

Abstract
A method for the catalytic, enantioselective, intermolecular, 1,2-sulfenoamination of alkenes is described. Functionalization is achieved through the intermediacy of an enantioenriched, configurationally stable thiiranium ion generated by Lewis base activation of a readily available sulfur electrophile. A diverse set of anilines and benzylamines react with different styrenes to afford products in good yield and stereoselectivity. Downstream manipulation of the products is facilitated by deprotonation of the amines to enable carbon–sulfur bond cleavage.
The transformation of simple alkenes into chiral, non-racemic, vicinally functionalized building blocks serves as an important strategy for the production of value added chemicals. In the past 30 years, numerous approaches to alkene difunctionalization such as epoxidation, dihydroxylation, aminohydroxylation and halofunctionalization have been reported.1–5 On the other hand, the sulfenofunctionalization of alkenes proceeding through racemic thiiranium ion intermediates has been known since the early 1960s, but enantioselective variants are significantly less developed. Despite their high reactivity, thiiranium ions are configurationally stable at low temperature and readily intercepted stereospecifically with various nucleophiles to afford anti-sulfenofunctionalized products.6
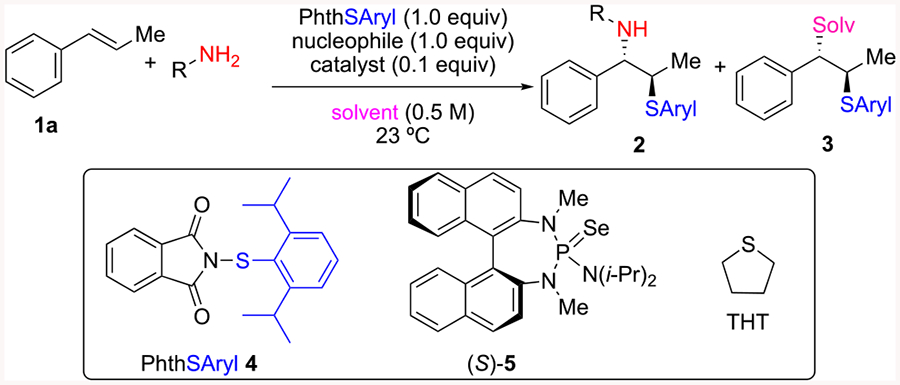
As part of an ongoing program on the enantioselective functionalization of alkenes, we have exploited the activation of group 16 Lewis acids by Lewis bases to access seleno- and sulfenofunctionalized products.7 Association of a Lewis basic selenophosphoramide catalyst 5 and group 16 Lewis acid 4 (“Sulfenylating Agent”) results in a highly electrophilic donor–acceptor complex.8 This complex is intercepted by an unactivated alkene to form an enantioenriched, configurationally stable thiiranium ion which subsequently undergoes stereospecific nucleophilic opening. A number of nucleophiles including alcohols, phenols, amines, anilines, arenes and alkenes serve effectively in intramolecular proceses.9–13 In addition, a recent report from these laboratories demonstrated the sulfenocyclization of polyenes enabled by the use of hexafluoroisopropyl alcohol (HFIP) as the solvent.14 The utility of protic solvents has since been further applied to the carbosulfenylation of alkenylboronates.15 Although intramolecular functionalization has proven successful, previous attempts to achieve intermolecular functionalization with similar nucleophiles have met with limited success largely owing to the use of a strong acid (MsOH) which leads to decomposition of starting material. We reasoned the use of HFIP, which obviates the need for strong acids, could enable successful intermolecular sulfenofunctionalization.
The intermolecular functionalization of thiiranium ions, pioneered in 1982 by Trost, involves formation of a racemic thiiranium ion (generated from dimethyl(methylthio)sulfonium tetrafluoroborate (DMSTF)), which then undergoes invertive opening (Scheme 1A).16 A variety of nitrogen-based nucleophiles afford the sulfenoamination products including amines, azide, and nitrite nucleophiles to provide the amino, azido, and nitro sulfides, respectively. Brownbridge subsequently reported the treatment of alkenes with phenylsulfenamides in dichloromethane to access 1,2-aminothiols in moderate yields and, when in the presence of acetonitrile, the corresponding amidines were formed (Scheme 1B).17 In the intervening years a number of methods have been disclosed to access racemic 1,2-aminothiols including trifluoromethylthioamination of alkenes catalyzed by diaryl selenides reported by Zhao et al. and a highly efficient electrochemical oxy- and aminosulfenylation of alkenes by Yuan et al.18,19
Scheme 1.

Intermolecular Sulfenoamination by Interception of Thiiranium Ions
Indeed, strategies to access enantiomerically enriched 1,2-amino thiols have largely relied on the desymmetrization of aziridines.20 In contrast, the formation and functionalization of enantioenriched thiiranium ions has been significantly less explored. In 1994 Rayner reported the synthesis of benzoxazines by the intramolecular capture of thiiranium ions generated from enantiomerically enriched sulfenyl sulfonium salts.21 Although the products are obtained in acceptable yield, enantioinduction was poor, ostensibly from substrate-mediated background reaction. In the same year, Pasquato et al. disclosed the first enantioselective intermolecular sulfenoamination achieved through an enantioenriched thiiranium ion intermediate (Scheme 1C).22 A stoichiometric quantity of a chiral dinaphtho[2,1-c:1′,2′-e][1,2]dithiin sulfenylating agent produces enantioenriched thiiranium ions which undergo capture with acetonitrile in a Ritter-type process to provide the corresponding sulfeno acetamides in good yield and enantioselectivity. Herein, we report a catalytic, enantioselective, intermolecular sulfenoamination of alkenes enabled by the stereospecific capture of enantioenriched thiiranium ions.
Orienting experiments (Table 1) initially employed HFIP (0.5 M), (E)-2-methylstyrene 1a, sulfenylating agent 48b (1.0 equiv), catalyst (S)-5 (0.1 equiv) or tetrahydrothiophene (THT, 0.1 equiv) and p-tolulenesulfonamide (TsNH2, 1.0 equiv) as the nucleophile. Unfortunately, only the oxysulfenylated product 3 corresponding to solvent incorporation was observed (entry 1). The same result was observed when the comparatively less nucleophilic nonafluoro-tert-butyl alcohol (9F-t-BuOH) was employed (entry 2). The use of mixed solvent systems (dichloromethane/HFIP, dichloromethane/9F-t-BuOH and toluene/9F-t-BuOH) failed to provide the desired products, resulting in the generation of undesired side products (entries 3–5). At this point it was deemed prudent to survey several amines that could intercept the thiiranium ion more readily than HFIP. Nucleophiles such as tert-butyl carbamate and 2-aminobenzothiazole again afforded the oxysulfenylated product (entries 6 and 7). Gratifyingly, p-anisidine was identified as an effective nucleophile and no solvent incorporation was observed (entry 8).
Table 1.
Reaction Optimization
 | |||||
|---|---|---|---|---|---|
| entry | nucleophile | Lewis base | solvent | 2 | 3 |
| 1 | TsNH2 | (S)-5 | HFIP | 0% | 76%a |
| 2 | TsNH2 | (S)-5 | 9F-t-BuOH | 0% | 67%a |
| 3 | TsNH2 | (S)-5 | HFIP/ CH2CI2 | 0% | 0%b |
| 4 | TsNH2 | THT | 9F-t-BuOH/ CH2CI2 | 0% | 0%b |
| 5 | TsNH2 | THT | 9F-t-BuOH/ toluene | 0% | 0%b |
| 6 | 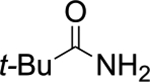 |
THT | HFIP | 0% | 81%c |
| 7 | 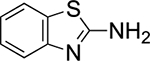 |
THT | HFIP | 0% | 79%c |
| 8 | 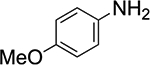 |
(S)-5 | HFIP | 73%a | 0% |
Yield of isolated products.
Starting material remaining and side products were formed.
Yield obtained by 1H NMR analysis against an internal standard.
Reaction conditions were further refined through the use of Design of Experiment software.23 Four reaction factors were examined in this protocol: overall concentration, equivalents of both sulfenylating agent and p-anisidine, as well as catalyst loading. A total of 25 experiments were conducted and the results were analyzed by 1H NMR spectroscopy and a response surface was generated. A slight excess of sulfenylating agent (1.2 equiv) and p-anisidine (1.6 equiv) were required because sulfenylation of the p-anisidine to form the corresponding thiohydroxylamine was identified as a parasitic pathway. Additionally, high concentration was key to minimize the formation of any oxysulfenylated product. Lastly, reactions run for 24 h at ambient temperature maintain high yields with catalyst loading as low as 5 mol %.
Under the optimized conditions, a number of anilines containing both electron rich and electron withdrawing functional groups were evaluated (Table 2). 4-Haloanilines were competent, affording the corresponding aminofunctionalized products 2a–2c as single diastereomers in good yield and 98:2 enantiomeric ratio. Absolute configuration was confirmed by single crystal X-ray diffraction24 of 2c and corresponds to the selectivity models previously described.8 2-Iodoaniline was incorporated in good yield and high enantioselectivity was maintained. Electron-rich anilines were also incorporated giving products 2e and 2f in good yield. Owing to the mild conditions afforded by HFIP, acid-labile protecting groups are compatible as demonstrated by synthesis of pivalate 2g in 84% yield and carbamate 2h in 83% yield. Of note is the ability of electron deficient anilines to outcompete theformation of oxysulfenylated product. 4-Aminoacetopheone and benzocaine were cleanly incorporated to afford products 2i and 2j respectively in good yield. 4-Aminophenylboronic acid pinacol ester provided the desired product 2k in 82% yield and thereby provides an additional functional group for subsequent cross coupling reactions. 2-Aminopyridine was also competent affording 2l in good yield and selectivity.
Table 2.
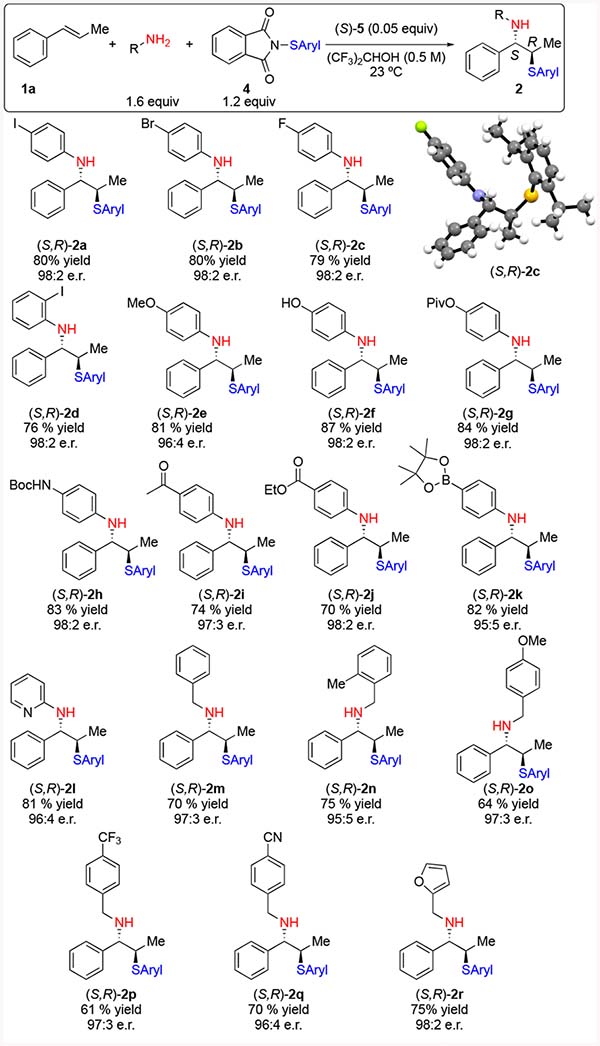
|
All reactions performed on a 1.00 mmol scale.
Yield of isolated, analytically pure product.
Enantiomeric ratio determined by CSP-HPLC (high performance liquid chromatography) or CSP SFC analysis.
The method was extended to a number of benzylic amine nucleophiles. Benzylamine was incorporated efficiently as was 2-methylbenzylamine to give the corresponding products 2m and 2n in 70% and 75% yield respectively with no appreciable loss in enantioselectivity. Benzylamines containing the inductively withdrawing and resonance withdrawing trifluoromethyl and 4-cyano substituents reacted efficiently to give 2p and 2q. 2-Furfurylmethylamine was smoothly incorporated in 75% yield and 98:2 e.r. Incorporation of aliphatic amines was largely unsuccessful owing to their increased Brønsted basicity (leading to protonation under these conditions) resulting in the formation of the oxysulfenylation product exclusively.
With respect to the olefin scope (Table 3), styrene, which had previously given poor results in the presence of strong acid, was readily functionalized in 80% yield and 96:4 e.r. Additionally, products 7b and 7c arising from 2-fluorostyrene and 2-methylstyrene respectively were obtained in synthetically useful yields however, both suffered from slight degradation in selectivity. 3-Methylbutenylbenzene reacted to afford 7d in good yield and selectivity. Attempts to functionalize α-methylstyrene afforded the desired sulfenofunctionalized product, however in racemic form. This outcome likely arises from the intermediacy of a stabilized, open carbocation rather than by stereospecific ring opening of a thiiranium ion. Similarly, anethole is also capable of efficient cation stabilization which might result in a mixture of diastereomers. Gratifyingly, anethole was competent in the reaction providing the desired product 7e in 74% yield and 95:5 e.r. Difunctionalized products containing extended aromatic systems (7f), e.g., N-tosyl substituted indole (7g), heterocycles (7h) and aliphatic alkenes (7i), were all produced in good yield and good to excellent selectivity.
Table 3.
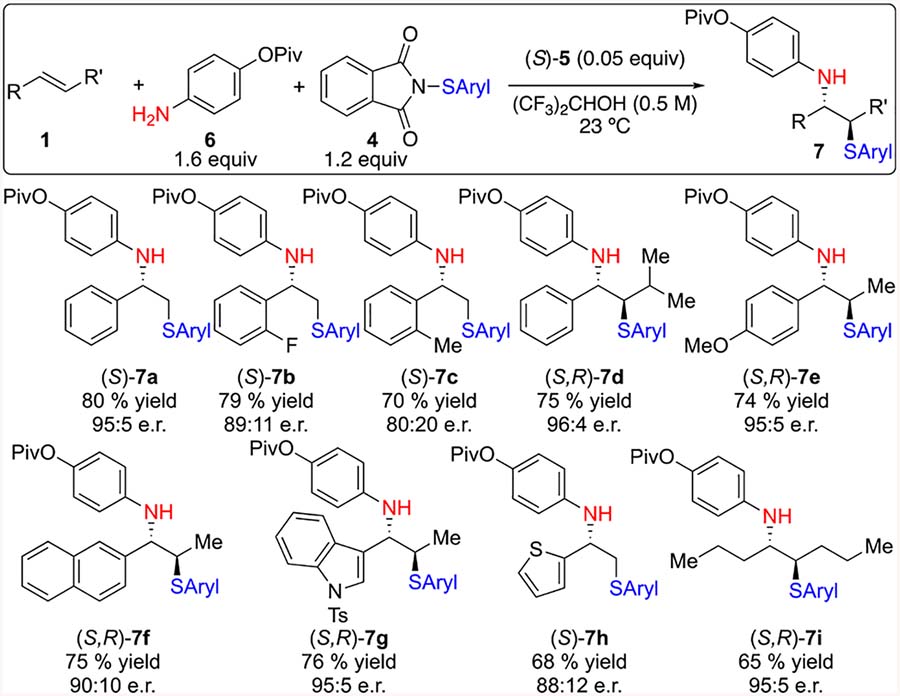
|
All reactions performed on a 1.00 mmol scale.
Yield of isolated, analytically pure product.
Enantiomeric ratio determined by CSP-HPLC or CSP SFC analysis.
A major objective of this project was the subsequent derivatization of the sulfenofunctionalized products. Although the increased steric bulk of the 2,6-diisopropylsulfenylating agent 4 affords superior enantioselectivities, this same property reduces reactivity and complicates subsequent functionalization. Initial attempts to affect C–S cleavage employed a variety of reducing agents that simply returned starting material even under forcing conditions. Treatment with lithium naphthalenide successfully affected C–S cleavage but resulted in spontaneous decomposition of the anion through β-elimination. To address this limitation, we were inspired by a report from Yus and co-workers, which describes the generation of stable β-amido alkyllithium species through a two-step, one-pot protocol by preforming the lithium amide, which serves to prevent β-elimination.25 This strategy has also been employed in the derivatization of β-oxyanionic alkyllithium reagents;26–31 however, to the best of our knowledge, no subsequent reports using this strategy in the derivatization of β-amido alkyllithiums have been disclosed.
By preforming the lithium amide (by deprotonation with n-BuLi) prior to treatment with lithium 4,4′-di-tert-butylbiphenylide (LiDTBB), the β-amido alkyllithium adduct i was successfully converted into a number of functionalized derivatives (Scheme 2). The desulfurized product 8 was formed in 87% yield by quenching the dianion with water whereas the 1,3-amino alcohol 9 was formed as a single diastereomer through capture with dimethylformamide and subsequent reduction with no loss in enantiomeric purity. Reaction of i with acetaldehyde proceeded in good yield to provide 10 as a mixture of C(2) diastereomers whereas treatment with dimethyl sulfate afforded the C-methylated product 11 in 84% yield. Aziridine 12 was accessed via the intermediate syn-2-chlorolithophenyl amide (by reaction with hexachloroethane) and subsequent invertive displacement. Finally, carbon dioxide was captured to afford the syn-amino acid 13, which was next cyclized to the β-lactam 14 after treatment with Mukaiyama’s reagent.32 The relative configurations of these adducts were established by comparison to literature values.33,34 Attempts to form the corresponding piperidine and azepine through capture of 1,3-dibromopropane and 1,4-dibromobutane were unsuccessful as numerous byproducts were observed while exhaustive allylation proceeded in poor yield and afforded the bis-allylated product in 55:45 d.r. Lastly, 15, derived from 4-aminophenol, was cleanly transformed to the primary amine through PIFA-mediated oxidation and subsequent hydrolysis.
Scheme 2.

Product Manipulations
From previous mechanistic investigations, a catalytic cycle is proposed in Figure 1. Activation of sulfenylating agent 4 by protonation with HFIP facilitates sulfur transfer to selenophosphoramide catalyst 5 to form cationic species ii. Subsequent transfer of the sulfenyl group to the alkene results in enantioselective thiiranium ion formation (species iii). Stereospecific, intermolecular nucleophilic capture and subsequent deprotonation affords the vicinally functionalized product and regenerates the catalyst.
Figure 1.

Proposed catalytic cycle.
In conclusion, an enantioselective, Lewis base-catalyzed, intermolecular sulfenoamination has been described. By employing HFIP as the solvent, both anilines and benzylic amines were employed as competent nucleophiles. The corresponding 1,2-sulfenoaminated products were obtained in good yields and excellent selectivities. A number of derivatives were accessed in high diastereoselectivity through a β-amido organolithium intermediate.
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (GM R35127010) for generous financial support. We also thank the UIUC SCS support facilities (microanalysis, mass spectrometry, X-ray and NMR spectroscopy) for their assistance. A.R. acknowledges the NIH-NIGMS CBI Training Grant (T32-GM070421) as well as the NSF for a Graduate Fellowship (GRFP).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07019.
Experimental Procedures, characterization data for all new compounds along with copies of spectra and chromatograms (PDF)
Data for 2c (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asymmetric Dihydroxylation. Chem. Rev 1994, 94 (8), 2483–2547. [Google Scholar]
- (2).Bodkin JA; McLeod D The Sharpless Asymmetric Aminohydroxylation. J. Chem. Soc. Perkin Trans 2002, 1 (24), 2733–2746. [Google Scholar]
- (3).Denmark SE; Kuester WE; Burk MT Catalytic, Asymmetric Halofunctionalization of Alkenes-A Critical Perspective. Angew. Chem., Int. Ed 2012, 51 (44), 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Landry ML; Burns NZ Catalytic Enantioselective Dihalogenation in Total Synthesis. Acc. Chem. Res 2018, 51 (5), 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xia Q-H; Ge H-Q; Ye C-P; Liu Z-M; Su K-X Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation. Chem. Rev 2005, 105 (5), 1603–1662. [DOI] [PubMed] [Google Scholar]
- (6).Denmark SE; Vogler T Synthesis and Reactivity of Enantiomerically Enriched Thiiranium Ions. Chem. - Eur. J 2009, 15 (43), 11737–11745. [DOI] [PubMed] [Google Scholar]
- (7).Lewis Base Catalysis: A Platform for Enantioselective Addition to Alkenes Using Group 16 and 17 Lewis Acids (n → σ*) In Lewis Base Catalysis in Organic Synthesis; Vedejs E, Denmark SE, Eds.; Wiley: Weinheim, Germany, 2016; Vol. 3, pp 1153–1211. [Google Scholar]
- 8.(a) Denmark SE; Hartmann E; Kornfilt DJP; Wang H Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes. Nat. Chem 2014, 6 (12), 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartmann E; Denmark SE Structural, Mechanistic, Spectroscopic, and Preparative Studies on the Lewis Base Catalyzed, Enantioselective Sulfenofunctionalization of Alkenes. Helv. Chim. Acta 2017, 100, No. e1700158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Denmark SE; Kornfilt DJP; Vogler T Catalytic Asymmetric Thiofunctionalization of Unactivated Alkenes. J. Am. Chem. Soc 2011, 133 (39), 15308–15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Denmark SE; Kornfilt DJP Catalytic, Enantioselective, Intramolecular Sulfenofunctionalization of Alkenes with Phenols. J. Org. Chem 2017, 82 (6), 3192–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Denmark SE; Chi HM Lewis Base Catalyzed, Enantioselective, Intramolecular Sulfenoamination of Olefins. J. Am. Chem. Soc 2014, 136 (25), 8915–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Denmark SE; Chi HM Catalytic, Enantioselective, Intramolecular Sulfenoamination of Alkenes with Anilines. J. Org. Chem 2017, 82 (7), 3826–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Denmark SE; Jaunet A Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. J. Am. Chem. Soc 2013, 135 (17), 6419–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tao Z; Robb KA; Zhao K; Denmark SE Enantioselective, Lewis Base-Catalyzed Sulfenocyclization of Polyenes. J. Am. Chem. Soc 2018, 140 (10), 3569–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tao Z; Robb KA; Panger JL; Denmark SE Enantioselective, Lewis Base-Catalyzed Carbosulfenylation of Alkenylboronates by 1,2-Boronate Migration. J. Am. Chem. Soc 2018, 140 (46), 15621–15625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Trost BM; Shibata T Nucleophilic Attack on Olefins Initiated by Dimethyl(Methylthio)Sulfonium Fluoroborate (DMTSF). J. Am. Chem. Soc 1982, 104 (11), 3225–3228. [Google Scholar]
- (17).Brownbridge P Amidinosulphenylation of Alkenes. Tetrahedron Lett. 1984, 25 (34), 3759–3762. [Google Scholar]
- (18).Luo J; Zhu Z; Liu Y; Zhao X Diaryl Selenide Catalyzed Vicinal Trifluoromethylthioamination of Alkenes. Org. Lett 2015, 17 (14), 3620–3623. [DOI] [PubMed] [Google Scholar]
- (19).Yuan Y; Chen Y; Tang S; Huang Z; Lei A Electrochemical Oxidative Oxysulfenylation and Aminosulfenylation of Alkenes with Hydrogen Evolution. Sci. Adv 2018, 4 (8), No. eaat5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang PA Organocatalyzed Enantioselective Desymmetrization of Aziridines and Epoxides. Beilstein J. Org. Chem 2013, 9, 1677–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Archer NJ; Rayner CM; Bell D; Miller D Synthetic Routes to Novel Homochiral Sulfenyl Sulfonium Salts and Their Use as Potential Enantioselective Sulfenylating Agents. Asymmetric Synthesis via Homochiral Thiiranium Ions. Synlett 1994, 1994 (8), 617–619. [Google Scholar]
- (22).Lucchini V; Modena G; Pasquato L Enantiopure Thiosulfonium Salts in Asymmetric Synthesis. Face Selectivity in Electrophilic Additions to Unfunctionalised Olefins. J. Chem. Soc., Chem. Commun 1994, 0 (13), 1565. [Google Scholar]
- (23).Stat-Ease. Design Expert9; Stat-Ease: Minneapolis, MN, 2015. [Google Scholar]
- (24).CCDC 1923219 contains the crystallographic data for compound (S,R)-2c. These data can be obtained free of charge from the Cambridge Crystallographic Data Center (www.ccdc.cam.a-c.uk).
- (25).Foubelo F; Gutiérrez A; Yus M β-Functionalised Organolithium Compounds through a Sulfur-Lithium Exchange. Tetrahedron Lett. 1997, 38 (27), 4837–4840. [Google Scholar]
- (26).Barluenga J; Flórez J; Yus M β-Substituted Organolithium Compounds; Direct Preparation and Reactivity. J. Chem. Soc., Chem. Commun 1982, 0 (20), 1153–1154. [Google Scholar]
- (27).Foubelo F; Yus M Functionalised Organolithium Compounds by Sulfur-Lithium Exchange. Chem. Soc. Rev 2008, 37 (12), 2620–2633. [DOI] [PubMed] [Google Scholar]
- (28).Malathong V; Rychnovsky SD Polyol Synthesis with β-Oxyanionic Alkyllithium Reagents: Syntheses of Aculeatins A, B, and D. Org. Lett 2009, 11 (18), 4220–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Huckins JR; De Vicente J; Rychnovsky SD Synthesis of the C1–C52 Fragment of Amphidinol 3, Featuring a β-Alkoxy Alkyllithium Addition Reaction. Org. Lett 2007, 9 (23), 4757–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Paterson I; Miller NA Total Synthesis of the Marine Macrolide (+)-Neopeltolide. Chem. Commun 2008, 70 (39), 4708–4710. [DOI] [PubMed] [Google Scholar]
- (31).Kartika R; Gruffi TR; Taylor RE Concise Enantioselective Total Synthesis of Neopeltolide Macrolactone Highlighted by Ether Transfer. Org. Lett 2008, 10 (21), 5047–5050. [DOI] [PubMed] [Google Scholar]
- (32).Huang H; Iwasawa N; Mukaiyama T A convenient method for the construction of β-lactam compounds from β-amino acids using 2-chloro-1-methylpyridinium iodide as condensing reagent. Chem. Lett 1984, 13, 1465–1466. [Google Scholar]
- (33).Yadav LDS; Rai A; Rai VK; Awasthi C Novel Aziridination of α-Halo Ketones: An Efficient Nucleophile-Induced Cyclization of Phosphoramidates to Functionalized Aziridines. Tetrahedron Lett. 2008, 49 (4), 687–690. [Google Scholar]
- (34).Isoda M; Sato K; Funakoshi M; Omura K; Tarui A; Omote M; Ando A Diastereoselective Synthesis of Syn-β-Lactams Using Rh-Catalyzed Reductive Mannich-Type Reaction of α,β-Unsaturated Esters. J. Org. Chem 2015, 80 (16), 8398–8405. [DOI] [PubMed] [Google Scholar]