

Abstract
Objective:
To describe an unusual case of familial male precocious puberty (FMPP) characterized by periodic remission compared to a series of boys with typical testotoxicosis.
Methods:
Medical records of boys with FMPP followed at our institution from 2001–2017 were reviewed. Variables analyzed included age, family history, physical exam, hormone levels, bone age, and treatment.
Results:
A boy of age 2 years 10 months presented with growth acceleration and masturbatory behaviors. On exam, he had 6-mL testes, an enlarged phallus (10.5 × 2.5 cm), and Tanner 2 pubic hair. Testosterone was 242 ng/dL (normal level, ≤30 ng/dL). Genetic testing revealed an Asp578Gly luteinizing hormone receptor mutation confirming FMPP. Anastrozole 1 mg and bicalutamide 50 mg daily were started. During 7.5 years of follow-up, two periods of spontaneous remission occurred lasting >3 years and 10 months, respectively. Both were characterized by prepubertal testosterone levels (10 to 28 ng/dL) and arrested pubertal development off therapy. Relapses were marked by elevated testosterone, growth acceleration, and pubertal progression. Ten additional boys aged 3.46 ± 0.72 years with FMPP were identified, one of whom also had an Asp578Gly mutation. Average testosterone at presentation was 335 ± 193 ng/dL (range, 146 to 778 ng/dL) and average bone age/chronologic age was 2.02 ± 0.47. All were treated with bicalutamide and anastrozole or letrozole.
Conclusion:
We report a case of intermittent FMPP in contrast to a series of boys with a characteristic clinical course. To our knowledge, a similar case has not previously been reported. Our case expands the clinical spectrum of this rare condition.
INTRODUCTION
Familial male precocious puberty (FMPP), also known as testotoxicosis, is an autosomal dominant disorder that causes peripheral precocious puberty (PP) and affects only males. FMPP is caused by an activating mutation of the luteinizing hormone (LH) receptor, resulting in autonomous testosterone production by testicular Leydig cells (1,2). Affected boys typically present before 4 years of age with PP, growth acceleration, and advanced skeletal maturation (3). While the severity of the disease may vary (4,5), to our knowledge, intermittent spontaneous and complete remission in a boy with FMPP has never been described. The objective of this report is to describe an unusual case of FMPP characterized by periodic remission of PP compared to a series of boys with typical testotoxicosis followed at our institution.
METHODS
Following Institutional Review Board approval, medical records of children followed for FMPP in the endocrine clinic at Riley Hospital for Children in Indianapolis, Indiana, for the previous 16 years (2001–2017) were identified. Data collected included age at presentation, family history, testosterone, LH, and follicle-stimulating hormone (FSH) levels, bone age, and Tanner stage at presentation and follow-up visits. Testosterone was measured using a chemiluminescent immunoassay. Data on treatment regimens and final height outcomes, when available, were also included.
CASE REPORT
A boy of age 2 years 10 months presented with PP, growth acceleration, and masturbatory behaviors. He had no family history of FMPP. On exam, he had 6-mL testes, an enlarged phallus (10.5 × 2.5 cm), and Tanner 2 pubic hair. Bone age (BA) was 5.5 years (BA/chronologic age [CA], 1.97). Gonadotropins were undetectable, testosterone was 242 ng/dL (normal, ≤30 ng/dL), 17-hydroxyprogesterone, and β-human chorionic gonadotropin were normal. A gonadotropin-releasing hormone analogue stimulation test revealed basal and peak stimulated LH and FSH levels of 0.9 and 0.4 mIU/mL and 0.5 and 2.6 mIU/mL, respectively. Genetic testing revealed an Asp578Gly activating LH receptor mutation (6). The third-generation aromatase inhibitor anastrozole and the androgen receptor blocker bicalutamide were started at daily oral doses of 1 mg and 50 mg, respectively. During 7.5 years of follow-up, two periods of spontaneous remission occurred, which lasted >3 years in one instance and 10 months in the other. Both were characterized by prepubertal testosterone levels (10 to 28 ng/dL), a decrease in growth velocity to 4 cm/year, and arrested pubertal development. Treatment with anastrozole and bicalutamide was discontinued completely at 6 years and restarted during the relapse at 7.3 years. As seen in Figure 1, relapses were marked by elevated testosterone and growth acceleration up to 11 cm/year. Pubertal progression and bone age advancement were also noted concurrent with re-activation of the disease. At 9 years 4 months of age, central puberty was noted. At the last follow-up appointment at 10.25 years, BA was 13 years, giving a height prediction of 179.3 cm (midparental height, 182 ± 5 cm), at which point treatment was stopped. Table 1 illustrates the age, growth velocity percentiles, and serum testosterone levels at each clinic visit.
Fig. 1.
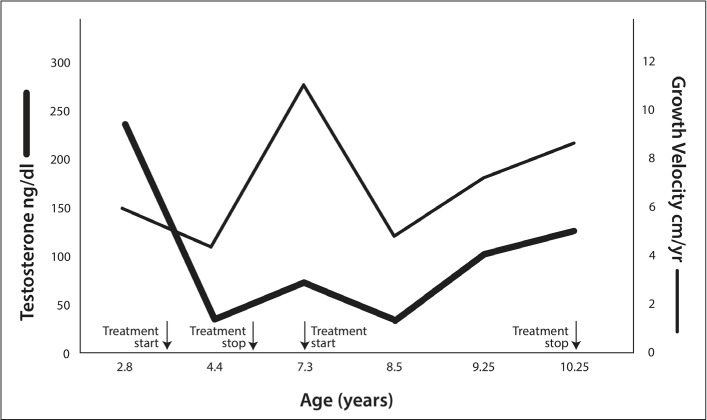
Serum testosterone and growth velocity during follow-up of our case. Serum testosterone fell to 21 ng/dL by the first follow-up visit. Testosterone levels remained relatively low between 21 and 58 ng/dL until the first relapse at 7.3 years. Treatment was discontinued between age 6 years and the time of the first relapse, after which the patient went into remission again at 8 years, during which his testosterone decreased to 28 ng/dL. Approximately 10 months later, clinical progression was again noted, and central puberty ensued.
Table 1.
Age, Growth Velocity Percentiles, and Testosterone Levels at Each Visit in the Case
| Age (years) | Visit number | Growth velocity percentile | Testosterone level (ng/dL) |
|---|---|---|---|
| 2.8 | Initial visit | NA | 242 |
| 3.3 | 1 | 25th | 29 |
| 3.8 | 2 | 20th | 38 |
| 4.4 | 3 | 10th | 10 |
| 6 | 4 | 65th | 28 |
| 7.3 - off treatment | 2 | >97th | 69 |
| 8.5 - on treatment | 3 | 20th | 28 |
| 9.25 | 4 | >97th | 100 |
| 10.25 | Last visit | >97th | 126 |
Ten additional boys aged 3.46 ± 0.72 years (range, 2.5 to 4.25 years) with FMPP were identified. Average testosterone at presentation was 335 ± 193 ng/dL (range, 146 to 778 ng/dL), and average BA/CA was 2.02 ± 0.47 (range, 1.37 to 2.8). All except 3 boys (70%) had pubertal-sized testes measuring between 4 and 8 mL, 100% had a fully pubertal penis, 6 (60%) had pubic hair ranging between Tanner stages 2 and 4, and 1 subject (10%) had body odor. Eight patients (80%) had a family history of FMPP (brother in 6 patients, father in 1, and maternal uncle in 1). Genetic testing was performed in 2 patients, revealing an Ile542Leu mutation in 1 patient and his father and an Asp578Gly mutation in another. Treatment consisting of an aromatase inhibitor and an androgen receptor blocker was started at an average age of 3.52 ± 0.68 years (range, 2.3 to 4.25 years). Eight patients were also treated with leuprolide acetate at an average age of 5.45 ± 1.05 years (range, 3.3 to 6.7 years) due to the development of secondary central PP. Two patients have reached adult height, and 5 patients are still on treatment. In contrast to our unusual case, serum testosterone levels in these 10 subjects remained elevated throughout their treatment course. Baseline and follow-up characteristics as well as the treatment regimens used in this cohort are summarized in Table 2.
Table 2.
Summary of the Clinical Characteristics of the Cohort
| Patient | Age at dx (years) | Testosterone at dx (ng/dL) | Testosterone levels over time ng/dL (range) | Testicular volume at dx (mL) | Family hx of PP | BA/CA at dx | Treatment regimen | Age at CPP dx (years) | Age at cessation of treatment (years) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.16 | 572 | 125–572 | 3 | Yes (Father) | 2.16 | Anastrozole-Bicalutamide | 5.41 | 11.9 |
| 2 | 2.5 | 174 | 174–599 | 3–4 | Yes (Brother & MGF) | 2.4 | Anastrozole-Bicalutamide | 6 | Ongoing |
| 3 | 3.7 | 442 | 442–778 | 8 | Yes (Brother & MGF) | 1.89 | Anastrozole-Bicalutamide | 6 | Ongoing |
| 4 | 2.5 | 128 | 128–730 | 2 | Yes (Brother & MGF) | 1.74 | Letrozole Bicalutamide Tamoxifen | None | Ongoing |
| 5 | 4 | 225 | 225–394 | 2 | Yes (Brother) | 2.75 | Anastrozole-Bicalutamide | 4.5 | Ongoing |
| 6 | 4.08 | 146 | 146–359 | 6 | No | 1.71 | Anastrozole-Bicalutamide | 5.5 | Ongoing |
| 7 | 2.9 | 329 | 277–329 | 2 | No | 1.37 | Anastrozole-Bicalutamide | None | Lost to follow-up |
| 8 | 4.25 | 299 | 299–419 | 6 | No | 2.3 | Anastrozole-Bicalutamide | 5.9 | Lost to follow-up |
| 9 | 2.33 | 280 | 92–309 | 5 | Yes (Maternal uncle) | 1.9 | Spironolatone Anastrozole | 3.33 | 13.25 |
| 10 | 4.41 | 255 | 255–423 | 6 | No | 2.04 | Letrozole Bicalutamide Tamoxifen | 6.75 | Lost to follow-up |
Abbreviations: BA = bone age; CA = chronologic age; CPP = central precocious puberty; dx = diagnosis; hx = history; MGF = maternal grandfather.
DISCUSSION
FMPP is an extremely rare form of peripheral PP in boys that results from a gain-of-function mutation in the LH receptor and subsequent ligand-independent testosterone biosynthesis (1–3). The most common mutation causing FMPP is the Asp578Gly missense mutation that was identified in our unusual case and in another of our cohort (6). This mutation is in the sixth transmembrane helix of the receptor, a known site of G-protein activation (5,6). Although several other mutations have been identified, no specific phenotype-genotype correlations have been described.
Standard of care for the treatment of boys with FMPP involves combination therapy consisting of an anti-androgen such as bicalutamide and an anti-estrogen such as the potent third-generation aromatase inhibitors anastrozole or letrozole (7–9). However, neither of these medications target the underlying pathophysiology of the condition, and thus, extreme elevations of testosterone persist or, in view of the mechanism of action of these drugs, are even amplified during treatment. The paradoxical decrease in serum testosterone to prepubertal values twice in the treatment course of our patient is clear evidence of amelioration of the disease process itself rather than indicating a response to therapy.
At least 3 individuals have been reported in whom a genetically proven LH receptor gene mutation was present but did not result in the typical clinical course, including in the father of a child treated at our center (4). In all cases, a history of PP was reported, but a nonprogressive clinical course apparently ensued and treatment was never required. These cases suggest the presence of modifying factors that have the potential to significantly modify the expression of the LH receptor mutation in testotoxicosis.
Our case is entirely unique in that a complete but transient remission of PP occurred not once but twice during the course of ~8 years of follow-up. To our knowledge, such a clinical sequence of events in a patient with FMPP has never previously been described. What factors might be responsible for triggering mutant LH receptor re-activation and/or quiescence are a matter of speculation. There are other rare conditions in which autonomous cellular hyper-function exhibits an “on and off” phenomenon, such as the intermittent development of large unilateral ovarian cysts in girls with McCune-Albright syndrome. Sensitivity to the differential basal temperatures present in different tissues has been hypothesized to be the cause of simultaneous loss and gain of function in a patient with a Gs-alpha mutation causing both pseudohypoparathyroidism and testotoxicosis (10). How these examples may relate to our patient with FMPP is unclear. Additional in-depth genetic analyses and/or in vitro functional studies at the cellular level would be informative.
CONCLUSION
In conclusion, we report a fascinating case of FMPP marked by periodic spontaneous remission in comparison to a case series of boys with a typical natural history and disease trajectory. Our case expands the clinical spectrum of this rare condition.
Abbreviations:
- BA
bone age
- FMPP
familial male precocious puberty
- LH
luteinizing hormone
- PP
precocious puberty
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Shenker A, Laue L, Kosugi S, Merendino JJ, Jr, Minegishi T, Cutler GB., Jr. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652–654. doi: 10.1038/365652a0. [DOI] [PubMed] [Google Scholar]
- 2.Schedewie HK, Reiter EO, Beitins IZ et al. Testicular Leydig cell hyperplasia as a cause of familial sexual precocity. J Clin Endocrinol Metab. 1981;52:271–278. doi: 10.1210/jcem-52-2-271. [DOI] [PubMed] [Google Scholar]
- 3.Rosenthal SM, Grumbach MM, Kaplan SL. Gonadotropin-independent familial sexual precocity with premature Leydig and germinal cell maturation (familial testotoxicosis): effects of a potent luteinizing hormone-releasing factor agonist and medroxyprogesterone acetate therapy in four cases. J Clin Endocrinol Metab. 1983;57:571–579. doi: 10.1210/jcem-57-3-571. [DOI] [PubMed] [Google Scholar]
- 4.Kreher N, Pescovitz OH, Delameter P, Tiulpakov A, Hochberg Z. Treatment of familial male limited precocious puberty with bicalutamide and anastrazole. J Pediatr. 2006;149:416–420. doi: 10.1016/j.jpeds.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 5.Jeha GS, Lowenthal ED, Chan WY, Wu SM, Karaviti LP. Variable presentation of precocious puberty associated with the D564G mutation of the LHCGR gene in children with testotoxicosis. J Pediatr. 2006;149:271–274. doi: 10.1016/j.jpeds.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Laue L, Chan WY, Hsueh AJ et al. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc Natl Acad Sci U S A. 1995;92:1906–1910. doi: 10.1073/pnas.92.6.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiter EO, Mauras N, McCormick K et al. Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testostoxicosis: a phase II, open-label pilot study (BATT) J Pediatr Endocrinol Metab. 2010;23:999–1009. doi: 10.1515/jpem.2010.161. [DOI] [PubMed] [Google Scholar]
- 8.Lenz AM, Shulman D, Eugster EA et al. Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics. 2010;126:e728–e733. doi: 10.1542/peds.2010-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leschek EW, Flor AC, Bryant JC, Jones JV, Barnes KM, Cutler GB., Jr. Effect of antiandrogen, aromatase inhibitor, and gonadotropin-releasing hormone analog on adult height in familial male precocious puberty. J Pediatr. 2017;190:229–235. doi: 10.1016/j.jpeds.2017.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liri T, Herzmark P, Nakamoto JM, Van Dop C, Bourne HR. Rapid GDP release from Gs alpha in patients with gain and loss of endocrine function. Nature. 1994;371:164–168. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]