
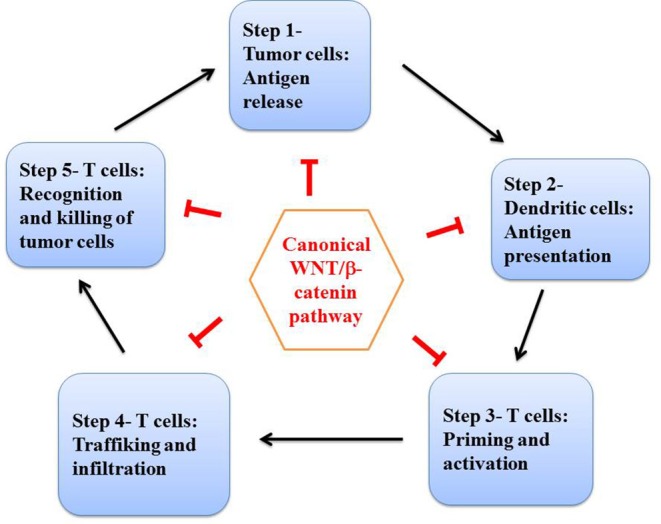
The cancer immune cycle. Adapted and simplified schema of the tumor immune cycle proposed by Chen and Mellman (5) and by Wang et al. (12). Numerous studies have shown that the canonical WNT signaling is involved at each step of the tumor immune cycle: step 1 (tumor antigene release), step 2 (antigene presentation), step 3 (priming and activation), and steps 4 and 5 (trafficking, infiltration, and tumor cell elimination).
Trafficking of T cells and their infiltration into tumor tissues lead to the antigen release and elimination of cancer cells (66), following a self-propagating cyclic process the final aim of which is to kill cancer cells. Negative regulators of the tumor-immune cycle by using antibodies against cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death protein1 (PD-1) (67) ameliorate survival rates of patients suffering from metastatic melanoma (68). The canonical WNT signaling is responsible for the onset and advancement of many cancers and is a well-established molecular target for cancer therapy (69–71). Numerous mutations in genes encoding the canonical WNT pathway have been reported in colorectal cancers presenting mutations of APC and β-catenin genes (69, 70, 72). Dysfunction of the canonical WNT signaling has been demonstrated in tumor immunology (73, 74). TCF1 is the endpoint of the canonical WNT signaling (Figure 2) and is a T lymphocyte-specific transcription factor which plays an important role in the development of T cells (75). TCF1 is involved in maturation and secondary expansion of memory CD8+ T cells and activates CD8+ effector T cells (76).
In cancers, the immune contexture that is partly characterized by the functional orientation of tumor-infiltrating immune cells influences the outcome of patients. Moreover, in metastatic sites, it reflects the corresponding primary cancer (77). The immune-suppressive mesenchymal stem cells (MSCs) have been shown to be an important source of myofibroblasts in vivo. We have recently reported in vitro that the immune-suppressive capabilities of MSCs are not altered after their differentiation into myofibroblasts (78). In MSCs, involvement of the canonical WNT signaling promotes metastatic growth and chemo-resistance of cholangiocarcinoma (79).
WNT/β-Catenin Signaling and Dendritic Cells (DCs)
DCs have tumor antigens on the major histocompatibility complex molecules and prime effector T cells. Antigens are released from cancer cells before encountering DCs, then priming and activation of CD4+ and CD8+ T cells follow. Before priming effector T cells, DCs differentiate into CD103+ DCs that are important for recruitment of effector T cells into tumors (80, 81). Activating the mutated β-catenin pathway initiates the gene expression of interferon regulatory factor 8 (IRF8) that leads to differentiation and expansion of CD103+ DCs (82). Moreover, activation of β-catenin releases CXCL9/10 in CD103+ DCs and inhibits infiltration of effector T cells (81).
WNT/β-Catenin Signaling and CD8+ T Cells
In the tumor-immune cycle, peripheral naïve CD8+ T cells differentiate into effector T cells and destroy cancer cells rapidly (81). CD8+ T cells are activated and primed by DCs, and then infiltrate tumors to kill cancer cells (83). During tumor development, cancer cells avoid action of the immune cycle by inhibiting CD8+ T cell infiltration (84). Mature naïve CD8+ T cells are activated by APC and then proliferate in spleen and lymph nodes (5). Upregulation of the WNT/β-catenin pathway induces apoptosis of mature naïve CD8+ T cells partially through to the target gene c-MYC (85).
WNT/β-Catenin Pathway and T Helper (Th) Cells
Th cells are subdivided into Th1, Th2, and Th17 and they help the CD8+ T cell anti-tumor response through T cell cytokine release (86). Th cells inhibit cancer cells by activating CD27, CD137, and 4-1BB as well as favoring the synthesis of memory CD8+ T cells (87). The WNT/β-catenin signaling plays a role in the development and function of Th cells (88–90). It also inhibits Th cells and impairs the anti-tumor immunity. Abnormal WNT/β-catenin pathway impairs the immunity induced by CD4+ T cells. Auto-immune encephalomyelitis activates the canonical WNT pathway that impairs infiltration of CD4+ T cells. This can be restored by inhibiting the WNT/β-catenin signaling (90). β-catenin is upregulated in colorectal tumor, and the immunity induced by intra-tumoral CD4+ T cells is decreased (89).
WNT/β-Catenin Signaling and Regulatory T (Treg) Cells
Treg cells support immunosuppression and tolerance to immune stress by inhibiting the effector T (Teff) cell response (91, 92). The canonical WNT signaling is critical for the Treg cell development and exerts a negative regulation on CD4+ and CD8+ T cells. Infiltration of Treg cells into tumors might be due to the inhibition of the anti-tumor immune response. In cancer, down-regulation of the protein kinase CK2, a negative regulator of canonical WNT pathway, inhibits the Treg-mediated immune suppression (93, 94).
WNT/β-Catenin Pathway and Tumor Cells
At the last step of the immune cycle, cancer cells are recognized and destroyed by effector T cells. To escape an immune effect, tumor cells express a negative regulatory molecule PD-L1 and generate mutant tumor antigens (95, 96). cMYC, a target of the canonical WNT pathway binds the promoters of the CD47 and PD-L1 genes. cMYC inhibition ameliorates the anti-tumor response (97). Upregulation of the WNT/β-catenin signaling in tumor cells induces T cell exclusion and resistance to immunotherapy. Blockade of the canonical WNT signaling decreases the expression of both CD47 and PD-L1 and improves the anti-tumor immune response (85). In transgenic mouse melanoma, β-catenin stabilization results in a decrease of both T-cell priming and CD8+ T cell infiltration (80). Cancer cells remain in a quiescent state after inhibition of the canonical WNT signaling by DKK1 (98). Although cancer immunotherapy by blocking cancer driver genes can benefit patients (99–102), there are several types of resistance to cancer immunotherapy (primary, adaptative, and acquired) (84). Targeting the canonical WNT pathway could help to overcome the immune-resistance.
WNT/β-Catenin and TGF-β1 Pathways, Tumor Stroma and Myofibroblasts
The canonical WNT pathway is tightly involved in the development and maintenance of the tumor stroma. The transforming growth factor-β1 (TGF-β1) and myofibroblasts (103) play a key role in this process. TGF-β1 activates the canonical WNT signaling (Figures 3, 6) and induces the differentiation of fibroblasts into myofibroblasts (31, 33). Myofibroblasts represent key constituents of the tumor stroma which is active in the cancer development as well as in maintaining the tissue architecture. In cancer stroma, myofibroblasts represent the predominant sub-population of cancer-associated cells (104, 105). Myofibroblasts, epithelial cells and connective tissue cells provide favorable conditions for tumor invasion, with loss of epithelial properties and transition to epithelial-mesenchymal characteristics (EMT) (106) that greatly influences the invasive carcinoma advancement and in which the canonical WNT signaling plays a crucial role.
Figure 6.

Canonical WNT/β-catenin pathway, Warburg glycolysis, and cancer. Schema of the canonical WNT pathway under aerobic glycolysis in cancer. In the presence of WNT ligands (“on state”), β-catenin accumulates in the cytosol and then translocates to the nucleus to bind TCF-LEF co-transcription factors. This induces the WNT-response gene transcription (CYCLIN D1, cMYC, PDK, MCT-1, fibronectin, etc). Glucose itself activates the canonical WNT pathway. PDK inhibits the PDH complex in mitochondria. Thus, pyruvate cannot be fully converted into acetyl-CoA and cannot enter the TCA cycle. cMYC activates LDH-A which converts cytosolic pyruvate into lactate. MCT-1 favors lactate extrusion out of the cytosol which favors angiogenesis. cMYC increases glutamine entry into both the cytosol and mitochondria. cMYC-induced glutamine enhances aspartate and nucleotide synthesis.
In activated cancer stroma cells, there are neo-formations of contractile stress fibers containing α-smooth muscle actin (α-SMA) and non-muscle myosin (NMMIIA). These cells are named myofibroblasts. In myofibroblasts, there is co-existence of fibroblastic morphological characteristics, such as a well-developed endoplasmic reticulum and α-SMA forming contractile actin microfilament bundles. Proto-myofibroblasts are low contractile cells and when mechanical stress and TGF-β1 are present, they can differentiate into myofibroblasts that express α-SMA and NMMIIA, which are incorporated in stress fibers. This renders myofibroblasts highly contractile (13, 107).
In cancers, mechanical and chemical conditions generating myofibroblasts promote the cancer progression. The stromal extracellular matrix (ECM) contains several molecules such as collagen, elastin, fibronectin, hyaluronic acid, proteoglycans, glycoproteins etc. The cancer stroma presents several similarities with skin healing (108, 109). In cancers, stromal cells become over-activated, proliferate, and upregulate the ECM production. ECM protects growth factors from degradation and can modulate the activity of growth factors and cytokines (109–111). The main factors are the fibroblast growth factor, platelet-derived growth factor, tumor necrosis factor, epidermal growth factor, hepatocyte growth factor, and vascular endothelial growth factor.
Cancer cells release microparticles derived from the plasma which contribute to activate tumor-associated myofibroblasts. Cancer exosomes differentiate fibroblasts into myofibroblasts (112). In cancers, hyperpermeable vessels allow leakage of proteins from the plasma, fibrinogen and fibronectin into the stroma. Myofibroblasts derive from different precursor cells. EMT represents also a mechanism of myofibroblast generation during tumor development (113). In the tumor stroma, pericytes could acquire contractile myofibroblast properties (114, 115). Bone-marrow (BM)-derived MSCs and blood-circulating fibrocytes could be also sources for cancer-associated myofibroblasts. BM-derived cells can differentiate into myofibroblasts in tumor development (116, 117). MSCs target to the stroma environment of epithelial cancers (115, 118–121). In tumor-conditioned medium, BM-MSCs differentiate into myofibroblasts containing α-SMA (122).
With mechanical stress, TGF-β1, the most important myofibrogenic growth factor, plays a central role in myofibroblast activation and by upregulating the canonical WNT signaling (Figures 2, 3). In all circumstances in which TGF-β1 is upregulated, the canonical WNT pathway is overactivated. TGF-β1 is involved in tumor formation (123) and development (124–126). TGF-β1 induces ECM deposition and acts in various epithelial-mesenchymal interactions (127). Latency and activation of TGF-β1 is regulated by the latent TGF-β1 binding protein-1 (LTBP-1) (128, 129). In cancers, cells induce proangiogenic effects and favor EMT (124–126). TGF-β1 disrupts cell-cell junctions between vascular endothelial cells and favors extravasation of metastasizing cancer cells (130). In tumor cells, the TGF-β1 blockade decreases metastasis (131, 132). In the activated tumor stroma, myofibroblasts stimulate the synthesis and activation of TGF-β1 and generate an autocrine forward loop (133).
TGF-β1 is synthesized together with the latency-associated peptide (LAP) that is cleaved in the cell and linked to TGF-β1. This leads to the small latent complex (SLC) (134). In compliant low contractile proto-myofibroblasts, TGF-β1, and LAP form the small latent complex (SLC) (14, 134). Myofibroblasts secrete SLC which is covalently bound with LTBP-1 (134, 135). LTBP-1 belongs to the family of the fibrillin proteins and stores the latent TGF-β1 in the ECM. Integrins activate the latent TGF-β1 (136), which contains RGD for integrin binding in LAP (137–139). These mechanisms involve several αv integrins (140–142). Myofibroblasts exert a tension on the ECM-stored latent TGF-β1 through integrins. Traction force exerted on SLC induces a conformational change in LAP that releases activated TGF-β1. Stiff ECM favors differentiation into a myofibroblast phenotype (13) by activating the latent TGF-β1. Tumor-associated stroma is stiffer than the surrounding normal soft connective tissue (143–147). Activated cancer-associated fibroblasts produce several growth factors and cytokines that promote tumor progression such as FGF, EGF, TGF-β1, HGF, VEGF, stroma-derived factor-1, and the pro-inflammatory cytokines IL-1, IL-6, IL-8, and CXCL14 (148–150). They also provide conditions leading to the tumor progression (147). Increased stiffness of cancer-associated myofibroblasts modifies epithelial cells toward an invasive and proliferative phenotype.
Canonical WNT/β-Catenin Signaling, Warburg Glycolysis, and Cancer
Upregulation of the canonical WNT pathway plays a pivotal role in metabolism and particularly in the aerobic glycolysis (Figure 6). The Warburg effect (15) denotes that cancer cells, even under aerobic conditions, favor the cytosolic glycolysis pathway rather than mitochondrial phosphorylation. The last product of cytosolic glycolysis is pyruvate, which is then converted into lactate, hence the term “aerobic glycolysis” given to this phenomenon. In terms of ATP production, aerobic glycolysis is much less efficient than mitochondrial oxidative phosphorylation due to the shunt of the tricarboxylic acid (TCA) cycle. In the aerobic glycolysis, one glucose molecule leads to the formation of only 2 ATP molecules, instead of 38 ATP molecules in mitochondrial oxidative phosphorylation. In cancer development, the role of the canonical WNT pathway in metabolism is now better understood (151, 152). Activation of the canonical WNT signaling via TCF/ LEF induces an activation of the aerobic glycolysis, through upregulation of glycolytic enzymes such as pyruvate dehydrogenase kinase 1 (PDK1), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDH-A), monocarboxylate transporter 1 (MCT-1), and glucose transporter (Glut). Activation of PDK1 is required for the Warburg aerobic glycolysis. PDK1 phosphorylates the pyruvate dehydrogenase complex (PDH) which is then inhibited and therefore prevents the conversion of pyruvate into acetyl-CoA in mitochondria, blocking the pyruvate access to oxidative phosphorylation (153). In this manner, the WNT-driven Warburg metabolism favors the use of glucose for cancer cell proliferation (22). Upregulation of the canonical WNT pathway leads to β-catenin translocation in the nucleus where β-catenin interacts with TCF/LEF. This results in activation of the β-catenin target genes (PDK1, MTC-1, cMYC, CYCLIN D1, COX- 2, AXIN- 2) (7, 8, 21, 22) (Figures 2, 3, 6). Upregulation of the canonical WNT pathway leads to cell proliferation, EMT, migration and angiogenesis (11, 20, 154–156). In various cancers, PDK-1 is upregulated (22, 157–159), so that the pyruvate conversion into acetyl-CoA in mitochondria is decreased and the WNT-driven Warburg aerobic glycolysis induced in spite of the availability of oxygen. In the cytosol, pyruvate is converted into lactate through LDH-A activation. Upregulation of both LDH-A and MCT-1 leads to lactate formation and the secretion of lactate outside of the cell. This induces vascularization (160) and anabolic production of biomass for nucleotide synthesis (161, 162). Furthermore, PDK-1 and−2 create conditions conductive to angiogenesis (163, 164). Inhibition of the canonical WNT signaling decreases the PDK-1 level via the transcription regulation and reduces in vivo tumor growth (22). cMYC, a target gene of β-catenin activates the aerobic glycolysis and glutaminolysis, induces the uptake of glutamine into the cell and mitochondria, activates LDH-A and activates aspartate synthesis that finally leads to nucleotide synthesis (165, 166). cMYC also stimulates the hypoxia-inducible factor-α (HIF-1α) which in turn regulates PDK-1 (167). In carcinogenesis, HIF-1α activates the Warburg aerobic glycolysis (168). In this process, a part of the pyruvate is converted into acetyl-Co-A which enters the TCA cycle, and is converted into citrate. This leads to the synthesis of proteins and lipids. Cellular accumulation of metabolic intermediates such as glycine, aspartate, serine, and ribose, allows de novo synthesis of nucleotides (Figure 6), contributing to cell growth and proliferation. Lactate also induces angiogenesis. Importantly, aerobic glycolysis is also induced in response to TGF-β1 (169) and glucose consumption is increased in cancer cells. High glucose concentration regulates tumor-related processes. Glucose itself directly influences the canonical WNT signaling (170). High glucose levels enhance the nuclear translocation of β-catenin in response to canonical WNT activation. In cancer cells, glucose-induced β-catenin acetylation increases canonical WNT signaling. Stimulation of the canonical WNT pathway leads to activation of HIF-1α causing metabolic remodeling (154, 171) and accentuates the Warburg effect. Thus, cancer cells use the Warburg effect at all oxygen levels (172). The increase in lactate production and the activation of HIF-1α by the upregulated canonical WNT signaling are associated with the increase of angiogenesis and poor prognosis of cancers (173). Lactate released from cancer cells, via the MCT-1 transporter allows entry of lactate anion into cancer endothelial cells. In normoxic endothelial cells, lactate activates HIF-1α in a positive feedback loop by blocking HIF-1α prolyl hydroxylation and then prevents HIF labeling by the von Hippel-Lindau protein (163, 173, 174).
Lactate released from the cell initiates a transformation of the microenvironment independently of hypoxia. This enables angiogenesis and activation of the NF-kappaB pathway and prevents cell death through antiapoptotic proteins facilitating survival of cancer cells in a hostile environment (175). Knockdown of PDK-1 reverts the Warburg phenotype, diminishes the normoxic HIF-1α expression, hypoxic cell survival, invasion, and inhibits cancer development. Glycolysis metabolites due to high PDK-1 expression may promote HIF-1α activation. This sustains a feed-forward loop for cancer development. If corrected, these metabolic abnormalities may represent new cancer therapies which can synergize with other cancer treatments.
PI3K-AKT Pathway in WNT-Driven Aerobic Glycolysis and Cancer
The serine/threonine kinase AKT or protein kinase B (PKB) is often stimulated in human cancers (176). AKT activation constitutes a hallmark of the Warburg effect in most cancers. In tumor cells, hyperactivation of phosphatidyl-inositol 3-kinase (PI3K)-protein kinase B (AKT) signaling is associated with an augmented rate of glucose metabolism. AKT is activated by PIP3. PI3K synthesizes phosphatidylinositol-3,4,5-triphosphate (PIP3) from PIP2. The PI3K-AKT signaling induces cell growth and proliferation, cell migration, and angiogenesis. This signaling is induced by the linking of extracellular ligands to a receptor tyrosine kinase (RTK) in the plasma membrane (Figure 6) and is upregulated in several cancers. The AKT signaling activates the canonical WNT pathway by decreasing the GSK-3β activity (inactive form: pGSK-3β). AKT activation increases the Warburg aerobic glycolysis in cancer. AKT increases the ATP production and enhances glycolytic metabolism by several mechanisms. It increases glucose uptake by increasing the Glu transporter expression. AKT activates mTORC1, which promotes HIF-1α under normoxic conditions and increases LDH activity. The PI3K-AKT signaling increases the hypoxia-inducible transcription factor (177). AKT phosphorylates and stimulates phosphofructokinase-2 (PFK2), which in turn induces activation of the phosphofructokinase-1 (PFK1) by the PFK2 product fructose-2,6-bisphosphate (Fru-2,6-P2), an activator of PFK1. PFK1 is an allosteric enzyme responsible for glycolytic oscillations and can lead to instability beyond which a new state can be created in time and in space. These oscillations are dissipative structures initially described by Goldbeter and Lefever (178) and behave far-from-equilibrium (60, 179, 180). Increase in activity of PFK-1 is characteristic of tumor cells and is induced by oncogenes.
Upregulation of the PI3K-AKT signaling is frequently associated with cancers, such as glioblastomas, breast, pancreatic, and ovarian cancers (181). AKT mRNA is increased in breast and prostate cancers. PI3K-AKT induces angiogenesis by activating VEGF in endothelial cells and acting on the endothelial nitric oxide synthase. This stimulates vasodilation and vascular remodeling (182). The PI3K-AKT pathway is inhibited by the phosphatase and tensin homolog (PTEN) (183) (Figure 6). PTEN is a PIP3-phosphatase and inhibits PI3K. As PI3K-AKT is a key signaling activated in cancer, PTEN appears to be an interesting target against the tumor progression.
Reverse Warburg Effect
The Warburg effect does not take into account the metabolic interactions between cancer cells and the microenvironment (184, 185). The reverse Warburg effect (186) is a new metabolic process describing interacting between cancer cells and neighboring cancer-associated fibroblasts (CAFs) or stroma cells (187). In several human tumor cells, aerobic glycolysis contributes <50% for energy production (188–191) and some cancer cells exhibit high rates of the mitochondrial oxidative phosphorylation system (OXPHOs) (192–194). Thus, OXPHOs and aerobic glycolysis are not always mutually exclusive. Heterogeneous cancer cells can show various metabolic phenotypes (195–198). The OXPHOs metabolic pathway does not invalidate the Warburg effect but rather exhibits the complex plasticity of cancer metabolism and is involved in proliferation, metastasis, angiogenesis, and drug resistance in cancer cells. Oxidative stress in the two-compartment metabolic coupling and variations in cellular electromagnetic field could initiate the reverse Warburg effect. Cancer cells secrete ROSs in the microenvironment, which induces oxidative stress in CAFs which is strongly correlated to the reverse Warburg effect. Oxidative stress mechanism drives CAFs-cancer cells metabolic coupling and favors tumor growth via genetic and metabolic mechanisms. Oxidative stress related loss of stromal caveolin-1 (Cav-1) increases the production of ROSs in tumor cells, which initiates the cascade of oxidative stress in CAFs via a positive feedback mechanism (187, 199, 200) and plays a role in cancer proliferation, recurrence and prognosis (201, 202). Although the reverse Warburg effect and the role of the canonical WNT signaling on the reverse Warburg effect are not totally understood to-day, in the future they could represent a new approach for cancer treatment.
Canonical WNT Signaling, Circadian Rhythms (CRs), and Cancer
CRs are endogenous rhythms that last ~24 h, due to a negative feedback produced by a protein acting on the expression of its own gene (6, 203, 204). In mammals, CRs involve several major factors such as brain and muscle aryl-hydrocarbon receptor nuclear translocator-like1 (BMAL1), circadian locomotor output cycles kaput (CLOCK), period 1 (PER1), period 2 (PER2), and period 3 (PER3) (205, 206). CRs are involved in hormone secretion, heart rate and blood pressure, sleep-awake, feeding patterns, energy metabolism, and body temperature. Importantly, disruption of CRs appears linked to cancer, leading to aberrant cellular proliferation and involving the canonical WNT signaling (23). Due to several connections between cellular metabolism and the circadian clock, abnormal metabolism described in cancers may be a consequence of disrupted CRs (207). A decreased BMAL1 activity changes the behavior of genes involved in the canonical WNT pathway (208). β-catenin induces PER2 degradation modifying the circadian clock gene expression in intestinal mucosa of ApcMin/+ mice (209). A deceased expression of PER1 and/or PER2 has been reported in numerous cancers: breast tumor (210), prostate tumor (211), pancreatic tumor (212), colorectal tumor (213), chronic myeloid leukemia (214), glioma (215, 216), and intestinal epithelial neoplastic transformation (209).
From a thermodynamic viewpoint, CRs operate far from equilibrium and are dissipative structures (56, 60, 217). By applying Schrödinger's concepts to the modern thermodynamics of physical, chemical and biological systems that behave far from equilibrium, Prigogine and his colleagues opened a new understanding for the description of dissipative structures which occupy a key role in the world of living beings. Cancers are exergonic processes in which heat flows from the cancer to its surroundings (218). In cancer cells, the entropy production rate increases and characterizes irreversible processes driven by changes in thermodynamics properties (61).
Canonical WNT Signaling, Inflammation, Fibrosis, and Cancer
Inflammation
Chronic inflammation contributes to cancer development (219, 220). The role of prostaglandin E2 (PGE2) by stimulating the canonical WNT pathway has been well-established. The crosstalk between PGE2 and the canonical WNT signaling indicates that chronic inflammation caused by a prolonged increase of PGE2 may upregulate the canonical WNT signaling leading to cell proliferation and cancer. PGE2 favors the transcription of β-catenin (221, 222) and leads to growth of cancer cells such as in colon cancer. Non-steroidal anti-inflammatory drugs (NSAIDs) improve the treatment of colorectal cancer (223), partly due to interference with the β-catenin signaling and by inhibiting the PGE2 synthesis. PGE2 regulates the canonical WNT activity of hematopoietic stem cells (HSCs) in the zebrafish. Inhibition of PGE2 synthesis blocks the WNT-induced alterations in HSCs. PGE2 alters the β-catenin degradation via the cAMP/PKA signaling. In stem cells, WNT upregulation requires PGE2 (224). Dimethyl-prostaglandin E2 increases HSCs in vivo and increases the canonical WNT pathway activity (225). Conversely the canonical WNT signaling activates interleukins (IL)-7R and IL-2Rβ. In neuroectodermal stem cells, PGE2 interacts with the canonical WNT pathway partly through PKA and PI3K (226). PGE2 upregulates several WNT-target genes such as Ctnnb1, Ptgs2, Ccnd1, Mmp9.
Cancer and Fibrosis: A Similar Pathway
Several intracellular processes involved in cancers are similar to those encountered in fibrosis, and particularly an activation of the canonical WNT /TGF-β1 signaling (227). Similarly to that observed in cancer stroma, activation of this pathway leads to differentiation of fibroblasts into myofibroblasts. In cancer (mammary carcinoma, epithelial cells in tumor mammary glands), fibrotic lesions (nodules of Dupuytrens, hypertrophic scars) (228), and normal placental stem villi (229), the main myosin molecular motor in myofibroblasts is the non-muscle myosin IIA (NMMIIA). Contractile NMMIIA kinetics are particularly slow (230) and their entropy production rate is dramatically low (231). These contractile myofibroblasts allow skin retraction in wound healing (232–234). Patients suffering of cancer often receive a therapy by external ionizing radiations that leads to damages in both tumor cells and health tissues. Radiations induce numerous skin changes including inflammation and late radiation-induced fibrosis (RIF). RIF are characterized by fibroblast proliferation and myofibroblast differentiation, DNA damage, inflammation by stimulation of the NF-kappaB pathway (32). The over-expression of TGF-β1 and the upregulated canonical WNT signaling are involved in the molecular mechanism underlying the RIF.
Conclusions and Perspectives
It is currently well-demonstrated that the canonical WNT signaling intervenes at every stages in development of many cancers. When activated, this signaling induces and initiates several periodic or oscillating phenomena, such as the cell division cycle, the immune cycle, numerous circadian rhythms, and phosphofructokinase oscillations that are dissipative structures. This is because β-catenin modifies the expression of a considerable number of genes involved in one or others of these decisive areas of the cancer pathogeny. Thus, CYCLIN D1 and cMYC influence the cell division cycle. TGF-β1 also plays an important role activating the canonical WNT signaling directly or indirectly, and by acting on both the PI3K-AKT and GSK-3β pathways. Activation of the canonical WNT signaling affects many enzymes or transporters, resulting in Warburg glycolysis, which diverts pyruvate metabolism to lactate, a useful pathway for protein synthesis in tumor cells. All these different functions of the canonical WNT give it an eminent status as target immunotherapies against cancer.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Christophe Locher, Director of the Clinical Research Center, Grand Hôpital de l'Est Francilien (GHEF), Meaux, France and Mr. Vincent Gobert, Administrative Manager of the Clinical Research Center, Meaux, France. Prof. Brian Keogh has substantially improved the English language in the manuscript.
Glossary
Abbreviations
- APC
adenomatous polyposis coli
- AEC
alveolar epithelial cells
- BMAL1
circadian brain and muscle aryl-hydrocarbon receptor nuclear translocator-like1
- CR
circadian rhythm
- CTGF
connective tissue growth factor
- CTLA-4
cytotoxic T lymphocyte-associated antigen-4
- DCs
Dendritic Cells
- DKK1
dickkopf-1
- DSH
disheveled
- EGF
epidermal growth factor
- EMT
epithelial-mesenchymal transition
- FGF
fibroblast growth factor
- FZD
Frizzled
- Glu
glucose transporter
- GSK-3β
glycogen synthase kinase-3β
- HGF
hepatocyte growth factor
- HIF
hypoxia-inducible transcription factor
- IRF8
interferon regulatory factor 8
- LDH
lactate dehydrogenase
- LAP
latency-associated peptide
- LTBP-1
latent TGF-β1 binding protein-1
- CLOCK
locomotor output cycles kaput
- LRP5/6
low-density lipoprotein receptor-related protein 5/6
- MCT-1
monocarboxylate lactate transporter-1
- NMMIIA
non-muscle myosin IIA
- PER
period
- PPARγ
peroxisome proliferator-activated receptor gamma
- PI3K
phosphatidyl-inositol 3-kinase
- PDGF
platelet-derived growth factor
- PTEN
phosphatase and tensin homolog
- PFK
phosphofructokinase
- AKT
protein kinase B
- PD-1
programmed cell death protein1
- PDH
pyruvate dehydrogenase complex
- PDK
pyruvate dehydrogenase kinase
- PKM2
pyruvate kinase M2
- RTK
receptor tyrosine kinase
- Treg cell
Regulatory T cell
- SLC
small latent complex
- SMA
smooth muscle actin
- ECM
extracellular matrix
- TCF/LEF
T-cell factor/lymphoid enhancer factor
- TGF
Transforming Growth Factor
- Th
T helper
- TCA
tricarboxylic acid
- TNF-β
tumor necrosis factor beta
- VEGF
vascular endothelial growth factor.
Footnotes
Funding. The authors thank the Grand Hôpital de l'Est Francilien (GHEF) for its valuable support in making the necessary research facilities available for this study.
References
- 1.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. (1982) 31:99–109. 10.1016/0092-8674(82)90409-3 [DOI] [PubMed] [Google Scholar]
- 2.Murray AW. Cyclin synthesis and degradation and the embryonic cell cycle. J Cell Sci Suppl. (1989) 12:65–76. 10.1242/jcs.1989.Supplement_12.7 [DOI] [PubMed] [Google Scholar]
- 3.Murray AW. Cell biology: the cell cycle as a cdc2 cycle. Nature. (1989) 342:14–5. 10.1038/342014a0 [DOI] [PubMed] [Google Scholar]
- 4.Nurse P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem. (2002) 3:596–603. [DOI] [PubMed] [Google Scholar]
- 5.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. 10.1016/j.immuni.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 6.Savvidis C, Koutsilieris M. Circadian rhythm disruption in cancer biology. Mol Med. (2012) 18:1249–60. 10.2119/molmed.2012.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. (1998) 281:1509–12. 10.1126/science.281.5382.1509 [DOI] [PubMed] [Google Scholar]
- 8.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. (1999) 96:5522–7. 10.1073/pnas.96.10.5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. (1999) 398:422–6. 10.1038/18884 [DOI] [PubMed] [Google Scholar]
- 10.Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signalling. EMBO J. (2012) 31:2705–13. 10.1038/emboj.2012.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. (2006) 127:469–80. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- 12.Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. (2018) 39:648–58. 10.1016/j.tips.2018.03.008 [DOI] [PubMed] [Google Scholar]
- 13.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. (2010) 43:146–55. 10.1016/j.jbiomech.2009.09.020 [DOI] [PubMed] [Google Scholar]
- 14.Otranto M, Sarrazy V, Bonte F, Hinz B, Gabbiani G, Desmouliere A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes Migrat. (2012) 6:203–19. 10.4161/cam.20377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 16.Goldbeter A. Mechanism for oscillatory synthesis of cyclic AMP in Dictyostelium discoideum. Nature. (1975) 253:540–2. 10.1038/253540a0 [DOI] [PubMed] [Google Scholar]
- 17.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. (2002) 296:1644–6. 10.1126/science.1071549 [DOI] [PubMed] [Google Scholar]
- 18.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. (2004) 5:691–701. 10.1038/nrg1427 [DOI] [PubMed] [Google Scholar]
- 19.Nusse R. Wnt signaling in disease and in development. Cell Res. (2005) 15:28–32. 10.1038/sj.cr.7290260 [DOI] [PubMed] [Google Scholar]
- 20.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. (2012) 149:1192–205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 21.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. (2009) 10:468–77. 10.1038/nrm2717 [DOI] [PubMed] [Google Scholar]
- 22.Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. (2014) 33:1454–73. 10.15252/embj.201488598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lecarpentier Y, Claes V, Duthoit G, Hebert JL. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front Physiol. (2014) 5:429 10.3389/fphys.2014.00429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerhold DL, Liu F, Jiang G, Li Z, Xu J, Lu M, et al. Gene expression profile of adipocyte differentiation and its regulation by peroxisome proliferator-activated receptor-gamma agonists. Endocrinology. (2002) 143:2106–18. 10.1210/endo.143.6.8842 [DOI] [PubMed] [Google Scholar]
- 25.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. (2002) 16:2793–801. 10.1210/me.2002-0020 [DOI] [PubMed] [Google Scholar]
- 26.Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. (2009) 5:442–7. 10.1038/nrrheum.2009.137 [DOI] [PubMed] [Google Scholar]
- 27.Lu D, Carson DA. Repression of β-catenin signaling by PPAR γ ligands. Eur J Pharmacol. (2010) 636:198–202. 10.1016/j.ejphar.2010.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. (2006) 26:5827–37. 10.1128/MCB.00441-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Djouadi F, Lecarpentier Y, Hebert JL, Charron P, Bastin J, Coirault C. A potential link between PPAR signaling and the pathogenesis of arrhythmogenic right ventricular cardiomyopathy (ARVC). Cardiovasc Res. (2009) 84:83–90. 10.1093/cvr/cvp183 [DOI] [PubMed] [Google Scholar]
- 30.Lecarpentier Y, Claes V, Vallee A, Hebert JL. Thermodynamics in cancers: opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin Transl Med. (2017) 6:14. 10.1186/s40169-017-0144-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallee A, Lecarpentier Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/beta-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol. (2018) 9:745. 10.3389/fimmu.2018.00745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallee A, Lecarpentier Y, Guillevin R, Vallee JN. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR gamma in radiation-induced fibrosis. Oncotarget. (2017) 8:90579–604. 10.18632/oncotarget.21234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lecarpentier Y, Schussler O, Claes V, Vallée A. The myofibroblast: TGFβ-1, a conductor which plays a key role in fibrosis by regulating the balance between PPARγ and the canonical WNT Pathway. Nucl Recep Res. (2017) 4:101299 10.11131/2017/101299 [DOI] [Google Scholar]
- 34.Fu M, Zhang J, Lin Y, Zhu X, Zhao L, Ahmad M, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma (PPAR gamma) gene expression by transforming growth factor beta in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochem J. (2003) 370(Pt 3):1019–25. 10.1042/bj20021503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phan SH. The myofibroblast in pulmonary fibrosis. Chest. (2002) 122(6 Suppl.):286S−9S. 10.1378/chest.122.6_suppl.286S [DOI] [PubMed] [Google Scholar]
- 36.Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. (1994) 145:114–25. [PMC free article] [PubMed] [Google Scholar]
- 37.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. (2000) 257:180–9. 10.1006/excr.2000.4869 [DOI] [PubMed] [Google Scholar]
- 38.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. (2005) 166:1321–32. 10.1016/S0002-9440(10)62351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. (2009) 119:213–24. 10.1172/JCI36940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willis BC, Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L525–34. 10.1152/ajplung.00163.2007 [DOI] [PubMed] [Google Scholar]
- 41.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. (2006) 3:377–82. 10.1513/pats.200601-004TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldmann T, Zissel G, Watz H, Dromann D, Reck M, Kugler C, et al. Human alveolar epithelial cells type II are capable of TGFbeta-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir Res. (2018) 19:138. 10.1186/s12931-018-0841-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massague J. G1 cell-cycle control and cancer. Nature. (2004) 432:298–306. 10.1038/nature03094 [DOI] [PubMed] [Google Scholar]
- 44.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. (2000) 275:21797–800. 10.1074/jbc.C000023200 [DOI] [PubMed] [Google Scholar]
- 45.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. (2002) 111:241–50. 10.1016/S0092-8674(02)01014-0 [DOI] [PubMed] [Google Scholar]
- 46.Nusse R. Wnt signaling and stem cell control. Cell Res. (2008) 18:523–7. 10.1038/cr.2008.47 [DOI] [PubMed] [Google Scholar]
- 47.Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ. Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene. (1994) 9:3635–45. [PubMed] [Google Scholar]
- 48.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. (1998) 12:3499–511. 10.1101/gad.12.22.3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. (2003) 12:381–92. 10.1016/S1097-2765(03)00287-9 [DOI] [PubMed] [Google Scholar]
- 50.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. (2004) 101:9085–90. 10.1073/pnas.0402770101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. (2007) 6:24. 10.1186/1476-4598-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hassanian SM, Ardeshirylajimi A, Dinarvand P, Rezaie AR. Inorganic polyphosphate promotes cyclin D1 synthesis through activation of mTOR/Wnt/beta-catenin signaling in endothelial cells. J Thromb Haemos. (2016) 14:2261–73. 10.1111/jth.13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sansom OJ, Reed KR, van de Wetering M, Muncan V, Winton DJ, Clevers H, et al. Cyclin D1 is not an immediate target of beta-catenin following Apc loss in the intestine. J Biol Chem. (2005) 280:28463–7. 10.1074/jbc.M500191200 [DOI] [PubMed] [Google Scholar]
- 54.Zhang J, Gill AJ, Issacs JD, Atmore B, Johns A, Delbridge LW, et al. The Wnt/β-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum Pathol. (2012) 43:1044–50. 10.1016/j.humpath.2011.08.013 [DOI] [PubMed] [Google Scholar]
- 55.Gerard C, Goldbeter A. Temporal self-organization of the cyclin/Cdk network driving the mammalian cell cycle. Proc Natl Acad Sci USA. (2009) 106:21643–8. 10.1073/pnas.0903827106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldbeter A. Computational approaches to cellular rhythms. Nature. (2002) 420:238–45. 10.1038/nature01259 [DOI] [PubMed] [Google Scholar]
- 57.Olmeda D, Castel S, Vilaro S, Cano A. Beta-catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell. (2003) 14:2844–60. 10.1091/mbc.e03-01-0865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, et al. Cell cycle control of wnt receptor activation. Dev Cell. (2009) 17:788–99. 10.1016/j.devcel.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 59.Hadjihannas MV, Bernkopf DB, Bruckner M, Behrens J. Cell cycle control of Wnt/beta-catenin signalling by conductin/axin2 through CDC20. EMBO Rep. (2012) 13:347–54. 10.1038/embor.2012.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prigogine I, Nicolis G, Babloyantz A. Nonequilibrium problems in biological phenomena. Ann NY Acad Sci. (1974) 231:99–105. 10.1111/j.1749-6632.1974.tb20557.x [DOI] [PubMed] [Google Scholar]
- 61.Kondepudi D, Prigogine I. Modern Thermodynamics From Heat Engines to Dissipative Structures. New York, NY: Wiley & Sons; (1999). [Google Scholar]
- 62.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci USA. (1974) 71:1286–90. 10.1073/pnas.71.4.1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pourquie O. The segmentation clock: converting embryonic time into spatial pattern. Science. (2003) 301:328–30. 10.1126/science.1085887 [DOI] [PubMed] [Google Scholar]
- 64.Goldbeter A, Pourquie O. Modeling the segmentation clock as a network of coupled oscillations in the Notch, Wnt and FGF signaling pathways. J Theor Biol. (2008) 252:574–85. 10.1016/j.jtbi.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 65.Coventry BJ, Ashdown ML, Quinn MA, Markovic SN, Yatomi-Clarke SL, Robinson AP. CRP identifies homeostatic immune oscillations in cancer patients: a potential treatment targeting tool? J Transl Med. (2009) 7:102. 10.1186/1479-5876-7-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. (2017) 168:487–502.e15. 10.1016/j.cell.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 Blockade enhances T-cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin Cancer Res. (2017) 23:454–65. 10.1158/1078-0432.CCR-16-1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med. (2017) 23:60–8. 10.1038/nm.4219 [DOI] [PubMed] [Google Scholar]
- 71.Tammela T, Sanchez-Rivera FJ, Cetinbas NM, Wu K, Joshi NS, Helenius K, et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature. (2017) 545:355–9. 10.1038/nature22334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. (2017) 169:985–99. 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 73.Ganesh S, Shui X, Craig KP, Koser ML, Chopda GR, Cyr WA, et al. β-Catenin mRNA silencing and MEK inhibition display synergistic efficacy in preclinical tumor models. Mol Cancer Therap. (2018) 17:544–53. 10.1158/1535-7163.MCT-17-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-mediated β-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol Ther. (2018) 26:2567–79. 10.1016/j.ymthe.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van de Wetering M, Oosterwegel M, Dooijes D, Clevers H. Identification and cloning of TCF-1, a T lymphocyte-specific transcription factor containing a sequence-specific HMG box. EMBO J. (1991) 10:123–32. 10.1002/j.1460-2075.1991.tb07928.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. (2009) 15:808–13. 10.1038/nm.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Becht E, Giraldo NA, Dieu-Nosjean MC, Sautes-Fridman C, Fridman WH. Cancer immune contexture and immunotherapy. Curr Opin Immunol. (2016) 39:7–13. 10.1016/j.coi.2015.11.009 [DOI] [PubMed] [Google Scholar]
- 78.Lecarpentier Y, Schussler O, Sakic A, Rincon-Garriz JM, Soulie P, Bochaton-Piallat ML, et al. Human bone marrow contains mesenchymal stromal stem cells that differentiate in vitro into contractile myofibroblasts controlling T lymphocyte proliferation. Stem Cells Int. (2018) 2018:6134787. 10.1155/2018/6134787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang W, Zhong W, Yuan J, Yan C, Hu S, Tong Y, et al. Involvement of Wnt/β-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget. (2015) 6:42276–89. 10.18632/oncotarget.5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. (2016) 30:324–36. 10.1016/j.ccell.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23.e4. 10.1016/j.ccell.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen SB, Smith NL, McDougal C, Pepper M, Shah S, Yap GS, et al. Beta-catenin signaling drives differentiation and proinflammatory function of IRF8-dependent dendritic cells. J Immunol. (2015) 194:210–22. 10.4049/jimmunol.1402453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. 10.1084/jem.20101158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. 10.1016/j.cell.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wong C, Chen C, Wu Q, Liu Y, Zheng P. A critical role for the regulated wnt-myc pathway in naive T cell survival. J Immunol. (2015) 194:158–67. 10.4049/jimmunol.1401238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. (2005) 22:371–83. 10.1016/j.immuni.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 87.Giuntoli RL, II, Lu J, Kobayashi H, Kennedy R, Celis E. Direct costimulation of tumor-reactive CTL by helper T cells potentiate their proliferation, survival, and effector function. Clin Cancer Res. (2002) 8:922–31. [PubMed] [Google Scholar]
- 88.Sorcini D, Bruscoli S, Frammartino T, Cimino M, Mazzon E, Galuppo M, et al. Wnt/β-Catenin Signaling Induces Integrin α4β1 in T cells and promotes a progressive neuroinflammatory disease in mice. J Immunol. (2017) 199:3031–41. 10.4049/jimmunol.1700247 [DOI] [PubMed] [Google Scholar]
- 89.Sun X, Liu S, Wang D, Zhang Y, Li W, Guo Y, et al. Colorectal cancer cells suppress CD4+ T cells immunity through canonical Wnt signaling. Oncotarget. (2017) 8:15168–81. 10.18632/oncotarget.14834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lengfeld JE, Lutz SE, Smith JR, Diaconu C, Scott C, Kofman SB, et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci USA. (2017) 114:E1168–77. 10.1073/pnas.1609905114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. (2017) 27:109–18. 10.1038/cr.2016.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
- 93.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci. (2009) 66:1850–7. 10.1007/s00018-009-9153-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N, et al. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat Immunol. (2015) 16:267–75. 10.1038/ni.3083 [DOI] [PubMed] [Google Scholar]
- 95.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. 10.1038/nm730 [DOI] [PubMed] [Google Scholar]
- 96.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. (2004) 64:1140–5. 10.1158/0008-5472.CAN-03-3259 [DOI] [PubMed] [Google Scholar]
- 97.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. 10.1126/science.aac9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. (2016) 165:45–60. 10.1016/j.cell.2016.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. 10.1126/science.271.5256.1734 [DOI] [PubMed] [Google Scholar]
- 100.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. 10.1002/j.1460-2075.1992.tb05481.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci USA. (1997) 94:8099–103. 10.1073/pnas.94.15.8099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. 10.1016/S1074-7613(00)80089-8 [DOI] [PubMed] [Google Scholar]
- 103.Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. (1971) 27:549–50. 10.1007/BF02147594 [DOI] [PubMed] [Google Scholar]
- 104.Orimo A, Weinberg RA. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol Ther. (2007) 6:618–9. 10.4161/cbt.6.4.4255 [DOI] [PubMed] [Google Scholar]
- 105.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. (2006) 5:1640–6. 10.4161/cbt.5.12.3354 [DOI] [PubMed] [Google Scholar]
- 106.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical wnt signaling. Cold Spring Harb Perspect Biol. (2010) 2:a002915. 10.1101/cshperspect.a002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. (2002) 3:349–63. 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- 108.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. 10.1056/NEJM198612253152606 [DOI] [PubMed] [Google Scholar]
- 109.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. (2008) 9:628–38. 10.1038/nrm2455 [DOI] [PubMed] [Google Scholar]
- 110.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. (2009) 326:1216–9. 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. (2002) 295:2387–92. 10.1126/science.1067100 [DOI] [PubMed] [Google Scholar]
- 112.Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. (2010) 70:9621–30. 10.1158/0008-5472.CAN-10-1722 [DOI] [PubMed] [Google Scholar]
- 113.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. (2002) 2:442–54. 10.1038/nrc822 [DOI] [PubMed] [Google Scholar]
- 114.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev cell. (2011) 21:193–215. 10.1016/j.devcel.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 115.Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE. (2012) 7:e30563. 10.1371/journal.pone.0030563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ishii G, Sangai T, Ito T, Hasebe T, Endoh Y, Sasaki H, et al. In vivo and in vitro characterization of human fibroblasts recruited selectively into human cancer stroma. Int J Cancer. (2005) 117:212–20. 10.1002/ijc.21199 [DOI] [PubMed] [Google Scholar]
- 117.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. (2004) 64:8492–5. 10.1158/0008-5472.CAN-04-1708 [DOI] [PubMed] [Google Scholar]
- 118.Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. Int J Hematol. (2007) 86:8–16. 10.1532/IJH97.06230 [DOI] [PubMed] [Google Scholar]
- 119.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Instit. (2004) 96:1593–603. 10.1093/jnci/djh299 [DOI] [PubMed] [Google Scholar]
- 120.Hung SC, Deng WP, Yang WK, Liu RS, Lee CC, Su TC, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. (2005) 11:7749–56. 10.1158/1078-0432.CCR-05-0876 [DOI] [PubMed] [Google Scholar]
- 121.Menon LG, Picinich S, Koneru R, Gao H, Lin SY, Koneru M, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. (2007) 25:520–8. 10.1634/stemcells.2006-0257 [DOI] [PubMed] [Google Scholar]
- 122.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. (2008) 68:4331–9. 10.1158/0008-5472.CAN-08-0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. (1998) 8:1243–52. 10.1016/S0960-9822(07)00533-7 [DOI] [PubMed] [Google Scholar]
- 124.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. (2006) 6:506–20. 10.1038/nrc1926 [DOI] [PubMed] [Google Scholar]
- 125.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys acta. (2007) 1775:21–62. 10.1016/j.bbcan.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 126.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. (2010) 10:415–24. 10.1038/nrc2853 [DOI] [PubMed] [Google Scholar]
- 127.Silberstein GB, Strickland P, Coleman S, Daniel CW. Epithelium-dependent extracellular matrix synthesis in transforming growth factor-beta 1-growth-inhibited mouse mammary gland. J Cell Biol. (1990) 110:2209–19. 10.1083/jcb.110.6.2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sweetwyne MT, Murphy-Ullrich JE. Thrombospondin1 in tissue repair and fibrosis: TGF-β-dependent and independent mechanisms. Matrix Biol. (2012) 31:178–86. 10.1016/j.matbio.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. (2012) 113:410–8. 10.1002/jcb.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Azuma H, Ehata S, Miyazaki H, Watabe T, Maruyama O, Imamura T, et al. Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J Natl Cancer Instit. (2005) 97:1734–46. 10.1093/jnci/dji399 [DOI] [PubMed] [Google Scholar]
- 131.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. (2008) 133:66–77. 10.1016/j.cell.2008.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. (2007) 12:313–27. 10.1016/j.ccr.2007.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. (2010) 107:20009–14. 10.1073/pnas.1013805107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. (2003) 116(Pt 2):217–24. 10.1242/jcs.00229 [DOI] [PubMed] [Google Scholar]
- 135.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. (2004) 41:233–64. 10.1080/10408360490460933 [DOI] [PubMed] [Google Scholar]
- 136.Hinz B, Gabbiani G. Fibrosis: recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. (2010) 2:78. 10.3410/B2-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Derynck R, Jarrett JA, Chen EY, Goeddel DV. The murine transforming growth factor-beta precursor. J Biol Chem. (1986) 261:4377–9. [PubMed] [Google Scholar]
- 138.Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci. (2011) 36:47–54. 10.1016/j.tibs.2010.08.002 [DOI] [PubMed] [Google Scholar]
- 139.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature. (2011) 474:343–9. 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. (2008) 87:601–15. 10.1016/j.ejcb.2008.01.012 [DOI] [PubMed] [Google Scholar]
- 141.Nishimura SL. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. (2009) 175:1362–70. 10.2353/ajpath.2009.090393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheppard D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. (2005) 24:395–402. 10.1007/s10555-005-5131-6 [DOI] [PubMed] [Google Scholar]
- 143.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. (2005) 8:241–54. 10.1016/j.ccr.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 144.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. (2009) 28:113–27. 10.1007/s10555-008-9173-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol. (2005) 15:329–41. 10.1016/j.semcancer.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 146.Andersen ES, Christensen PB, Weis N. Transient elastography for liver fibrosis diagnosis. Eur J Intern Med. (2009) 20:339–42. 10.1016/j.ejim.2008.09.020 [DOI] [PubMed] [Google Scholar]
- 147.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol. (2011) 12:308–19. 10.1038/nrm3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev. (2012) 31:195–208. 10.1007/s10555-011-9340-x [DOI] [PubMed] [Google Scholar]
- 149.Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. (2010) 316:2713–22. 10.1016/j.yexcr.2010.04.032 [DOI] [PubMed] [Google Scholar]
- 150.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. (2009) 9:239–52. 10.1038/nrc2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. (2000) 103:311–20. 10.1016/S0092-8674(00)00122-7 [DOI] [PubMed] [Google Scholar]
- 152.Thompson CB. Wnt meets Warburg: another piece in the puzzle? EMBO J. (2014) 33:1420–2. 10.15252/embj.201488785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, et al. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Progr Nucleic Acid Res Mol Biol. (2001) 70:33–75. 10.1016/S0079-6603(01)70013-X [DOI] [PubMed] [Google Scholar]
- 154.Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. (2005) 179:56–65. 10.1159/000084509 [DOI] [PubMed] [Google Scholar]
- 155.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. (2005) 5:744–9. 10.1038/nrc1694 [DOI] [PubMed] [Google Scholar]
- 156.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. (2008) 8:387–98. 10.1038/nrc2389 [DOI] [PubMed] [Google Scholar]
- 157.Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. (2006) 66:632–7. 10.1158/0008-5472.CAN-05-3260 [DOI] [PubMed] [Google Scholar]
- 158.Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer. (2008) 98:1975–84. 10.1038/sj.bjc.6604356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Baumunk D, Reichelt U, Hildebrandt J, Krause H, Ebbing J, Cash H, et al. Expression parameters of the metabolic pathway genes pyruvate dehydrogenase kinase-1 (PDK-1) and DJ-1/PARK7 in renal cell carcinoma (RCC). World J Urol. (2013) 31:1191–6. 10.1007/s00345-012-0874-5 [DOI] [PubMed] [Google Scholar]
- 160.Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, et al. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid Redox Signal. (2007) 9:1115–24. 10.1089/ars.2007.1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 162.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. (2008) 283:22700–8. 10.1074/jbc.M801765200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, et al. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. (2013) 32:1638–50. 10.1038/onc.2012.198 [DOI] [PubMed] [Google Scholar]
- 165.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. (2008) 105:18782–7. 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. (2010) 70:859–62. 10.1158/0008-5472.CAN-09-3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. (2007) 27:7381–93. 10.1128/MCB.00440-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. (2002) 277:23111–5. 10.1074/jbc.M202487200 [DOI] [PubMed] [Google Scholar]
- 169.Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem. (2015) 290:25427–38. 10.1074/jbc.M115.646984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chocarro-Calvo A, Garcia-Martinez JM, Ardila-Gonzalez S, De la Vieja A, Garcia-Jimenez C. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol Cell. (2013) 49:474–86. 10.1016/j.molcel.2012.11.022 [DOI] [PubMed] [Google Scholar]
- 171.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. (1995) 92:5510–4. 10.1073/pnas.92.12.5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Constant JS, Feng JJ, Zabel DD, Yuan H, Suh DY, Scheuenstuhl H, et al. Lactate elicits vascular endothelial growth factor from macrophages: a possible alternative to hypoxia. Wound Repair Regener. (2000) 8:353–60. 10.1111/j.1524-475X.2000.00353.x [DOI] [PubMed] [Google Scholar]
- 173.Vallee A, Guillevin R, Vallee JN. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev Neurosci. (2018) 29:71–91. 10.1515/revneuro-2017-0032 [DOI] [PubMed] [Google Scholar]
- 174.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. (2001) 292:468–72. 10.1126/science.1059796 [DOI] [PubMed] [Google Scholar]
- 175.Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. (2011) 71:2550–60. 10.1158/0008-5472.CAN-10-2828 [DOI] [PubMed] [Google Scholar]
- 176.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. (2009) 19:25–31. 10.1016/j.semcancer.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. (2011) 4:51. 10.3389/fnmol.2011.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Goldbeter A, Lefever R. Dissipative structures for an allosteric model. Application to glycolytic oscillations. Biophys J. (1972) 12:1302–15. 10.1016/S0006-3495(72)86164-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Prigogine I, Nicolis G. Biological order, structure and instabilities. Q Rev Biophys. (1971) 4:107–48. 10.1017/S0033583500000615 [DOI] [PubMed] [Google Scholar]
- 180.Lecarpentier Y, Claes V, Hebert JL. PPARs, Cardiovascular metabolism, and function: near- or far-from-equilibrium pathways. PPAR Res. (2010) 2010:783273. 10.1155/2010/783273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. (2002) 14:381–95. 10.1016/S0898-6568(01)00271-6 [DOI] [PubMed] [Google Scholar]
- 182.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. (2007) 129:1261–74. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Georgescu MM. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer. (2010) 1:1170–7. 10.1177/1947601911407325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Martinez-Outschoorn UE, Lisanti MP. Tumor microenvironment: introduction. Semin Oncol. (2014) 41:145. 10.1053/j.seminoncol.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 185.Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat Med. (2016) 22:1351–7. 10.1038/nm.4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. (2017) 8:57813–25. 10.18632/oncotarget.18175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. (2014) 25:47–60. 10.1016/j.semcancer.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 188.Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. (2004) 313:459–65. 10.1016/j.bbrc.2003.11.136 [DOI] [PubMed] [Google Scholar]
- 189.Li H, Li X, Ge X, Jia L, Zhang Z, Fang R, et al. MiR-34b-3 and miR-449a inhibit malignant progression of nasopharyngeal carcinoma by targeting lactate dehydrogenase A. Oncotarget. (2016) 7:54838–51. 10.18632/oncotarget.10761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hernandez-Resendiz I, Roman-Rosales A, Garcia-Villa E, Lopez-Macay A, Pineda E, Saavedra E, et al. Dual regulation of energy metabolism by p53 in human cervix and breast cancer cells. Biochim Biophys Acta. (2015) 1853:3266–78. 10.1016/j.bbamcr.2015.09.033 [DOI] [PubMed] [Google Scholar]
- 191.Rodriguez-Enriquez S, Gallardo-Perez JC, Marin-Hernandez A, Aguilar-Ponce JL, Mandujano-Tinoco EA, Meneses A, et al. Oxidative phosphorylation as a target to arrest malignant neoplasias. Curr Med Chem. (2011) 18:3156–67. 10.2174/092986711796391561 [DOI] [PubMed] [Google Scholar]
- 192.Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. (2014) 5:592–602. 10.1007/s13238-014-0082-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M, et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. (2013) 4:e504. 10.1038/cddis.2013.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mandujano-Tinoco EA, Gallardo-Perez JC, Marin-Hernandez A, Moreno-Sanchez R, Rodriguez-Enriquez S. Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim Biophys Acta. (2013) 1833:541–51. 10.1016/j.bbamcr.2012.11.013 [DOI] [PubMed] [Google Scholar]
- 195.Lee M, Yoon JH. Metabolic interplay between glycolysis and mitochondrial oxidation: the reverse Warburg effect and its therapeutic implication. World J Biol Chem. (2015) 6:148–61. 10.4331/wjbc.v6.i3.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, et al. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer. (2007) 97:646–53. 10.1038/sj.bjc.6603922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ertel A, Tsirigos A, Whitaker-Menezes D, Birbe RC, Pavlides S, Martinez-Outschoorn UE, et al. Is cancer a metabolic rebellion against host aging? In the quest for immortality, tumor cells try to save themselves by boosting mitochondrial metabolism. Cell Cycle. (2012) 11:253–63. 10.4161/cc.11.2.19006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle. (2010) 9:2412–22. 10.4161/cc.9.12.11989 [DOI] [PubMed] [Google Scholar]
- 199.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. (2010) 9:2423–33. 10.4161/cc.9.12.12048 [DOI] [PubMed] [Google Scholar]
- 200.Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. (2010) 9:1960–71. 10.4161/cc.9.10.11601 [DOI] [PubMed] [Google Scholar]
- 201.Martinez-Outschoorn UE, Curry JM, Ko YH, Lin Z, Tuluc M, Cognetti D, et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4. Cell Cycle. (2013) 12:2580–97. 10.4161/cc.25510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. The reverse Warburg effect in osteosarcoma. Oncotarget. (2014) 5:7982–3. 10.18632/oncotarget.2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Goodwin BC. Oscillatory behavior in enzymatic control processes. Adv Enzyme Regul. (1965) 3:425–38. 10.1016/0065-2571(65)90067-1 [DOI] [PubMed] [Google Scholar]
- 204.Hardin PE, Hall JC, Rosbash M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature. (1990) 343:536–40. 10.1038/343536a0 [DOI] [PubMed] [Google Scholar]
- 205.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. (1998) 280:1564–9. 10.1126/science.280.5369.1564 [DOI] [PubMed] [Google Scholar]
- 206.Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA. (1998) 95:5474–9. 10.1073/pnas.95.10.5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE. (2010) 5:e8561. 10.1371/journal.pone.0008561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med. (2012) 18:1768–77. 10.1038/nm.2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yang X, Wood PA, Ansell CM, Ohmori M, Oh EY, Xiong Y, et al. Beta-catenin induces beta-TrCP-mediated PER2 degradation altering circadian clock gene expression in intestinal mucosa of ApcMin/+ mice. J Biochem. (2009) 145:289–97. 10.1093/jb/mvn167 [DOI] [PubMed] [Google Scholar]
- 210.Winter SL, Bosnoyan-Collins L, Pinnaduwage D, Andrulis IL. Expression of the circadian clock genes Per1 and Per2 in sporadic and familial breast tumors. Neoplasia. (2007) 9:797–800. 10.1593/neo.07595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu J, et al. A role for the clock gene per1 in prostate cancer. Cancer Res. (2009) 69:7619–25. 10.1158/0008-5472.CAN-08-4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Suzuki T, Sato F, Kondo J, Liu Y, Kusumi T, Fujimoto K, et al. Period is involved in the proliferation of human pancreatic MIA-PaCa2 cancer cells by TNF-alpha. Biomed Res. (2008) 29:99–103. 10.2220/biomedres.29.99 [DOI] [PubMed] [Google Scholar]
- 213.Mostafaie N, Kallay E, Sauerzapf E, Bonner E, Kriwanek S, Cross HS, et al. Correlated downregulation of estrogen receptor beta and the circadian clock gene Per1 in human colorectal cancer. Mol Carcinogenes. (2009) 48:642–7. 10.1002/mc.20510 [DOI] [PubMed] [Google Scholar]
- 214.Yang MY, Yang WC, Lin PM, Hsu JF, Hsiao HH, Liu YC, et al. Altered expression of circadian clock genes in human chronic myeloid leukemia. J Biol Rhythms. (2011) 26:136–48. 10.1177/0748730410395527 [DOI] [PubMed] [Google Scholar]
- 215.Fujioka A, Takashima N, Shigeyoshi Y. Circadian rhythm generation in a glioma cell line. Biochem Biophys Res Commun. (2006) 346:169–74. 10.1016/j.bbrc.2006.05.094 [DOI] [PubMed] [Google Scholar]
- 216.Xia HC, Niu ZF, Ma H, Cao SZ, Hao SC, Liu ZT, et al. Deregulated expression of the Per1 and Per2 in human gliomas. Canadian J Neurol Sci. (2010) 37:365–70. 10.1017/S031716710001026X [DOI] [PubMed] [Google Scholar]
- 217.Prigogine I. Life and physics. New perspectives. Cell Biophys. (1986) 9:217–24. 10.1007/978-1-4612-4834-7_13 [DOI] [PubMed] [Google Scholar]
- 218.Atkins PW. Physical Chemistry, 5th Edn. Oxford; Melbourne, VIC; Tokio: Oxford University Press; (1990). [Google Scholar]
- 219.Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA. (2012) 109:2796–801. 10.1073/pnas.1104303108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. (2007) 19:209–16. 10.1016/j.coi.2007.01.001 [DOI] [PubMed] [Google Scholar]
- 221.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. (2005) 280:26565–72. 10.1074/jbc.M413056200 [DOI] [PubMed] [Google Scholar]
- 222.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. (2005) 310:1504–10. 10.1126/science.1116221 [DOI] [PubMed] [Google Scholar]
- 223.Ricchi P, Zarrilli R, Di Palma A, Acquaviva AM. Nonsteroidal anti-inflammatory drugs in colorectal cancer: from prevention to therapy. Br J Cancer. (2003) 88:803–7. 10.1038/sj.bjc.6600829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. (2009) 136:1136–47. 10.1016/j.cell.2009.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Li L, Kim HT, Nellore A, Patsoukis N, Petkova V, McDonough S, et al. Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/β-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J. (2014) 4:e178. 10.1038/bcj.2013.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wong CT, Ahmad E, Li H, Crawford DA. Prostaglandin E2 alters Wnt-dependent migration and proliferation in neuroectodermal stem cells: implications for autism spectrum disorders. Cell Commun Signal. (2014) 12:19. 10.1186/1478-811X-12-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vallee A, Lecarpentier Y, Vallee JN. Thermodynamic aspects and reprogramming cellular energy metabolism during the fibrosis process. Int J Mol Sci. (2017) 18:E2537. 10.3390/ijms18122537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chiavegato A, Bochaton-Piallat ML, D'Amore E, Sartore S, Gabbiani G. Expression of myosin heavy chain isoforms in mammary epithelial cells and in myofibroblasts from different fibrotic settings during neoplasia. Virchows Archiv. (1995) 426:77–86. 10.1007/BF00194701 [DOI] [PubMed] [Google Scholar]
- 229.Matsumura S, Sakurai K, Shinomiya T, Fujitani N, Key K, Ohashi M. Biochemical and immunohistochemical characterization of the isoforms of myosin and actin in human placenta. Placenta. (2011) 32:347–55. 10.1016/j.placenta.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 230.Lecarpentier Y, Claes V, Lecarpentier E, Guerin C, Hebert JL, Arsalane A, et al. Ultraslow myosin molecular motors of placental contractile stem villi in humans. PLoS ONE. (2014) 9:e108814. 10.1371/journal.pone.0108814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lecarpentier Y, Claes V, Hebert JL, Krokidis X, Blanc FX, Michel F, et al. Statistical mechanics of the human placenta: a stationary state of a near-equilibrium system in a linear regime. PLoS ONE. (2015) 10:e0142471. 10.1371/journal.pone.0142471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR, Majno G. Granulation tissue as a contractile organ. A study of structure and function. J Exp Med. (1972) 135:719–34. 10.1084/jem.135.4.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gabbiani G. The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res. (1981) 54:183–94. [PubMed] [Google Scholar]
- 234.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. (2007) 170:1807–16. 10.2353/ajpath.2007.070112 [DOI] [PMC free article] [PubMed] [Google Scholar]