

Abstract
Systemic lupus erythematosus (SLE) is a devastating autoimmune disease in which hyperactive T cells play a critical role. Understanding molecular mechanisms underlying the T cell hyperactivity will lead to identification of specific therapeutic targets. Serine/arginine-rich splicing factor 1 (SRSF1) is an essential RNA-binding protein that controls posttranscriptional gene expression. We have demonstrated that SRSF1 levels are aberrantly decreased in T cells from patients with SLE and that they correlate with severe disease, yet the role of SRSF1 in T cell physiology and autoimmune disease is largely unknown. Here we show that T cell–restricted Srsf1-deficient mice develop systemic autoimmunity and lupus-nephritis. Mice exhibit increased frequencies of activated/effector T cells producing proinflammatory cytokines, and an elevated T cell activation gene signature. Mechanistically, we noted increased activity of the mechanistic target of rapamycin (mTOR) pathway and reduced expression of its repressor PTEN. The mTOR complex 1 (mTORC1) inhibitor rapamycin suppressed proinflammatory cytokine production by T cells and alleviated autoimmunity in Srsf1-deficient mice. Of direct clinical relevance, PTEN levels correlated with SRSF1 in T cells from patients with SLE, and SRSF1 overexpression rescued PTEN and suppressed mTORC1 activation and proinflammatory cytokine production. Our studies reveal the role of a previously unrecognized molecule, SRSF1, in restraining T cell activation, averting the development of autoimmune disease, and acting as a potential therapeutic target for lupus.
Keywords: Autoimmunity, Immunology
Keywords: Cytokines, Lupus, T cells

Introduction
Systemic lupus erythematosus (SLE) is a chronic debilitating multi-system autoimmune disease that disproportionately afflicts women, causing significant morbidity and mortality (1, 2). Failure of the immune system to recognize self-antigens leads to inappropriate immune responses, systemic autoimmunity, and organ damage. The identification of novel molecules and mechanisms underlying the pathogenic immune response in SLE will guide the development of appropriate therapeutic targets. While autoreactive B cells, autoantibodies, and immune complexes are important in disease pathogenesis (3), and elevated type I interferons (IFNs) have been implicated in disease (4, 5), therapeutic strategies targeting B cells and type I IFN signaling in SLE have shown limited success. In this regard, the role of T cells in disease pathogenesis is beginning to be understood, and therapeutic strategies targeting T cells are emerging (6). Aberrantly activated effector T (Teff) cells and cytokines that orchestrate not only germinal center B cell responses for autoantibody production (7–9) but also promote inflammatory infiltration in target organs (10, 11), are critical players in disease pathogenesis. These Teff cells and cytokines are found both in lupus-prone mice and in patients with SLE (12–15), and therefore are potential promising therapeutic targets. Yet the molecules and mechanisms which control the activated T cell phenotype in SLE are poorly understood.
A key pathway involved in the activation of T cells is the phosphoinositide-3 kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR) pathway. The mTOR pathway determines cell fate decisions of T cell activation, differentiation, proliferation, and cell death (16). Resting quiescent T cells have low levels of mTOR activity, while mTOR complex 1 (mTORC1) activation leads to increased proportions of activated/effector cells and a decreased pool of naive cells (17). Recently, mTOR activation has emerged as a central regulator of T cell dysfunction in systemic autoimmune diseases and as a promising therapeutic target for SLE (15, 18). Increased activity of the mTORC1 pathway in T cells is known to promote the aberrant production of cytokines IFN-γ, IL-17, and IL-4 (19, 20), and contribute to systemic autoimmunity (9). Furthermore, activation of mTORC1 is associated with altered aberrantly activated T cell responses in patients with SLE (21, 22). The phosphatase and tensin homolog (PTEN), a lipid phosphatase, negatively regulates the PI3K/Akt signaling pathway (23) and inhibits the activity of the mTOR pathway (24). PTEN-deficient T cells are hyperresponsive to TCR stimulation (25), and Pten deletion in T cells in mice leads to autoimmune lymphoproliferative disease and aberrant production of cytokines (26, 27). PTEN expression is decreased in B cells from patients with SLE and contributes to B cell hyperactivity (28). However, it is still unknown whether PTEN is altered in T cells from patients with SLE and whether it contributes to T cell hyperactivity.
Serine arginine-rich splicing factor 1 (SRSF1), previously known as splicing factor 2/alternative splicing factor (SF2/ASF), is the prototype member of the highly conserved serine arginine (SR) family of RNA-binding proteins. SRSF1 controls posttranscriptional gene expression via pre-mRNA alternative splicing, mRNA stability, and translation (29). While SRSF1 is an essential ubiquitous molecule that controls genes involved in cell survival and proliferation (30), little is known of its role in the immune system. T cells from patients with SLE display hyperactive T cell receptor (TCR)/CD3+ signaling and proinflammatory cytokine production (1, 31). This overactive lupus T cell phenotype is associated with a reduced expression of the CD3 zeta (ζ) chain (32), attributed in part to defective alternative splicing of its 3′ UTR (33). Using discovery approaches, we found that SRSF1 binds to the 3′ UTR of the CD3ζ mRNA in human T cells (34, 35). We showed that SRSF1 promotes normal expression of CD3ζ by limiting expression of an unstable isoform of its 3′ UTR (35) and that SRSF1 levels decrease upon T cell activation (36). We demonstrated that SRSF1 expression is aberrantly decreased in T cells from patients with SLE and correlates with severe disease (37)(38). In addition, decreased SRSF1 expression levels have been linked to the defective expression of multiple autoantigen transcripts in the organ-specific autoimmune inflammatory myositis (39). Thus, studies in humans suggest that the reduced expression of SRSF1 correlates with an activated T cell phenotype and the expression of autoimmune disease. However, evidence of a causal in vivo relationship among SRSF1, T cell dysfunction, and autoimmune disease is still lacking.
Here we show that mice that lack Srsf1 in mature T cells develop systemic autoimmunity and lupus nephritis with increased proportions of activated/effector T cell populations and proinflammatory cytokine-producing T cells. Mechanistically, we show that T cells from Srsf1-deficient mice exhibit increased mTOR activity and reduced expression of its negative regulator PTEN, whereas rapamycin and overexpression of PTEN normalize the inflammatory cytokine production. Importantly, PTEN levels are reduced in patients with SLE with lower SRSF1 expression and SRSF1 rescues PTEN levels, suppresses mTORC1 activity, and reduces proinflammatory cytokine production by T cells from patients with SLE. Our studies demonstrate that SRSF1 is a previously unrecognized molecule that restrains T cell activation and controls the development of autoimmune disease.
Results
Srsf1-cKO mice developed autoimmunity and lupus nephritis.
Because germline Srsf1 deletion is embryonic lethal (40), we generated conditional distal.Lckcre.Srsf1fl/fl (Srsf1-cKO hereafter) mice to study the role of SRSF1 in T cells (Supplemental Figure 1, A and B; supplemental material available online with this article; https://doi.org/10.1172/JCI127949DS1). At birth, Srsf1-cKO mice were viable with normal external features and body weight (Supplemental Figure 1C). As expected (the distal Lck promoter is expressed late in thymic development and mainly in mature T cells), T cell development was normal in Srsf1-cKO mice (Supplemental Figure 1, C–F). We asked whether the Srsf1-cKO mice develop signs of autoimmunity. We measured autoantibodies in the sera of Srsf1-cKO mice using microarray analyses of serum IgG reactivity against a 128-autoantigen panel, which includes the lupus-associated autoantigens dsDNA, histones, and a number of nuclear, membrane, phospholipid, and cytoplasmic protein autoantigens (Figure 1A). Serum from a lupus-prone MRL/lpr mouse, which spontaneously develops an autoimmune lupus-like disease, was included as positive control. We found that Srsf1-cKO mice develop a wide array of lupus-associated autoantibodies at levels comparable to those seen in MRL/lpr mice (Figure 1A). We confirmed by ELISA the presence of antinuclear (ANA) antibodies in sera from Srsf1-cKO mice (Supplemental Figure 2A). In aged mice (12–18 months), we observed a lymphoproliferative phenotype (Figure 1B and Supplemental Figure 2B). We also found that T cells from Srsf1-cKO mice exhibit increased proliferative capacity as assessed by Ki67 staining (Figure 1C). The kidneys of aged Srsf1-cKO mice showed histopathologic evidence of lupus nephritis with glomerulonephritis, mesangial and endocapillary proliferation, and interstitial inflammatory cell infiltration (Figure 1D) with significantly higher histopathology scores compared with control mice (Figure 1E). Immunohistochemistry staining showed T cell infiltration in kidneys of Srsf1-cKO mice (Supplemental Figure 2C). Srsf1-cKO mice developed significant proteinuria compared with WT mice (Figure 1F). These results demonstrate that the loss of SRSF1 in T cells leads to systemic autoimmunity and lupus-like disease.
Figure 1. Srsf1-cKO mice develop systemic autoimmunity and lupus nephritis.

(A) Heatmaps show autoantibody microarray data from sera of 12-week-old WT and Srsf1-cKO mice (n = 3 each) and 1 MRL/lpr mouse, analyzed for IgG reactivity against a 128-autoantigen panel. (B) Spleen images from aged (12- to 18-month-old) WT and Srsf1-cKO mice. Data are representative of at least 4 independent experiments. (C) Flow cytometry plots and graphs show Ki67 staining on live-gated CD4+ and CD8+ T cells from spleen of WT and Srsf1-cKO mice (2- to 18-month-old, n = 11 [WT], n = 8 [KO]). (D) Representative light microscopy images of H&E-stained paraffin sections of kidneys from aged (9- to 18-month-old) mice. Original magnification ×40. Arrows represent mesangial and endocapillary proliferation (upper) and interstitial inflammatory cell infiltration (lower). (E) Graph shows semiquantitative scores for kidney histopathology (n = 12 [WT], n = 9 [KO]). (F) Graph shows proteinuria from WT and Srsf1-cKO mice (n = 7 each). Mann-Whitney U test (E); unpaired t test (C and F); *P < 0.05, ***P < 0.005.
T cells from Srsf1-cKO mice exhibited a hyperactive phenotype and produced proinflammatory cytokines.
Examination of peripheral lymphoid T cells from spleen revealed significantly increased proportions of activated CD69hi cells (Figure 2, A and B), and effector/memory CD44hiCD62Llo and CD44hiCD62Lhi populations in the CD4 and CD8 compartments (Figure 2, C and D). In parallel, we recorded a significant reduction in the naive T cell pool in both CD4 and CD8 compartments (Figure 2E). Further, we found increased proportions of IL-17–, IFN-γ–, and IL-4–producing CD4+ T cells, and IFN-γ–producing CD8+ T cells from Srsf1-cKO mice (Figure 2F). In addition, we found that the frequencies of CD4+ CXCR5+PD1+ T follicular helper (Tfh) cells were increased in Srsf1-cKO mice (Figure 2G). We also detected the increased presence of spontaneous germinal centers in the spleens from Srsf1-cKO mice (Supplemental Figure 2D). We found that the frequencies of GL7+Fas+ germinal center B cells were increased in Srsf1-cKO mice (Supplemental Figure 2E). In addition, IgD– memory/plasma B cells were also increased in Srsf1-cKO mice (Supplemental Figure 2F). There was no difference in the frequencies and absolute numbers of B cells, CD11b+ cells, and CD11c+ cells in spleen of control WT and Srsf1-cKO mice (Supplemental Figure 3, A and B). The frequencies of CD4+CD25+FoxP3+ Treg cells in Srsf1-cKO mice were comparable with those of control mice (Supplemental Figure 4, A and B) and suppressed the proliferation of conventional T (Tconv) cells normally in in vitro Treg suppression assays for 3 days (Supplemental Figure 4, C and D). These results show that loss of SRSF1 induces the development of activated, inflammatory cytokine-producing T cells.
Figure 2. T cells from Srsf1-cKO mice have a hyperactive phenotype and produce proinflammatory cytokines.

Spleen cells were isolated from WT and Srsf1-cKO mice and analyzed by flow cytometry. (A) Plots show CD69 expression gated on live CD4+ T cells. (B) Graph shows percent of CD69+CD4+ T cells (n = 13 [WT], n = 14 [KO]; < 20-week-old mice). (C) Plots show CD62L and CD44 staining gated on live CD4+ and CD8+ T cells in spleen (D) Graphs show percent of activated effector/ memory subsets (CD44hiCD62Lhi and CD44hiCD62Llo) of CD4+ and CD8+ T cells (CD4+: n = 11 [WT], n = 13 [KO]; CD8+: n = 18 [WT], n = 16 [KO]; < 20-week-old mice). (E) Graphs show percentage of naive (CD44loCD62Lhi) CD4+ and CD8+ T cells (CD4+: n = 11 [WT]; n = 13 [KO]; CD8+: n = 18 [WT], n = 16 [KO]; < 20-week-old mice). (F) Spleen cells were stimulated for 4 hours with PMA, ionomycin with monensin followed by surface and intracellular cytokine staining for flow cytometry. Plots show IL-17, IL-4, and IFN-γ staining gated on live CD4+ and CD8+ T cells. Graphs below show percentage of cytokine-producing CD4+ and CD8+ T cells from spleen (IL-17: n = 8 each; IL-4: n = 6–7; CD4+ IFN-γ: n = 13 each; CD8+ IFN-γ: n = 3–4; CD4+: < 20-week-old mice; CD8+: 9- to 40-week-old mice). (G) Splenocytes from WT or Srsf1-cKO mice were stained for PD1 and CXCR5 by flow cytometry. Plots show cells gated on live CD4+ T cells. Graph below shows percentage of Tfh cells (n = 20 [WT]; n = 21 [KO]; 10- to 28-week-old mice). Unpaired t test, *P < 0.05, **P < 0.005.
Transcriptomics analysis of CD4+ Teff cells from Srsf1-cKO mice revealed an elevated T cell activation gene signature.
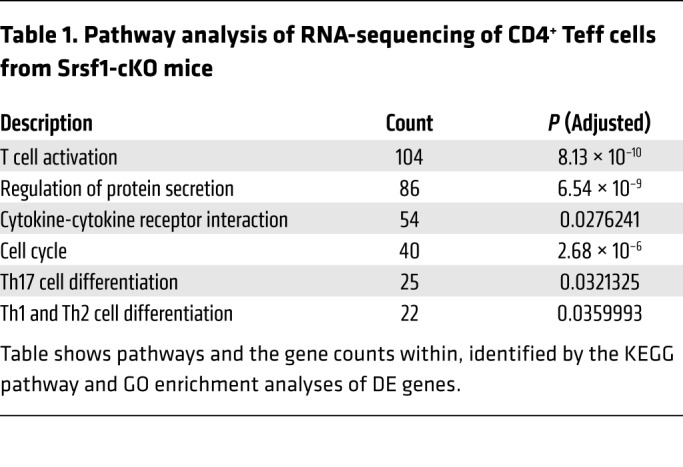
Because the Srsf1-cKO mice develop autoimmune inflammatory disease, and exhibit increased proportions of proinflammatory Teff cells, we asked whether SRSF1 controls the genes and pathways involved in T cell activation and differentiation. We performed transcriptomics analysis by RNA-sequencing of CD4+ Teff cells derived by stimulation of naive CD4+ T cells with anti-CD3 and anti-CD28 for 72 hours, from 7-week-old control WT or Srsf1-cKO mice with 3 mice per group (Figure 3 and Table 1). At the fold change greater than 1, and a P value less than 0.05, 890 genes were upregulated and 1021 genes downregulated in Srsf1-cKO mice compared with control mice, whereas at the fold change greater than 2 cutoff, and a P value less than 0.05, 312 genes were upregulated and 300 genes were downregulated in Srsf1-cKO mice compared with normal mice (Figure 3A). Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses revealed the top pathways to include genes associated with T cell activation, regulation of protein secretion, cytokine-cytokine receptor interaction, cell cycle, Th17 differentiation, and Th1 and Th2 differentiation (Table 1). Cluster analysis revealed predominantly genes involved in T cell activation, protein secretion, cytokine production, ribosome assembly, cell cycle, and chromatin modifications (Figure 3B). The expression levels of multiple cytokines including Ifng, Il17, Il4, and Il21 were found to be increased (Figure 3C). Because IL-21 is known to play important roles in Tfh differentiation, the increased Il21 expression may contribute to the observed increase in Tfh cells in Srsf1-cKO mice. These data indicate that SRSF1 controls genes involved in T cell activation, differentiation, and cytokine production, and the loss of SRSF1 leads to unrestrained activation and proinflammatory phenotype of T cells.
Figure 3. Transcriptomics analysis of CD4+ Teff cells from Srsf1-cKO mice reveals an elevated T cell activation gene signature.

(A) CD4+ Teff cells were generated as follows. Naive CD4+ T cells were sorted from spleens of WT and Srsf1-cKO mice (n = 3 each), and stimulated with anti-CD3 (0.5 μg/mL) and anti-CD28 (1.0 μg/mL) for 72 hours. RNA-sequencing data analysis shows differentially expressed (DE) genes with fold change (FC) differences at P < 0.05. (B) GO terms enrichment map of DE genes. The size of the red circles indicates the number of genes within a given pathway, and the color represents the P values relative to the other displayed terms. Outlines (added manually) indicate groups of similar GO terms. (C) Heatmap showing average expression of DE cytokine genes in WT and Srsf1-cKO mice.
Table 1. Pathway analysis of RNA-sequencing of CD4+ Teff cells from Srsf1-cKO mice.
T cells from Srsf1-cKO mice displayed increased activation of the mTORC1 pathway and reduced expression of PTEN.
It is known that Pten-deficient mice exhibit features of T cell hyperactivity with increased proinflammatory cytokines and autoimmunity (27). Further, activation of the mTOR pathway is associated with hyperactive T cell responses with inflammatory cytokine production in patients with SLE (15, 21, 22). In our transcriptomics analysis from Srsf1-cKO mice, a number of genes of the PI3K/AKT/mTOR pathway were differentially expressed, and the expression of its repressor PTEN was found to be downregulated (Figure 4A). Given these findings, we examined the activity of the mTOR pathway in the Srsf1-cKO mice. We found increased expression of phosphorylated S6 (pS6) protein levels in CD4+ Teff cells derived by stimulation of naive CD4+ T cells with anti-CD3 and anti-CD28 for 72 hours, indicating an increased activity of the mTORC1 pathway (Figure 4B). It was previously reported that SRSF1 regulates the splicing of S6K1, an mTORC1 downstream signaling molecule, and modulates the mTOR pathway in cell lines (41). We found that deletion of Srsf1 in T cells results in the decrease of the short isoform of S6K1 (Supplemental Figure 5), consistent with previous reports. Although this short isoform of S6K1 associates with activation of the mTORC1 pathway (42), in the present study we found that serine-threonine kinase AKT, upstream of S6K1, is activated in CD4+ Teff cells from Srsf1-cKO mice (Figure 4C). Therefore, we hypothesized that decreased PTEN levels result in the activation of mTOR pathway. We confirmed the decreased mRNA (Figure 4D) and protein (Figure 4E) expression levels of PTEN in T cells from Srsf1-cKO mice. Because SRSF1 controls gene expression at the posttranscriptional level, which is frequently mediated through the 3′ UTR, we investigated the effect of SRSF1 on the 3′ UTR activity of PTEN. Cotransfection of a PTEN 3′ UTR–luciferase construct along with an Srsf1-expression vector in HEK293T cells revealed an increase in PTEN 3′ UTR activity (Figure 4F). Transfection of the PTEN 3′ UTR into freshly isolated total T cells from Srsf1-cKO mice led to decreased 3′ UTR activity, which was rescued by the overexpression of SRSF1 (Figure 4G). These results show that SRSF1 controls the expression of PTEN and loss of SRSF1 leads to reduced PTEN expression and increased activity of the mTOR pathway.
Figure 4. T cells from Srsf1-cKO mice exhibit increased activation of the mTORC1 pathway and reduced expression of PTEN.

(A) Fold expression of the mTOR pathway genes from RNA-sequencing data. (B–D) Naive CD4+ T cells were isolated from spleens of mice, and stimulated with anti-CD3 (0.5 μg/mL) and anti-CD28 (1.0 μg/mL) for 72 hours. (B) Total protein was immunoblotted for pS6 and total S6. Data are from 1 representative of 3 independent experiments. Graph shows relative densitometry quantitation of pS6 and S6 (n = 3 each). (C) After 72 hours, additional stimulation with anti-CD3 (10 μg/mL) and anti-CD28 (10 μg/mL) for 5 minutes was performed. Total protein was immunoblotted for p-AKT and total AKT. Data are from 1 representative of 4 independent experiments. Graph shows relative densitometry quantitation of p-AKT and AKT (n = 4 each). (D) Total RNA was isolated and reverse transcribed. Pten expression was measured by real-time qPCR and normalized to housekeeping gene cyclophilin A (n = 5 each). (E) Total protein was isolated from total T cells and immunoblotted for PTEN and β-actin. Data are from 1 representative of 4 independent experiments. Graph shows densitometric quantitation of PTEN normalized to β-actin (n = 4 each). (F) HEK293T cells were transfected with a pGL3–PTEN 3′ UTR luciferase plasmid and cotransfected with either a pcDNA3.1 empty vector (EV) or an Srsf1 (pSrsf1) plasmid, and included an internal control pRL-TK luciferase plasmid. Cells were lysed and dual luciferase assays performed. Graph shows relative luciferase units normalized to the internal luciferase control from n = 4 in 4 independent experiments. (G) T cells from spleens of mice were transfected with a PTEN-3′ UTR luciferase plasmid and either EV or pSrsf1 plasmid. Twenty-four hours later, cells were lysed and luciferase activity measured as in F (n = 7). Unpaired t test (B–E); 1-way ANOVA with Bonferroni’s correction (F and G); *P < 0.05, **P < 0.005, ***P < 0.0005; mean ± SEM.
Rapamycin treatment reduced proinflammatory cytokine production by T cells and alleviated autoimmunity in Srsf1-cKO mice.
Recently, hyperactivity of the mTOR pathway has been associated with aberrant T cell responses in SLE (15, 43), and targeted inhibition of this pathway with the lipophilic macrolide antibiotic rapamycin has led to improved T cell function and shown efficacy in a recent clinical trial in patients with active SLE (44). To assess whether the increased mTORC1 activation of T cells from Srsf1-cKO mice contributes to their proinflammatory phenotype and autoimmune phenotype, we examined the effect of rapamycin. Treatment of spleen cells from Srsf1-cKO mice ex vivo with rapamycin reduced the production of proinflammatory cytokines IL-17 (Figure 5, A and B) and IFN-γ (Figure 5, C and D) from CD4+ T cells. Furthermore, to assess the effects of rapamycin on disease in vivo, we treated Srsf1-cKO mice with rapamycin and evaluated the effect on features of autoimmunity. We observed reduced levels of IL-17 cytokine production ex vivo (Figure 5E), and decreased proportions of CD4+ PD1+CXCR5+ Tfh cells in the spleens from cKO mice (Figure 5F). Importantly, we observed a reduction in the levels of serum dsDNA and histone autoantibodies (Figure 5G). In addition, treatment with rapamycin reduced T cell infiltration into the kidneys (Figure 5H). These data indicate that deficiency of SRSF1 leads to increased activation of the mTOR pathway, the ensuing proinflammatory function of T cells, and systemic autoimmunity, which can be corrected by rapamycin.
Figure 5. Rapamycin treatment reduces proinflammatory cytokine production by T cells and alleviates autoimmunity in Srsf1-cKO mice.

(A–D) Spleen cells from WT or Srsf1-cKO mice were cultured for 4 hours with PMA plus ionomycin in the presence of monensin. Rapamycin (1 nM or 10 nM) was added for the duration of cultures. Cells were collected, surface stained, fixed, and permeabilized for intracellular cytokine staining. (A and C) Plots show IL-17 and IFN-γ intracellular staining gated on live CD4+ T cells. (B) Graphs show data from n = 8 mice in 7 independent experiments. (D) Graph shows data from n = 15 mice each in 10 independent experiments. (E–H) Rapamycin (2 mg/kg) or PBS was administered to WT and Srsf1-cKO mice by intraperitoneal injection once every 2 days for 4 weeks (F and G) or every day for 1 week (E and H). (E) Spleen cells were cultured with anti-CD3 (2 μg/mL) and anti-CD28 (2 μg/mL) for 24 hours and IL-17A measured in the supernatants by ELISA. Data are shown as fold values normalized to controls (n = 5 mice in 2 independent experiments). (F) Spleen cells were analyzed by flow cytometry, and graph shows the frequency of Tfh cells in spleen (n = 5–6 mice in 2 independent experiments). (G) Serum was collected and autoantibodies measured by ELISA (n = 5–6 mice in 2 independent experiments). (H) Cells from kidneys were analyzed by flow cytometry, and graph shows the frequency of T cells in kidneys (n = 5 mice in 2 independent experiments). One-way ANOVA with Bonferroni correction (B, D, E, and F); 2-tailed unpaired t test (G and H); *P < 0.05, **P < 0.005; mean ± SEM.
PTEN overexpression reduced proinflammatory cytokine production by T cells from Srsf1-cKO mice.
To confirm that SRSF1 modulates T cell activation via regulation of PTEN expression, PTEN was overexpressed by transient transfections in T cells from WT and Srsf1-cKO mice, followed by stimulation with PMA and ionomycin for 4 hours. We confirmed the overexpression of PTEN at the mRNA and protein levels (Supplemental Figure 6, A and B). We found that cytokine production is decreased by PTEN overexpression in T cells from Srsf1-cKO mice but not in WT T cells (Figure 6, A and B). Of note, transfection by electroporation is toxic to cells and accounts for the overall reduced frequencies of IFN-γ–producing cells (Figure 6) compared with untransfected cells (Figure 5C and Supplemental Figure 6C). These data indicate that deficiency of SRSF1 leads to decreased PTEN expression, increased activation of the mTOR pathway and the ensuing proinflammatory function of T cells (Supplemental Figure 7) which contribute to systemic autoimmune disease.
Figure 6. PTEN overexpression reduces proinflammatory cytokine production by T cells from Srsf1-cKO mice.

(A) T cells were isolated from spleens of WT or Srsf1-cKO mice and transfected by electroporation with empty vector (EV) or PTEN overexpression plasmid (pPTEN). Sixteen hours later, cells were stimulated with PMA plus ionomycin in the presence of monensin for 4 hours and cells were collected, surface stained, fixed, and permeabilized for intracellular cytokine staining. (B) Graphs show average data from n = 9 mice in 5 independent experiments. Two-tailed paired t test, *P < 0.05, mean ± SEM.
PTEN is decreased in T cells from patients with SLE with low SRSF1 levels.
Although PTEN expression is decreased and contributes to the hyperactivity of B cells in patients with SLE (28), its role in T cells in SLE has not been studied. Given our findings of increased mTOR activity and the decreased expression and regulatory role of PTEN in T cells from the Srsf1-cKO mice (Figure 4, Figure 5, and Figure 6), we asked whether PTEN expression levels are altered in T cells from patients with SLE. Therefore, we assessed PTEN and SRSF1 protein expression levels in T cells from patients with SLE and compared with age-, race-, and sex-matched healthy control individuals. We found that the expression of PTEN was decreased in T cells from patients with SLE who had decreased expression levels of SRSF1, compared with those from healthy individuals (Figure 7A). Importantly, we observed a linear correlation between PTEN and SRSF1 expression levels in T cells from patients with SLE (R2 = 0.226, P = 0.0163, Figure 7B). These results suggest that the low SRSF1 levels may contribute to the reduced expression of PTEN in T cells from patients with SLE.
Figure 7. PTEN is decreased in T cells from patients with SLE and correlates with SRSF1 levels.

SRSF1 overexpression increases PTEN, suppresses mTORC1 activity, and reduces proinflammatory cytokine production. (A and B) Peripheral blood T cells were isolated from patients with SLE and normal (N) healthy control individuals. Total protein was immunoblotted for PTEN, SRSF1, and β-actin. (A) Data are from 1 representative of 10 independent experiments. Graph shows relative quantitation by densitometry (n = 14 SRSF1 low and n = 12 SRSF1 normal patients). (B) Graph shows a linear correlation between relative PTEN and SRSF1 expression. (C) Peripheral blood T cells were isolated from patients with SLE and transfected with empty vector (EV) or SRSF1 overexpression plasmid (pSrsf1). At 16 to 18 hours after transfections, SLE T cells were stimulated with anti-CD3, anti-CD28, and crosslinker for 5 minutes. Total protein was immunoblotted for PTEN. Data are from 1 representative of 5 independent experiments (n = 10). Graph shows relative quantitation by densitometry. (D) Total protein was immunoblotted for pS6 and total S6 in stimulated SLE T cells after transfection as described in C. Data are from 1 representative of 4 independent experiments (n = 4). Graph shows relative quantitation by densitometry. (E) At 16 to 18 hours after transfection, cells were stimulated with PMA and ionomycin in the presence of monensin. After 4 hours, cytokine production was analyzed by flow cytometry. Graphs show quantitation of cytokine data (n = 8). Paired t test, *P < 0.05, mean ± SEM. The mTOR pathway via the regulation of PTEN expression. In T cells from KO mice and patients with SLE, reduced SRSF1 levels contribute to reduced PTEN levels and increased activity of the mTOR pathway. Rapamycin blocks the mTORC1 pathway and reduces proinflammatory cytokines.
SRSF1 overexpression increased PTEN levels, suppressed mTORC1 activity, and reduced proinflammatory cytokine production in T cells from patients with SLE.
To assess whether SRSF1 overexpression would increase PTEN expression and reduce mTORC1 activity and the aberrant cytokine production by T cells from patients with SLE, we transiently transfected T cells from patients with SLE with an Srsf1-expression vector followed by stimulation with anti-CD3 and anti-CD28. We found that SRSF1 overexpression increased PTEN protein levels (Figure 7C), reduced activation of the mTORC1 pathway as evidenced by a reduction in pS6 expression levels (Figure 7D), and led to reduced frequencies of IL-17– and IFN-γ–producing CD4+ T cells (Figure 7E). These findings show that decreased SRSF1 expression contributes to mTORC1 activation and proinflammatory cytokine production in T cells from patients with SLE through the control of PTEN.
Discussion
In this study, we demonstrate that selective deletion of SRSF1 in T cells in mice leads to aberrantly activated proinflammatory cytokine-producing Teff cells, leading to systemic autoimmunity and lupus nephritis (Figure 1 and Figure 2). We report that SRSF1 controls the expression of genes involved in T cell activation, differentiation, and cytokine signaling (Figure 3). At the mechanistic level, we demonstrate that SRSF1 regulates the 3′ UTR of the mRNA of the mTOR inhibitor PTEN and limits its expression, causing increased mTOR activity (Figure 4). Rapamycin treatment corrects proinflammatory cytokine production in T cells and alleviates autoimmunity in Srsf1-deficient mice (Figure 5). Of clinical relevance is our finding that T cells from patients with SLE with low expression levels of SRSF1 had decreased levels of PTEN, which correlate with decreased expression levels of SRSF1 (Figure 7). Further, restitution of SRSF1 levels corrected the proinflammatory cytokine production by hyperactive SLE T cells (Figure 7). Thus, we uncover SRSF1 as what we believe is a novel molecular brake in the control of T cell hyperactivity and the prevention of autoimmune disease. Our findings highlight an important role for SRSF1 in the immune system and in the control of immune-mediated disease. SRSF1 deficiency unrestrains T cell activation, unleashes the production of proinflammatory cytokines IL-17 and IFN-γ by CD4+ Teff cells, and leads to activation of B cells and inflammatory infiltration in target organs, leading to systemic autoimmunity and organ inflammation. Increased activity of the mTOR pathway contributes to the proinflammatory phenotype, which is rescued by rapamycin administration.
The mTOR pathway is a recently emerging crucial therapeutic target in autoimmune diseases, including SLE (15, 43). Increased mTORC1 activity has been demonstrated in T cells from patients with SLE and correlates with expansion of proinflammatory cytokine-producing T cells (21, 22). Treatment of lupus-prone MRL/lpr mice with rapamycin, an inhibitor of mTORC1 signaling, improved disease parameters (45). Importantly, a recent clinical trial of rapamycin in patients with SLE has shown efficacy in patients with active SLE (44). Here, we find that SRSF1 deficiency in mice results in increased activation of mTORC1 pathway, which parallels the observed increased mTORC1 activity (15) and the decreased SRSF1 expression in patients with SLE (37).
Although PTEN is known to suppress the mTOR pathway and the expression of PTEN is decreased in B cells from patients with SLE (28), the role of PTEN in T cells from patients with SLE remains unknown. We previously showed that the expression levels of SRSF1 are decreased in SLE T cells and inversely correlate with disease activity, suggesting the important role of SRSF1 in SLE T cells (37). Our current study extends this concept with a number of crucial new findings. Here we show a linear correlation between PTEN and SRSF1 in T cells from patients with SLE, and that patients with lupus with low SRSF1 expression levels exhibit lower expression levels of PTEN. Furthermore, the overexpression of SRSF1 rescues PTEN expression in lupus T cells (Figure 7). This is the first evidence showing the reduction of PTEN in T cells in a subset of patients with SLE. Moreover, the overexpression of SRSF1 in lupus T cells reduces mTOR activity and the production of proinflammatory cytokines, suggesting that the SRSF1-PTEN-mTOR pathway may be a novel therapeutic target in patients with SLE.
T cell quiescence is enforced by molecules able to restrain T cell activation, and the expression of such molecules generally subsides upon TCR stimulation to enable effector differentiation programs. These genes include, among others, the Kruppel-like factor (KLF) genes, Schlafen-2, Tsc2, and the Forkhead box (Foxo) family (46). T cells from the Srsf1-cKO mice preferentially expressed genes involved in the Foxo1 signaling pathway, cell cycle, T cell activation, cytokine-cytokine receptor interaction, and Th differentiation programs. Therefore, it appears that SRSF1 may be involved in the maintenance of resting T cell quiescence. In agreement with these findings is our previous finding that high SRSF1 protein levels in resting human T cells decreased following TCR stimulation due to degradation by the ubiquitin-proteasome pathway (36). In contrast, deficiency or decreased expression of SRSF1 as recorded in our Srsf1-cKO mice and in patients with SLE, respectively, contributes to T cell activation, loss of tolerance, autoimmunity, and organ inflammation. Of direct clinical relevance is the observation that T cells from patients with SLE exhibit a hyperactive phenotype and functional defects (13), which are linked mechanistically to decreased SRSF1 expression (37).
A limitation of our study is that the T cell–restricted deletion of Srsf1 is under the distal Lck promoter, which is expressed late during thymic development, as opposed to the proximal Lck promoter, which is expressed early in the double-negative thymocytes. Therefore, the role of SRSF1 in early thymic development is bypassed, which may differentially influence T cell physiology and function. Also, Srsf1 is absent from birth and does not address the possibility of altering T cell function should its levels decrease later in life. Deletion of Srsf1 in adult mice may result in differential physiological and pathophysiological outcomes. SRSF1 levels are decreased in T cells from patients with SLE but are not completely absent. Therefore, while the homozygous Srsf1-cKO mice are used in proof-of-concept studies here, the heterozygous (distal.Lckcre.Srsf1flox/wt mice) Srsf1-cKO mice, which also develop some features of T cell activation and signs of autoimmune disease (unpublished observations), may serve as a better model for human disease in future studies. Because SRSF1 regulates target genes via various mechanisms, it is possible that other pathways also contribute to T cell activation and autoimmunity. In addition, although the frequency of Tregs and functional results from short-term in vitro suppression assays are comparable between WT and Srsf1-cKO mice, further studies (including generating Treg conditional Srsf1-cKO mice) are required to assess precisely the role of SRSF1 in Tregs. Furthermore, the differences observed in the distribution of activated/memory T cell populations within the CD4 and CD8 compartments imply a potentially differential role of SRSF1 in CD4+ and CD8+ T cells (Figure 2, C–E). Besides these limitations, our patient numbers in the human studies are small, and a larger cohort of patients with SLE is needed to systematically define the clinical characteristics of patients stratified based on molecular levels of SRSF1 and PTEN. In addition, whether T cells from patients with SLE with and without low SRSF1 expression exhibit differential response to rapamycin needs to be examined. Increased activation of the mTOR pathway is associated with altered metabolism in T cells in mice and in patients with SLE (47, 48). It would be interesting to assess whether Srsf1-deficient T cells exhibit such defects.
In conclusion, we have uncovered what we believe is a previously unrecognized role of SRSF1 in T cell physiology and demonstrated the impact of its absence on the expression of autoimmune disease. Loss of SRSF1 limits the expression of the mTOR repressor PTEN, leading to aberrant T cell activation and the expression of systemic autoimmunity. Our results suggest that prevention of the loss of SRSF1 should be of therapeutic value to autoimmune diseases.
Methods
Human subjects.
Patients with SLE, all fulfilling the American College of Rheumatology classification criteria (49), were recruited at the rheumatology clinic at Beth Israel Deaconess Medical Center (BIDMC), Boston, MA (Supplemental Table 1). Age-, race-, and sex-matched healthy individuals were recruited as controls. Peripheral blood was drawn from patients by venipuncture.
Mice.
C57BL/6J (stock 000664), B6.129S4-Srsf1-flox (stock 018020), and B6.dLck.Cre (stock 012837) mice were purchased from The Jackson Laboratory. In most experiments, sex-matched littermates were used as WT controls. All mice were maintained in the specific pathogen–free animal facility at BIDMC.
Antibodies and reagents.
The following flow cytometry antibodies were from Biolegend: anti–mouse-CD4 (clone GK1.5), CD8a (clone 53-6.7), CD11b (clone M1/70), CD11c (clone N418), CD19 (clone 6D5), CD25 (clone PC61), CD44 (clone IM7), B220 (clone RA3-6B2), CD62L (clone MEL-14), CD69 (clone H1.2F3), CD90.2 (clone 53-2.1), TCR-β (clone H57-597), IgD (clone 11-26c.2a), IL-4 (clone 11B11), IL-17A (clone JC11-18F10.1), IFN-γ (clone XMG1.2), CXCR5 (clone L138D7), Ki67 (clone 16A8), CD127 (clone A019D5), PD-1 (clone 29F.1A12), GL7 (clone GL7), purified anti–mouse-CD3 (clone 145-2C11), CD28 (clone 37.51), CD16/32 (Fc block), anti–human CD3 (clone OKT3), CD4 (clone OKT4), CD8 (clone SK1), and IFN-γ (clone 4S.B1). Brilliant Violet 421–conjugated streptavidin, Zombie Aqua Fixable Viability kit mouse IL-17A ELISA kit, and carboxyfluorescein succinimidyl ester (CFSE) were also from Biolegend. Anti-mouse Foxp3 (clone FJK-16s) antibody, mouse regulatory T cell staining kit, and anti–human IL-17A antibody (clone eBIO64DEC17) were from eBioscience. Anti–mouse Fas (clone Jo2) antibody, Cytofix/Cytoperm, Perm/Wash buffer, and monensin were from BD Biosciences. ACK lysing buffer was from Thermo Fisher Scientific. SRSF1 rabbit polyclonal antibody for flow cytometry was from LifeSpan Biosciences and monoclonal antibody for Western blot (clone 96) was from Thermo Fisher Scientific. Anti-S6 ribosomal protein phospho-Ser 235 and Ser 236 (clone D57.2 2E), anti-S6 ribosomal protein (clone 5G10), anti-PTEN (clone 138G6), anti–phospho-AKT (Ser473; clone D9E), anti–phospho-AKT (Thr308; clone 244F9), and anti-AKT (clone C67E7) antibodies were from Cell Signaling Technology. Phorbol myristic acid (PMA), ionomycin, mitomycin C, and β-actin antibody (clone AC-74) were from Sigma-Aldrich. Goat anti–rabbit IgG horseradish peroxidase (HRP) and goat anti–mouse IgG HRP antibodies were from Thermo Fisher Scientific. Rapamycin and goat anti–mouse IgG were from Millipore. HEK293T cells were purchased from American Type Culture Collection (ATCC). The pcDNA3.1-SRSF1 expression plasmid was a gift from James Manley (Columbia University, NY), and the pGL3-PTEN 3′ UTR plasmid was a gift from Jeffrey Rosen (Addgene plasmid 28104). The pcDNA3-FLAG PTEN plasmid was a gift from Jaewhan Song (Addgene plasmid 78777).
Cell isolation and tissue processing.
Spleens and lymph nodes were homogenized using a syringe plunger and mesh cell strainer. After perfusion, kidneys were homogenized and digested in HBSS medium with 1.5 mg/mL collagenase B (Boehringer Mannheim) at 37°C for 30 minutes. RBC lysis was performed with ACK lysing buffer. All cell cultures were in RPMI complete medium (RPMI plus 10% FBS plus penicillin and streptomycin antibiotics). Blood samples from mice were collected by tail vein incision and capillary tube collection, and serum was separated by centrifugation. Urine samples were collected from mice in individual metabolic cages and urine protein was measured by Multistix (Siemens). Human T cells from peripheral blood were isolated using the Rosette Sep human T cell enrichment cocktail (STEMCELL Technologies). For histopathology, tissues were immediately fixed in 10% formalin overnight, processed in an automated tissue processor, embedded into paraffin blocks, and sent to the BIDMC histopathology core for sectioning and slide staining with H&E. Slides were evaluated blind for histopathology scoring, utilizing semiquantitative scoring criteria for lupus nephritis (50).
Flow cytometry.
Zombie aqua viability dye was used for live/dead cell staining. Surface staining was performed in FACS staining buffer (PBS plus 2% FBS) on ice for 20 minutes with Fc block. For cytokine production, cells from mice were stimulated for 4 hours in culture medium with PMA (100 ng/mL), ionomycin (1 μM), and monensin (1 μL/mL). Human T cells were stimulated with PMA (10 ng/mL), ionomycin (400 ng/mL), and monensin (1 μL/mL) for 4 hours. Cells were surface stained followed by fixation and permeabilization. Appropriate antibodies were used for intracellular staining for cytokines or Foxp3 and Ki67. Flow cytometry data were acquired on a BD LSRII or CytoFLEX LX and analyzed with FlowJo software. All procedures were performed according to the manufacturer’s instructions.
Autoantibody detection.
Sera from 12-week-old WT and Srsf1-cKO mice and one 16-week-old MRL/lpr mouse (as positive control) were analyzed for the presence of autoantibodies using the Autoantigen Microarray Super Panel (128-antigen panel) at the Genomics and Microarray Core facility at the University of Texas, Southwestern Medical Center (Dallas, TX). Heatmaps were prepared using R software. Antinuclear antibody (ANA) in serum was measured using an ELISA kit (Alpha Diagnostic International) following the manufacturer’s instructions. Briefly, the serum was diluted and assayed for antigen reactivity on precoated plates followed by incubation for 60 minutes. Bound IgG was detected with anti–mouse IgG HRP antibody and developed with a tetramethylbenzidine (TMB) substrate. Plates were read at 450 nm on a spectrophotometer. To detect anti–double stranded (ds)DNA and anti–histone antibodies, Immulon II plates (Dynatech) precoated with BSA were coated individually with 50 μg/mL calf thymus DNA (Sigma-Aldrich) or 50 μg/mL calf thymus histone (Sigma-Aldrich). The serum was diluted (1:500) and assayed for autoantigen reactivity against the antigens described above by incubation overnight at 4°C. Bound IgG was detected with a goat polyclonal HRP–anti-mouse IgG detection antibody (SouthernBiotech) and visualized at 450 nm using a TMB substrate (Sigma-Aldrich).
mRNA expression and RT-PCR.
Total RNA was isolated using the RNeasy mini kit (Qiagen) and reverse-transcribed into cDNA using the RNA to cDNA premix (Clontech). Real-time PCR amplification was carried out with SYBR Green I using a LightCycler 480 (Roche) and the following program: initial denaturation at 95°C for 5 minutes; 40 cycles of amplification (denaturation at 95°C for 15 seconds, annealing at 60°C for 15 seconds, extension at 72°C for 30 seconds); one cycle of melting curves (95°C for 15 seconds, 65°C for 2 minutes, and 97°C continuous), and a final cooling at 37°C. Threshold cycle values were used to calculate relative mRNA expression by the ΔCt relative quantification method. Primer sequences are listed in Supplemental Table 2.
RNA sequencing.
Naive CD4+ T cells were isolated from spleens of 7-week-old WT and Srsf1-cKO mice (n = 3 each) and stimulated with anti-CD3 (0.5 μg/mL) and anti-CD28 (1 μg/mL) for 72 hours. Cells were restimulated with PMA (100 ng/mL) and ionomycin (1 μM) for 4 hours. Total RNA was extracted using the RNeasy mini kit (Qiagen) and submitted for sequencing to the Molecular Biology Core Facility (MBCF) at the Dana-Farber Cancer Institute (DFCI). Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits according to the manufacturer’s protocols. Samples were sequenced on an Illumina NextSeq500 run with single-end 75-bp reads. Data analyses were performed by the MBCF at DFCI and the Harvard T.H. Chan School of Public Health Biostatistics Core. The data were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE 136286.
Western blot.
To detect pS6, naive CD4+ T cells were isolated from spleens of 9- to 10-week-old WT and Srsf1-cKO mice and stimulated with anti-CD3 (0.5 μg/mL) and anti-CD28 (1 μg/mL) for 72 hours. Human T cells were stimulated with soluble anti-CD3 (5 μg/mL), anti-CD28 (2.5 μg/mL) and cross linker goat anti–mouse IgG (10 μg/mL) for 5 minutes. Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Boston Bioproducts). Lysates were electrophoresed on NuPAGE 4%–12% Bis-Tris gels (Life Technologies) and transferred to PVDF membrane. Membranes were blocked with 5% (wt/vol) nonfat milk in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 1 hour, incubated with primary antibody (1:1000, or 1:10,000 for β-actin antibody) in 5% milk in TBS-T or Hikari solution A (Nacalai Tesque) at 4°C overnight or at room temperature for 2 hours for β-actin antibody. Membranes were washed 3 times with TBS-T, incubated with HRP-conjugated secondary antibody (1:2000 for ECL detection reagents or 1:4000 for ECL prime detection reagents; GE Healthcare) for 1 hour, washed 3 times with TBS-T, developed with ECL reagents, and visualized by a Fujifilm LAS-4000 imager or Bio-Rad ChemiDoc imager. Densitometry was performed using Quantity One software (Bio-Rad).
Transfections.
HEK293T cells were cultured in DMEM complete medium. One day prior to transfection, 0.3 × 106 cells were seeded in a 12-well plate. Cells were cotransfected with the pGL3-PTEN 3′ UTR luciferase plasmid and either pcDNA3.1 empty vector (EV) or pcDNA3.1-Srsf1 vector using Lipofectamine (Invitrogen). All transfections included a pRL-TK Renilla luciferase plasmid as internal control. Human T cells and mouse T cells were transfected using the Amaxa human T cell nucleofector kit and the mouse T cell nucleofector kit, respectively, following the manufacturer’s instructions. Briefly, 3 × 106 to 6 × 106 cells were resuspended in 100 μL nucleofector solution, plasmid DNA was added, and cells were transferred into a cuvette and electroporated using the U-014 program for human cells or the X-001 program for mouse cells in the nucleofector device. Cells were immediately rescued into prewarmed medium and cultured overnight.
Luciferase assays.
Cells were transfected as described above. After 24 hours in culture, cells were collected, lysed in passive lysis buffer (Promega), and luciferase activity was quantified on a luminometer using the dual-luciferase assay system (Promega) according to the manufacturer’s instructions.
Immunofluorescence and immunohistochemistry staining.
For immunofluorescence staining, spleens were embedded in OCT tissue media (Tissue-Tek) and frozen on dry ice. Frozen sections (7-μm thickness) were fixed to slides in ice-cold acetone for 15 minutes and air dried for 30 seconds. The sections were blocked with 10% horse serum for 30 minutes at room temperature, then stained for 30 minutes at room temperature in a humidified chamber with fluorescence-conjugated anti-IgD (catalog 405717; Biolegend), anti-CD4 (catalog 100533; Biolegend), anti-B220 (catalog 103207; Biolegend), and biotinylated PNA (catalog B-1075; Vector Laboratories). Biotinylated PNA was detected using by streptavidin–Alexa Fluor 350 (catalog S-11249; Life Technologies). For immunohistochemistry staining, slides of formalin-fixed, paraffin-embedded kidneys were incubated with purified anti–mouse CD3 antibody (clone 100201; Biolegend) overnight at 4°C after antigen retrieval. The primary antibody was detected with rabbit anti–rat HRP secondary antibody (clone ab6734, Abcam).
In vitro suppression assays.
Total T cells were isolated from spleens by magnetic assisted cell sorting (MACS), using the Pan T cell isolation kit (Miltenyi Biotech). Conventional CD4+ T cells (Tconv) (CD4+CD25–CD127hi) and Treg cells (CD4+CD25+CD127lo) were sorted by flow cytometry (FACSAria II, BD Bioscience). Tconv cells were labeled with CFSE and cocultured with Treg cells at increasing ratios for 3 days in the presence of mitomycin C–treated splenocytes and anti-CD3 and anti-CD28, and proliferation of Tconv was analyzed by flow cytometry.
Rapamycin treatment.
Rapamycin was dissolved in DMSO and diluted in PBS. For in vitro culture treatment experiments, a final concentration of 1 nM or 10 nM rapamycin was added for the duration of cell cultures. For in vivo treatment experiments, rapamycin (2 mg/kg dissolved in DMSO, diluted in 150 μL PBS) or PBS (equal volume DMSO in 150 μL PBS) was administered to mice by intraperitoneal injection once every 2 days for 4 weeks. After 4 weeks of treatment, mice were euthanized and spleen cells were analyzed by flow cytometry, and sera was collected to measure autoantibodies by ELISA. In some experiments, mice were treated with rapamycin every day for 1 week. Kidneys were collected to analyze T cell infiltration by flow cytometry. Spleen cells were cultured with anti-CD3 (2 μg/mL) and anti-CD28 (2 μg/mL) for 24 hours and IL-17A was measured in supernatants by ELISA (Biolegend).
Statistics.
Student’s 2-tailed t test, Mann-Whitney U test, and 1-way ANOVA were used to calculate statistical significance among groups. A P value of less than 0.05 was considered significant. In all graphs, data represent mean ± SEM.
Study approval.
For human studies, written informed consent was obtained from all participants and all studies were approved by the institutional review board (Committee on Clinical Investigations) at BIDMC. All animal studies were approved by the Institutional Animal Care and Use Committee at BIDMC.
Author contributions
VRM conceptualized the study. TK, HL, DC, and VRM designed and performed the experiments, and analyzed and interpreted data. TK, HL, GCT, and VRM prepared the manuscript.
Supplementary Material
Acknowledgments
We thank Andrew Gillooly, Michael Mosho, and Melissa Carr-Reynolds for excellent technical assistance; Suzanne Krishfield for recruiting patients with SLE and healthy individuals; Donna Farber and Martin Flajnik for critical reading of the manuscript; and Amir Sharabi and Abel Suarez-Fueyo for helpful discussions. This work was funded by NIH grants K01AR060781 and R01AR068974 to VRM, R01AI42269 to GCT, and an SICPA Foundation grant to DC.
Version 1. 09/05/2019
In-Press Preview
Version 2. 11/04/2019
Electronic publication
Version 3. 12/02/2019
Print issue publication
Funding Statement
R01AR068974, K01AR060781
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(12):5411–5423.https://doi.org/10.1172/JCI127949.
Contributor Information
Takayuki Katsuyama, Email: tkatsuya@bidmc.harvard.edu.
Hao Li, Email: hli13@bidmc.harvard.edu.
Denis Comte, Email: denis.comte@gmail.com.
George C. Tsokos, Email: gtsokos@bidmc.harvard.edu.
Vaishali R. Moulton, Email: vmoulton@bidmc.harvard.edu.
References
- 1.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Yen EY, Singh RR. Brief report: lupus-an unrecognized leading cause of death in young females: a population-based study using nationwide death certificates, 2000-2015. Arthritis Rheumatol. 2018;70(8):1251–1255. doi: 10.1002/art.40512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front Immunol. 2018;9:427. doi: 10.3389/fimmu.2018.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Crow MK, Olferiev M, Kirou KA. Targeting of type I interferon in systemic autoimmune diseases. Transl Res. 2015;165(2):296–305. doi: 10.1016/j.trsl.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo MS, Tsokos GC. Recent developments in systemic lupus erythematosus pathogenesis and applications for therapy. Curr Opin Rheumatol. 2018;30(2):222–228. doi: 10.1097/BOR.0000000000000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caielli S, et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med. 2019;25(1):75–81. doi: 10.1038/s41591-018-0254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu HC, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 9.Yi W, et al. The mTORC1-4E-BP-eIF4E axis controls de novo Bcl6 protein synthesis in T cells and systemic autoimmunity. Nat Commun. 2017;8(1):254. doi: 10.1038/s41467-017-00348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amarilyo G, Lourenço EV, Shi FD, La Cava A. IL-17 promotes murine lupus. J Immunol. 2014;193(2):540–543. doi: 10.4049/jimmunol.1400931. [DOI] [PubMed] [Google Scholar]
- 11.Koga T, Ichinose K, Tsokos GC. T cells and IL-17 in lupus nephritis. Clin Immunol. 2017;185:95–99. doi: 10.1016/j.clim.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi SC, Morel L. B cell contribution of the CD4+ T cell inflammatory phenotypes in systemic lupus erythematosus. Autoimmunity. 2017;50(1):37–41. doi: 10.1080/08916934.2017.1280028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katsuyama T, Tsokos GC, Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. 2018;9:1088. doi: 10.3389/fimmu.2018.01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med. 2017;23(7):615–635. doi: 10.1016/j.molmed.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–182. doi: 10.1038/nrrheum.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol. 2014;5:686. doi: 10.3389/fimmu.2014.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12(9):888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perl A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann N Y Acad Sci. 2015;1346(1):33–44. doi: 10.1111/nyas.12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang K, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. 2013;39(6):1043–1056. doi: 10.1016/j.immuni.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol. 2014;192(9):4134–4144. doi: 10.4049/jimmunol.1301859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai ZW, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–2246. doi: 10.4049/jimmunol.1301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 24.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckler JL, Walsh PT, Porrett PM, Choi Y, Turka LA. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J Immunol. 2006;177(7):4262–4266. doi: 10.4049/jimmunol.177.7.4262. [DOI] [PubMed] [Google Scholar]
- 26.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/- mice. Science. 1999;285(5436):2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki A, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14(5):523–534. doi: 10.1016/S1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 28.Wu XN, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. 2014;6(246):246ra99. doi: 10.1126/scitranslmed.3009131. [DOI] [PubMed] [Google Scholar]
- 29.Howard JM, Sanford JR. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA. 2015;6(1):93–110. doi: 10.1002/wrna.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Manley JL. New talents for an old acquaintance: the SR protein splicing factor ASF/SF2 functions in the maintenance of genome stability. Cell Cycle. 2005;4(12):1706–1708. doi: 10.4161/cc.4.12.2210. [DOI] [PubMed] [Google Scholar]
- 31.Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest. 2015;125(6):2220–2227. doi: 10.1172/JCI78087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nambiar MP, et al. Reconstitution of deficient T cell receptor zeta chain restores T cell signaling and augments T cell receptor/CD3-induced interleukin-2 production in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48(7):1948–1955. doi: 10.1002/art.11072. [DOI] [PubMed] [Google Scholar]
- 33.Chowdhury B, et al. Stability and translation of TCR zeta mRNA are regulated by the adenosine-uridine-rich elements in splice-deleted 3’ untranslated region of zeta-chain. J Immunol. 2006;177(11):8248–8257. doi: 10.4049/jimmunol.177.11.8248. [DOI] [PubMed] [Google Scholar]
- 34.Moulton VR, Kyttaris VC, Juang YT, Chowdhury B, Tsokos GC. The RNA-stabilizing protein HuR regulates the expression of zeta chain of the human T cell receptor-associated CD3 complex. J Biol Chem. 2008;283(29):20037–20044. doi: 10.1074/jbc.M710434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moulton VR, Tsokos GC. Alternative splicing factor/splicing factor 2 regulates the expression of the zeta subunit of the human T cell receptor-associated CD3 complex. J Biol Chem. 2010;285(17):12490–12496. doi: 10.1074/jbc.M109.091660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moulton VR, Gillooly AR, Tsokos GC. Ubiquitination regulates expression of the serine/arginine-rich splicing factor 1 (SRSF1) in normal and systemic lupus erythematosus (SLE) T cells. J Biol Chem. 2014;289(7):4126–4134. doi: 10.1074/jbc.M113.518662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc Natl Acad Sci USA. 2013;110(5):1845–1850. doi: 10.1073/pnas.1214207110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.et al. Decreased expression of Serine/arginine-rich splicing factor 1 in T cells from patients with active systemic lupus erythematosus accounts for reduced expression of RasGRP1 and DNA methyltransferase 1. Arthritis Rheumatol. 2018;70(12):2046–2056. doi: 10.1002/art.40585. [DOI] [PubMed] [Google Scholar]
- 39.Xiong Z, et al. Alternative splicing factor ASF/SF2 is down regulated in inflamed muscle. J Clin Pathol. 2006;59(8):855–861. doi: 10.1136/jcp.2005.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120(1):59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14(3):185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-Hur V, et al. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013;3(1):103–115. doi: 10.1016/j.celrep.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the mechanistic target of rapamycin in SLE: explosion of evidence in the last five years. Curr Rheumatol Rep. 2016;18(12):73. doi: 10.1007/s11926-016-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai ZW, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. 2018;391(10126):1186–1196. doi: 10.1016/S0140-6736(18)30485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 1994;37(2):289–297. doi: 10.1002/art.1780370219. [DOI] [PubMed] [Google Scholar]
- 46.Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends Immunol. 2012;33(5):224–230. doi: 10.1016/j.it.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perl A. Review: Metabolic control of immune system activation in rheumatic diseases. Arthritis Rheumatol. 2017;69(12):2259–2270. doi: 10.1002/art.40223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin Y, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan EM, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 50.Austin HA, Muenz LR, Joyce KM, Antonovych TT, Balow JE. Diffuse proliferative lupus nephritis: identification of specific pathologic features affecting renal outcome. Kidney Int. 1984;25(4):689–695. doi: 10.1038/ki.1984.75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.