

Abstract
Transient dative substrate–Ti interactions have been found to play a key role in controlling the regioselectivity of alkyne insertion and [2+2] cycloaddition in Ti-catalyzed [2+2+1] pyrrole synthesis and Ti-catalyzed alkyne hydroamination. TMS-protected alkynes with pendent Lewis basic groups can invert the regioselectivity of TMS-protected alkyne insertion, leading to the selective formation of highly substituted 3-TMS pyrroles. The competency of various potential directing groups was investigated, and it was found that the directing-group effect can be tuned by modifying the catalyst Lewis acidity, the directing-group basicity, or the directing-group tether length. Dative directing-group effects are unexplored with Ti catalysts, and this study demonstrates the potential power of dative substrate-Ti interactions in tuning selectivity.
Keywords: directing-group effects, multicomponent reactions, pyrroles, titanium, alkynes
Graphical Abstract
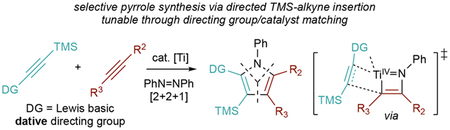
INTRODUCTION
Highly substituted pyrroles are important components of many bioactive natural products, drug candidates, and FDA-approved drugs.1–4 Recently, we have reported several multicomponent formal [2+2+1] pyrrole syntheses from alkynes and diazenes or azides (Figure 1).5–9 These methods provide facile, modular access to highly substituted pyrrole cores that are otherwise challenging to synthesize.
Figure 1.
Top: general overview of directing groups in Ti catalyzed/mediated chemistry; Bottom: Ti-catalyzed [2+2+1] pyrrole synthesis and methods for chemo- and regioselective alkyne heterocoupling.
Attaining regio- and chemoselectivity can be inherently difficult [2+2+1] cycloaddition reactions such as our Ti-catalyzed pyrrole synthesis or the Pauson–Khand reaction.10,11 Previous studies demonstrated that the selectivity of Ti-catalyzed alkyne coupling is driven by intrinsic alkyne stereoelectronic properties, and this substrate-driven selectivity results in a statistical mixture of regioisomers.5 More recently, we discovered that the heterocoupling of TMS-protected alkynes can occur with exceptional regioselectivity, yielding 2-TMS pyrroles (Figure 1, top).8 These reactions are highly selective because of the electronic properties of the TMS-protected alkynes: they do not easily undergo the [2+2] cycloadditions with Ti═NR imidos, but they rapidly (because of their electron-richness) and regioselectively (because of the thermodynamic α-Si effect8,12–19) insert into azatitanacyclobutenes. This protocol can access all possible penta- and tetrasubstituted pyrrole substitution patterns except for 1,2,3,5-tetrasubstitution.
An alternative strategy for regiocontrol is to use heteroatom-based directing groups to enforce selectivity. For example, sulfur and nitrogen-substituted alkenes have been used in the cobalt catalyzed [2+2+1] Pauson–Khand reaction to direct the product regioselectivity.20–22 This method has been applied further to the formal synthesis of Prostaglandin A2.23 Removable directing groups have also been used in the Pauson–Khand reaction, notably by using silicon-tethered pyridinyl vinyl silanes which could go through facile hydrolytic desilylation.24,25
Pendent alkoxide directing groups have been used extensively in Ti-catalyzed and mediated reactions to engender regio-, chemo-, and enantioselective transformations26. For example, Sharpless’ catalytic asymmetric alkene epoxidation relies on alkoxide directing groups to engender enantiofacial selectivity,27 while more recently Micalizio has used alkoxides to direct alkyne insertion regioselectivity in Ti-mediated reductive coupling reactions (Figure 1, middle),18,19,28–31 and Burns has used alkoxides to direct stereoselective alkene dihalogenation and haloazidation reactions.32–35 Alkoxide-directed coupling reactions have also served as a platform for heterocycle syntheses, such as pyridines and indolizidines.36,37
To exert complete regiocontrol over multisubstituted pyrrole synthesis, we have undertaken a study of pendent directing-group effects in Ti-catalyzed [2+2+1] pyrrole synthesis (Figure 1, bottom), envisioning that it would be possible to reverse the regiochemical course of the reaction. In contrast to anionic directing groups, the use of dative directing groups in titanium catalysis is unexplored. However, we envisioned that transient dative donor interactions could promote chemo- and regioselective reactions while also undergoing the facile ligand exchange needed for productive redox catalysis. We herein report that TMS-protected alkynes with pendent Lewis basic groups can invert the regioselectivity of TMS-protected alkyne insertion in Ti-catalyzed [2+2+1] pyrrole synthesis. This demonstrates that a dative directing-group effect can override the substrate’s intrinsic selectivity. These reactions lead to the formation of 3-TMS-substituted pyrroles, which can be used in complex molecule synthesis.
RESULTS AND DISCUSSION
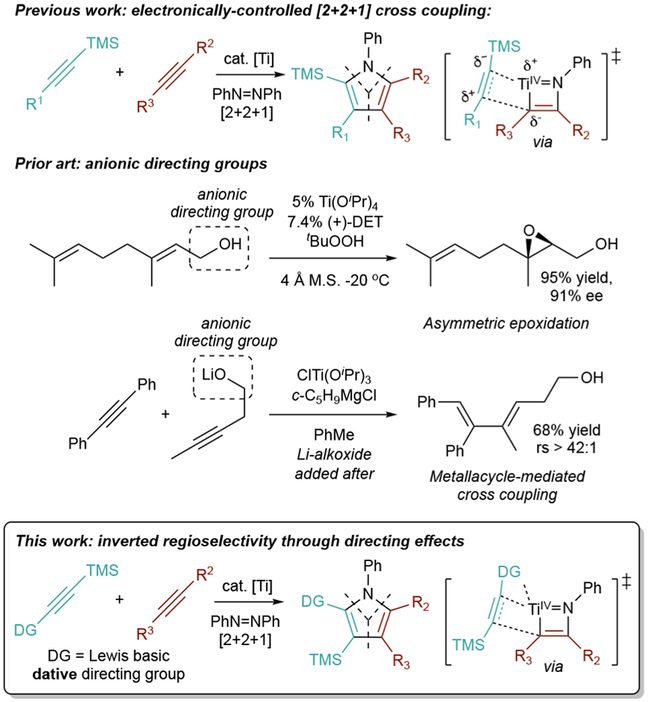
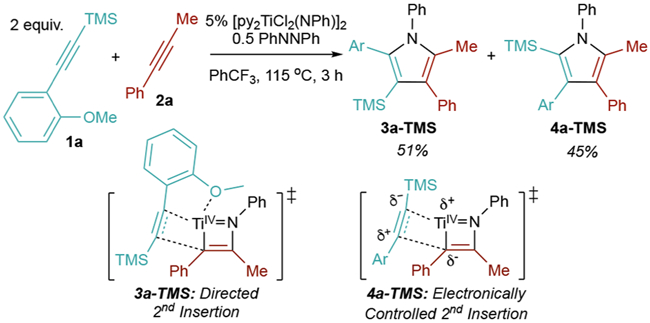
Unlike other TMS-protected alkynes, the [2+2+1] heterocouplng of ((2-methoxyphenyl)ethynyl)trimethylsilane (1a) with phenylpropyne (2a) was remarkably unselective for 4a-TMS, giving a 1.1:1 ratio of 3a-TMS:4a-TMS (Table 1, entry 1).8 We speculated that the increased yield of 3a-TMS was a result of the o-OMe group precoordinating to Ti and directing second alkyne insertion in a manner opposite the electronic properties of the alkyne (Figure 2).
Table 1.
Optimization of Reaction Conditions to Promote Directed Alkyne Insertiona
 | ||||||
|---|---|---|---|---|---|---|
| entry | T (°C) |
X (equiv) |
time (h) |
Y (%) |
yield of 3ab (%) |
ratio of 3a/4ab |
| 1c | 115 | 2 | 2 | 10 | 51 | 1.1 |
| 2 | 115 | 2 | 1 | 10 | 76 | 5.5 |
| 3 | 80 | 2 | 1 | 10 | 81 | 7.2 |
| 4 | 60 | 2 | 2 | 10 | 79 | 8.8 |
| 5 | 60 | 1 | 2 | 10 | 67 | 9.3 |
| 6 | 45 | 2 | 24 | 10 | 74 | 10.5 |
| 7 | 45 | 1 | 24 | 10 | 74 | 10.3 |
A mixture of 0.1–0.2 mmol 1a, 0.1 mmol 2a, 0.225 mmol PhNNPh, 0.0025–0.01 mmol (THF)3TiI2(NPh), and 0.01 Ph3CH (internal standard) in 0.5 mL of CF3Ph were heated at desired temperature and time.
Yield and selectivity are determined by 1H NMR and reported with respect to 2a.
Reported in ref 8 with [py2TiCl2(NPh)]2 as catalyst.
Figure 2.
Initial observation of a directing-group effect: o-Me substituted aryl alkynes (1a) have poor selectivity for electronically preferred 2nd insertion product 4a-TMS.
Thus, we sought to find conditions that favored the directed 3-TMS product 3a-TMS. First, [py2TiCl2(NPh)]2 was replaced by the more Lewis acidic catalyst THF3TiI2(NPh)38,39 to promote stronger coordination of the o-OMe group. This increased the 3a-TMS:4a-TMS ratio to 5.5:1 (Table 1, entry 2). Lowering the reaction temperature further improved the selectivity, giving regioisomeric ratios >10.5:1 (Table 1, entries 2–6). At lower temperatures, dissociation of the o-OMe is likely slowed, resulting in formation of the kinetically preferred metallacycle leading to 3a-TMS over 4a-TMS.8 In our previous studies of TMS-alkyne heterocouplings, chemoselectivities were good at a 1:1 ratio of the two alkynes, but from a practical perspective, we found it better to use an excess of the TMS-alkyne in order to make product separations more simple. However, here reducing the equivalents of 1a had little effect on yield or selectivity, indicating that the directing-group effect can more effectively outcompete 2a homocoupling (entries 5 and 7). Thus, the conditions in entry 6 were deemed optimized.
The directing effect was explored on a range of TMS-protected alkynes with pendent Lewis basic functionality (Table 2). The optimized conditions from Table 1 were used (Condition A), along with the prior conditions for TMS-alkyne heterocoupling catalysis8 (Condition B).
Table 2.
Scope of Directing-Group Effects in Ti-Catalyzed [2+2+1] Pyrrole Synthesis
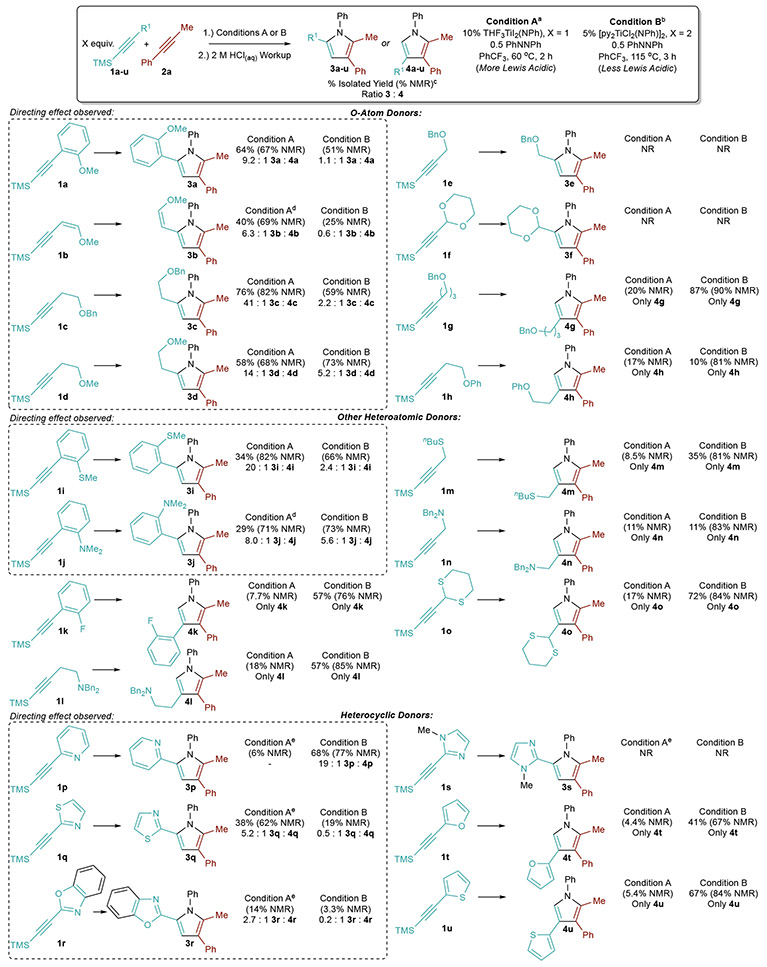
|
Condition A: A mixture of 0.5 mmol 1 (1.1 equiv), 0.5 mmol 2 (1.1 equiv), 0.225 mmol PhNNPh (0.5 equiv) and 0.05 mmol THF3I2Ti(NPh) (0.1 equiv) in 2.5 mL CF3Ph was heated for 2 h. Reactions were quenched with 2 M HCl in MeOH;
Condition B: A mixture of 1.0 mmol 1 (2.2 equiv), 0.5 mmol 2 (1.1 equiv), 0.225 mmol PhNNPh (0.5 equiv) and 0.025 mmol [py2Cl2TiNPh]2 (0.05 equiv) in 2.5 mL CF3Ph was heated for 3 h. Reactions were quenched with 2 M HCl in MeOH;
Isolated and/or NMR yield of major regioisomer drawn and are reported with respect to 2a.
Reaction run at 80 °C;
Reaction run at 115 °C.
First, substrates containing O atom donors (1a–1h) were tested. Substrates that can form five-membered chelates upon second insertion such as 1a, vinyl methyl ether 1b, and homopropargylic ethers 1c and 1d are excellent directing groups with the more Lewis acidic catalyst THF3TiI2(NPh) (Condition A), giving high regioselectivity and yields of pyrroles 3a–3d. Directing-group effects are observed whether the 2-C linker is sp2 (1a, 1b) or sp3 (1c, 1d) hybridized. Substrates 1b–1d are significantly less selective with the less Lewis acidic catalyst [py2TiCl2(NPh)]2, although in the case of 1d, acceptable regioselectivity for 3d is still obtained. Phenyl ether 1h, which also contains a 2-C linker, manifests no directing-group effect—presumably due to a combination of the weaker aryl ether donor O and increased rotational degrees of freedom with respect to 1a.
In contrast to 2-C linkers, shorter (1-C) linkers such as propargylic ethers (1e) and acetals (1f) do not yield productive reactivity due to C–O bond decomposition.40 Longer (3-C) linkers (1g) display no directing-group effect, but are still competent for heterocoupling to the 2-unsubstituted pyrrole (4g).
Next, other Lewis basic heteroatom donors were examined. Arenes with an ortho –SMe (1i) or –NMe2 (1j) couple with high regioselectivity to the directed products 3i and 3j. The less basic and softer –SMe group (1i) directed to higher regioselectivity (20:1) than harder –OMe (1a, 9.3:1) and –NMe2 (1j, 8.0:1), which is unexpected due to the preference of TiIV for harder ligands. Given the sensitivity to chelate size observed with O atom donors, the increased regioselectivity in 1i may be a function of the S atomic size and resulting chelate angles of the S-containing ring.
Tertiary aryl amine 1j displays good directing ability, although aliphatic pendent tertiary amines (1l, 1n) do not. This may be due to an increase in tether flexibility coupled with the sterics of the 3° amine. Nonetheless, these substrates do not inhibit catalysis and are selective for heterocoupling to products 4l and 4n. Aryl fluorides (1k), alkyl thioethers (1m), and thioacetals (1o) give high yields of the nondirected products 4k, 4m, and 4o with [py2TiCl2(NPh)]2, which is more functional-group tolerant than THF3TiI2(NPh) owing to reduced Lewis acidity.
Heterocyclic substituent reactivity is strongly dependent on the heterocycle basicity.41 N-Methylimidazole (1s) is too basic and inhibits catalysis for both THF3TiI2(NPh) and [py2TiCl2(NPh)]2 reactions through coordination to Ti. Pyridine (1p), which is a reasonably strong heterocyclic base but less basic than 1s, inhibits catalysis with the more Lewis acidic THF3TiI2(NPh), but acts as a directing group and yields high regioselectivity (19:1) with [py2TiCl2(NPh)]2. Thiazole (1q), which is less basic than pyridine, acts as a selective directing group (5.2:1) with THF3TiI2(NPh), but not with the less Lewis-acidic [py2TiCl2(NPh)]2 (0.5:1). Benzoxazole (1r), which is similar to thiazole 1q in basicity, yields similar selectivities but poor yields due to C–O bond cleavage, similar to other propargylic O-functional groups (1e, 1f). Very weakly basic furan (1t) and thiophene (1u) pendants are not strong enough to serve as directing groups with either catalyst but still yield normal heterocoupled products 4t and 4u.
Internal alkyne 1w is also suitable for directed heterocoupling, giving 3w in moderate yield (Figure 3). This takes advantage of the extreme sensitivity of [2+2] cycloaddition to substrate sterics as well as the directing-group effect in second alkyne insertion. However, the window for success is narrow: less sterically encumbered alkynes (1v) predominantly homocouple due to their higher reactivity, while more sterically encumbered internal alkynes (1x) are too hindered even for second insertion and thus yield mainly pyrrole products of PhCCMe homocoupling.
Figure 3.
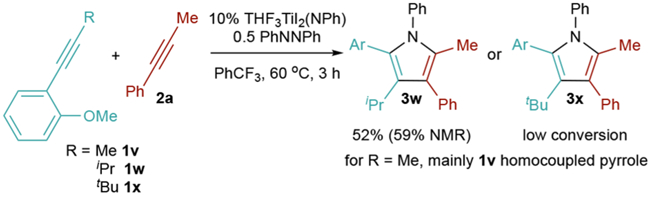
Directing-group effects on internal alkyne heterocoupling.
Other internal alkynes can also serve as [2+2] partners along with directing-group-tethered TMS alkynes. For example, reaction of 3-hexyne (2b) maintains good chemo- and regioselectivity to form 3ab, albeit with a longer reaction time (Figure 4, top). Additionally, internal alkynes with appended directing groups such as 1v—which typically would undergo rapid homocoupling (vide supra)—can be heterocoupled with TMS alkynes with pendent strong directing groups such as 1p to form products such as 3pv (Figure 4, bottom). Unfortunately, all heterocoupling attempts with terminal alkynes have failed; these alkynes appear to be too reactive and instead undergo either alkyne trimerization or pyrrole formation via homocoupling.
Figure 4.
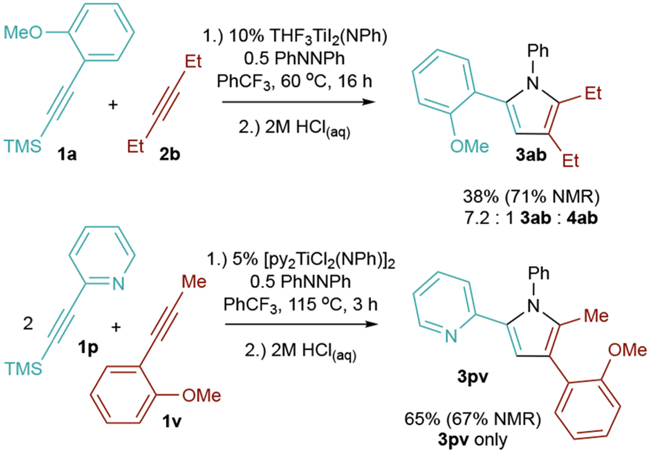
Directed heterocoupling with other internal alkyne [2+2] partners.
[2+2+1] directed heterocoupling can enable facile synthesis of pyrrole-containing EP1 antagonist derivatives (Figure 5).3,42–44 These moieties are typically synthesized through classical Paal-Knorr syntheses, and require multistep pyrrole construction followed by postfunctionalization of the pyrrole core to introduce more intricate substitution patterns42,43 such as the 2-methyl-1,3,5-triaryl core of 5y. Benzyl aryl ethers like that in 5y do not serve as directing groups in the [2+2+1] reaction, but o-Me derivative 1y is a deprotectable surrogate. Directed [2+2+1] heterocoupling of 1y with 2a and (p-BrPh)2N2 gives methoxy-protected 3y, which can be deprotected to phenol 4y prior to benzylation to 5y in 22% overall yield (Figure 5). The advantage of this protocol is that the [2+2+1] reaction is widely tolerant of substitution on both azobenzene7 and alkyne8 fragments, allowing access to diverse structures with relative ease.
Figure 5.
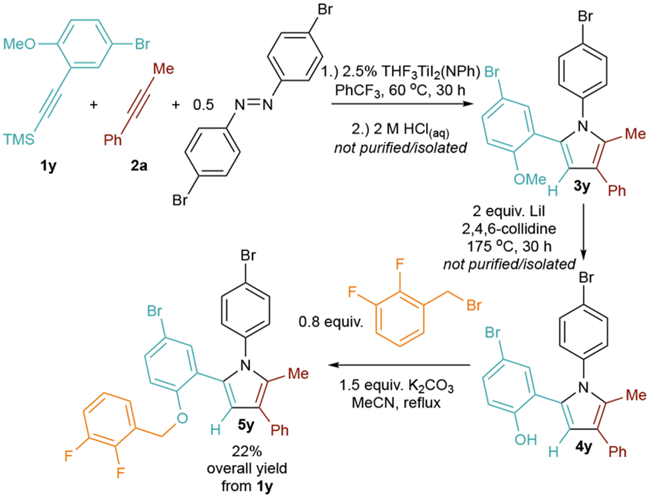
Synthesis of an EP1 antagonist derivative 5y.
In addition to alkyne insertion, directing-group effects can also be seen during the [2+2] cycloaddition step (Figure 6). For example, reaction of 1z under catalytic conditions results in exclusive formation of 6z. In contrast, the reaction of 1-phenylpropyne gives a mixture of all 3 possible regioisomers.5,7,45–47 Since 1z has little steric or electronic bias for [2+2] cycloaddition or insertion, it is likely that the –OMe group directs the [2+2] cycloaddition. Similarly, reaction of 1v results exclusively in formation of 6v.
Figure 6.
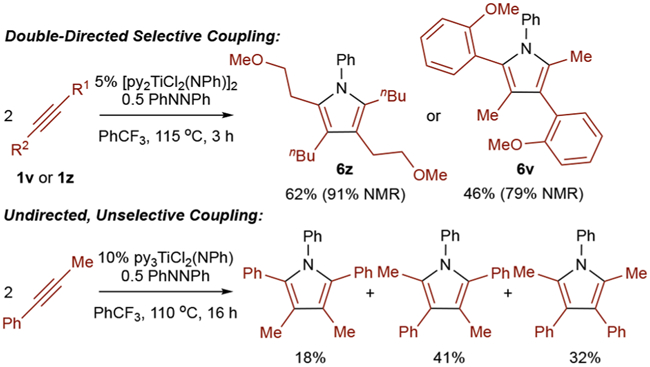
Substrate-directed regioselective pyrrole formation via alkyne homocoupling.
Having established that directing groups can influence the regiochemistry of [2+2] cycloaddition, the possibility of performing substrate-directed Ti-catalyzed alkyne hydroamination was explored.48 Reaction of electronically unbiased substrate 1z with aniline resulted in selective formation of 7z, after directed [2+2] cycloaddition and protonolysis of the metallacycle by aniline (Figure 7). This reaction represents the first example of Ti-catalyzed directed alkyne hydroamination, and it yields electronically unbiased dissymmetric ketones that are difficult to synthesize selectively via Wacker oxidation of internal alkenes.49–54 It is notable that weakly Lewis-basic dialkyl ether of 1z is capable of directing selective alkyne [2+2] cycloaddition despite stoichiometric quantities of aniline.
Figure 7.
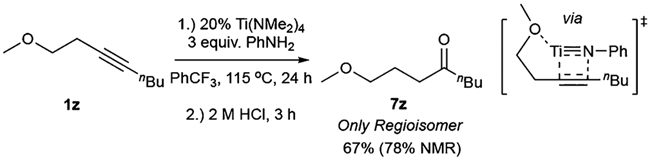
Substrate-directed regioselective alkyne hydroamination.
CONCLUSIONS
In summary, we have shown that TMS-protected alkynes with pendent Lewis basic groups can invert the regioselectivity of TMS-protected alkyne insertion, leading to selective formation of highly substituted 3-TMS pyrroles. This method provides a complementary approach to regioselective [2+2+1] pyrrole formation and also demonstrates how dative substrate—Ti interactions can influence—and, importantly, tune—regioselectivity through directing-group effects, which was previously unexplored in Ti catalysis.
The inverse regioselectivity highly depends on the coordinating strength of the functional groups, the tether length, and the Lewis acidity of the Ti catalyst. These effects work in concert, providing a high degree of tunability of the pyrrole regiochemistry. Balancing the directing-group Lewis basicity with the catalyst Lewis acidity is critical for catalysis: too strong of an acid–base pairing inhibits catalysis, while too weak of a pairing results in formation of 2-TMS pyrrole 4 instead of the 3-TMS pyrrole 3. Further, we have shown that dative directing-group effects can also play a role in other Ti-catalyzed reactions such as alkyne hydroamination, which leads to the selective formation of electronically unbiased imines/ketones via directed [2+2] alkyne/Ti═NR cycloaddition. This work will provide a basis for the development of other, selective Ti-catalyzed reactions that can exploit transient dative interactions, expanding directed Ti catalysis beyond classical alkoxide directing groups.
EXPERIMENTAL SECTION
General Considerations.
All air- and moisture-sensitive compounds were manipulated in a glovebox under a nitrogen atmosphere. The solvent, PhCF3, was predried in a Pure Process Technology Solvent System and passed through activated basic alumina before being used. All high-boiling liquid reagents were freeze pump-thawed three times, brought into the glovebox, diluted in hexanes or diethyl ether, passed through activated basic alumina, pumped dry in vacuo and checked by 1H NMR in CDCl3 to ensure the dryness. [(py)2TiCl2NPh)]2,55 (THF)3TiI2(NPh)8, ((2-methoxyphenyl)ethynyl)trimethylsilane (1a), 2-((trimethylsilyl)ethynyl)pyridine (1p)8, trimethyl(thiophen-2-ylethynyl)silane (1u)8, 1-methoxy-2-(prop-1-yn-1-yl)benzene (1v)8, and (E)-1,2-bis(4-bromophenyl)diazene5 were prepared according to reported procedure. 1-Phenyl-1-propyne (2a) and 3-hexyne (2b) were purchased from Sigma-Aldrich. Azobenzene was purchased from TCI Chemicals and purified by dissolving it in hexanes and washing with water. The organic layer was collected, dried over Na2SO4, and the solvent was removed in vacuo.
1H, 13C, 1H–13C and 1H–15N HMBC, NOESY, and No-D 1H NMR spectra were collected on Bruker Avance III HD NanoBay 400 MHz, Bruker Avance III HD 500 MHz, or Varian Inova 500 MHz spectrometers. High-resolution mass spectra were collected on Agilent 7200 GC/QTOF-MS, Bruker BioTOF II ESI/TOF-MS, or AB-Sciex 4800 MALDI/TOF-MS. Chemical shifts are reported with references of residual protio-solvent impurity: 1H (s, 7.16 ppm for C6D5H; s, 7.27 for ppm of CHCl3), 13C (t, 128.39 ppm for C6D6; t, 77.23 ppm for CDCl3). No-D NMR spectrum were referenced to the proton signal of the internal standard triphenylmethane (Ph3CH, ppm = 5.0) at a delay time = 30 s and acquisition time = 5 s.
Example Reaction Condition Optimization (Table 1).
Example for Entry 2.
In a glovebox, to an NMR tube was added PhNNPh (8.2 mg, 0.045 mmol, 0.5 equiv), (THF)3I2TiNPh (6.1 mg, 0.01 mmol, 0.11 equiv), ((2-methoxyphenyl)ethynyl)trimethylsilane (1a) (20.4 mg, 0.1 mmol, 1.1 equiv), phenylpropyne (2a) (11.6 mg, 0.1 mmol, 1.1 equiv), triphenylmethane (4.9 mg, 0.02 mmol, 0.22 equiv) as the internal standard, and 0.5 mL of PhCF3. The NMR tube was sealed and brought out of the glovebox, and a t = 0 h No-D 1H NMR spectrum was taken. The NMR tube was heated at 115 °C in an oil bath. No-D 1H NMR spectra were taken every 15 min until the full consumption of PhNNPh. Yields are reported with respect to 2a.
Example Synthesis and Isolation of Multisubstituted Pyrroles via Heterocoupling Reactions (Table 2, Condition A).
Example for ((2-Methoxyphenyl)ethynyl)- trimethylsilane (1a).
In a glovebox, to a 7 mL vial was added PhNNPh (41.0 mg, 0.225 mmol, 0.5 equiv), (THF)3I2TiNPh (30.5 mg, 0.05 mmol, 0.11 equiv), ((2-methoxyphenyl)-ethynyl)trimethylsilane (1a) (102 mg, 0.5 mmol, 1.1 equiv), phenylpropyne (2a) (58.0 mg, 0.5 mmol, 1.1 equiv), and 2.5 mL of PhCF3. The reaction was then sealed with a Teflon screw cap and heated at 60 °C outside of the glovebox in an oil bath for 2 h. The reaction was diluted by 2 mL of ethyl acetate, quenched by 2 mL of 2 M HCl in methanol for 1 h, washed with DI water, dried over Na2SO4, and concentrated prior to purification. The product 3a was isolated by silica gel chromatography using 5% EA/Hex eluent to give a paleyellow oil. (120 mg, 64% corrected yield). Yields are reported with respect to 2a.
Example Synthesis and Isolation of Multisubstituted Pyrroles via Heterocoupling Reactions (Table 2, Condition B).
Example for (5-(Benzyloxy)pent-1-yn-1-yl)- trimethylsilane (1g).
In a glovebox, to a 7 mL vial was added PhNNPh (41.0 mg, 0.225 mmol, 0.5 equiv), [(py)2Cl2TiNPh]2 (18.4 mg, 0.025 mmol, 0.055 equiv), (5-(benzyloxy)pent-1-yn-1-yl)trimethylsilane (1g) (246 mg, 0.1 mmol, 2.2 equiv), phenylpropyne (2a) (58.0 mg, 0.5 mmol, 1.1 equiv), and 2.5 mL of PhCF3. The reaction was then sealed with a Teflon screw cap and heated at 115 °C outside of the glovebox in an oil bath for 3 h. The reaction was diluted by 2 mL of ethyl acetate, quenched by 2 mL of 2 M HCl in methanol for 1 h, washed with DI water, dried over Na2SO4, and concentrated prior to the purification.The product 4g was isolated by silica gel chromatography using 2% EA/Hex eluent to give a colorless oil (166 mg, 87% yield). Yields are reported with respect to 2a.
Example No-D 1H NMR Determination of Pyrrole Regioisomeric Ratios (Table 2, Condition A).
Example for ((2-Methoxyphenyl)ethynyl)trimethylsilane (1a).
In a glovebox, to an NMR tube was added PhNNPh (8.2 mg, 0.045 mmol, 0.5 equiv), (THF)3I2TiNPh (6.1 mg, 0.01 mmol, 0.11 equiv), ((2-methoxyphenyl)ethynyl)trimethylsilane (1a) (20.4 mg, 0.1 mmol, 1.1 equiv), phenylpropyne (2a) (11.6 mg, 0.1 mmol, 1.1 equiv), triphenylmethane (4.9 mg, 0.02 mmol, 0.22 equiv) as the internal standard, and 0.5 mL of PhCF3. The NMR tube was sealed and brought out of the glovebox, and a t = 0 h No-D 1H NMR spectrum was taken. The NMR tube was heated at 60 °C in an oil bath for 2 h. The NMR tube was then cooled to ambient temperature, and a t = 2 h No-D 1H NMR spectrum was taken. Yields are reported with respect to 2a.
Example No-D 1H NMR Determination of Pyrrole Regioisomeric Ratios (Table 2, Condition B).
Example for (5-(Benzyloxy)pent-1-yn-1-yl)trimethylsilane (1g).
In a glovebox, to an NMR tube was added PhNNPh (8.2 mg, 0.045 mmol, 0.5 equiv), [(py)2Cl2TiNPh]2 (6.1 mg, 0.005 mmol, 0.055 equiv), (5-(benzyloxy)pent-1-yn-1-yl)trimethylsilane (1g) (49.2 mg, 0.2 mmol, 2.2 equiv), phenylpropyne (2a) (11.6 mg, 0.1 mmol, 1.1 equiv), triphenylmethane (4.9 mg, 0.02 mmol, 0.22 equiv) as the internal standard, and 0.5 mL of PhCF3. The NMR tube was sealed and brought out of the glovebox, and a t = 0 h No-D 1H NMR spectrum was taken. The NMR tube was heated at 115 °C in an oil bath for 3 h. The NMR tube was then cooled to ambient temperature, and a t = 3 h No-D 1H NMR spectrum was taken. Yields are reported with respect to 2a.
Synthesis of 5-(5-Bromo-2-((2,3-difluorobenzyl)oxy)- phenyl)-1-(4-bromophenyl)-2-methyl-3-phenyl-1H-pyrrole (5y).
Reaction of 1y with 2a to form 3y.
In a glovebox, to a 20 mL of vial was added 283 mg of 1y (1 eq., 1 mmol), 116 mg of 2a (1 eq., 1 mmol), 166 mg of (E)-1,2-bis(4-bromophenyl)diazene (0.5 eq., 0.5 mmol), 15 mg of (THF)3I2TiNPh (2.5%, 0.025 mmol), and 5 mL of PhCF3. The reaction was then sealed with a Teflon screw cap and heated in the glovebox for 30 h at 60 °C. After being cooled down to ambient temperature, the reaction was removed from the glovebox, 10 mL of hexanes was added to the reaction, and the mixture was filtered through a fine frit. The filtrate was concentrated in vacuo and mixed with 10 mL of 2 M HCl in methanol. After the mixture was stirred for 2 h, it was diluted by 20 mL of DI water and extracted by hexanes (15 mL × 3). The organic layer was washed with brine, dried by MgSO4, and concentrated in vacuo to get a yellow solid, 3y, that was carried on in the reaction as-is.
Deprotection of 3y to 4y.
In a glovebox, the entirety of 3y was dissolved in 2 mL of 2,4,6-collidine and transferred to a 7 mL vial, followed by the addition of 266 mg LiI (2 eq., 2 mmol). The vial was sealed and heated outside of the glovebox at 175 °C for 30 h. The reaction was diluted by 20 mL of methanol and 20 mL of DI water and then acidified by 20 mL of 2 M HCl in methanol. The mixture was extracted by hexanes (15 mL × 6), and the organic layer was washed with brine and dried by MgSO4. Removal of the volatiles yielded the crude oily product 4y, which was directly used in the next step.
Benzylation of 4y to 5y.
Next, 0.2 g of K2CO3 (1.44 equiv, 1.44 mmol), 0.1 mL 2,3-difluorobenzyl bromide (0.78 equiv, 0.78 mmol) and 3 mL MeCN were added. The mixture was stirred at reflux for 3 h. The mixture was diluted by 10 mL of DI water and extracted by hexanes (20 mL × 3). The collected organic layer was washed with brine, dried by MgSO4, and concentrated. The only purification of the final product was carried out via silica gel chromatography using 12% DCM/Hex eluent to give a white solid 5y (135 mg, 22% overall yield with respect to 1y). 1H NMR (400 MHz, C6D6): δ 7.62–7.60 (m,2H), 7.35 (t, 3JHH =7.8 Hz, 2H), 7.24 (dd, 3JHH = 7.5 Hz, 4JHH = 1.7 Hz, 1H), 7.18–7.15 (m, 1H), 6.97–6.93 (m, 2H), 6.90 (td, 3JHH = 7.9 Hz, 4JHH = 1.7 Hz, 1H), 6.77–6.71 (m,3H), 6.65–6.58 (m, 3H), 6.54 (tdd, 3JHH = 7.9, 5.0 Hz, 4JHH = 1.4 Hz, 1H), 6.39 (d, 3JHH = 8.0 Hz, 1H), 4.57 (s, 2H, –O–CH2–Ar), 2.08 (s, 3H, 2-NC4–CH3) ppm. 13C NMR (101 MHz, C6D6): δ 156.0, 150.7 (dd, 1JCF = 249 Hz, 2JCF = 12.4 Hz), 148.3 (dd, 1JCF = 249 Hz, 2JCF = 13.2 Hz), 138.9, 137.8, 132.3, 131.9, 130.3, 129.8, 128.9, 128.9, 128.6, 127.5 (d, 3JCF = 10.9 Hz), 126.6, 125.9, 124.4 (dd, 2JCF = 6.7 Hz, 3JCF = 4.7 Hz), 124.0 (app t, 2,3JCF = 3.1 Hz), 123.8 (d, 3JCF = 1.1 Hz), 121.5, 120.8, 116.6, 116.4, 113.2, 111.8, 63.6 (dd, 3JCF = 4.5 Hz, 4JCF = 3.0 Hz), 12.7 ppm. MALDI-HRMS: calcd for C30H21NOBr2F2 [M-Br+], 528.0775; found, 528.0305.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by the National Institutes of Health (1R35GM119457) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow). Equipment for the Chemistry Department NMR facility was supported through a grant from the National Institutes of Health (S10OD011952) with matching funds from the University of Minnesota.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b04669.
Full experimental procedures, characterization data, and NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Fan H; Peng J; Hamann MT; Hu J-F Lamellarins and Related Pyrrole-Derived Alkaloids from Marine Organisms. Chem. Rev 2008, 108, 264–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Baumann M; Baxendale IR; Ley SV; Nikbin N An Overview of the Key Routes to the Best Selling 5-Membered Ring Heterocyclic Pharmaceuticals. Beilstein J. Org. Chem 2011, 7, 442–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bhardwaj V; Gumber D; Abbot V; Dhiman S; Sharma P Pyrrole: A Resourceful Small Molecule in Key Medicinal Hetero-Aromatics. RSC Adv 2015, 5, 15233–15266. [Google Scholar]
- (4).Khajuria R; Dham S; Kapoor KK Active Methylenes in the Synthesis of a Pyrrole Motif: An Imperative Structural Unit of Pharmaceuticals, Natural Products and Optoelectronic Materials. RSC Adv. 2016, 6, 37039–37066. [Google Scholar]
- (5).Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nat. Chem 2016, 8, 63–68. [DOI] [PubMed] [Google Scholar]
- (6).Davis-Gilbert ZW; Tonks IA Titanium Redox Catalysis: Insights and Applications of an Earth-abundant Base Metal. Dalton Trans. 2017, 46, 11522–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2+2+1] Pyrrole Synthesis from Alkynes and Azobenzene. J. Am. Chem. Soc 2018, 140, 7267–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem. Int. Ed 2018, 57, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pearce AJ; See XY; Tonks IA Oxidative Nitrene Transfer from Azides to Alkynes via Ti(ii)/Ti(iv) Redox Catalysis: Formal [2+2+1] Synthesis of Pyrroles. Chem. Commun 2018, 54, 6891–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fager-Jokela E; Muuronen M; Patzschke M; Helaja J Electronic Regioselectivity of Diarylalkynes in Cobalt-Mediated Pauson–Khand Reaction: An Experimental and Computational Study with Para- and Meta-Substituted Diarylalkynes and Norbornene. J. Org. Chem 2012, 77, 9134–9147. [DOI] [PubMed] [Google Scholar]
- (11).Fager-Jokela E; Muuronen M; Khaizourane H; Vázquez-Romero A; Verdaguer X; Riera A; Helaja J Regioselectivity of Intermolecular Pauson–Khand Reaction of Aliphatic Alkynes: Experimental and Theoretical Study of the Effect of Alkyne Polarization. J. Org. Chem 2014, 79, 10999–11010. [DOI] [PubMed] [Google Scholar]
- (12).Stockis A; Hoffmann R Metallacyclopentanes and Bisolefin Complexes. J. Am. Chem. Soc 1980, 102, 2952–2962. [Google Scholar]
- (13).Skibbe V; Erker G Zur Bildung Metallacyclischer Fünfringverbindungen: Regioselektivität der Addition von Alkinen an Zirconocenkomplexe. J. Organomet. Chem 1983, 241, 15–26. [Google Scholar]
- (14).Negishi E; Holmes SJ; Tour JM; Miller JA; Cederbaum FE; Swanson DR; Takahashi T Novel Bicyclization of Enynes and Diynes Promoted by Zirconocene Derivatives and Conversion of Zirconabicycles into Bicyclic Enones via Carbonylation. J. Am. Chem. Soc 1989, 111, 3336–3346. [Google Scholar]
- (15).Lefeber C; Ohff A; Tillack A; Baumann W; Kempe R; Burlakov VV; Rosenthal U Darstellung und Regioselektive Reaktionen des Phosphinfreien Zirconocen-Alkin-Komplexes Cp2Zr-(THF)(tBuC2SiMe3). J. Organomet. Chem 1995, 501, 189–194. [Google Scholar]
- (16).List AK; Koo K; Rheingold AL; Hillhouse GL Preparation of Trimethylsilyl Alkyne Complexes of Bis-(pentamethylcyclopentadienyl)zirconium, (η5-C5Me5)2Zr-(RC≡CSiMe3), and Their Regioselective Reactions with Nitrous Oxide. Inorg. Chim. Acta 1998, 270, 399–404. [Google Scholar]
- (17).Gao Y; Shirai M; Sato F Synthesis of Substituted Pyrroles from an Alkyne, an Imine and Carbon Monoxide via an Organotitanium Intermediate. Tetrahedron Lett. 1996, 37, 7787–7790. [Google Scholar]
- (18).Mizoguchi H; Micalizio GC Synthesis of Angularly Substituted trans-Fused Decalins through a Metallacycle-Mediated Annulative Cross-Coupling Cascade. Angew. Chem. Int. Ed 2016, 55, 13099–13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jeso V; Aquino C; Cheng X; Mizoguchi H; Nakashige M; Micalizio GC Synthesis of Angularly Substituted Trans-Fused Hydroindanes by Convergent Coupling of Acyclic Precursors. J. Am. Chem. Soc 2014, 136, 8209–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Krafft ME Regiocontrol in the Intermolecular Cobalt-Catalyzed Olefin-Acetylene Cycloaddition. J. Am. Chem. Soc 1988, 110, 968–970. [Google Scholar]
- (21).Krafft ME; Juliano CA; Scott IL; Wright C; McEachin MD The Directed Pauson-Khand Reaction. J. Am. Chem. Soc 1991, 113, 1693–1703. [Google Scholar]
- (22).Krafft ME; Juliano CA Studies on the Use of Bidentate Ligands in the Directed Pauson-Khand Reaction. J. Org. Chem 1992, 57, 5106–5115. [Google Scholar]
- (23).Krafft ME; Wright C A Formal Total Synthesis of PGA2 Using the Directed Pauson-Khand Reaction. Tetrahedron Lett. 1992, 33, 151–152. [Google Scholar]
- (24).Itami K; Mitsudo K; Yoshida J. i. A Pyridylsilyl Group Expands the Scope of Catalytic Intermolecular Pauson–Khand Reactions. Angew. Chem., Int. Ed 2002, 41, 3481–3484. [DOI] [PubMed] [Google Scholar]
- (25).Itami K; Mitsudo K; Fujita K; Ohashi Y; Yoshida J.-i. Catalytic Intermolecular Pauson–Khand-Type Reaction: Strong Directing Effect of Pyridylsilyl and Pyrimidylsilyl Groups and Isolation of Ru Complexes Relevant to Catalytic Reaction. J. Am. Chem. Soc 2004, 126, 11058–11066. [DOI] [PubMed] [Google Scholar]
- (26).Hoveyda AH; Evans DA; Fu GC Substrate-Directable Chemical Reactions. Chem. Rev 1993, 93, 1307–1370. [Google Scholar]
- (27).Gao Y; Klunder JM; Hanson RM; Masamune H; Ko SY; Sharpless KB Catalytic Asymmetric Epoxidation and Kinetic Resolution: Modified Procedures Including in situ Derivatization. J. Am. Chem. Soc 1987, 109, 5765–5780. [Google Scholar]
- (28).Ryan J; Micalizio GC An Alkoxide-Directed Carbometalation of Internal Alkynes. J. Am. Chem. Soc 2006, 128, 2764–2765. [DOI] [PubMed] [Google Scholar]
- (29).Micalizio GC; Mizoguchi H The Development of Alkoxide-Directed Metallacycle-Mediated Annulative Cross-Coupling Chemistry. Isr. J. Chem 2017, 57, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Umemura S; McLaughlin M; Micalizio GC Convergent Synthesis of Stereodefined exo-Alkylideneγ-Lactams from β-Halo Allylic Alcohols. Org. Lett 2009, 11, 5402–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McLaughlin M; Takahashi M; Micalizio GC An Alkoxide-Directed Intermolecular [2+2+1] Annulation: A Three-Component Coupling Reaction for the Synthesis of Tetrasubstituted αβ-Unsaturated γ-Lactams. Angew. Chem. Int. Ed 2007, 46, 3912–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hu DX; Seidl FJ; Bucher C; Burns NZ Catalytic Chemo-, Regio-, and Enantioselective Bromochlorination of Allylic Alcohols. J. Am. Chem. Soc 2015, 137, 3795–3798. [DOI] [PubMed] [Google Scholar]
- (33).Landry ML; Hu DX; McKenna GM; Burns NZ Catalytic Enantioselective Dihalogenation and the Selective Synthesis of (–)-Deschloromytilipin A and (–)-Danicalipin A. J. Am. Chem. Soc 2016, 138, 5150–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Landry ML; Burns NZ Catalytic Enantioselective Dihalogenation in Total Synthesis. Acc. Chem. Res 2018, 51, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Seidl FJ; Min C; Lopez JA; Burns NZ Catalytic Regio- and Enantioselective Haloazidation of Allylic Alcohols. J. Am. Chem. Soc 2018, 140, 15646–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yang D; Micalizio GC A Convergent Stereoselective Synthesis of Quinolizidines and Indolizidines: Chemoselective Coupling of 2-Hydroxymethyl-Substituted Allylic Silanes with Imines. J. Am. Chem. Soc 2009, 131, 17548–17549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen MZ; Micalizio GC Three-Component Coupling Sequence for the Regiospecific Synthesis of Substituted Pyridines. J. Am. Chem. Soc 2012, 134, 1352–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Billow BS; McDaniel TJ; Odom AL Quantifying Ligand Effects in High-Oxidation-State Metal Catalysis. Nat. Chem 2017, 9, 837. [DOI] [PubMed] [Google Scholar]
- (39).DiFranco SA; Maciulis NA; Staples RJ; Batrice RJ; Odom AL Evaluation of Donor and Steric Properties of Anionic Ligands on High Valent Transition Metals. Inorg. Chem 2012, 51, 1187–1200. [DOI] [PubMed] [Google Scholar]
- (40).Okamoto S Synthetic Reactions Using Low-valent Titanium Reagents Derived from Ti(OR)4 or CpTiX3 (X = O-i-Pr or Cl) in the Presence of Me3SiCl and Mg. Chem. Rec 2016, 16, 857–872. [DOI] [PubMed] [Google Scholar]
- (41).Lide DR CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, 2005. [Google Scholar]
- (42).Hall A; Atkinson S; Brown SH; Chessell IP; Chowdhury A; Clayton NM; Coleman T; Giblin GMP; Gleave RJ; Hammond B; Healy MP; Johnson MR; Michel AD; Naylor A; Novelli R; Spalding DJ; Tang SP Structure–Activity Relationships of 1,5-Biaryl Pyrroles as EP1 Receptor Antagonists. Bioorg. Med. Chem. Lett 2006, 16, 3657–3662. [DOI] [PubMed] [Google Scholar]
- (43).Hall A; Atkinson S; Brown SH; Chessell IP; Chowdhury A; Giblin GMP; Goldsmith P; Healy MP; Jandu KS; Johnson MR; Michel AD; Naylor A; Sweeting JA Discovery of Novel, Non-Acidic 1,5-Biaryl Pyrrole EP1 Receptor Antagonists. Bioorg. Med. Chem. Lett 2007, 17, 1200–1205. [DOI] [PubMed] [Google Scholar]
- (44).Hall A; Brown SH; Chessell IP; Chowdhury A; Clayton NM; Coleman T; Giblin GMP; Hammond B; Healy MP; Johnson MR; Metcalf A; Michel AD; Naylor A; Novelli R; Spalding DJ; Sweeting J 1,5-Biaryl Pyrrole Derivatives as EP1 Receptor Antagonists: Structure–Activity Relationships of 4- and 5-Substituted Benzoic Acid Derivatives. Bioorg. Med. Chem. Lett 2007, 17, 732–735. [DOI] [PubMed] [Google Scholar]
- (45).Haak E; Bytschkov I; Doye S Intermolecular Hydroamination of Alkynes Catalyzed by Dimethyltitanocene. Angew. Chem., Int. Ed 1999, 38, 3389–3391. [PubMed] [Google Scholar]
- (46).Khedkar V; Tillack A; Beller M A Dramatic Effect of Aryloxo Ligands on the Titanium-Catalyzed Hydroamination of Alkynes. Org. Lett 2003, 5, 4767–4770. [DOI] [PubMed] [Google Scholar]
- (47).Ochi N; Nakao Y; Sato H; Sakaki S {2+2} Cycloaddition of Alkyne with Titanium–Imido Complex: Theoretical Study of Determining Factor of Reactivity and Regioselectivity. J. Phys. Chem. A 2010, 114, 659–665. [DOI] [PubMed] [Google Scholar]
- (48).Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- (49).Zhu C; Cook SP A Concise Synthesis of (+)-Artemisinin. J. Am. Chem. Soc 2012, 134, 13577–13579. [DOI] [PubMed] [Google Scholar]
- (50).DeLuca RJ; Edwards JL; Steffens LD; Michel BW; Qiao X; Zhu C; Cook SP; Sigman MS Wacker-Type Oxidation of Internal Alkenes using Pd(Quinox) and TBHP. J. Org. Chem 2013, 78, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Morandi B; Wickens ZK; Grubbs RH Practical and General Palladium-Catalyzed Synthesis of Ketones from Internal Olefins. Angew. Chem., Int. Ed 2013, 52, 2944–2948. [DOI] [PubMed] [Google Scholar]
- (52).Morandi B; Wickens ZK; Grubbs RH Regioselective Wacker Oxidation of Internal Alkenes: Rapid Access to Functionalized Ketones Facilitated by Cross-Metathesis. Angew. Chem., Int. Ed 2013, 52, 9751–9754. [DOI] [PubMed] [Google Scholar]
- (53).Lerch MM; Morandi B; Wickens ZK; Grubbs RH Rapid Access to β-Trifluoromethyl-Substituted Ketones: Harnessing Inductive Effects in Wacker-Type Oxidations of Internal Alkenes. Angew. Chem., Int. Ed 2014, 53, 8654–8658. [DOI] [PubMed] [Google Scholar]
- (54).Carlson AS; Calcanas C; Brunner RM; Topczewski JJ Regiocontrolled Wacker Oxidation of Cinnamyl Azides. Org. Lett 2018, 20, 1604–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).See XY; Beaumier EP; Davis-Gilbert ZW; Dunn PL; Larsen JA; Pearce AJ; Wheeler TA; Tonks IA Generation of TiII Alkyne Trimerization Catalysts in the Absence of Strong Metal Reductants. Organometallics 2017, 36, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.