

Abstract
Manganese (Mn) participates in a variety of distinct physiological processes, including acting as a cofactor for several enzymes and metalloenzymes, in addition to playing a role in immune function, endocrine function, hematopoiesis, and oxidative stress regulation. Mn homeostasis is tightly regulated via intestinal absorption and hepatobiliary and intestinal excretion. In this issue of the JCI, Mercadante and colleagues explored the role of the metal transporter Slc30a10 in vivo using a mouse model system. The authors used whole-body and tissue-specific gene knockouts to show that Slc30a10 is paramount for Mn excretion in the liver and small intestines. These findings provide further insights into mechanisms for Mn homeostasis as well as potential targets for addressing Mn-associated disorders or environmental exposures.
Manganese homeostasis
Manganese (Mn) is an essential metal typically acquired from the diet and water supply (1, 2). Over the past decade, discoveries in human genetics have placed a greater focus on the importance of understanding Mn homeostasis. First, several single-gene Mendelian disorders of Mn homeostasis have been described (Table 1). In 2012, the first report of a disease now referred to as hypermanganesemia with dystonia 1 (HMNDYT1) (OMIM #613280) detailed two consanguineous families with autosomal-recessive mutations in the Mn exporter SLC30A10 (encoding the protein ZNT10) (3, 4). Subsequently, a report describing a similar phenotype, hypermanganesemia with dystonia 2 (HMNDYT2) (OMIM #617013), described autosomal-recessive mutations in the Mn importer SLC39A14 (encoding the protein ZIP14) (5). Furthermore, severe hypomanganesemia was attributed to autosomal-recessive mutations in the Mn importer SLC39A8 (encoding the protein ZIP8). In this condition, chronically low serum Mn is associated with seizures, developmental delays, deafness, and liver disease and is characterized by impaired activity of Mn-dependent glycosylation enzymes (congenital disorder of glycosylation type IIn [CDG2N] [OMIM #616721]) (6).
Table 1. Single-gene Mendelian disorders of Mn homeostasis.

In addition to these rare single-gene disorders, which have been highly informative regarding key roles of Mn transporters, Mn homeostasis has been implicated in complex diseases and traits through GWAS. In particular, the coding variant rs13107325 (Ala391Thr) in SLC39A8 has been associated with reduced blood pressure and hypertension risk (7), lower HDL cholesterol levels (8), higher BMI (9), increased schizophrenia risk (10, 11), and increased alcohol intake and alcohol use disorder risk (12), among many other traits (13). This variant has an 8% minor allele frequency in individuals of European ancestry. The corresponding protein was reported to have modestly reduced function (14) and reduced expression in the human liver. Notably, in a GWAS of blood Mn levels (15), the only two genome-wide significant loci were SLC39A8 (the lead SNP was the same Ala391Thr coding variant and was associated with lower blood Mn) and noncoding SNPs on chromosome 1 near SLC30A10. Thus, genetic variation influences Mn homeostasis over a wide range, from subtle effects that influence pleiotropic phenotypic variation and risk of disease, to more dramatic effects that cause major disease due to Mn excess or deficiency.
Intestinal absorption is one point of regulation of Mn homeostasis, and less than 5% of ingested Mn is absorbed. The divalent metal transporter 1 (DMT1), encoded by the gene SLC11A2, likely facilitates this intestinal absorption (1, 16). Conversely, Mn is eliminated through biliary excretion (16). In fact, hepatic regulation of biliary Mn excretion may regulate whole-body Mn homeostasis. We previously showed that SLC39A8, an importer of Mn, is expressed on the canalicular membrane of hepatocytes and serves to “reclaim” Mn from the bile back into the hepatocyte, thereby defending against Mn depletion (17). Indeed, although SLC39A8 is expressed in multiple tissues, mice with a liver-specific Slc39a8 knockout had decreased blood and tissue Mn levels that were similar to those seen with whole-body Slc39a8 knockout and consistent with the key role of hepatic SLC39A8 in Mn homeostasis (Figure 1) (17).
Figure 1. Model for Mn homeostasis with a focus on excretion pathways.
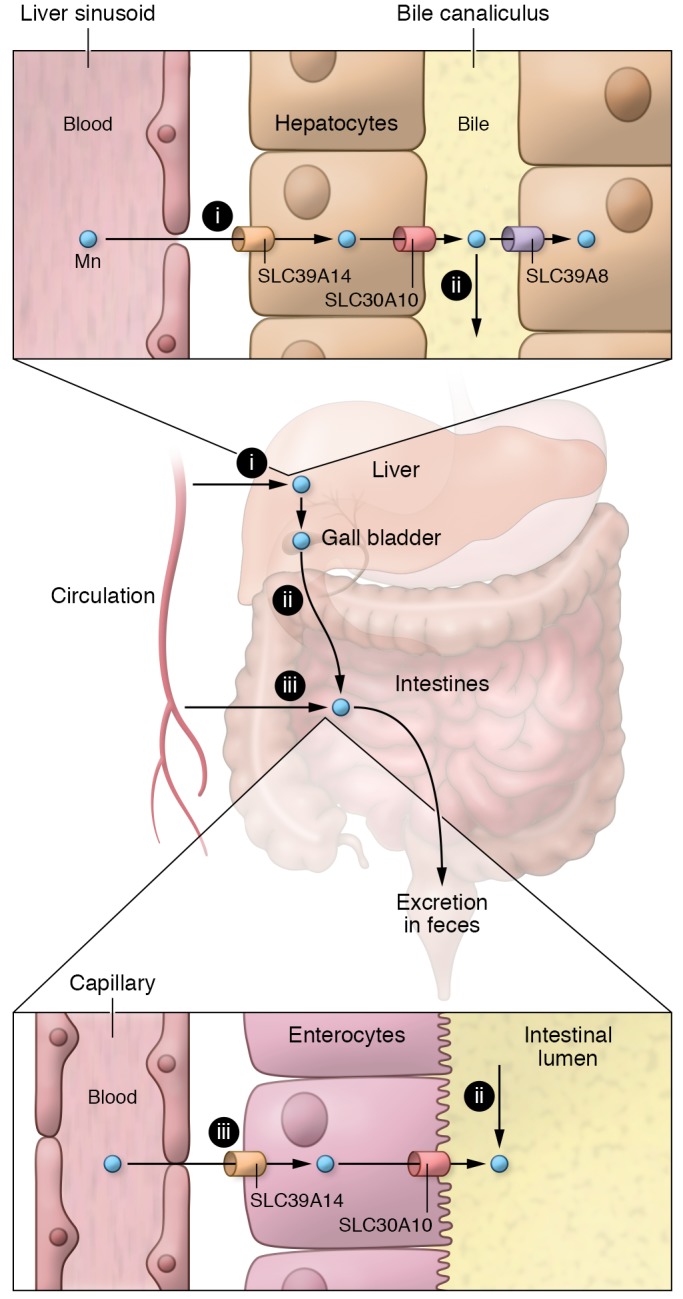
Systemic Mn homeostasis is largely regulated by pathways of Mn excretion in hepatocytes and enterocytes. (i) Mn uptake from blood by hepatocytes is mediated by SLC39A14 at the basolateral membrane. Mn excretion from hepatocytes into bile is mediated by SLC30A10 at the canalicular membrane. (ii) Mn reuptake from bile into hepatocytes is mediated by SLC39A8 also at the canalicular membrane. Biliary Mn ultimately arrives in the intestinal lumen and, if not reabsorbed (not shown), is excreted in the feces. (iii) In addition, Mn is taken up from blood by enterocytes via SLC39A14 at the basolateral membrane and effluxed into the lumen by SLC30A10 at the apical surface. This figure does not show absorption of Mn via uptake pathways from the gut lumen into enterocytes and blood.
SLC30A10 is essential for manganese excretion
In the current issue of the JCI, Mercadante et al. provide data revealing the critical role of the Mn exporter SLC30A10, thus advancing our understanding of biliary Mn excretion (18). Mice deficient in whole-body Slc30A10 showed elevated blood and tissue Mn levels and, because of impaired biliary excretion of Mn, had biliary Mn levels that were inappropriately low for the tissue Mn excess. The authors showed that Slc30a10 was localized to the canalicular membrane of hepatocytes and that hepatocyte-specific deletion of Slc30a10 resulted in the virtual absence of biliary excretion following Mn injection, confirming that hepatocyte Slc30a10 is essential for biliary Mn excretion. Interestingly, although liver-specific deletion of Slc30a10 also virtually eliminated biliary Mn excretion, it resulted in only a modest increase in tissue Mn, indicating that Slc30a10 expression in other tissues, or other factors, may compensate for the reduced biliary Mn excretion. They went on to show that intestinal enterocyte Slc30a10 also contributes to Mn excretion; however, even the double-knockout (hepatocyte/enterocyte) mice failed to show the same degree of tissue Mn excess seen in the whole-body knockout mice. This report establishes the key physiologic role of SLC30A10 in vivo in mediating biliary and intestinal excretion of Mn and defending against whole-body Mn excess (Figure 1) (18).
SLC39A14 (ZIP14) is another Mn importer expressed in both hepatocytes and enterocytes (Figure 1). In both humans and mice, deficiency of SLC39A14 is associated with hypermagnesemia and Mn toxicity (5, 19). In the liver, SLC39A14 is localized to the basolateral membrane of hepatocytes and is the major transporter responsible for importing Mn into the liver; hepatocyte-specific Slc39a14 ablation in mice reduced liver Mn but had no effect on blood Mn levels (20). SLC39A14 is also expressed on the basolateral membrane of enterocytes, where it imports Mn from blood to enterocytes and facilitates its fecal excretion. Enterocyte-specific deletion increased blood Mn and caused tissue Mn overload, indicating that intestinal SLC39A14 is quantitatively more important for Mn homeostasis (20). Notably, SLC39A14 was not identified as a significant locus in the GWAS for blood Mn, suggesting that modest genetic variation in its expression or function does not substantially influence Mn homeostasis in humans, in contrast to SLC39A8 and SLC30A10.
Thus, SLC30A10 and SLC39A8 appear to act as the primary regulators of biliary Mn excretion and systemic Mn homeostasis, with SLC30A10 exporting Mn into the bile and SLC39A8 reclaiming it from the bile (Figure 1). In this view, it is perhaps no surprise that these two genes were the top genetic loci associated with blood Mn levels on GWAS (15). These common variant studies indicate that subtle changes in expression or function of these two transporters can influence Mn homeostasis and blood concentrations. In the case of the highly pleiotropic SLC39A8, reduced Mn concentrations due to the Ala391Thr allele appear to directly influence a wide variety of phenotypic traits and disease risks. Altered Mn homeostasis may influence phenotypes through modulation of the activity of certain Mn-dependent enzymes including transferases, hydrolases, lyases, isomerases, and oxidoreductases (2). For example, Slc39a8 deletion in mice resulted in a significant decrease in the tissue activity of arginase, a well-recognized Mn-dependent enzyme, which could explain the association of this variant with reduced blood pressure (17). Furthermore, both humans and mice with genetically reduced SLC39A8 activity have lower levels of protein N-glycosylation due to reduced activity of certain Mn-dependent glycosyltransferases (6, 17), which could affect a variety of phenotypic traits. A hypothesis — not yet confirmed — is that the pleiotropic effects of modestly genetically reduced SLC39A8 activity lead to reduced blood and tissue Mn concentrations, which in turn cause reduced activity of specific Mn-dependent enzymes in specific tissues. It remains a mystery why genetic variants at the SLC30A10 locus that are also associated with blood Mn levels lack the pleiotropic associations with phenotypes and disease risk that SLC39A8 variants show. The common variants at SLC30A10 may have a quantitatively reduced effect on Mn concentrations and thus less of an effect on phenotype, or there could be tissue-specific effects of genetic variation on these transporters.
One of the important frontiers in the area of Mn-related phenotypic traits and disease is understanding the relationship between systemic Mn homeostasis and the role of Mn in the brain. The Mendelian conditions of deficiency in SLC30A10 or SLC39A14 (Mn excess) or SLC39A8 (Mn deficiency) have major neurobehavioral and brain phenotypes (Table 1). Furthermore, the SLC39A8 Ala391Thr coding variant associated with lower blood Mn is associated with schizophrenia, alcohol use disorder, and reduced cognitive performance. In addition to their effects on Mn excretion and systemic Mn homeostasis, all three of these genes are expressed in the brain as well. As Mn overload occurs in the brain as a result of loss of SLC30A10 or SLC39A14, neither of these proteins can be responsible for Mn uptake into the brain. Conversely, the systemic Mn deficiency associated with loss of SLC39A8 may be exacerbated in the brain if SLC39A8 also plays a key role in Mn uptake into the brain. Many questions remain: How is Mn transported across the blood-brain barrier? Do blood Mn concentrations directly influence Mn concentrations in the brain? What cell types in the brain express these Mn transporters and what is their function in the brain? Does variation in brain Mn concentrations influence specific Mn-dependent enzymes in the brain, thereby influencing phenotypic traits and risk of disease? Notably, in the Mendelian condition of SLC39A8 deficiency, which is characterized by severely reduced Mn concentrations, clinical improvement was noted with Mn supplementation (21). Could Mn supplementation ameliorate some of the disease risk associated with genetically reduced SLC39A8 activity, such as the risk for schizophrenia or alcohol use disorder? Substantial work is required to understand the role of systemic and local Mn homeostasis in brain function, cognition, and neurobehavioral and psychiatric disorders.
In summary, the seemingly esoteric topic of Mn homeostasis has turned out to be of critical importance to human biology and disease, as revealed by rare single-gene disorders that result in markedly dysregulated Mn homeostasis, and through genetic variation in Mn homeostasis that leads to a variety of phenotypes and risk of disease. Continued investigation into the regulation of Mn homeostasis is likely to lead directly to therapeutic hypotheses around the prevention and treatment of a number of human conditions associated with altered Mn homeostasis.
Acknowledgments
We would like to acknowledge Nicholas Hand for helpful comments and the Leducq Foundation Transatlantic Center of Excellence for funding support.
Version 1. 11/04/2019
Electronic publication
Version 2. 12/02/2019
Print issue publication
Footnotes
Conflict of interest: DJR is a founder and equity owner of VascularStrategies and Staten Biotechnology; serves on scientific advisory boards for Alnylam, Novartis, Pfizer, and Verve; has received compensation for consulting for Akcea in the past year; and has received research funding from Biogen and Corvidia in the past year.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(12):5082–5085. https://doi.org/10.1172/JCI133120.
See the related article at Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity.
Contributor Information
Nathan Katz, Email: nathanka@mail.med.upenn.edu.
Daniel J. Rader, Email: rader@mail.med.upenn.edu.
References
- 1.Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med. 2005;26(4-5):353–362. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Yang X. The essential element manganese, oxidative stress, and metabolic diseases: links and interactions. Oxid Med Cell Longev. 2018;2018:7580707. doi: 10.1155/2018/7580707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuschl K, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90(3):457–466. doi: 10.1016/j.ajhg.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quadri M, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90(3):467–477. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuschl K, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. doi: 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JH, et al. SLC39A8 deficiency: a disorder of manganese transport and glycosylation. Am J Hum Genet. 2015;97(6):894–903. doi: 10.1016/j.ajhg.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehret GB, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet. 2016;48(10):1171–1184. doi: 10.1038/ng.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardiñas AF, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50(3):381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kranzler HR, et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun. 2019;10(1):1499. doi: 10.1038/s41467-019-09480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48(7):709–717. doi: 10.1038/ng.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, et al. A blood pressure-associated variant of the SLC39A8 gene influences cellular cadmium accumulation and toxicity. Hum Mol Genet. 2016;25(18):4117–4126. doi: 10.1093/hmg/ddw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng E, et al. Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum Mol Genet. 2015;24(16):4739–4745. doi: 10.1093/hmg/ddv190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth JA. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol Res. 2006;39(1):45–57. doi: 10.4067/s0716-97602006000100006. [DOI] [PubMed] [Google Scholar]
- 17.Lin W, et al. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest. 2017;127(6):2407–2417. doi: 10.1172/JCI90896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mercadante CJ, et al. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J Clin Invest. 2019;129(12):5442–5461. doi: 10.1172/JCI129710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aydemir TB, et al. Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci. 2017;37(25):5996–6006. doi: 10.1523/JNEUROSCI.0285-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheiber IF, Wu Y, Morgan SE, Zhao N. The intestinal metal transporter ZIP14 maintains systemic manganese homeostasis. J Biol Chem. 2019;294(23):9147–9160. doi: 10.1074/jbc.RA119.008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JH, et al. SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genet Med. 2018;20(2):259–268. doi: 10.1038/gim.2017.106. [DOI] [PubMed] [Google Scholar]