

Abstract
Ankylosing spondylitis (AS) is a type of axial inflammation. Over time, some patients develop spinal ankylosis and permanent disability; however, current treatment strategies cannot arrest syndesmophyte formation completely. Here, we used mesenchymal stem cells (MSCs) from AS patients (AS MSCs) within the enthesis involved in spinal ankylosis to delineate that the HLA-B27–mediated spliced X-box–binding protein 1 (sXBP1)/retinoic acid receptor-β (RARB)/tissue-nonspecific alkaline phosphatase (TNAP) axis accelerated the mineralization of AS MSCs, which was independent of Runt-related transcription factor 2 (Runx2). An animal model mimicking AS pathological bony appositions was established by implantation of AS MSCs into the lumbar spine of NOD-SCID mice. We found that TNAP inhibitors, including levamisole and pamidronate, inhibited AS MSC mineralization in vitro and blocked bony appositions in vivo. Furthermore, we demonstrated that the serum bone-specific TNAP (BAP) level was a potential prognostic biomarker to predict AS patients with a high risk for radiographic progression. Our study highlights the importance of the HLA-B27–mediated activation of the sXBP1/RARB/TNAP axis in AS syndesmophyte pathogenesis and provides a new strategy for the diagnosis and prevention of radiographic progression of AS.
Keywords: Autoimmunity, Bone Biology
Keywords: Bone disease, Rheumatology

Introduction
Ankylosing spondylitis (AS) is a type of chronic arthritis characterized by inflammatory spondylitis, peripheral arthritis, and enthesitis. Typically, it occurs in young adult males and has a strong association with human leukocyte antigen-B27 (HLA-B27) (1). In addition to chronic spinal inflammation, the heterotopic formation of new bone frequently starts from the enthesis, where the ligament or tendon connects to bone, in one-third of patients within 2 years, resulting in ossification and ankylosis of adjacent vertebral bodies (“syndesmophytes”) that can lead to permanent disability (2, 3). Despite treatments such as NSAIDs, anti–TNF-α, or IL-17A inhibitor that inhibit inflammation, there are no treatment modalities that completely arrest syndesmophyte formation (4–11). The lack of treatments is associated with our incomplete understanding of the causative pathological roles of stromal activation, lack of a proper platform for drug screening, and absence of good biomarkers to select a high-risk group of patients who are most likely to exhibit radiographic progression. Although the accelerated osteogenesis of AS patient–derived bone marrow (BM) mesenchymal stem cells (MSCs) from hips or bone-derived precursors from facet joints has been reported (12–15), the role of MSCs from the enthesis or nidus of syndesmophytes, and the underlying genetic pathways involved, are largely unknown. Herein, we isolated MSCs from the enthesis involved in the spinal ankylosis of AS patients (AS MSCs) and found that the genetic HLA-B27–mediated spliced X-box–binding protein 1 (sXBP1)/retinoic acid receptor-β (RARB)/tissue-nonspecific alkaline phosphatase (TNAP) pathway contributed to the syndesmophyte formation of AS. In addition, we established a novel in vivo disease model that mimics the pathological bony apposition induced by AS MSCs, which might be a useful platform for the drug exploration of AS. We demonstrated that the repurposing drug pamidronate, with dual effects on TNAP inhibition and osteoporosis prevention, is a prospective candidate for blocking syndesmophyte formation. In a clinical setting, we used serum from AS patients of Western and Eastern cohorts, and found that serum bone-specific TNAP levels may be a prognostic biomarker to select high-risk AS patients with a propensity for radiographic progression. Our findings delineate that HLA-B27 mediates stromal activation in the pathogenesis of syndesmophyte formation, which might help the development of a new therapeutic strategy to prevent spinal ankylosis in AS in the future.
Results
AS MSCs derived from the enthesis involved in spinal ankylosis exhibit accelerated mineralization in a Runt-related transcription factor 2–independent manner.
Multipotent MSCs have the capacity for self-renewal and differentiation into multiple mesenchymal lineages, including osteoblasts, adipocytes, and chondrocytes (16). Here, we established an ex vivo cell culture model from MSCs within the enthesis involved in spinal ankylosis of 3 AS patients (AS MSCs) to study the activation of the stromal progenitor cells in AS. We compared these cells with MSCs from non-AS controls who underwent traumatic surgery (control MSCs) at a similar site (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI125212DS1). These cells adopted a fibroblastic-like morphology (Supplemental Figure 1A) and expressed surface markers characteristic of MSCs (Supplemental Figure 1B). They possessed trilineage differentiation potential as assayed by specific standard induction methods (Supplemental Figure 1C and ref. 17).
AS MSCs and control MSCs were cultured in defined osteogenic induction media to assess their osteogenic potential. Remarkably, alizarin red S (ARS) staining for calcium deposition and von Kossa staining for phosphates showed a significantly accelerated rate of mineralization in the 3 AS MSCs compared with the 3 control MSCs (Figure 1, A and B, and Supplemental Figure 2). Generally, the deposition of calcium phosphate crystals suggests the differentiation of MSCs into osteoblasts regulated by Runt-related transcription factor 2 (Runx2) (18). The Wingless (Wnt) and bone morphogenetic protein (BMP) (19, 20) pathways are important for normal MSCs’ commitment to osteoblastogenesis through Runx2. However, we found that neither of these pathways contributed to the accelerated mineralization in the ex vivo AS MSCs culture or in vivo BM specimens from AS patients (Supplemental Figure 3). In addition, although accelerated mineralization was observed in AS MSCs, Runx2 expression was comparable between AS MSCs and control MSCs (Figure 1C). Furthermore, Runx2 knockdown in AS MSCs by 2 independent shRNAs (Figure 1D) did not affect the accelerated mineralization (Figure 1, E and F). Likewise, the expressions of Runx2 downstream osteoblastic markers, including osteoadherin, osteocalcin, and collagen 1A1 (Col1), were not higher in AS MSCs compared with control MSCs after osteogenic induction (Figure 1, G–I). These results suggest that Runx2-independent acceleration in mineralization, but not mature osteoblastogenesis, may contribute to the pathological phenotype of AS MSCs after osteogenic induction.
Figure 1. Runx2-independent accelerated mineralization in AS MSCs.

(A) ARS staining of enhanced mineralization in AS MSCs cultured under osteogenic conditions at the indicated days compared with control MSCs. (B) Quantification of ARS staining showing the differential rate in mineralization between AS MSCs and control MSCs at the indicated days. (C) RT-qPCR of Runx2 mRNA levels in AS MSCs and control MSCs at days 0 and 7 under osteogenic induction. (D–F) MSCs were transduced with lentiviral vectors carrying 2 independent shRNAs against Runx2 (shRunx2) or control shRNA (shCtrl) under osteogenic conditions. (D) RT-qPCR showing the knockdown efficiency by shRunx2 in AS MSCs and control MSCs at day 7 under osteogenic induction, normalized to the value of control MSCs expressing shCtrl. (E) ARS staining showing the effects of Runx2 knockdown on the mineralization of AS MSCs and control MSCs with quantification (F) at day 18 under osteogenic induction. (G) Immunofluorescence staining of AS MSCs and control MSCs at day 14 under osteogenic induction with DAPI (blue) and osteoadherin-specific antibody (green). (H and I) RT-qPCR of collagen 1A1 (H) and osteocalcin (I) mRNA levels in AS MSCs and control MSCs at days 0 and 7 under osteogenic induction. All experiments were done in the AS patient group using AS MSCs (derived from A1, A2, and A3 with experimental triplicates) and in the control group using control MSCs (derived from C1, C2, and C3 with experimental triplicates). Data are shown as the mean ± SEM. *P < 0.05; **P < 0.01; ****P < 0.0001 by 1-way ANOVA, followed by Tukey’s honestly significant difference (HSD) test. Representative images from AS (A1) MSCs and control (C3) MSCs are shown in E and G. Scale bars: 200 μm (A and E); 20 μm (G).
Enhanced expression of TNAP is essential for abnormal mineralization in AS MSCs.
To investigate further the regulatory mechanism of accelerated mineralization in AS MSCs, we analyzed gene expressions between AS MSCs and control MSCs after osteogenic induction at days 0, 3, and 7 by microarray analyses. One hundred fifty-three genes and 109 genes were upregulated and downregulated, respectively (consistently >2-fold in AS MSCs at 3 time points) in comparison with the control MSCs, after osteogenic induction (Supplemental Tables 2 and 3). The distribution of the 10 most significant terms in the biological process ontology was obtained by Gene Ontology (GO) analysis (Supplemental Figure 4A). Results of the Ingenuity Pathway Analysis (IPA) of gene networks involved in osteogenesis pathways are shown in Figure 2A. Further validation of these genes involved in osteogenesis revealed that elevation of tissue-nonspecific alkaline phosphatase (TNAP) expression (Supplemental Figure 4, B–R, and Figure 2, B and C) and elevation of alkaline phosphatase (ALP) activity (Supplemental Figure 5A) were most closely linked with accelerated mineralization in AS MSCs compared with control MSCs, both before and after osteogenic induction. ALP is a large superfamily of ubiquitous ectoenzymes that catalyze dephosphorylation and transphosphorylation reactions. They include 4 isoenzymes — TNAP and placental, germ cell, and intestinal ALP — encoded by separate genes. Among them, TNAP is encoded by the ALPL gene and distributed in liver/bone/kidney tissues with alternative splicing transcript variants. It hydrolyzes the anti-mineral factor pyrophosphate into procalcifying inorganic phosphate to promote mineralization (21–23).
Figure 2. Enhanced expression of TNAP is essential for abnormal mineralization in AS MSCs.

(A) IPA of differentially expressed genes involved in osteogenesis between AS MSCs and control MSCs. (B and C) TNAP mRNA (B) and protein levels (C) in AS and control MSCs at the indicated days after osteogenic induction. (D) ARS staining of AS MSCs or control MSCs treated with TNAP inhibitors (100 μM levamisole, 100 μM beryllium sulfate, or 1 μg/mL pamidronate) under osteogenic induction with quantification (E). (F and G) TNAP mRNA (F) and protein levels (G) were suppressed by 2 shRNAs against TNAP in AS MSCs at day 7 under osteogenic induction. (H) ARS staining of AS MSCs expressing shTNAP or shCtrl under osteogenic induction with quantification (I). (J) ARS staining of control MSCs transduced with a control vector (pLAS2w) or vector overexpressing TNAP (pLAS2w-TNAP) with quantification (K). (L) Immunoblot shows TNAP protein expression in control MSCs transduced with pLAS2w or pLAS2w-TNAP. (M) ARS staining of AS MSCs and control MSCs cultured in GM with or without BGP at day 18 with quantification (N). All statistical data in the AS patient group and control group are from AS MSCs (A1, A2, and A3) and control MSCs (C1, C2, and C3), respectively, with 3 experimental repeats. Data are the mean ± SEM. **P < 0.01; ****P < 0.0001 by 2-tailed Student’s t test (2 groups) or 1-way ANOVA, followed by Tukey’s HSD test. Representative images from AS (A1) MSCs and control (C3) MSCs are shown in D, H, and M. Scale bars: 200 μm (D, H, J, and M).
To determine the role of TNAP in the abnormal mineralization of AS MSCs, we treated osteogenic cultures with uncompetitive (levamisole or pamidronate) (22, 23) and competitive (beryllium sulfate) (24) TNAP inhibitors. Notably, accelerated mineralization in AS MSCs was blocked effectively by TNAP inhibitors (Figure 2, D and E). A similar reduction of accelerated mineralization in AS MSCs was observed when the expression of TNAP was silenced by 2 independent shRNAs against TNAP (Figure 2, F–I, and Supplemental Figure 5B). Moreover, TNAP overexpression via lentiviral transduction in control MSCs showed enhanced mineralization (Figure 2, J–L). To demonstrate whether spontaneous mineralization occurred in AS MSCs, we cultured MSCs in growth medium (GM) in the presence of β-glycerophosphate (BGP; a substrate of TNAP for mineralization) (ref. 21 and Figure 2, M and N). As expected, MSCs cultured in GM did not calcify. However, AS MSCs exhibited spontaneously accelerated mineralization in the presence of BGP, suggesting that TNAP upregulation in AS MSCs was sufficient to enhance spontaneous mineralization in the presence of its substrate only. Similar results were demonstrated in 3 additional AS MSCs (Supplemental Table 4 and Supplemental Figure 6). Taken together, these results demonstrate that the enhanced expression of TNAP is essential for accelerated and spontaneous mineralization in AS MSCs.
TNAP blockade inhibits new bony appositions induced by AS MSCs in NOD-SCID mice.
Next, we established an AS MSC–based in vivo disease model to mimic pathological bony appositions, which was used to test the therapeutic potential of TNAP inhibitors. For this purpose, AS MSCs delivered in fibrin were implanted in the eroded right lamina of the lumbar spine segment (L4–5) after the decortication procedure to promote new bone ingrowth (25), which mimicked the pathophysiological scenario of microtrauma in AS. Entheses are regarded as sites of microtrauma resulting from increased biomechanical stress, in which new bone appositions tend to fill in the erosion of the cortical bone, joining the deeper bone in the eroded ends of ligaments (26, 27). Notably, NOD-SCID mice implanted with AS MSCs, but not control MSCs, developed new bony appositions (Figure 3A). Spinal tissues from implanted sites showed that AS MSCs formed bone-like tissues bridging with host bone, which consisted of bone-forming cells wrapped with osteoid, osteocytes within lacunae, and osteochondral-like tissues. In contrast, the implanted control MSCs only formed fibrous-like tissues, without bone-like or osteochondral tissues (Figure 3B). The time sequence of the development of new bony appositions in NOD-SCID mice implanted with AS MSCs is shown in Supplemental Figure 7. Furthermore, ectopic bony appositions induced by the implantation of AS MSCs were blocked when TNAP was silenced by shRNAs in contrast to control shRNA-transduced AS MSCs (Figure 3C). Consistently, the oral administration of TNAP inhibitors largely abrogated new bony appositions in NOD-SCID mice implanted with AS MSCs (Figure 3D), but did not alter the overall bone mineral density (Supplemental Figure 8). The quantitative volumes of new bony appositions between groups are shown in Figure 3E. Moreover, the administration of drugs did not induce whole-body weight loss (Supplemental Figure 9) or other systemic toxicity. These results suggest that TNAP blockade inhibits new bony appositions induced by AS MSCs in NOD-SCID mice.
Figure 3. TNAP blockade inhibits new bony appositions induced by AS MSCs in NOD-SCID mice.

(A, C, and D) Representative images of lumbar spine micro-CT of NOD-SCID mice implanted with AS MSCs (derived from A1, A2, and A3 with triplicates in each group) or control MSCs (derived from C1, C2, and C3 with triplicates in each group) in the artificial cortical defect of the right lamina of lumbar spine segment L4–5 (A); with AS MSCs (derived from A1, A2, and A3 with triplicates in each group) transduced with shCtrl or shTNAP (C); or with AS MSCs (derived from A1, A2, and A3 with triplicates in each group) plus daily oral administration of H2O (n = 9), levamisole (10 mg/kg) (n = 9), beryllium sulfate (7.5 mg/kg) (n = 9), or pamidronate (0.3 mg/kg) (n = 9) (D). Images were taken 3 weeks after implantation. Longitudinal view over L4–6 (left), longitudinal view at high magnification over L4 (middle), and cross-sectional view over L4 (right) are shown. New bony appositions are highlighted by a red rectangle (middle) and white arrow (right). (B) Representative H&E staining images showing MSC-implanted sites (arrowhead-dotted areas) in the spinal tissues. AS MSCs (A1) formed new woven bone apposition (asterisks) bridging with host bone (HB), with some osteochondral-like tissues surrounding the newly formed bone (white arrow), while control MSCs (C3) formed fibrous-like tissues (black arrow) in direct contact with host bone. Higher-magnification views of areas in red rectangles in upper panels are shown in lower panels (scale bars: 200 μm). (E) The quantitative volumes of new bony appositions (mm3) between groups (A, C, and D). Data are the mean ± SEM (n = 9 in E). ****P < 0.0001 by 2-tailed Student’s t test (2 groups) or 1-way ANOVA, followed by Tukey’s HSD test.
TNAP in BM and serum from AS patients is elevated significantly compared with that from controls, and serum bone-specific TNAP levels are associated with radiographic severity in AS patients.
Next, we examined whether the enhanced expression of TNAP could be detected in the BM and peripheral blood of AS patients. Immunohistochemical (IHC) staining of BM specimens (Supplemental Table 5) revealed the increased expression of TNAP in the BM of AS patients compared with normal individuals and non-AS patient controls (Figure 4A and Supplemental Figure 10, A and B). IHC double staining showed that most TNAP-positive cells were of the monocyte (CD68+) or myeloid (myeloperoxidase-positive) lineages, or were MSCs (CD44+) (Figure 4, B–D, and Supplemental Figure 10, C–E). These results imply that the enhanced expression of TNAP is involved in the accelerated mineralization of ex vivo AS MSC cultures, as is present in the immune cells/stromal cells of in vivo BM specimens from AS patients.
Figure 4. TNAP in the BM from AS patients is enhanced significantly in comparison with controls.

(A) IHC staining of the BM from AS patients (A1 and A2) and normal controls (C4 and C5) with a TNAP-specific antibody. Inset represents high magnification of the boxed area. (B–D) Double IHC staining of BM sections from AS patients (A1 and A2) with TNAP antibody (in brown) and the indicated second primary antibodies (in blue). The colocalizations of TNAP/CD68 (monocyte lineage) (B), TNAP/myeloperoxidase (MPO) (myeloid lineage) (C), and TNAP/CD44 (MSC) (D) were visualized as dark purple. Inset represents high magnification of the boxed area; top image is the microscopic image, and bottom image is the composite pseudocolored image by spectral unmixing technique with a spectral library (staining with TNAP antibody in brown and secondary antibody in blue). The colocalizations of TNAP/CD68, TNAP/MPO, and TNAP/CD44 are presented as turquoise. Scale bars: 100 μm (A–D).
To determine whether enhanced TNAP levels could be detected in the peripheral blood of AS patients, we measured the serum levels of bone-specific TNAP (BAP) to exclude interference of TNAP from the liver and kidney using samples from 2 separate cohorts: one was a Taiwanese cohort that included 104 AS patients and 50 healthy controls, and the other was a British cohort with 184 AS patients. Their demographic characteristics and the comparative differences between the subgroups/cohorts are shown in Supplemental Tables 6–8. The percentages of AS patients with increased serum BAP levels (>20.2 μg/L) in the Taiwanese and British cohorts were 9.61% and 25.54%, respectively. Overall, serum BAP levels were significantly higher in AS patients than in healthy controls (12.954 ± 5.538 μg/L and 6.296 ± 2.110 μg/L; P < 0.001) in the Taiwanese cohort, while serum BAP levels in AS patients were 17.607 ± 8.055 μg/L in the British cohort.
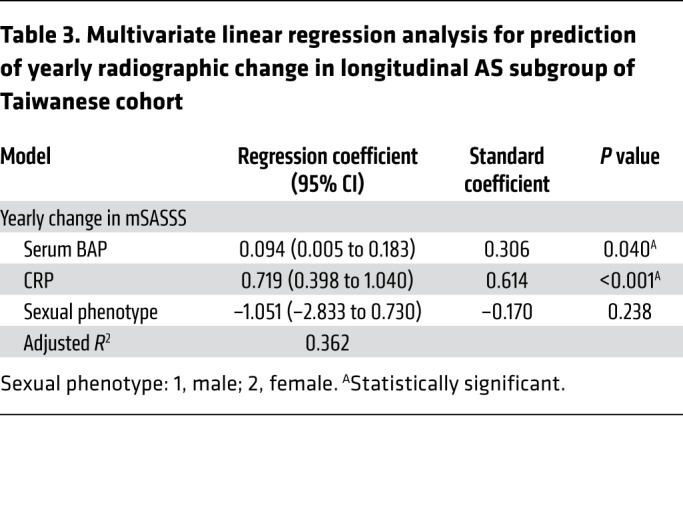
We then tested whether serum BAP levels were correlated with clinical parameters. Radiographic severity was measured by the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS). Remarkably, there were significant positive correlations between serum BAP levels and radiographic severity in AS patients (P < 0.05 for both cohorts by mSASSS) (Table 1). Likewise, to clarify the relative contributions of various risk factors that might be used to predict radiographic severity in AS patients, multivariate regression analyses were performed. The results showed that serum BAP levels, disease duration, and C-reactive protein (CRP) were independent risk factors in the Taiwanese cohort; while in the British cohort, serum BAP levels, disease duration, and male sexual phenotype were independent risk factors (Table 2). Notably, serum BAP levels and disease duration were the 2 common predictors for radiographic severity, regardless of the cohort analyzed. Next, we performed the retrospective subgroup analysis of 37 AS patients in the Taiwanese cohort who had a longitudinal follow-up of radiographic change (Supplemental Table 9). In this longitudinal AS subgroup analysis, we found that serum BAP levels and CRP were 2 independent risk factors for the prediction of yearly radiographic progression in AS patients (Table 3). To summarize, the analyses of Taiwanese and British cohorts validated our experimental findings regarding the importance of TNAP in AS, and also demonstrated that the serum BAP level may be a prognostic biomarker to predict a high-risk group of AS patients with a propensity for radiographic progression.
Table 1. Correlations between serum levels of BAP and clinical parameters of AS patients in Taiwanese cohort and British cohort.
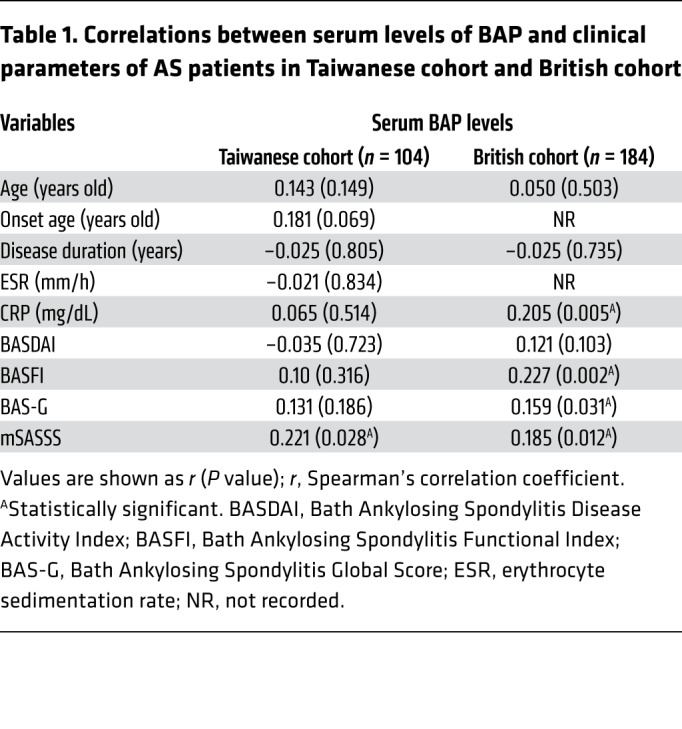
Table 2. Multivariate linear regression analysis for prediction of radiographic severity in AS patients of Taiwanese and British cohorts.
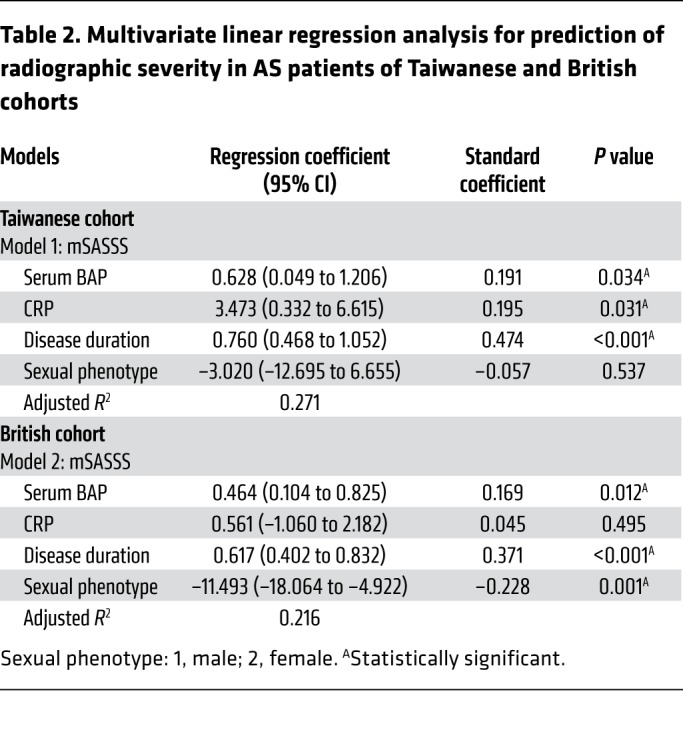
Table 3. Multivariate linear regression analysis for prediction of yearly radiographic change in longitudinal AS subgroup of Taiwanese cohort.
Upregulation of the transcription factor RARB promotes the expression of TNAP in AS MSCs.
To investigate further the upstream regulatory mechanism of enhanced TNAP expression in AS MSCs, we analyzed the microarray data using the category of transcription factors with at least a 2-fold change at 2 or more time points in AS MSCs compared with control MSCs. There were 36 differentially expressed genes in this category (Figure 5A). We also used online software (PROMO and MALGEN on the ALGGEN server) to predict the binding of 44 transcription factors to the ALPL promoter (Supplemental Figure 11A). By combining these 2 computational analyses, we identified 2 transcription factors present in both analyses, retinoic acid receptor-β (RARB) and leukocyte-enhancing factor 1 (LEF1), which are potential regulators of TNAP expression in AS MSCs. Quantitative reverse transcriptase PCR and immunoblot analyses (Figure 5, B–E) revealed that only RARB expression was increased in AS MSCs under osteogenic induction. More importantly, TNAP expression (Figure 5, F and G) and ALP activity (Supplemental Figure 11D) were reduced in MSCs depleted of RARB by 2 independent shRNAs (Supplemental Figure 11, B and C), consistent with a previous report showing that the RARB–retinoic X receptor heterodimer binds to a retinoic acid–responsive element in the promoter region of ALPL in a human osteoblastic osteosarcoma line (28). Furthermore, the enhanced mineralization in AS MSCs was inhibited by RARB knockdown (Figure 5, H and I). These data documented that RARB is an upstream transcription factor that promotes enhanced TNAP in AS MSCs.
Figure 5. RARB is an upstream regulator that promotes TNAP expression in AS MSCs.

(A) Differential gene expression of transcription factors between AS MSCs and control MSCs under osteogenic induction at the indicated days. (B–E) Expression of transcription factors RARB and LEF1 in AS and control MSCs measured by RT-qPCR (B and C) and immunoblotting (D and E). (F and G) RT-qPCR (F) and immunoblot analyses (G) showing reduced TNAP expression in AS or control MSCs transduced with 2 shRNAs against RARB at day 7 under osteogenic induction compared with the effects of shCtrl. (H) ARS staining of mineralization in AS or control MSCs expressing shRARB or shCtrl under osteogenic induction with quantification (I). All experiments were done in the AS patient group using AS MSCs (derived from A1, A2, and A3 with experimental triplicates) and/or in the control group using control MSCs (derived from C1, C2, and C3 with experimental triplicates). Data are the mean ± SEM. *P < 0.05; **P < 0.01; ****P < 0.0001 by 1-way ANOVA, followed by Tukey’s HSD test. Representative immunoblots from AS (A1) MSCs and control (C3) MSCs are shown in G and H. Scale bar: 200 μm (H).
HLA-B27 mediates the upregulation of the RARB/TNAP axis in AS MSCs.
Because AS is strongly associated with HLA-B27 (1), we investigated whether this genetic trait contributed to the upregulated RARB/TNAP axis in AS MSCs. First, we performed HLA-B knockdown in AS MSCs by using 2 independent shRNAs (Supplemental Figure 12, A and B). Interestingly, the enhanced mineralization in AS MSCs was inhibited by the 2 independent shRNAs against HLA-B (Figure 6, A and B), accompanied by the decreased expressions of RARB and TNAP (Figure 6C). Notably, HLA-B27 overexpression via the lentiviral transduction of control MSCs (Figure 6D and Supplemental Figure 12C) induced enhanced mineralization (Figure 6, E and F) and increased the protein levels of RARB and TNAP (Figure 6G). These results demonstrated the causative relationship of the genetic HLA-B27 trait in triggering abnormal RARB/TNAP axis to promote enhanced mineralization of AS MSCs.
Figure 6. HLA-B27 mediates the upregulation of the RARB/TNAP axis in AS MSCs.

(A) ARS staining of mineralization in AS MSCs transduced with shHLA-B or shCtrl under osteogenic induction with quantification (B). (C) Immunoblot analyses showing the expression of RARB and TNAP in AS MSCs expressing shHLA-B or shCtrl at day 7 under osteogenic induction. (D) Immunoblot showing HLA-B27 expressions of control MSCs transduced with pLAS2w or pLAS2w-HLA-B27. (E) ARS staining of control MSCs transduced with control lentiviral vector (pLAS2w) or a vector expressing HLA-B27 (pLAS2w-HLA-B27) with quantification (F). (G) Immunoblot analyses showing RARB and TNAP expressions in control MSCs transduced with pLAS2w or pLAS2w-HLA-B27. All experiments done in the AS patient group and the control group are from AS MSCs (A1, A2, and A3) and control MSCs (C1, C2, and C3), respectively, with at least 2–3 experimental repeats. Data are the mean ± SEM. ***P < 0.001; ****P < 0.0001 by 2-tailed Student’s t test (2 groups) or 1-way ANOVA, followed by Tukey’s HSD test. Representative images from AS (A1) MSCs are shown in C. Scale bars: 200 μm (A and E).
The HLA-B27–mediated activation of the p-IRE1/sXBP1 pathway upregulates the RARB/TNAP axis in AS MSCs.
Results from the HLA-B27–transgenic rat model suggested that misfolding of the HLA-B27 heavy chain (HC), whose formation of dimer structure in disulfide-linked complexes was dependent on the unpaired cysteine at position 67 (Cys67), can result in upregulated endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), and lead to AS development (29–31). We thus examined whether HLA-B27 misfolding and UPR pathways participated in the upregulation of the RARB/TNAP axis in AS MSCs. We used the conformation-specific antibody HC10 to immunoprecipitate the free unfolded or misfolded MHC class I HCs that were not assembled with β2-microglobulin (32), subsequently performing immunoblotting analysis with HLA-B27–specific antibody. Our results showed that there were substantial accumulations of disulfide-linked HLA-B27 HC dimers (misfolded) and HLA-B27 HC monomers (unfolded) in AS MSCs (Figure 7A), which were not observed in control MSCs. Moreover, HC10-reactive HCs were associated with the expression of the ER chaperone binding immunoglobulin protein (BiP), a global marker of UPR (30), in AS MSCs, but not in control MSCs. We then investigated the molecules involved in the 3 main UPR pathways, the PERK/activating transcription factor 4 (ATF4)/CCAAT enhancer–binding protein–homologous protein (CHOP) pathway, the inositol-requiring 1 (IRE1)/spliced X-box–binding protein 1 (sXBP1) pathway, and the activating transcription factor 6 (ATF6) pathway (ref. 33 and Supplemental Figure 13, A–F). Increased mRNA levels of ATF4 and sXBP1 were observed in AS MSCs compared with control MSCs. Because ATF4 is a downstream target of PERK, we treated AS MSCs with a PERK inhibitor to determine the association of the PERK/ATF4/CHOP pathway in the enhanced mineralization of AS MSCs. However, the enhanced mineralization of AS MSCs was not affected by treatment with a PERK inhibitor (Supplemental Figure 13, G and H). Phosphorylated IRE1 (p-IRE1) catalyzes the excision of introns from unspliced XBP1 (uXBP1) mRNA, resulting in a frameshift in the XBP1 coding sequence into sXBP1, which is more transcriptionally active (31). We found that the expression of p-IRE1/sXBP1 was elevated in AS MSCs compared with control MSCs (Figure 7, B and C). Notably, the knockdown of HLA-B in AS MSCs decreased the expression of p-IRE1/sXBP1 (Figure 7D), while HLA-B27 overexpression in control MSCs enhanced the expression of p-IRE1/sXBP1 (Figure 7E). These data suggest that the p-IRE1/sXBP1 axis is the downstream target of HLA-B27 in AS MSCs. Moreover, prediction of transcription factor binding by online software (PROMO and MALGEN on the ALGGEN server) revealed that XBP1 may bind to the RARB promoter. A chromatin immunoprecipitation (ChIP) assay further revealed the significant binding of sXBP1 at multiple regions spanning fragments 1, 2, 3, 6, and 7 in the RARB promoter in AS MSCs (Figure 7F). Consistently, knockdown of XBP1 by 2 independent shRNAs in AS MSCs under osteogenic induction (Supplemental Figure 14, A–C) caused reduced expression of RARB/TNAP (Figure 7G) as well as the abolished mineralization of AS MSCs (Figure 7, H and I). Additionally, we overexpressed HLA-B7, which does not have unpaired Cys67 in its extracellular domain and thus does not misfold in control MSCs (refs. 29–31 and Supplemental Figure 15, A and D). As expected, no significant enhancement of the p-IRE1/sXBP1/RARB/TNAP axis and mineralization was found (Supplemental Figure 15, B–E). These data indicate that HLA-B27 misfolding mediates the activation of the p-IRE1/sXBP1/RARB/TNAP pathway and plays an important role in accelerating the mineralization of AS MSCs.
Figure 7. The HLA-B27–mediated activation of the p-IRE1/sXBP1 pathway promotes the upregulation of the RARB/TNAP axis in AS MSCs.

(A) Unfolded or misfolded HCs were immunoprecipitated (IP) from lysates of AS MSCs and control MSCs with HC10 antibody, followed by immunoblotting with HLA-B27 and anti-BiP antibodies. In AS MSCs, monomers of HLA-B27 HCs and dimers of disulfide-linked HLA-B27 HCs are indicated by an arrow and an arrowhead, respectively. (B and C) Immunoblot showing the expressions of p-IRE1 (B) and sXBP1 (C) protein in AS and control MSCs at day 7 under osteogenic induction. (D) Immunoblot showing the expressions of p-IRE1 and sXBP1 in AS MSCs transduced with shHLA-B or shCtrl. (E) Immunoblot showing the expressions of p-IRE1 and sXBP1 protein in control MSCs transduced with pLAS2w-HLA-B27 or pLAS2w. (F) ChIP assay showing sXBP1 binding at fragments (Frags) 1, 2, 3, 6, and 7 within 1400 bp upstream of the RARB transcriptional start site in AS MSCs. RARB 3′-untranslated region (3′UTR) was used as the negative control locus. (G) Immunoblot showing the expressions of RARB and TNAP in AS MSCs expressing shXBP1 or shCtrl. (H) ARS staining of mineralization in AS MSCs transduced with shXBP1 or shCtrl under osteogenic induction with quantification (I). All experiments done in the AS patient group and control group are from AS MSCs (A1, A2, and A3) and control MSCs (C1, C2, and C3), respectively, with at least 2–3 experimental repeats. Data are the mean ± SEM. **P < 0.05; ***P < 0.001; ****P < 0.0001 by 2-tailed Student’s t test (2 groups) or ****P < 0.0001 by 1-way ANOVA, followed by Tukey’s HSD test. Representative immunoblots from AS (A1) MSCs are shown in G.
Discussion
Currently, there is no treatment to efficiently arrest the spinal ankylosis of AS patients. Current treatment strategies have mostly focused on the amelioration of inflammation (4–11). Although NSAIDs, long-term TNF-α blocker, or IL-17A inhibitor (4–11) slow down the rate of syndesmophyte formation, there is still an unmet need for new strategies to resolve this problem. Further understanding of the stromal pathway activation in AS is of high priority.
By studying AS MSCs derived from the enthesis involved in spinal ankylosis, we demonstrate that HLA-B27 misfolding mediates the activation of the p-IRE1/sXBP1/RARB/TNAP pathway in AS MSCs, which contributes to the pathogenesis of syndesmophyte formation. We further explored the therapeutic application of this pathway to develop a new strategy to arrest AS spinal ankylosis. Currently, ER stress and the UPR triggered by the misfolding of HLA-B27 molecules was proposed as 1 of the 3 major mechanistic hypotheses for AS pathogenesis (34, 35). Prior mechanistic studies using HLA-B27–transgenic rats suggested that the upregulation of UPR via sXBP1 in macrophages was involved in AS development through the IL-23/IL-17 axis (36, 37). However, whether UPR is relevant to AS patients is controversial, because macrophages from AS patients exhibited greater IL-23 production in response to lipopolysaccharide, but had no significant UPR induction (38). Here, we demonstrate that HLA-B27 misfolding upregulates p-IRE1/sXBP1 in the UPR pathway in AS MSCs.
Interestingly, accelerated mineralization in AS MSCs is Runx2-independent, indicating that syndesmophyte formation in AS may not reflect the standard osteoblast-mediated bone remodeling. Here, we used a standard cocktail of dexamethasone, ascorbic acid, and BGP to induce MSCs toward osteogenic differentiation (39), in which dexamethasone induces Runx2 expression, ascorbic acid leads to the increased secretion of Col1, and BGP is a phosphate source for hydroxyapatite formation. This osteogenic induction system induced osteogenesis in control MSCs and AS MSCs. Runx2 and Col1 were induced in control MSCs and AS MSCs after osteogenic differentiation at day 7, compared with their primitive status without osteogenic induction. However, the osteoblastogenesis of AS MSCs was not as prominent as that in control MSCs, which was associated with their lower expression of extracellular matrix factors such as Col1/osteoadherin. This abnormal osteogenesis of AS MSCs was characterized by the enhancement of TNAP, which promotes prominent extracellular matrix mineralization. Nevertheless, whether the calcium phosphate complexes formed in the abnormal osteogenesis of AS MSCs are mature hydroxyapatite with a mineral/matrix ratio close to that of bone remains to be investigated. Notably, pathological entheses in AS patients are collagen-rich tissues. These unique collagen-rich entheses might serve as scaffolds to support pathological mineral depositions. In addition, compared with the differences in previous reports showing enhanced Runx2-mediated osteogenesis in AS patient–derived BM MSCs from hips or bone precursors from facet joints under osteogenic induction (12–15), our findings via enthesis-derived MSCs from the diseased niche of spinal ankylosis indicate that different sources of stromal cells may reflect different microenvironmental stimuli and determine the distinctive nature of stromal activation. Further study of the diseased MSCs and microenvironmental niche interaction might allow us to clarify the developmental process of syndesmophyte formation in situ.
In addition, we established a novel in vivo disease model that mimics the pathological bony apposition induced by AS MSCs, which might be a useful platform for drug screening for AS. Entheses in AS are thought to be sites of microtrauma resulting from increased biomechanical stress (26, 27). In this model, we implanted AS MSCs embedded in fibrin over the site of cortical erosions after a decortication procedure, which mimicked the repetitive microtrauma in the syndesmophyte formation of AS. We also used a commercial fibrin sealant to provide an ideal 3D porous scaffold, which presented a better physiological environment for bone formation than a 2D culture system and has favorable properties for promoting bone engineering, including desirable porosity for new bone ingrowth and capillary infiltration (40, 41). In addition, NOD-SCID mice are immunodeficient, which is ideal for the implantation of AS MSCs without immune rejection. This model might help us focus on the pathological alterations of AS MSCs, which have not been investigated in an in vivo model. Because the immunodeficiency trait of this model excludes immunity in the pathogenesis of ectopic calcification, the significance of the effects of a TNF-α blocker or IL-17A inhibitor was not assessed in this model. Nevertheless, we identified 2 approved drugs (levamisole and pamidronate) that may be repurposed for drug development in blocking new bone formation in AS. Levamisole has been used as an anthelmintic agent for several years (42) and was reported to marginally reduce the inflammation of AS patients by acting on T regulatory cells (43–45). The clinical application of levamisole in blocking syndesmophyte formation through TNAP inhibition has not been explored in AS patients. However, previous studies reported that the consumption of 50–200 mg/d of levamisole caused agranulocytosis by inducing leukocyte agglutinating antibodies in 0.08%–5% of the patient population, especially HLA-B27+ patients (46, 47). Therefore, the prospective development of levamisole derivatives with reduced toxicity is important. Given that TNAP is also essential for bone homeostasis, the long-term effect of a TNAP inhibitor on the interference of bone remodeling should be considered. Although the bone mineral density in our animal model was not affected by levamisole treatment, the development of more specific TNAP inhibitors selectively targeting the specified stromal cells in AS patients deserves further investigation.
Another critical prospect would be to develop a dual-purpose drug with effects on TNAP inhibition and osteoporosis prevention; nitrogen-containing bisphosphates (such as pamidronate) are worthy of exploration for this purpose. While bisphosphates have been the prescription drug of choice for osteoporosis treatment, functioning by inhibiting osteoclastic bone resorption, they also inhibit TNAP enzymes through the chelation of Mg2+/Zn2+ with their bone hook structure (24). Interestingly, our data identified that pamidronate not only blocked the enhanced mineralization of AS MSCs under osteogenic induction, but also inhibited new bony apposition in our AS MSC–based animal model. Although previous studies reported that the clinical application of pamidronate controlled AS disease activity through its antiinflammatory effects (48–51) such as the modulation of proinflammatory cytokines in macrophage/monocyte lineages, only one study suggested that it lowered serum BAP levels (50). To date, there are no available data indicating that it blocks syndesmophyte formation in AS through TNAP inhibition. Therefore, our experimental findings revealed for the first time to our knowledge that pamidronate is an attractive target for AS drug development with dual effects on preventing spinal fusion and osteoporosis. Collectively, although the equivalent human dosages of levamisole/pamidronate required to block syndesmophyte formation in AS should be examined in future studies, the effective therapeutic dosages used in this AS MSC–based mouse model (levamisole 10 mg/kg/d or pamidronate 0.3 mg/kg/d, orally) are lower than the dosages used to treat mice with parasitic infections (20 mg/kg/d, orally) (52) or osteoporosis (5 mg/kg/d, orally) (53).
Based on these findings, we collected 2 separate cohorts from Taiwan and England to test whether serum TNAP levels may be used to screen AS patients with a high risk of developing radiographic progression. We measured the serum levels of BAP to exclude the interference of TNAP derived from the liver and kidney. Our multivariate regression analysis showed that serum BAP levels, disease duration, and/or CRP were independent predictors of radiographic severity in AS. In addition, our longitudinal subgroup analysis also revealed that serum BAP and CRP levels were 2 predictors of yearly radiographic progression. Increased serum ALP levels were reported in 13%–47.5% of AS patients (54–57), and increased serum BAP levels in 40.0% of AS patients (56). However, few studies have reported that increased serum ALP levels, but not BAP levels as identified here, were correlated with radiographic severity (54). Our mechanistic studies uncovered the causative role of HLA-B27 in mediating the upregulation of TNAP, which triggered syndesmophyte formation. A further longitudinal survey should be undertaken to validate the usefulness of serum BAP level as a biomarker for the early identification of high-risk AS patients with a propensity for radiographic progression and test the effectiveness of early TNAP inhibitor treatment for preventing bony outgrowths.
There are still unresolved issues. First, in the BM of AS patients, most TNAP-positive cells are of myelocyte/monocyte lineages and MSCs. Although we found a diffuse enhancement of TNAP expression in MSCs of the BM of AS, it was reported that only the BM near cortical bone in AS showed localized enhancement of osteogenesis (58). This discrepancy might be explained by a report stating that the unique microenvironmental niche of BM favored the stemness of MSCs and prevented their spontaneous osteogenesis (59). Moreover, it has been proposed that biomechanical stress plays an important role in directing the lineage commitment of MSCs into osteogenesis (60). Because entheses are regions bearing high tensile forces (61), the biophysical characteristics of entheses might be a link between biomechanical stress and stromal activation (62) and explain why new bone formation mainly exists in enthesis-related lesions or cortical bones near the entheses. Nevertheless, how the complex interaction between biomechanical stress and enthesis MSCs triggers HLA-B27–mediated activation of the sXBP1/RARB/TNAP pathway deserves further investigation. Second, to examine the effects of autocrine cytokines on the enhanced mineralization of AS MSCs, we measured cytokines reported to have proinflammatory effects in AS (63, 64), including TNF-α, IL-1β, IL-6, IFN-γ, IL-17A, IL-22, IL-23, and IL-33. However, we did not find an enhancement of these cytokines produced by AS MSCs compared with control MSCs (Supplemental Figure 16). Although we did not check the levels of these cytokines in the entheses of our AS specimens, their presence in situ was identified in previous reports (65–68). Therefore, it is important to explore whether the interaction among diseased MSCs, cytokines, and specific immune cells, such as group 3 innate lymphoid cells present in entheses (69), triggers HLA-B27–mediated RARB/TNAP activation. Third, although our mechanistic studies used limited numbers of MSCs and should be validated using greater numbers of AS samples, our clinical data from 2 separate cohorts also indicated the importance of TNAP in the syndesmophyte formation of AS. Fourth, we speculated that the decortication procedure might provide osteogenic/vasogenic stimuli (70) to promote osteogenesis in AS MSCs because none of the in vitro osteogenic supplements was provided in our in vivo model. However, how tissue factors relevant to the decortication procedure and AS MSCs are triggered toward ectopic bony apposition in vivo should be clarified in the future. Finally, we did not explore the pathological mechanisms of AS MSCs from HLA-B27–negative AS patients, which should be explored in the future.
Taken together, the translational relevance of our study is highlighted by several findings: (a) Identification of “HLA-B27 misfolding” mediates the activation of the p-IRE1/sXBP1/RARB/TNAP pathway in AS MSCs within the enthesis, which is involved in spinal ankylosis and contributes to the pathogenesis of syndesmophyte formation. (b) Targeting the p-IRE1/sXBP1/RARB/TNAP pathway may elucidate a new strategy for blocking syndesmophyte formation; the dual effects of pamidronate indicate that it is a prospective candidate for drug development. (c) Establishment of an AS MSC–based animal model that mimics pathological bony apposition might be a useful platform for drug screening for AS therapy. (d) Serum BAP levels can be used as a biomarker to select high-risk AS patients with a propensity for radiographic progression. In addition to current antiinflammatory agents, our identification of TNAP as a therapeutic target may aid the development of a new strategy to protect AS patients from bony outgrowths in the future.
Methods
Study design.
The primary goals of the study were to investigate the pathological role of MSCs from the diseased niche of syndesmophytes and to determine whether genetic traits contributed to the altered osteogenesis pathways in AS. Herein, we isolated MSCs within the enthesis involved in the spinal ankylosis of AS patients who underwent wedge osteotomy (AS MSCs) and non-AS patients who underwent traumatic surgery at similar sites (control MSCs). We established ex vivo AS MSC cultures and developed an AS MSC–based in vivo disease model to mimic the pathological bony apposition, both of which were used for mechanistic studies and drug development. We also collected 2 separate cohorts from Taiwan and England to validate our experimental findings of the altered mechanism involved in the pathogenesis of syndesmophyte formation. We identified a useful biomarker that might help clinicians select a high-risk group of AS patients with a propensity for radiographic progression.
Isolation of MSCs within entheseal tissues affected by spinal ankylosis from AS patients and culture.
MSCs from the enthesis (bone, bone marrow, periosteum, supraspinatus, and interspinous ligaments) involved in spinal ankylosis from 3 AS patients (A1, A2, and A3) who had undergone spinal wedge osteotomy (AS MSCs) were isolated. MSCs from 3 non-AS individuals (C1, C2, and C3) who underwent traumatic surgery at similar sites (control MSCs) were used as controls. All individuals were Taiwanese (Supplemental Table 1). These specimens were minced finely and digested with 1 mg/mL collagenase D (Roche) after rinsing with growth medium (GM) containing α-minimum essential medium (α-MEM) (Gibco), 10% FBS (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich). After overnight incubation at 37°C, digested tissues were filtered through a 40-μm nylon filter to remove debris. Cells were collected by centrifugation and plated in culture dishes to allow attachment. Nonadherent cells were removed by changing of GM within 48 hours and then every 3 days. Upon reaching confluence, cells were harvested and subcultured. At passage 3, aliquots of primary MSCs were cryopreserved in liquid nitrogen. MSCs were reseeded at a density of 103 cells/cm2 in GM at 37°C under 5% CO2 atmosphere for expansion. The GM was changed twice per week, and subculture was performed at 90% confluence. The same passages (p3 to p5) of AS MSCs and control MSCs were compared.
Characterization of MSCs.
To characterize the trilineage differentiation potential of MSCs, they were treated under 3 culture conditions. For osteogenic induction, α-MEM was supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL ascorbate-2 phosphate (Sigma-Aldrich), 10 nmol/L dexamethasone (Sigma-Aldrich), and 10 mmol/L BGP (Sigma-Aldrich). After the appearance of the morphological features of differentiation, cells were fixed with 4% paraformaldehyde. Cells treated in the osteogenic culture were stained for alizarin red S (ARS; Sigma-Aldrich) or by a von Kossa staining kit (Abcam) for mineralization. Quantification of ARS staining was conducted by measurement of the optical density of extracted dye at 550 nm with a plate reader (Molecular Devices). Detailed information on the adipogenic and chondrogenic induction is provided in Supplemental Methods.
MSCs cultured under osteogenic induction with or without treatment.
MSCs were seeded at 8 × 104 cells per 6-well plate and grown in osteogenic induction medium at 37°C with 5% CO2, with or without levamisole (100 μM; Sigma-Aldrich), beryllium sulfate tetrahydrate (100 μM; Santa Cruz Biotechnology), pamidronate (1 μg/mL; Sigma-Aldrich), or PERK inhibitor (GSK2606414; 0.4, 4, or 20 nM; Merck Millipore). After morphological differentiation, cells were subjected to ARS staining.
Quantitative reverse transcriptase PCR.
Total RNA was extracted using the RNeasy Mini kit (Qiagen). First-strand cDNA was synthesized from 2 μg of total RNA using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative reverse transcriptase PCR (RT-qPCR) was carried out with a SYBR Green PCR Master Mix on a Prism 7300 Sequence Detection system (Applied Biosystems). The relative quantitation of marker genes was performed according to the ΔCt method using GAPDH as the internal control gene. The specific primers used for RT-qPCR are shown in Supplemental Table 10.
Immunoblotting and immunoprecipitation.
Cell lysates were prepared using M-PER Protein Extraction Reagent (Pierce) plus protease inhibitor cocktail (Halt, Pierce). Nuclear extracts used for immunoprecipitation were prepared by the protocol previously described (71). Two hundred micrograms of total cell lysates were incubated with 2 μg of mouse anti-HC10 antibody (catalog AM33035PU-N, Origene) at 4°C overnight and then reacted with Dynabeads Protein G (Thermo Fisher Scientific) for 3 hours to precipitate the protein-antibody complex. Immunoblot analysis was performed following previously published protocols (71). The primary antibodies were mouse anti–active dephosphorylated β-catenin (clone 8E7; 1:500; Merck Millipore), rabbit anti–phosphorylated (serine 463/465) Smad 1/5/8 (clone 41D10; 1:250; Cell Signaling Technology), rabbit anti-TNAP (clone EPR4477; 1:500; Abcam), rabbit anti-RARB (catalog ab53161; 1:500; Abcam), goat anti-LEF1 (clone N-17; 1:500; Santa Cruz Biotechnology), rabbit anti-IRE1 (catalog ab37073; 1:1000; Abcam), rabbit anti–phosphorylated (serine 724) IRE1 (catalog ab4818; 1:500; Abcam), rabbit anti-XBP1 (catalog ab37152; 1:2000; Abcam), anti–HLA-B27 (clone HLA-ABC-m3; 1:300; Origene), anti–HLA-B7 [clone EPR2623(2); 1:500; Abcam], rabbit anti-GAPDH (catalog GTX100118; 1:10,000; GeneTex), and mouse anti–β-actin (catalog MA5-15739; 1:10,000; Thermo Fisher Scientific). HRP-conjugated secondary antibodies used were goat anti-rat IgG, anti-rabbit IgG, anti-mouse IgG, and anti-goat IgG (all at 1:5000; Sigma-Aldrich). Immunoblotting experiments were done at least twice. All protein band densities were normalized to those of the GAPDH or β-actin loading control.
Immunofluorescence staining.
MSCs were cultured under osteogenic induction for 14 days, rinsed with PBS, and fixed in 4% paraformaldehyde, followed by permeabilization with 0.1% Triton X-100. Cells were blocked by 1% BSA in PBST. Mouse anti-osteoadherin antibody (clone 806001; 1:500; R&D) in blocking buffer was added and then incubated with secondary antibody (goat anti-mouse antibody conjugated with Alexa Fluor 488) (catalog A-210421; 1:400; Invitrogen). Cell nuclei were counterstained with DAPI (Sigma-Aldrich) in PBS. Fluorescent images were examined by a confocal microscope (Carl Zeiss LSM510) and captured by Zeiss EC Plan-Neofluar ×40/1.30 Oil DIC M27.
Microarray, Gene Ontology, and Ingenuity Pathway Analysis.
AS MSCs and control MSCs under osteogenic induction at days 0, 3, and 7 were harvested for microarray study according to the protocol provided by the manufacturer (3′ IVT Express kit, Thermo Fisher Scientific). The biotinylated cRNAs were hybridized to the GeneChip Human Genome U133 Plus 2.0 array (Affymetrix) at the GRC Microarray Core Facility, Academia Sinica. Data were acquired with GeneChip Scanner 3000 (Thermo Fisher Scientific) and analyzed by GeneSpring GX version 12 (Agilent Technologies). Genes expressed differentially in AS MSCs with at least a consistent 2-fold change at 3 time points compared with control MSCs are shown in Supplemental Tables 2 and 3. The distribution of enriched Gene Ontology (GO) terms was performed using the Biological Networks Gene Ontology program package with P ≤ 0.05. The 10 most significant terms in the biological process ontology were obtained to show the functional characteristics (Supplemental Figure 4A) of the given gene sets. Integrated analysis of gene networks involved in the osteogenesis pathway with reference to a database using Ingenuity Pathway Analysis (IPA) was performed (Figure 2A). In addition, differentially expressed genes in the category of transcription factors with at least a 2-fold change at 2 or more time points in AS MSCs compared with control MSCs were profiled, and the heatmap is shown in Figure 5A. The microarray data were deposited in the NCBI Gene Expression Omnibus database (GEO GSE134290).
Lentiviral transduction and siRNA transfection.
shRNA expression plasmids and bacteria clones for Runx2 (TRCN0000013653/00000412281), TNAP (TRCN0000052005/0000052007), RARB (TRCN0000021196/0000021198), XBP1 (TRCN0000019804/0000019808), and HLA-B (TRCN000057341/0000423192) were obtained from the RNAi Core Facility, Academia Sinica. The ALPL cDNA fragment was cut from plasmid obtained from the Mammalian Gene Collection of the Genome Research Center, National Yang-Ming University (clone 066116), and amplified by PCR with Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific). In some experiments, the HLA-B2705 cDNA (72) or HLA-B7 cDNA was assembled by cDNA synthesis by the Genomics Company or Tools Biotech Co. (both in Taipei, Taiwan), respectively. The ALPL fragment or the HLA-B2705 or HLA-B7 cDNA was inserted into a pLAS2w.Puro vector (RNAi Core Facility, Academia Sinica) by ligation at NheI and EcoRI sites (New England Biolabs). The procedure to prepare lentiviral vector and transduction was performed as previously described (73). Subconfluent MSCs were transduced with lentiviral vector at a multiplicity of 5 in the presence of 8 μg/mL Polybrene (Sigma-Aldrich). Twenty-four hours later, the culture medium was replaced with fresh GM containing puromycin (1 μg/mL; Gibco) to select transduced cells.
Animal models.
NOD.CB17-Prkdcscid/NcrCrlBltw (NOD-SCID) mice (8–10 weeks) were purchased from BioLASCO and maintained in a specific pathogen–free environment. Human MSCs were treated with BGP (10 mmol/L) in culture for 5 days, and embedded in fibrin (106 cells in 10 μL fibrin) (Baxter). To mimic the pathophysiological scenario of microtrauma in AS, we applied a decortication procedure over the cortical bone in the right lamina of the lumbar spine segment L4–5 by forceps friction 10 times to produce repetitive microtrauma, thus leading to cortical erosions. We next implanted AS MSCs embedded in fibrin over the site of cortical erosions. Mice were fed a 0.9% phosphate diet (Dyets) after surgery. Micro-CT (SkyScan 1076, Kontich) over lumbar spines was performed 3 weeks after implantation. Bone mineral density over the femur was measured by micro-CT in NOD-SCID mice following implantation with AS MSCs and daily oral administration of H2O, levamisole (10 mg/kg), beryllium sulfate tetrahydrate (7.5 mg/kg), or pamidronate (0.3 mg/kg) for 12 weeks. Quantitative volumes of new bony appositions were acquired by Bruker CT-Volume version 2.0 software.
Histology and IHC staining.
Lumbar spines of NOD-SCID mice were harvested 3 weeks after MSC implantation, decalcified with 5% formic acid (Sigma-Aldrich), fixed in 10% formalin, embedded in paraffin, and cut into 5-μm-thick sections for H&E staining. For IHC staining, human BM slices were obtained from 4 AS patients (A1, A2, A3, and A4), 2 healthy individuals (C4 and C5), and 1 non-AS patient control (C6) (Supplemental Table 5). Among them, the healthy controls (C4 and C5) were White, and their BM slides of lumbar spines were purchased from US Biomax. AS patients with spinal ankylosis had undergone spinal wedge osteotomy over lumbar spines; their BM specimens of lumbar spines were from surgical sites near entheseal tissues involved in spinal ankylosis. Non-AS patient control was the individual who underwent a BM biopsy of the hip for lymphoma workup and was free of lymphoma. AS patients and non-AS patient control were Taiwanese. BM sections were incubated with rabbit anti-TNAP (clone EPR4477; 1:200; Abcam), mouse anti–active dephosphorylated β-catenin (clone 8E7; 1:500; Merck Millipore), or rabbit anti–phosphorylated Smad 1/5/8 (serine 463/465) (catalog ab3848-1; 1:50; Merck Millipore) antibody followed by incubation with HRP-conjugated secondary antibody (Dako). Brown color was carried out by incubation with the chromogen DAB, and nuclei were counterstained with hematoxylin (all from Thermo Fisher Scientific).
For double staining, after color development of the first staining with rabbit anti-TNAP antibody (clone EPR4477; 1:200; Abcam) using HRP as described above, sections were incubated with a second primary antibody: rabbit anti-myeloperoxidase (anti-MPO) (catalog A0398; 1:1000; Dako) for myeloid lineages, mouse anti-CD68 (catalog M0876; 1:200; Dako) for monocyte lineages, or rabbit anti-CD44 (catalog ab157107; 1:200; Abcam) for MSCs, followed by incubation with ALP-conjugated secondary antibody (1:500; Southern Biotechnology). Blue color was carried out by incubation with nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate, toluidine salt (Roche).
To identify TNAP-positive cells, the colocalization of TNAP/MPO-positive, TNAP/CD68-positive, and TNAP/CD44-positive cells was determined using a spectral imaging technique with MetaMorphic Offline version 7.8.2.0 software. After spectral unmixing using the spectral library (staining with TNAP antibody in brown and the indicated second antibody in blue), composite pseudocolored images were created, in which the colocalization of TNAP/MPO, TNAP/CD68, and TNAP/CD44 was represented as turquoise.
Human peripheral blood samples and populations.
Two independent cohorts were recruited to study predictive biomarkers for high-risk AS patients with a propensity for radiographic progression. The “Taiwanese cohort” comprised 104 patients who fulfilled the modified New York criteria for AS diagnosis and 50 healthy controls from the Taipei Tzu Chi Hospital. A part of this population (37 AS patients who had retrospective follow-up of radiographic change) consisted of a longitudinal AS subgroup of the Taiwanese cohort. One hundred eighty-four AS patients in the “British cohort” were recruited at the Royal National Hospital for Rheumatic Diseases, UK. Clinical assessments and radiographic assessments were obtained on the same day. Clinical assessments included AS disease duration, serological tests (HLA-B27, C-reactive protein [CRP], or erythrocyte sedimentation rate [ESR]), and disease activity scoring with a validated version of the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI). Radiographic severity was assessed by the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS). The Bath Ankylosing Spondylitis Functional Index (BASFI) to determine the degree of functional limitation and the Bath Ankylosing Spondylitis Global Score (BAS-G) to assess the perceived effect of the disease on patients’ well-being were obtained. Details on BASDAI, mSASSS, BASFI, and BAS-G are provided in Supplemental Methods.
Measurements of serum BAP levels.
Human blood samples were collected, and serum was used for the measurement of BAP levels using the DiaSorin LIASON BAP OSTASE assay (1-step delayed-addition sandwich chemiluminescence immune assay) by the LIASON analyzer (DiaSorin). A reference range of serum BAP levels between 5.1 and 20.2 μg/L was considered normal according to the manufacturer.
Flow cytometry analysis and antibodies.
To analyze the cell surface expression of markers for MSCs, they were labeled with mouse anti-human antibodies (all from Ancell): CD31-PE (clone 158-2B3), CD34-PE (clone 43A1), CD44-PE (clone BU52), CD29-FITC (clone 4B7E), CD45-FITC (clone C11), and CD105-FITC (clone SN6/N1-3A1). In some experiments, AS MSCs were stained with mouse anti-human antibodies: HLA-B (clone JOAN-1, Abcam) or HLA-B7 (clone BB7.1, Bio-Rad) with goat anti-mouse IgG H&L (DyLight 488) secondary antibody (catalog ab96879, Abcam). To analyze the cell surface expression of HLA-B27, AS MSCs were stained with mouse anti–human HLA-B27–FITC (clone HLA-ABC-m3, GeneTex) antibody. Mouse isotype IgG1 antibodies (clone MOPC31C, Ancell) were used as control. Overall, 100,000 labeled cells were acquired and analyzed using a FACSCanto Flow Cytometer (BD Biosciences). FCS Express version 3.0 (De Novo Software) was used for data analyses.
ChIP assay.
The ChIP assay was performed according to the user’s manual for EZ-ChIP Kits (Merck Millipore). Briefly, 5 × 106 MSCs cultured in GM were trypsinized and fixed with 1% formaldehyde (Sigma-Aldrich), and formaldehyde was quenched by 125 mM glycine (Sigma-Aldrich). After lysis of cells and nuclear fractionation, the DNA was sheared by sonication using a Bioruptor NGS system (Diagenode). The protein-DNA complexes were purified by immunoaffinity capture with magnetic beads that bind to anti-sXBP1 (Abcam) or normal rabbit IgG (Bethyl). After elution of the complex and de–cross-linking of the protein-DNA complexes at 65°C, DNA was purified by RNase and protease K (Invitrogen) treatments. The enriched DNA was subjected to qPCR with specific primers amplifying genomic fragments containing putative sXBP1 sites on the RARB promoter, which were predicted by online software (PROMO and MALGEN on the Algorithmics and Genetics Group [ALGGEN; Polytechnic University of Catalonia, Barcelona, Spain] server), using sequences retrieved from the Ensembl genome browser (https://uswest.ensembl.org/Homo_sapiens/Gene/Sequence?db=core;g=ENSG00000077092;r=3:25174332-25597932). Data were expressed as the percentage of input DNA. The primer sequences used in qPCR are shown in Supplemental Table 10.
Other methods.
Detailed information on the measurement of BMP-2 and DKK-1, intracellular ALP enzyme activity, and cytokine release is provided in Supplemental Methods.
Statistics.
Data are shown as the mean ± SEM or mean ± SD. For MSCs or mouse experiments, comparisons between 2 or multiple groups were assessed using an unpaired, 2-tailed Student’s t test or an ANOVA test followed by Tukey’s honestly significant difference post hoc test. For human data, continuous values were analyzed by Mann-Whitney U test and categorical variables by Fisher’s exact test. Multivariate linear regression model was used to identify independent predictive factors of radiographic severity. Correlations between variables were determined by the Spearman’s rank correlation test. P values less than 0.05 were considered statistically significant. Data were analyzed by SPSS version 17 or GraphPad Prism version 6.01.
Study approval.
All human studies were approved by the Research Ethics Committees of Taipei Tzu Chi Hospital, Taipei Veterans General Hospital, Academic Sinica (all in Taipei, Taiwan), and the Royal National Hospital for Rheumatic Diseases (Bath, UK). All donors provided written informed consent before sampling in accordance with the Declaration of Helsinki. The animal experimental protocol was approved by the Animal Care and Utilization Committee of Taipei Tzu Chi Hospital.
Author contributions
CHL, SCH, and KIL conceived and designed the project. CHL, KHH, and SYY performed experiments. SR, CHC, CTC, IHC, JTC, TA, and JCCW provided human specimens or analytic methods. CHL, IYL, and IST analyzed data and participated in data interpretation. CHL, SCH, and KIL wrote the manuscript. WCT, SCH, and KIL supervised the research. All authors read and approved the manuscript.
Supplementary Material
Acknowledgments
We thank the Edanz Group (www.edanzediting.com/ac) for editing a draft of the manuscript. This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 103-2314-B-303-012-MY2; MOST 105-2314-B-303-019; MOST 104-2320-B001-016-MY3; MOST 107-2321-B-400-016), Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TCRD-TPE-MOST-103/104-05; TCRD-TPE-MOST-105-07), and Academia Sinica, Taiwan. We thank Taiwan Mouse Clinic (MOST 105-2325-B-001-010) for technical support in micro-CT scan experiments.
Version 1. 11/04/2019
Electronic publication
Version 2. 12/02/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(12):5357–5373.https://doi.org/10.1172/JCI125212.
Contributor Information
Chin-Hsiu Liu, Email: 2001windchild@gmail.com.
Chun-Hsiung Chen, Email: ch1106.chen@msa.hinet.net.
Kuo-Hsuan Hung, Email: kuninoki@gate.sinica.edu.tw.
Chung-Tei Chou, Email: ctchou1007@gmail.com.
Ing-Ho Chen, Email: ing.ho@msa.hinet.net.
Jui-Teng Chien, Email: jtchien1025@gmail.com.
I-Ying Lin, Email: iying@gate.sinica.edu.tw.
Shii-Yi Yang, Email: fadevictor@hotmail.com.
Takashi Angata, Email: angata@gate.sinica.edu.tw.
Wen-Chan Tsai, Email: d10153@ms14.hinet.net.
I-Shiang Tzeng, Email: istzeng@gmail.com.
Shih-Chieh Hung, Email: hung3340@gmail.com.
Kuo-I Lin, Email: kuoilin@gate.sinica.edu.tw.
References
- 1.Dougados M, Baeten D. Spondyloarthritis. Lancet. 2011;377(9783):2127–2137. doi: 10.1016/S0140-6736(11)60071-8. [DOI] [PubMed] [Google Scholar]
- 2.Tam LS, Gu J, Yu D. Pathogenesis of ankylosing spondylitis. Nat Rev Rheumatol. 2010;6(7):399–405. doi: 10.1038/nrrheum.2010.79. [DOI] [PubMed] [Google Scholar]
- 3.Benjamin M, Toumi H, Suzuki D, Hayashi K, McGonagle D. Evidence for a distinctive pattern of bone formation in enthesophytes. Ann Rheum Dis. 2009;68(6):1003–1010. doi: 10.1136/ard.2008.091074. [DOI] [PubMed] [Google Scholar]
- 4.Wanders A, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum. 2005;52(6):1756–1765. doi: 10.1002/art.21054. [DOI] [PubMed] [Google Scholar]
- 5.Poddubnyy D, et al. Effect of non-steroidal anti-inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Ann Rheum Dis. 2012;71(10):1616–1622. doi: 10.1136/annrheumdis-2011-201252. [DOI] [PubMed] [Google Scholar]
- 6.van der Heijde D, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis Rheum. 2008;58(5):1324–1331. doi: 10.1002/art.23471. [DOI] [PubMed] [Google Scholar]
- 7.van der Heijde D, et al. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with adalimumab for up to 2 years. Arthritis Res Ther. 2009;11(4):R127. doi: 10.1186/ar2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun J, et al. The effect of two golimumab doses on radiographic progression in ankylosing spondylitis: results through 4 years of the GO-RAISE trial. Ann Rheum Dis. 2014;73(6):1107–1113. doi: 10.1136/annrheumdis-2012-203075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baraliakos X, Haibel H, Listing J, Sieper J, Braun J. Continuous long-term anti-TNF therapy does not lead to an increase in the rate of new bone formation over 8 years in patients with ankylosing spondylitis. Ann Rheum Dis. 2014;73(4):710–715. doi: 10.1136/annrheumdis-2012-202698. [DOI] [PubMed] [Google Scholar]
- 10.Maas F, et al. Reduction in spinal radiographic progression in ankylosing spondylitis patients receiving prolonged treatment with tumor necrosis factor inhibitors. Arthritis Care Res (Hoboken) 2017;69(7):1011–1019. doi: 10.1002/acr.23097. [DOI] [PubMed] [Google Scholar]
- 11.Braun J, et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis. 2017;76(6):1070–1077. doi: 10.1136/annrheumdis-2016-209730. [DOI] [PubMed] [Google Scholar]
- 12.Xie Z, et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 2016;68(2):430–440. doi: 10.1002/art.39433. [DOI] [PubMed] [Google Scholar]
- 13.Xie Z, et al. MCP1 triggers monocyte dysfunctions during abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. J Mol Med. 2017;95(2):143–154. doi: 10.1007/s00109-016-1489-x. [DOI] [PubMed] [Google Scholar]
- 14.Jo S, et al. Accelerated osteogenic differentiation of human bone-derived cells in ankylosing spondylitis. J Bone Miner Metab. 2018;36(3):307–313. doi: 10.1007/s00774-017-0846-3. [DOI] [PubMed] [Google Scholar]
- 15.Jo S, et al. A novel role for bone-derived cells in ankylosing spondylitis: focus on IL-23. Biochem Biophys Res Commun. 2017;491(3):787–793. doi: 10.1016/j.bbrc.2017.07.079. [DOI] [PubMed] [Google Scholar]
- 16.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 17.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 18.Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 19.Kim JH, et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther Adv Musculoskelet Dis. 2013;5(1):13–31. doi: 10.1177/1759720X12466608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Deng C, Li YP. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8(2):272–288. doi: 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch. 2010;77(1):4–12. doi: 10.1272/jnms.77.4. [DOI] [PubMed] [Google Scholar]
- 22.Millán JL. Alkaline phosphatases: structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006;2(2):335–341. doi: 10.1007/s11302-005-5435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozlenkov A, Le Du MH, Cuniasse P, Ny T, Hoylaerts MF, Millán JL. Residues determining the binding specificity of uncompetitive inhibitors to tissue-nonspecific alkaline phosphatase. J Bone Miner Res. 2004;19(11):1862–1872. doi: 10.1359/JBMR.040608. [DOI] [PubMed] [Google Scholar]
- 24.Vaisman DN, McCarthy AD, Cortizo AM. Bone-specific alkaline phosphatase activity is inhibited by bisphosphonates: role of divalent cations. Biol Trace Elem Res. 2005;104(2):131–140. doi: 10.1385/BTER:104:2:131. [DOI] [PubMed] [Google Scholar]
- 25.Slappey G, Toribatake Y, Ganey TM, Ogden JA, Hutton WC. Guidelines to decortication in posterolateral spine fusion. J Spinal Disord. 1998;11(2):102–109. [PubMed] [Google Scholar]
- 26.La Cava G. Enthesitis; traumatic disease of insertions. J Am Med Assoc. 1959;169(3):254–255. doi: 10.1001/jama.1959.73000200002011a. [DOI] [PubMed] [Google Scholar]
- 27.Ball J. Enthesopathy of rheumatoid and ankylosing spondylitis. Ann Rheum Dis. 1971;30(3):213–223. doi: 10.1136/ard.30.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orimo H, Shimada T. Regulation of the human tissue-nonspecific alkaline phosphatase gene expression by all-trans-retinoic acid in SaOS-2 osteosarcoma cell line. Bone. 2005;36(5):866–876. doi: 10.1016/j.bone.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 29.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56(1):215–223. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 30.Tran TM, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172(8):5110–5119. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 31.Dangoria NS, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem. 2002;277(26):23459–23468. doi: 10.1074/jbc.M110336200. [DOI] [PubMed] [Google Scholar]
- 32.Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J Immunol. 2003;171(4):1918–1926. doi: 10.4049/jimmunol.171.4.1918. [DOI] [PubMed] [Google Scholar]
- 33.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13(3):374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 34.Colbert RA, Tran TM, Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol. 2014;57(1):44–51. doi: 10.1016/j.molimm.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen B, et al. Role of HLA-B27 in the pathogenesis of ankylosing spondylitis (Review) Mol Med Rep. 2017;15(4):1943–1951. doi: 10.3892/mmr.2017.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 37.DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60(9):2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng L, Lindstrom MJ, Smith JA. Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheum. 2011;63(12):3807–3817. doi: 10.1002/art.30593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langenbach F, Handschel J. Effects of dexamethasone, ascorbic acid and β-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res Ther. 2013;4(5):117. doi: 10.1186/scrt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noori A, Ashrafi SJ, Vaez-Ghaemi R, Hatamian-Zaremi A, Webster TJ. A review of fibrin and fibrin composites for bone tissue engineering. Int J Nanomedicine. 2017;12:4937–4961. doi: 10.2147/IJN.S124671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turnbull G, et al. 3D bioactive composite scaffolds for bone tissue engineering. Bioact Mater. 2018;3(3):278–314. doi: 10.1016/j.bioactmat.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janseen PA. The levamisole story. In: Jucker E, ed. Progress in Drug Research. New York, New York, USA: Springer-Verlag; 1976:1–66. [Google Scholar]
- 43.Rosenthal M. A critical review of the effect of levamisole in rheumatic diseases other than rheumatoid arthritis. J Rheumatol Suppl. 1978;4:97–100. [PubMed] [Google Scholar]
- 44.Christensen KD. Treatment of seronegative spondylarthritis with levamisole: a double-blind placebo-controlled study. Int J Immunopharmacol. 1979;1(2):147–150. doi: 10.1016/0192-0561(79)90018-3. [DOI] [PubMed] [Google Scholar]
- 45.Goldstein G. Mode of action of levamisole. J Rheumatol Suppl. 1978;4:143–148. [PubMed] [Google Scholar]
- 46.Drew SI, Carter BM, Nathanson DS, Terasaki PI. Levamisole-associated neutropenia and autoimmune granulocytotoxins. Ann Rheum Dis. 1980;39(1):59–63. doi: 10.1136/ard.39.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hodinka L, Géher P, Merétey K, Gyódi EK, Petrányi GG, Bozsóky S. Levamisole-induced neutropenia and agranulocytosis: association with HLA B27 leukocyte agglutinating and lymphocytotoxic antibodies. Int Arch Allergy Appl Immunol. 1981;65(4):460–464. doi: 10.1159/000232788. [DOI] [PubMed] [Google Scholar]
- 48.Toussirot E, Wendling D. Antiinflammatory treatment with bisphosphonates in ankylosing spondylitis. Curr Opin Rheumatol. 2007;19(4):340–345. doi: 10.1097/BOR.0b013e328133f57b. [DOI] [PubMed] [Google Scholar]
- 49.Santra G, Sarkar RN, Phaujdar S, Banerjee S, Siddhanta S. Assessment of the efficacy of pamidronate in ankylosing spondylitis: an open prospective trial. Singapore Med J. 2010;51(11):883–887. [PubMed] [Google Scholar]
- 50.Cairns AP, Wright SA, Taggart AJ, Coward SM, Wright GD. An open study of pulse pamidronate treatment in severe ankylosing spondylitis, and its effect on biochemical markers of bone turnover. Ann Rheum Dis. 2005;64(2):338–339. doi: 10.1136/ard.2004.022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maksymowych WP, et al. A six-month randomized, controlled, double-blind, dose-response comparison of intravenous pamidronate (60 mg versus 10 mg) in the treatment of nonsteroidal antiinflammatory drug-refractory ankylosing spondylitis. Arthritis Rheum. 2002;46(3):766–773. doi: 10.1002/art.10139. [DOI] [PubMed] [Google Scholar]
- 52.Chimwani DM, Britt DP. The efficacy of levamisole administered orally or parenterally against Heligmosomoides polygyrus in mice. J Helminthol. 1986;60(2):99–104. doi: 10.1017/S0022149X00008324. [DOI] [PubMed] [Google Scholar]
- 53.Clohisy DR, O’ Keefe PF, Ramnaraine ML. Pamidronate decreases tumor-induced osteoclastogenesis in osteopetrotic mice. J Orthop Res. 2001;19(4):554–558. doi: 10.1016/S0736-0266(00)00065-6. [DOI] [PubMed] [Google Scholar]
- 54.Kang KY, Hong YS, Park SH, Ju JH. Increased serum alkaline phosphatase levels correlate with high disease activity and low bone mineral density in patients with axial spondyloarthritis. Semin Arthritis Rheum. 2015;45(2):202–207. doi: 10.1016/j.semarthrit.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Kendall MJ, Lawrence DS, Shuttleworth GR, Whitfield AG. Haematology and biochemistry of ankylosing spondylitis. Br Med J. 1973;2(5860):235–237. doi: 10.1136/bmj.2.5860.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheehan NJ, Slavin BM, Kind PR, Mathews JA. Increased serum alkaline phosphatase activity in ankylosing spondylitis. Ann Rheum Dis. 1983;42(5):563–565. doi: 10.1136/ard.42.5.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siede WH, Seiffert UB, Merle S, Goll HG, Oremek G. Alkaline phosphatase isoenzymes in rheumatic diseases. Clin Biochem. 1989;22(2):121–124. doi: 10.1016/S0009-9120(89)80009-8. [DOI] [PubMed] [Google Scholar]
- 58.Bruijnen STG, et al. Bone formation in ankylosing spondylitis during anti-tumour necrosis factor therapy imaged by 18F-fluoride positron emission tomography. Rheumatology (Oxford) 2018;57(4):631–638. doi: 10.1093/rheumatology/kex448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen XD, Dusevich V, Feng JQ, Manolagas SC, Jilka RL. Extracellular matrix made by bone marrow cells facilitates expansion of marrow-derived mesenchymal progenitor cells and prevents their differentiation into osteoblasts. J Bone Miner Res. 2007;22(12):1943–1956. doi: 10.1359/jbmr.070725. [DOI] [PubMed] [Google Scholar]
- 60.Steward AJ, Kelly DJ. Mechanical regulation of mesenchymal stem cell differentiation. J Anat. 2015;227(6):717–731. doi: 10.1111/joa.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamin M, McGonagle D. The enthesis organ concept and its relevance to the spondyloarthropathies. Adv Exp Med Biol. 2009;649:57–70. doi: 10.1007/978-1-4419-0298-6_4. [DOI] [PubMed] [Google Scholar]
- 62.Jacques P, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis. 2014;73(2):437–445. doi: 10.1136/annrheumdis-2013-203643. [DOI] [PubMed] [Google Scholar]
- 63.Hreggvidsdottir HS, Noordenbos T, Baeten DL. Inflammatory pathways in spondyloarthritis. Mol Immunol. 2014;57(1):28–37. doi: 10.1016/j.molimm.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 64.Simone D, Al Mossawi MH, Bowness P. Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatology (Oxford) 2018;57(suppl 6):vi4–vi9. doi: 10.1093/rheumatology/key001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braun J, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995;38(4):499–505. doi: 10.1002/art.1780380407. [DOI] [PubMed] [Google Scholar]
- 66.Appel H, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther. 2011;13(3):R95. doi: 10.1186/ar3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Appel H, et al. In situ analysis of interleukin-23- and interleukin-12-positive cells in the spine of patients with ankylosing spondylitis. Arthritis Rheum. 2013;65(6):1522–1529. doi: 10.1002/art.37937. [DOI] [PubMed] [Google Scholar]
- 68.François RJ, Neure L, Sieper J, Braun J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann Rheum Dis. 2006;65(6):713–720. doi: 10.1136/ard.2005.037465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richard J. Group 3 innate lymphoid cells in human enthesis. Arthritis Rheum. 2017;69(9):1816–1822. doi: 10.1002/art.40150. [DOI] [PubMed] [Google Scholar]
- 70.Wohl GR, Towler DA, Silva MJ. Stress fracture healing: fatigue loading of the rat ulna induces upregulation in expression of osteogenic and angiogenic genes that mimic the intramembranous portion of fracture repair. Bone. 2009;44(2):320–330. doi: 10.1016/j.bone.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su ST, Ying HY, Chiu YK, Lin FR, Chen MY, Lin KI. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol Cell Biol. 2009;29(6):1421–1431. doi: 10.1128/MCB.01158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weiss EH, Kuon W, Dörner C, Lang M, Riethmüller G. Organization, sequence and expression of the HLA-B27 gene: a molecular approach to analyze HLA and disease associations. Immunobiology. 1985;170(5):367–380. doi: 10.1016/S0171-2985(85)80061-9. [DOI] [PubMed] [Google Scholar]
- 73.Lin KI, Calame K. Introduction of genes into primary murine splenic B cells using retrovirus vectors. Methods Mol Biol. 2004;271:139–148. doi: 10.1385/1-59259-796-3:139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.