

Abstract
Device‐encapsulated human stem cell‐derived pancreatic endoderm (PE) can generate functional β‐cell implants in the subcutis of mice, which has led to the start of clinical studies in type 1 diabetes. Assessment of the formed functional β‐cell mass (FBM) and its correlation with in vivo metabolic markers can guide clinical translation. We recently reported ex vivo characteristics of device‐encapsulated human embryonic stem cell‐derived (hES)‐PE implants in mice that had established a metabolically adequate FBM during 50‐week follow‐up. Cell suspensions from retrieved implants indicated a correlation with the number of formed β cells and their maturation to a functional state comparable to human pancreatic β cells. Variability in metabolic outcome was attributed to differences in number of PE‐generated β cells. This variability hinders studies on processes involved in FBM‐formation. This study reports modifications that reduce variability. It is undertaken with device‐encapsulated human induced pluripotent stem cell‐derived‐PE subcutaneously implanted in mice. Cell mass of each cell type was determined on intact tissue inside the device to obtain more precise data than following isolation and dispersion. Implants in a preformed pouch generated a glucose‐controlling β‐cell mass within 20 weeks in over 60% of recipients versus less than 20% in the absence of a pouch, whether the same or threefold higher cell dose had been inserted. In situ analysis of implants indicated a role for pancreatic progenitor cell expansion and endocrine differentiation in achieving the size of β‐ and α‐cell mass that correlated with in vivo markers of metabolic control. stem cells translational medicine 2019;8:1296&1305
Keywords: Diabetes, Induced pluripotent stem cells, Pancreatic differentiation, Cell transplantation, Progenitor cells
Device‐encapsulated human induced pluripotent stem cell‐derived pancreatic endoderm can generate a glucose‐controlling β‐cell mass in immune‐compromised mice. For a dose of 5 × 106 cells, this involves pancreatic progenitor cell differentiation as well as a cell mass increase following an initial cell loss. Measurement of these processes inside the device allows selection of conditions that help achieve metabolic control.
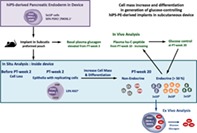
Significance Statement.
Human pluripotent stem cell‐derived pancreatic progenitors represent a candidate source for β‐cell replacement in type 1 diabetes. Subcutaneous implants in a device can generate insulin‐producing β cells in mice, capable of glucose control, but the outcome is variable. This study investigates formation of a β‐cell mass inside the devices to identify underlying mechanisms and conditions that reduce variability. A protocol modification resulted in more consistent formation of a glucose‐controlling β‐cell mass. The role of cell expansion and endocrine cell differentiation inside the devices was demonstrated. The methods and observations guide further development of this potential cell therapy.
Introduction
Type 1 diabetes causes a depletion in pancreatic β cells and hence a complete loss in cell‐regulated glucose control. β‐Cell implants prepared from human donor pancreases can restore endogenous insulin production and glucose control, but shortage of human donor organs limits implementation of this form of cell therapy. Human pluripotent stem cells may overcome this limitation as they represent a large‐scale cell source that can be differentiated into pancreatic cells in the laboratory, which were shown to generate insulin‐releasing implants in mice 1. Studies with human embryonic stem cell‐derived pancreatic endoderm (hES‐PE) have advanced this development to clinical translation 2, 3 (clinicalTrials.gov‐ViaCyte). The ability of hES‐PE to generate functional β‐cell implants in mice was demonstrated for nonencapsulated as well as for encapsulated grafts 1, 4, 5. This potential was also observed with human induced pluripotent stem cell‐derived pancreatic endoderm (hiPS‐PE) and with further differentiation stages containing insulin‐positive cells in the grafts 6, 7, 8, 9, 10.
We have assessed the functional β‐cell mass (FBM) that is formed in device‐encapsulated hES‐PE implants in the subcutis of mice, using a combination of in vivo and ex vivo markers 4, 5, 11. It was thus found that implants which resulted in plasma hu‐C‐peptide levels >6 ng/ml at post‐transplant (PT)‐week 20 achieved metabolic control from PT‐week 20 to 50 while this was not the case for recipients with lower levels. These implants presented the following characteristics when analyzed following retrieval at PT‐week 50: (a) more than 0.3 × 106 β cells at >50% endocrine purity and (b) a functional state of the β cells that was similar to that of isolated human pancreatic β cells in terms of secretory responses during perifusion, content in typical secretory vesicles, and nuclear NKX6.1‐PDX1‐MAFA coexpression 5. The processes leading to formation of this metabolically adequate FBM should now be investigated. To this end, the percent of recipients with adequate β‐cell mass formation needs first to be increased. In our prior study, only 26% achieved hu‐C‐peptide >6 ng/ml at PT‐week 20, the others exhibiting levels down to under 0.5 ng/ml, indicating a high variability in β‐cell mass formation, confirmed by PT‐week 50 counts of β‐cell numbers in retrieved and dispersed implants. This variability hinders studies on mechanisms, timing, and duration of the growth in β‐cell mass as it requires a higher number of recipients at start in order to establish a subgroup with similar rise to plasma hu‐C‐peptide levels that mark metabolic control. Moreover, it interferes with interpretation of in situ data during the first post‐transplant weeks when plasma hu‐C‐peptide is not yet detected and thus cannot serve as in vivo marker for the degree of β‐cell mass formation. To facilitate the investigation of early and late processes in β‐cell formation we wished to identify conditions that help establish PE‐generated β‐cell implants with less variability than in the model studied so far. Our study has been undertaken with hiPS‐PE instead of hES‐PE in our previous work and also applied another device for its encapsulation 9, 12. The similarity in 20‐week plasma hu‐C‐peptide profiles of comparable cell numbers at start and in variability of individual values supports use of this hiPS‐PE‐device combination to address the aim. Two modifications were tested for their ability to help reach plasma hu‐C‐peptide >6 ng/ml at PT‐week 20. The significance of this in vivo marker was evaluated by its correlation with glucose control and with the β‐cell mass as measured in situ on intact implants, a more precise method than previous measurements on retrieved and dispersed implants. The first modification consisted in increasing the number of cells in the device at start, the second in implanting the device in a preformed pouch, a method that was reported to improve outcome of nonencapsulated islet cell and hES‐PE implants 13, 14.
Materials and Methods
Transplantation of hiPS‐Derived PE in Mice
PE was derived from hiPS cells (produced by Fujifilm Cellular Dynamics, WI) by Nestlé Research (Lausanne, Switzerland) using a scalable and reproducible four‐stage differentiation protocol 9. Cryopreserved samples were shipped to Brussels. After thawing, aggregates were cultured for 72 hours in “stage 4” medium composed of Dulbecco's modified Eagle's medium high‐glucose with Glutamax, 1% penicillin/streptomycin and 0.5× B27, supplemented by 50 ng/ml Noggin, epidermal growth factor, and keratinocyte growth factor (R&D Systems, MN) and 30 ng/ml Heregulin‐β1 (tebu‐bio, France) 9. At end of culture, the preparation contained >90% living cells, the majority with nuclear PDX1 (Pancreatic and Duodenal Homeobox 1) positivity and 50% ± 5% being double‐positive for PDX1 and NKX6.1 (NK6 Homeobox 1) (average for three independent experiments; Supporting Information Fig. S1), similar to the characterization in 9. An average 10% of cells were single‐ or double‐hormone positive after staining for insulin, glucagon and somatostatin; they were negative for PDX1 and NKX6.1 (Supporting Information Fig. S1). Planar sheet macro‐devices were also provided by Nestlé Research. They are composed of an outer 77 μm polyester mesh and a hydrophilic polytetrafluoroethylene 0.45 μm‐porous (cell‐impermeable) inner membrane held together by a polypropylene frame 12. These devices have been shown to support long‐term survival of allogeneic myoblasts implanted in the subcutaneous space 12. They were loaded with 5 × 106 (Device 5M) or 15 × 106 (Device 15M) hiPS‐PE cells before subcutaneous implantation in 8–12 weeks old male Fox Chase SCID/Beige mice (CB17.Cg‐PrkdcscidLystbg‐J/Crl; Charles River, France). Devices with 5 × 106 cells were also tested after insertion in a preformed pouch (P‐Device 5M; Fig. 1). Mice were followed for circulating markers of implant function over 20 weeks PT with blood taken under basal condition (2 hours‐food removal) or following intraperitoneal glucose injection (3 g/kg body weight). Mice without implant were kept in parallel to collect control values on glycemia and plasma mouse C‐peptide levels; they had not undergone surgery. Plasma proinsulin and hu‐C‐peptide were measured by trefoil‐type time‐resolved fluorescence immunoassay 15, plasma glucagon by radioimmunoassay (Merck‐Millipore, Germany), mouse (m‐)C‐peptide by enzyme‐linked immunosorbent assay (Crystal Chem, IL), and blood glucose using Glucocard (Menarini Diagnostics, Italy). These levels were also determined in a subgroup of recipient and control mice following alloxan injection (Sigma–Aldrich, MO; 50 mg/kg body weight intravenously) at PT‐week 20, which is expected to destroy the mouse pancreatic β cells. Procedures were approved by the Ethical Committee of VUB and carried out according to European Community Council Directive (86/609/EEC). As in our previous studies we use the term “graft” for cells before transplantation and “implant” for the tissue after implantation. Device‐encapsulated implants were retrieved for in situ (PT‐weeks 2–20) and ex vivo analysis (PT‐week 20).
Figure 1.
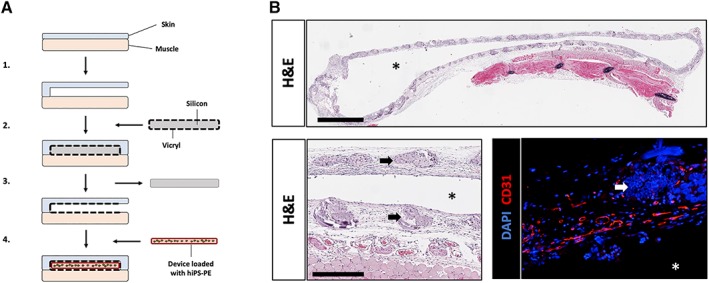
Pretreatment of implant site by forming tissue pouch in subcutaneous space. (A): Medical‐grade silicon sheets (Invotec, FL) are cut according to the size of our devices and sewn into Vicryl bags (Ethicon, NJ) using surgical wire. A subcutaneous space is created by separating the skin and muscle layers before inserting the silicon‐Vicryl pocket and stitching the wound. After 5 weeks of healing, the silicon sheet can be easily retrieved from the pocket without any tissue attachment. A device loaded with human induced pluripotent stem cell‐derived pancreatic endoderm cells is then inserted in the location of the silicon, before closing the wound. (B): Histology of PT‐week 5 pouches shows engraftment and vascularization of the Vicryl mesh (Hematoxylin–Eosin; scale bar = 2 mm on top, 500 μm on lower left; and immunofluorescent staining for CD31, lower right; scale bar: 250 μm). Stars indicate the lumen of the tissue pouch. Arrows indicate remnants of Vicryl fibers (lower left).
In Situ Histological Analysis of hiPS‐PE Implants
Retrieved devices were fixed in 4% neutral‐buffered formaldehyde, processed, embedded in paraffin, and completely sectioned (5 μm). Sections were stained for cellular reactivity with guinea pig anti‐insulin, rabbit anti‐glucagon (each 1/1,000, in house‐produced), rat anti‐somatostatin (1/100, Abcam, U.K.), rabbit anti‐vimentin (1/100, Dako, Denmark), mouse anti‐CK19 (1/20, Dako), and rabbit anti‐CD31 (1/100, Abcam). Cells were characterized for nuclear positivity after incubation with goat anti‐PDX1 (1/100, R&D Systems, MN), mouse anti‐NKX6.1 (1/100, Hybridoma Bank, NIH, IA), rabbit anti‐MAFA (V‐Maf Musculoaponeurotic Fibrosarcoma Oncogene Homolog A) (1/1,000, a kind gift of Dr. A. Rezania), and mouse anti‐KI67 (1/100, Dako) or rabbit anti‐KI67 (1/100, Acris, Germany). Secondary antibodies were Cyanine‐ or AlexaFluor‐conjugated F(ab′)2 fragments of affinity‐purified antibodies allowing multiple labelling (1/500, Jackson ImunoResearch, U.K.). Nuclei were stained by DAPI (Sigma–Aldrich). Digital images were analyzed with SmartCapture 3 software (DSUK, U.K.). Total volumes of cell types were determined semiautomatically according to an adaptation of a morphometric method originally established in rat pancreases 16 and described in Supporting Information Figure S2. For general histology, sections were stained with Hematoxylin & Eosin or Masson's Trichrome, images acquired with an Aperio CS2 (Leica Biosystems, Germany) and visualized with Pathomation software (Belgium).
Ex Vivo Analysis of hiPS‐PE‐Derived Cells Isolated from Retrieved Devices
At PT‐week 20, implanted devices were retrieved and opened to isolate tissue content and disperse it to an aggregate suspension (<500 μm diameter) using type XI collagenase (1 mg/ml, Sigma–Aldrich). After washing, samples were taken for analysis of cell number (NucleoCounter YC‐100, ChemoMetec, Denmark), composition and functions as previously described 4, 5. Cell composition following dispersion was determined by manual count in immunocytochemistry on cells fixed in glutaraldehyde and embedded in araldite. Sections were stained using the same antibodies as described above. Insulin secretory responsiveness to glucose was determined in a perifusion assay. A sample with 500–700 × 103 cells was loaded on a P2‐Bio‐gel column (Bio‐Rad, CA) and perifused for 60‐minutes at 2.5 mmol/l glucose (basal), before pulsing with 5, 10, or 20 mmol/l glucose, with or without 10 nmol/l glucagon 4. Perifusate was collected in 1 minute fractions and assayed for insulin 4. Data were expressed per 103 β cells as determined in the sample that was added to the column.
Statistical Analysis
Results are expressed as means ± SD. Statistical analysis was performed using Prism 5.0 (GraphPad Software, CA) for comparison by one‐way analysis of variance with Tukey's post hoc test or Student's t test (statistical significance at p < .05).
Results
Effect of Implantation in Preformed Pouch on Generation of Human β Cells in Device‐Encapsulated hiPS‐PE over 20 Weeks
Plasma hu‐C‐peptide was used as in vivo marker for the generation of β cells in hiPS‐PE implants. The peptide was not detected in mice without device (<0.1 ng/ml detection limit). As in previous studies 4, 5, a glucose‐induced level >0.5 ng/ml was set as criterium for the presence of functioning human β cells. This was the case for the average levels in the Device 5M group from PT‐week 10 onward, progressively increasing up to PT‐week 20 (Fig. 2A). They were similar for devices containing threefold more cells at start (Device 15M) but significantly higher when devices with 5M cells were implanted in a preformed pouch (P‐Device 5M; Fig. 2A). As for hES‐PE implants 5, individual values within each group varied widely. Increasing cell number from 5 to 15 million cells did not increase the percent recipients that reached the aimed in vivo marker (hu‐C peptide >6 ng/ml; <20% in each group) but implants in a pouch achieved it in over 60% of recipients.
Figure 2.
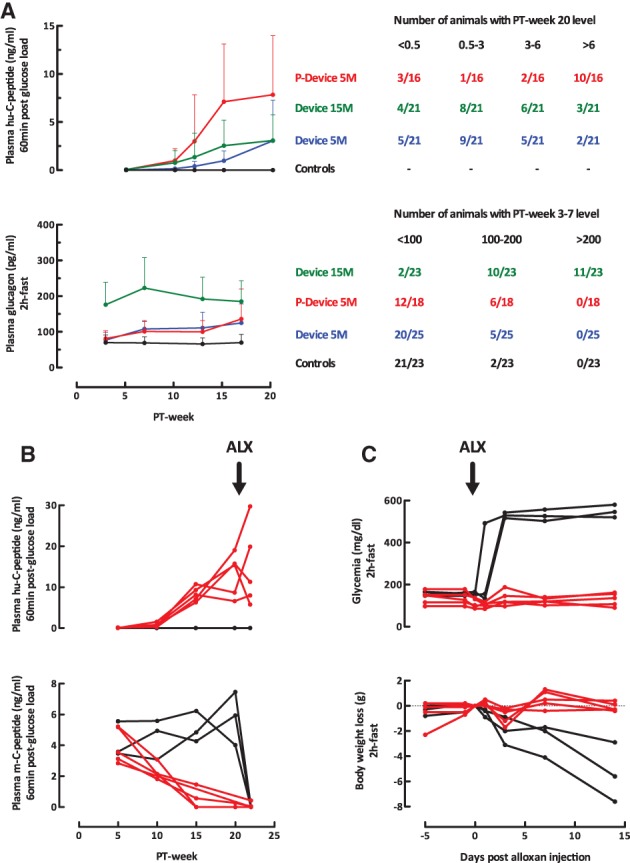
In vivo markers of implant function in recipients of device‐encapsulated human induced pluripotent stem cell‐derived pancreatic endoderm. (A): Plasma human (hu‐)C‐peptide levels (60 minutes postglucose load, 3 g/kg body weight, intraperitoneal) and plasma glucagon levels (basal, after 2 hours fast). Left part shows averages ± SD over the 20‐week follow‐up period of subgroups with 5 or 15 million cells at start with or without preformed pouch (P). Plasma hu‐C‐peptide <0.1 ng/ml (assay limit of detection) was considered as zero. Right part tabulates number of animals per subgroup according to the change in hormone levels. (B, C): Recipients of P‐Device with plasma hu‐C‐peptide levels >6 ng/ml (n = 5; red curves) and age‐matched controls (n = 3; black curves) were injected with alloxan (50 mg/kg BW) at post‐transplant week 20 and followed for plasma hu‐C‐peptide and mouse (m‐)C‐peptide levels (60 minutes postglucose load), basal glycemia, (2 hours fast) and body weight.
Recipients of device‐encapsulated hiPS‐PE implants exhibited higher fasting plasma glucagon levels (Fig. 2A), as was also the case for device‐encapsulated hES‐PE implants 5. The increase was noticed from PT‐week 3 onward and remained present until the end of the study. Levels were comparable for implants with 5 × 106 cells whether placed in a preformed pouch or not, but significantly higher with 15 × 106 cells at start. For this higher cell dose, the majority of recipients presented levels >200 pg/ml at PT‐week 3–7, which was never achieved in the other groups or in mice without implant (Fig. 2A). The higher plasma glucagon levels were not correlated with higher plasma hu‐C‐peptide levels at PT‐week 20; they also occurred in recipients with hu‐C‐peptide <0.5 ng/ml throughout follow‐up.
Basal and Stimulated Glucose‐Responsiveness of Device‐Encapsulated hiPS‐PE Implants Associated to Glucose Control in Recipients
Recipients exhibiting functioning human β cells at PT‐week 20 (hu‐C‐peptide >0.5 ng/ml at minute 60 postglucose injection) were analyzed for their plasma hu‐C‐peptide levels at basal glycemia and following glucose injection, an index for their glucose responsiveness (Table 1). Those in Device 5M group exhibited a response, but this was only statistically significant at minute 60, at which time glycemia was still elevated as in mice without implant. This was also the case in the Device 15M group, with similar values. In the P‐Device 5M, they presented higher hu‐C‐peptide levels than those in the Device 5M group, basal as well as after glucose injection. Both minute 15 and minute 60 values were statistically higher than basal with a reduction in glycemia to fasting values at minute 60 (Table 1). These observations indicate that the β‐cell mass in pouch‐implants is secretory active at basal glycemia in mice (150 mg/dl) and rapidly responsive to a rise in glycemia. It is responsible for the correction in glycemia at minute 60 since the pancreatic β‐cell mass of these mice is suppressed as shown by the plasma mouse C‐peptide levels (<0.5 ng/ml 60 minutes after glucose injection): this suppression progressively developed over the preceding weeks (red curves in Fig. 2B) as opposed to values >3 ng/ml in mice without implant (black curves in Fig. 2B).
Table 1.
Metabolic parameters at PT‐weeks 17–20 in recipients of functioning hiPS‐PE implants
| Plasma hu‐C‐peptide (ng/ml) postglucose load | Glycemia (mg/dl) postglucose load | Plasma glucagon (pg/ml) | |||||
|---|---|---|---|---|---|---|---|
| 0 minutes | 15 minutes | 60 minutes | 0 minutes | 15 minutes | 60 minutes | 2 hours‐fasting | |
| Device SC 5 × 106 cells | 0.7 ± 0.7 | 1.9 ± 2.4 | 4.0 ± 4.5†† | 161 ± 15° | 501 ± 62††† | 311 ± 107††† ‡‡‡ | 144 ± 50°°° |
| Device SC 15 × 106 cells | 0.7 ± 0.5 | 1.8 ± 1.8 | 3.8 ± 2.5††† ‡‡ | 145 ± 18** | 495 ± 80††† | 302 ± 106††† ‡‡‡ | 195 ± 57* °°° |
| P‐device SC 5 × 106 cells | 2.4 ± 1.0*** | 5.9 ± 3.0*** † | 9.6 ± 5.3** ††† ‡ | 149 ± 30 | 419 ± 165** ††† | 165 ± 78*** °° ‡‡‡ | 153 ± 88° |
| Controls | NA | NA | NA | 147 ± 19 | 473 ± 83††† | 287 ± 103††† ‡‡‡ | 70 ± 23 |
Glycemia, plasma human C‐peptide, and glucagon levels measured at PT‐weeks 17 (glucagon) and 20 (glycemia, C‐peptide). Measurements were performed before (0 minutes), 15 minutes, or 60 minutes postglucose load (3 g/kg body weight, intraperitoneal) in 2 hours‐fasted animals.
Statistical differences at a given time point by Student's t test:
Versus Device SC 5 × 106 cells:
p < .05.
p < .01.
p < .001.
Versus Controls:
p < .05.
p < .05.
p < .001.
Statistical differences between values at 0 minutes, 15 minutes, and 60 minutes within a given condition, by one‐way analysis of variance with Tukey's post hoc test:
15 minutes and 60 minutes versus 0 minutes:
p < .05.
p < .05.
p < .001.
15 minutes versus 60 minutes:
p < .05.
p < .05.
p < .001.
Abbreviations: hiPS‐PE, human induced pluripotent stem cell‐derived pancreatic endoderm; PT, post‐transplant; SC, subcutaneous.
The β‐cell mass in pouch‐implants maintained glucose control after injection of alloxan, without decline in hu‐C‐peptide levels and rise in glycemia (red curves in Fig. 2B, 2C). In mice without implant, alloxan abolished mouse C‐peptide release and caused hyperglycemia and body weight loss (black curves in Fig. 2B, 2C).
Correlation Between Glucose‐Induced Plasma Hu‐C‐Peptide Levels and Size of Formed β‐Cell and α‐Cell Mass in Devices with hiPS‐PE Implants
At PT‐week 20, glucose‐induced plasma hu‐C‐peptide levels correlated with total β‐cell volume as measured in the device (Fig. 3A). They were also strongly correlated with α‐cell volume, and weakly with that of nonendocrine CK19‐positive cells (Fig. 3A). When total cell volume measured in the device was compared with that in the initial grafts, a wide variability was noticed. For hu‐C‐peptide levels <0.5 ng/ml, it was lower than at start (Fig. 3A) and/or mainly consisted of hormone‐negative cells (Fig. 4) that were vimentin (VIM)‐ or CK19‐positive. For levels >0.5 ng/ml, it was higher (7/9 examined implants; Fig. 3A) and consisted for a large proportion of single‐hormone positive cells (Fig. 4B).
Figure 3.
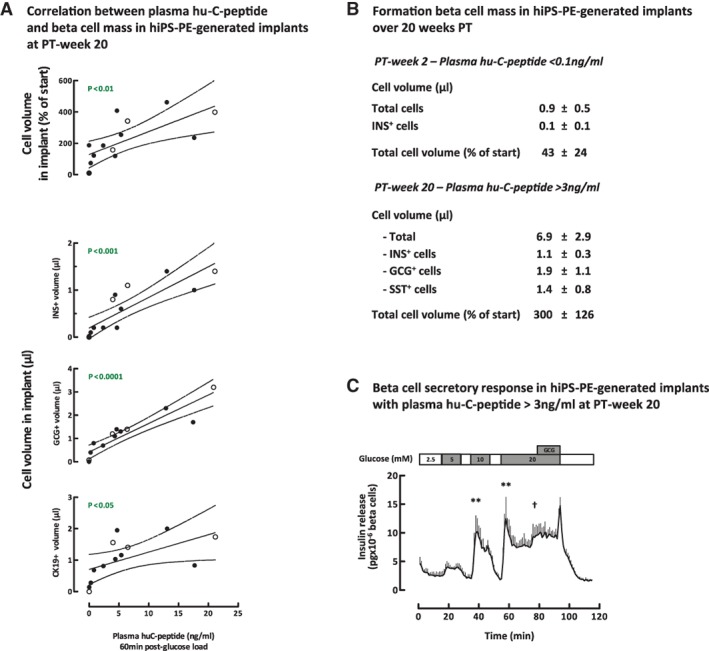
Correlation between glucose‐induced plasma hu‐C‐peptide levels and pancreatic endocrine cell mass in device‐encapsulated human induced pluripotent stem cell‐derived pancreatic endoderm (hiPS‐PE) implants at post‐transplant (PT)‐week 20. (A): In situ cell mass measurements (as volume) in devices at PT‐week 20 with initially 5 × 106 hiPS‐PE cells are plotted against the corresponding glucose‐induced plasma hu‐C‐peptide levels. Linear regression analysis indicates high correlations for the volumes of insulin‐ (μl) and of glucagon‐ (μl) positive cells and lower correlations for the volume of CK19‐positive cells (μl) and total cell volume (expressed as percent of initial volume). (B): In situ total and hormone‐positive cell mass in devices of pouch‐implants with plasma hu‐C‐peptide >3 ng/ml at PT‐week 20. Comparison with values at PT‐week 2 (no hu‐C‐peptide detected). (C): ex vivo β‐cell secretory response of pouch‐implants retrieved from mice with plasma hu‐C‐peptide >3 ng/ml. Glucose concentrations are indicated on top. Curve shows insulin release at baseline (2.5 mmol/l glucose) and at higher glucose concentration in absence and presence of glucagon (10 nmol/l). Data are expressed as pg × 10−3 β cells × minute−1. Statistical differences in first‐phase insulin release peaks at 5–10–20 mmol/l glucose versus baseline, by one‐way analysis of variance with Tukey's post hoc test: **, p < .01. Student's t test of second‐phase release at 20 mmol/l glucose with or without glucagon: †, p < .05.
Figure 4.
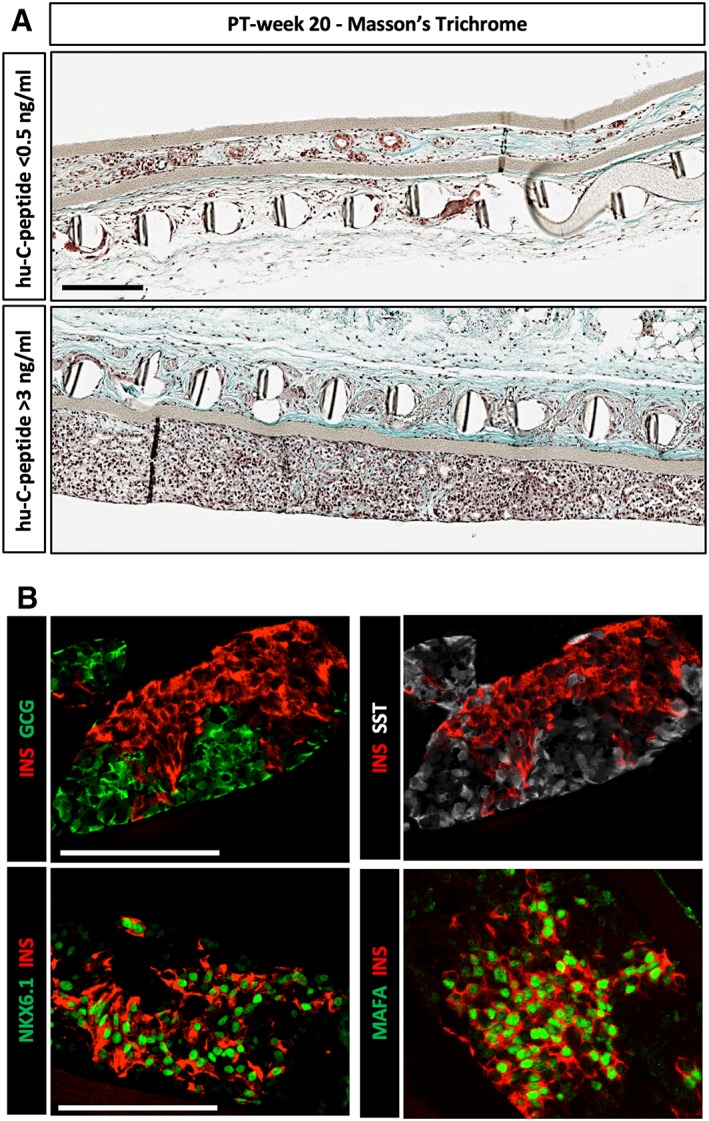
Pancreatic endocrine cells in device‐encapsulated human induced pluripotent stem cell‐derived pancreatic endoderm (hiPS‐PE) implants at post‐transplant (PT)‐week 20. (A): In situ histological analysis of device‐implants at PT‐week 20 and stained with Masson's trichrome, illustrating cell recovery and phenotype in implants from recipients without or with glucose‐stimulated plasma hu‐C‐peptide (<0.5 ng/ml, top; >3 ng/ml, bottom). Scale bar: 100 μm. (B): In situ analysis of device‐implants in pouch with plasma hu‐C‐peptide levels >3 ng/ml at PT‐week 20. Presence of single‐hormone positive cells staining for insulin, glucagon, or somatostatin (top). Most insulin‐positive cells exhibit strong nuclear expression of transcription factors PDX1, NKX6.1 and MAFA (bottom). Scale bars: 100 μm.
In pouch‐implants with plasma hu‐C‐peptide >3 ng/ml at PT‐week 20, average total cell volume was threefold higher than in grafts at start. When compared with cell volume at PT‐week 2, a sevenfold increase seemed to have occurred between these time points, following a cell loss during the first 2 weeks postimplantation (Fig. 3B). At PT‐week 20, no double‐hormone positive cells were noticed and 64% of cells were single‐hormone positive (Fig. 3B). The volume of insulin‐positive cells was 40% smaller than that of the associated glucagon‐positive cell population and also slightly smaller than the somatostatin‐positive cell volume. Insulin‐positive cells exhibited nuclear coexpression of PDX1, NKX6.1, and MAFA (Fig. 4B). As in hES‐PE implants 4, 5, the hormone‐negative subpopulation consisted of CK19‐ and VIM‐positive cells.
The newly formed β cells were examined for their secretory function following retrieval of the implants at PT‐week 20 (Fig. 3C). After dispersion, implant aggregates were perifused at varying glucose concentrations and insulin was measured in the perifusate. Their responsiveness to changes in glucose concentration appeared slightly higher than that previously reported for device‐encapsulated hES‐PE at PT‐week 20 4 but lower than that at PT‐week 50 5. Insulin was immediately released when basal glucose (2.5 mmol/l) was elevated to 5, 10, or 20 mmol/l, with rapid suppression upon return to the basal condition. At 20 mmol/l, second‐phase release was amplified by glucagon (Fig. 3C), indicating responsiveness of the formed β cells to the α‐cell hormone. No differences were observed in insulin secretory capacity of β cells retrieved from implants with different plasma hu‐C‐peptide levels, either with or without pouch.
Identification of Cells in Proliferative Activity in Device‐Encapsulated hiPS‐PE Implants
The observed growth in cell mass between PT‐weeks 2 and 20 led us to search for cells in proliferative activity. At PT‐week 2, all analyzed implants—whether with or without pouch—were mainly composed of PDX1+/NKX6.1+/SOX9+ positive cells, with <10% hormone‐positive cells, similar to grafts at start. However, their PDX1+/NKX6.1+ cells were strongly CK19‐positive while this staining was weak and spotty in the initial aggregates (Fig. 5). A large proportion of these CK19+/PDX1+/NKX6.1+ were now organized as an epithelial cell layer around a lumen, with VIM expression at their basal pole and CK19 at their apical pole (Fig. 5). A smaller fraction with weaker CK19+ staining occurred clustered between the duct‐like structures and was found associated with hormone‐positive cells (Fig. 5), some double‐positive, and mostly glucagon‐positive and NKX6.1‐negative. KI67‐positivity was almost exclusively found in the PDX1+/NKX6.1+/CK19+ cells, indicating that 13% of these cells were in proliferative activity (Fig. 6). Proliferation rates in these CK19+ cells tended to be higher in pouch implants but the difference was not statistically significative.
Figure 5.
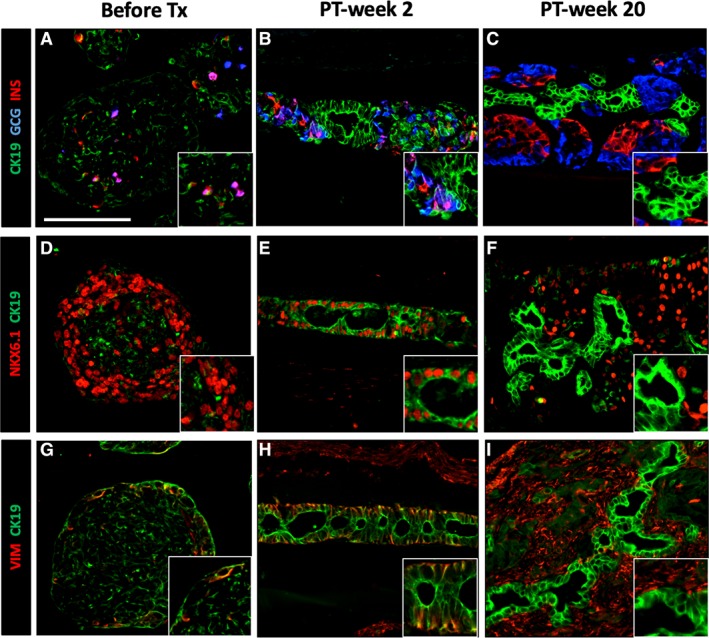
Transient CK19‐expression in pancreatic progenitor cells in human induced pluripotent stem cell‐derived pancreatic endoderm (hiPS‐PE) implants. In situ histological analysis of device‐encapsulated hiPS‐PE implants at post‐transplant (PT)‐week 2 and 20, comparison with start preparation. The preparation at start contained 45%–55% PDX1+/NKX6.1+/hormone‐negative cells that are considered as pancreatic progenitors; they exhibited a weak and spotty CK19‐positivity (A, D, G). At PT‐week 2 (B, E, H), the majority of PDX1+/NKX6.1+ cells stained strongly positive for cytokeratin‐19 (CK19) with vimentin‐positivity (VIM) at their basal pole; they formed an epithelial cell layer around small cysts Adjacent cell clusters contained weakly CK19+ cells associated to small numbers of hormone‐positive cells (insulin, glucagon). At PT‐week 20 (C, F, I), the epithelial cell layer was formed by CK19‐positive that were negative for PDX1, NKX6.1, and VIM, while adjacent cell clusters contained PDX1+/NKX6.1+/insulin‐positive cells. Scale bar: 100 μm.
Figure 6.
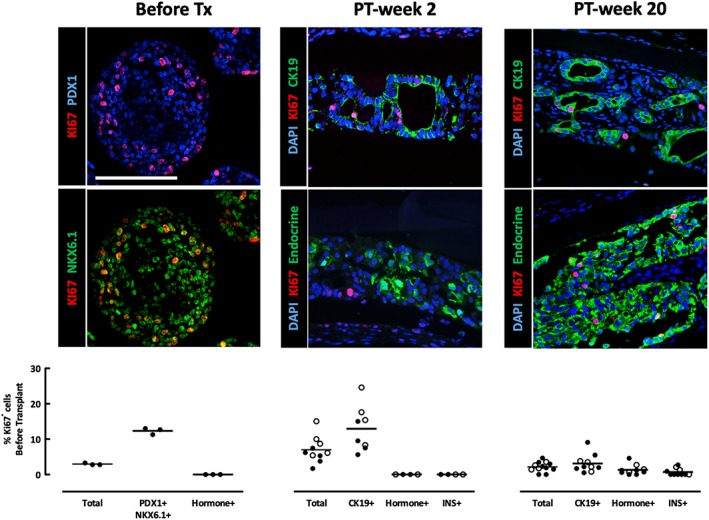
Identification of cells in proliferating activity in device‐encapsulated human induced pluripotent stem cell‐derived pancreatic endoderm (hiPS‐PE) implants. In situ analysis of device‐encapsulated hiPS‐PE implants for the presence and identification of KI67‐positive cells, and comparison with start preparation. Cell types were identified on basis of their staining for CK19 (cytokeratin‐19), insulin (INS), vimentin (VIM), and pancreatic endocrine (insulin plus glucagon plus somatostatin) markers. Scale bar: 100 μm. In the start preparation 96% ± 5% of KI67+ cells were located in PDX1+/NKX6.1+ pancreatic progenitor cells which exhibited 12% ± 1% KI67‐positive cells. At PT‐week 2, implants from the P‐Device condition (empty circles) tended to have a higher proliferation rate than the Device condition (filled circles; 9.1% ± 3.8% vs. 4.9% ± 2.2%, respectively), but this difference was not statistically significant. Virtually all KI67+ cells (100% ± 1%) were identified as CK19+ cells present in epithelial cell layers. At PT‐week 20, most KI67+ cells remained located in the CK19+ cell population of the epithelial cell layers, but the percentage of CK19+/KI67+ cells was significantly lower than at PT‐week 2 (2.4% ± 1.5%) but higher than the percentage of INS+/KI67+ cells (0.7% ± 1.0%).
At PT‐week 20, all implants—whether with or without pouch—still presented CK19+ cells in epithelial layers around a lumen but these were now NKX6.1−/VIM−; they represented the major fraction of cells in proliferative activity but with a lower and variable percentage (0.5%–9%). Between these duct‐like structures, clusters of single‐hormone positive cells were observed, including PDX1+/NKX6.1+/insulin (INS)+ cells with low and variable KI67‐positivity (0%–3%).
Discussion
This study shows that subcutaneous implants of device‐encapsulated hiPSC‐PE can generate a FBM that controls glycemia in mice. It thus reproduces the outcome that we previously reported for hES‐PE using another device 5. Glucose control by the hiPS‐PE‐generated β cells was demonstrated by plasma hu‐C‐peptide levels that responded to basal and stimulated glucose levels while mouse pancreatic β cells were suppressed. This now remained the case after destroying the mouse pancreatic β cells with alloxan to which human β cells are resistant 17. Metabolic control had developed within 20 weeks post‐transplantation in recipients with glucose‐induced plasma hu‐C‐peptide levels >6 ng/ml. Both in vivo markers were correlated with the size of the formed human β‐cell mass. This outcome was observed in the majority of recipients of hiPS‐PE devices which was not the case in our previous study with hES‐PE devices 5. This difference is not attributed to a difference in device or in quality of the preparation at start but to modifications in the procedure. Implants in a preformed pouch increased the percent with elevated hu‐C‐peptide levels within this follow‐up period. In addition, quantification of β‐cell mass on intact tissue inside the device is more precise provided that it is based on representative sampling as validated in the present study. This in situ approach will also help identify processes in the generation of the β‐cell mass and possible causes of failure.
Glucose‐controlling hiPS‐PE implants exhibited a β‐cell volume of 1.1 μl ± 0.3 μl at PT‐week 20, which is in the range of the β‐cell volume measured in the normal mouse pancreas (0.8–1.1 μl). Their β cells were single‐hormone positive, with nuclear positivity for PDX1, NKX6.1, and MAFA, markers of differentiation. When perifused ex vivo, they were shown to rapidly adjust their insulin release to increased or decreased glucose concentrations, a sign of functional maturation and a cellular basis for the observed in vivo hu‐C‐peptide responses. The amplitude of the in vivo secretory responses appears determined by the corresponding β‐cell volume in the implants as indicated by their close correlation.
Implants of device‐encapsulated hiPS‐PE in a preformed pouch reduced the number of recipients with low β‐cell mass at PT‐week 20. Pepper et al. showed that such site pretreatment facilitates survival and function of nonencapsulated pancreatic islet cells under the skin of mice, possibly by prevascularizing their microenvironment 13; they also found it to support development of in vivo β‐cell functions in nonencapsulated hES‐PE implants, but also of large cysts lined by PE‐generated epithelial cells 14, 18. In implants with metabolic control at PT‐week 20, we only noticed small cell lined cavities in the devices, with endocrine cells composing the major tissue fraction. Total cell volume was threefold higher than at start. This increase was not yet detected at PT‐week 2; on the contrary, total cell mass was lower than initially, possibly the result of cell loss during engraftment. Between PT‐weeks 2 and 20, it increased sevenfold comprising appearance of single‐hormone positive cells representing 64% of total cell volume at PT‐week 20.
The PDX1+/NKX6.1+/hormone‐negative pancreatic progenitor cells are considered as precursor for hormone‐positive cells and as drivers for cell expansion. In the start preparation they contained the majority of KI67‐positive cells and this was also the case in PT‐week 2 implants. At the latter time point, the PDX1+/NKX6.1+ population was however strongly CK19+ and organized as epithelial cell layer around small cavities. It also exhibited a VIM‐positivity at the basal pole of the cells, similar to that reported in proliferating duct cells of the mouse pancreas, and suggested to precede budding of newly formed β cells 19, 20. Our protocol was not designed to investigate such process but we did notice that the PDX1+/NKX6.1+/CK19+/VIM+ (KI67+) cell lining of the cavities had juxtaposed cell clusters containing weakly CK19+ cells and a small fraction of hormone‐positive cells, most expressing glucagon, with very few KI67+ cells. In the subsequent 18 weeks, both compartments increased in cell mass whereby the PDX1+/CK19+ epithelial cell layer became NKX6.1−/VIM− with less KI67 positive cells, and the neighboring cell clusters presented single‐hormone positive cells, including PDX1+/NKX6.1+/INS+ cells with few KI67+ cells. The model in which these data have been collected allows to investigate the processes between these two time points and the possible transition from epithelial cell layer to endocrine cell clusters.
On basis of the measured volumes of β‐cell and α‐cell population at PT week 20, and of the averages of individual β‐cell and α‐cell surface areas, we calculated that 106 β cells and 2 × 106 α cells had been formed since PT‐week 2; the number of somatostatin‐positive cells was not determined but their total mass suggests that it is not lower than that of the insulin‐positive cells. This endocrine cell formation occurs during a sevenfold expansion of total cell mass. It is at present unknown to which extent it depends on this cell amplification, and whether this is expressed as budding of endocrine progenitor cells from epithelia with replicating pancreatic progenitor cells, and/or whether it involves replication of newly formed endocrine cells. Formation of more α cells than β cells was also observed in device‐encapsulated hES‐PE implants at PT‐week 50 5; it may be induced by local influences such as hypoxia. The PE‐generated α cells have been shown to release glucagon under glucose‐dependence 4. They may thus exert positive paracrine effects on formation, maturation, and function of β cells 11, 21, 22, 23. They are also likely responsible for the higher plasma glucagon levels in recipients of hiPS‐PE or hES‐PE implants 5, occurring at an early stage, more than 5 weeks before hu‐C‐peptide was detected; implants with threefold higher cell number at start indeed resulted in a more pronounced elevation of plasma glucagon. It is so far unknown whether this elevation marks a local or systemic effect.
Conclusion
This study reports modifications that reduce variability in the formation of a FBM and thus facilitate studies on underlying mechanisms. Implants of device‐encapsulated hiPS‐PE cells in a preformed subcutaneous pouch were shown to generate, within 20 weeks, a glucose‐controlling β‐cell mass in the majority of recipient mice. This outcome was defined by in vivo markers that were applicable in normoglycemic mice. They correlated with β‐cell volume as measured in situ within intact implants. A dose of 5 × 106 cells at start with less than 10% mostly double‐hormone‐positive cells increased threefold over this period with more than 60% single hormone‐positive cells. In situ measurements of the mass of composing cell types indicated that the formed α‐cell mass was twofold larger than the formed β‐cell mass, raising the possibility of positive paracrine influences. Comparison with in situ analysis at PT‐week 2 indicated a role for pancreatic progenitor cell expansion and endocrine differentiation in achieving in vivo markers of metabolic control at PT‐week 20. Further studies using this model and markers can clarify their respective contribution and interactions.
Author Contributions
T.R.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; I.D.M., F.O.V.H., G.M.S., K.G.S., Z.L.: collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; B.K.: final approval of manuscript; M.R.C.K., C.H., N.B.: provision of study material, final approval of manuscript; D.G.P.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
T.R. is Ph.D. fellow of Research Foundation Flanders‐FWO. The other authors indicated no potential conflicts of interest.
Supporting information
Supplementary Figure 1 Cell composition of hiPS‐PE before transplantation.
Sections of fixed hiPS‐PE aggregates were stained for the nuclear markers PDX1 and NKX6.1 (Panel A1‐3) and for cytoplasmic positivity for the pancreatic hormones insulin (INS‐Panel A1, B1‐3), glucagon (GCG‐Panel B1,2) and somatostatin (SST‐Panel B1,3) (Scale bars: 100 μm). For each of these markers the percent positive cells was determined by manual count (>1,000 cells counted per section, one section per experiment, three independent experiments). A large proportion of cells exhibited double‐positivity for PDX1 and NKX6.1 without hormone‐positivity (50 ± 5%), an index for their pancreatic progenitor state. A smaller fraction was only positive for PDX1 and hormone‐negative (37 ± 4%). The hormone‐positive cells were negative for PDX1 and NKX6.1; they represented 11 ± 2% of all cells with a large proportion (3 ± 2%) being polyhormone positive.
Supplementary Figure 2 Cell mass measurements in device‐encapsulated implants
Total and (non‐)endocrine cell volumes were measured in device‐encapsulated implants. The method was adapted from that used to quantify beta cell mass in the rat pancreas (Chintinne et al. 2010).
(A) Retrieved devices were fixed in buffered formaldehyde and longitudinally cut in two parts before embedding in paraffin. They were completely sectioned, approximately 1,000 sections per device. Representative sections (1% of total implant; cfr. D) throughout the implant were immunostaining with DAPI (nuclei) or with antibodies to (non‐)endocrine cell markers. Stained cell areas were semi‐automatically quantified and values were extrapolated to calculate their total volume in the implant.
(B) Steps for acquisition, segmentation and quantification of cell areas in a section. Scale bar: 100 μm.
(C) Assessment of sample size to be analyzed in order to keep relative variation under 10%. Analysis of 1% of total implant surface was found sufficient.
(D) Validation of method to count the number of cells that had been loaded in a device. A strong and linear correlation was found between loaded cell number and DAPI volume in the device (μl).
(E) Nuclear size at PT‐week 2 was smaller than that at start and at PT‐week 20. The ratio of the average nuclear areas at start and at PT‐week 2 (0.89) was used as correction factor to calculate cell recovery at this time point.
Statistical difference between measurements at different time points calculated by 1‐way ANOVA with Tukey's post hoc test: * p < .05, ** p < .01.
(F) Similar average cross‐sectional areas of individual insulin‐ (INS) and glucagon‐ (GCG) positive cells at PT‐week 20.
Acknowledgments
We thank Dr. Rezania (when at BetaLogics, Venture, Janssen Research and Development, Raritan, NJ) for providing the anti‐MAFA antibody, and the Developmental Studies Hybridoma Bank (NIH, IA) for providing the anti‐NKX6.1 antibody. We are grateful to their collaborators at VUB‐Diabetes Research Center for their support, and the team of the Clinical Chemistry department of UZB for the peptide assays. This work was supported by grants from the European Commission (H2020 681070), Juvenile Diabetes Research Foundation (17‐2013‐296), and the Flemish Government (IWT130138).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kroon E, Martinson LA, Kadoya K et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose‐responsive insulin‐secreting cells in vivo. Nat Biotechnol 2008;26:443–452. [DOI] [PubMed] [Google Scholar]
- 2. Schulz TC, Young HY, Agulnick AD et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One 2012;7:e37004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agulnick A, Ambruzs D, Moorman A et al. Insulin‐producing endocrine cells differentiated in vitro from human embryonic stem cells function in macroencapsulation devices in vivo. Stem Cells Translational Medicine 2015;4:1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Motté E, Szepessy E, Suenens K et al. Composition and function of macroencapsulated human embryonic stem cell‐derived implants: Comparison with clinical human islet cell grafts. Am J Physiol Endocrinol Metab 2014;307:E838–E846. [DOI] [PubMed] [Google Scholar]
- 5. Robert T, De Mesmaeker I, Stangé GM et al. Functional beta cell mass from device‐encapsulated hESC‐derived pancreatic endoderm achieving metabolic control. Stem Cell Rep 2018;10:739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pagliuca FW, Millman JR, Gu M et al. Generation of functional human pancreatic β cells in vitro. Cell 2014;159:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rezania A, Bruin JE, Arora P et al. Reversal of diabetes with insulin‐producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014;32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 8. Russ HA, Parent AV, Ringler JJ et al. Controlled induction of human pancreatic progenitors produces functional beta‐like cells in vitro. EMBO J 2015;34:1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haller C, Piccand J, De Franceschi F et al. Macroencapsulated human iPSC‐derived pancreatic progenitors protect against STZ‐induced hyperglycemia in mice. Stem Cell Rep 2019;12:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Velazco‐Cruz L, Song J, Maxwell KG et al. Acquisition of dynamic function in human stem cell‐derived β cells. Stem Cell Rep 2019;12:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pipeleers D, Robert T, De Mesmaeker I et al. Concise review: Markers for assessing human stem cell‐derived implants as beta‐cell replacement in type 1 diabetes. Stem Cells Translational Medicine 2016;5:1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lathuilière A, Cosson S, Lutolf MP et al. A high‐capacity cell macroencapsulation system supporting the long‐term survival of genetically engineered allogeneic cells. Biomaterials 2014;35:779–791. [DOI] [PubMed] [Google Scholar]
- 13. Pepper AR, Gala‐Lopez B, Pawlick R et al. A prevascularized subcutaneous device‐less site for islet and cellular transplantation. Nat Biotechnol 2015;33:518–523. [DOI] [PubMed] [Google Scholar]
- 14. Pepper A, Pawlick R, Bruni A et al. Transplantation of human pancreatic endoderm cells reverses diabetes post transplantation in a prevascularized subcutaneous site. Stem Cell Rep 2017;8:1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Pauw PEM, Vermeulen I, Ubani OC et al. Simultaneous measurement of plasma concentrations of proinsulin and C‐peptide and their ratio with a trefoil‐type time‐resolved fluorescence immunoassay. Clin Chem 2008;54:1990–1998. [DOI] [PubMed] [Google Scholar]
- 16. Chintinne M, Stangé G, Denys B et al. Contribution of postnatally formed small beta cell aggregates to functional beta cell mass in adult rat pancreas. Diabetologia 2010;53:2380–2388. [DOI] [PubMed] [Google Scholar]
- 17. Eizirik DL, Pipeleers DG, Ling Z et al. Major species differences between humans and rodents in the susceptibility to pancreatic beta‐cell injury. Proc Natl Acad Sci USA 1994;91:9253–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pepper AR, Bruni A, Pawlick R et al. Post‐transplant characterization of long‐term functional hESC‐derived pancreatic endoderm grafts. Diabetes 2018;68:953–962. [DOI] [PubMed] [Google Scholar]
- 19. Ko SH, Suh SH, Kim BJ et al. Expression of the intermediate filament vimentin in proliferating duct cells as a marker of pancreatic precursor cells. Pancreas 2004;28:121–128. [DOI] [PubMed] [Google Scholar]
- 20. Inada A, Nienaber C, Katsuta H et al. Carbonic anhydrase II‐positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci USA 2008;105:19915–19919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pipeleers D, In't Veld P, Maes E et al. Glucose‐induced insulin release depends on functional cooperation between islet cells. Proc Natl Acad Sci USA 1982;79:7322–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keymeulen B, Korbutt G, De Paepe M et al. Long‐term metabolic control by rat islet grafts depends on the composition of the implant. Diabetes 1996;45:1814–1821. [DOI] [PubMed] [Google Scholar]
- 23. Keymeulen B, Anselmo J, Pipeleers D. Length of metabolic normalization after rat islet cell transplantation depends on endocrine cell composition of graft and on donor age. Diabetologia 1997;40:1152–1158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Cell composition of hiPS‐PE before transplantation.
Sections of fixed hiPS‐PE aggregates were stained for the nuclear markers PDX1 and NKX6.1 (Panel A1‐3) and for cytoplasmic positivity for the pancreatic hormones insulin (INS‐Panel A1, B1‐3), glucagon (GCG‐Panel B1,2) and somatostatin (SST‐Panel B1,3) (Scale bars: 100 μm). For each of these markers the percent positive cells was determined by manual count (>1,000 cells counted per section, one section per experiment, three independent experiments). A large proportion of cells exhibited double‐positivity for PDX1 and NKX6.1 without hormone‐positivity (50 ± 5%), an index for their pancreatic progenitor state. A smaller fraction was only positive for PDX1 and hormone‐negative (37 ± 4%). The hormone‐positive cells were negative for PDX1 and NKX6.1; they represented 11 ± 2% of all cells with a large proportion (3 ± 2%) being polyhormone positive.
Supplementary Figure 2 Cell mass measurements in device‐encapsulated implants
Total and (non‐)endocrine cell volumes were measured in device‐encapsulated implants. The method was adapted from that used to quantify beta cell mass in the rat pancreas (Chintinne et al. 2010).
(A) Retrieved devices were fixed in buffered formaldehyde and longitudinally cut in two parts before embedding in paraffin. They were completely sectioned, approximately 1,000 sections per device. Representative sections (1% of total implant; cfr. D) throughout the implant were immunostaining with DAPI (nuclei) or with antibodies to (non‐)endocrine cell markers. Stained cell areas were semi‐automatically quantified and values were extrapolated to calculate their total volume in the implant.
(B) Steps for acquisition, segmentation and quantification of cell areas in a section. Scale bar: 100 μm.
(C) Assessment of sample size to be analyzed in order to keep relative variation under 10%. Analysis of 1% of total implant surface was found sufficient.
(D) Validation of method to count the number of cells that had been loaded in a device. A strong and linear correlation was found between loaded cell number and DAPI volume in the device (μl).
(E) Nuclear size at PT‐week 2 was smaller than that at start and at PT‐week 20. The ratio of the average nuclear areas at start and at PT‐week 2 (0.89) was used as correction factor to calculate cell recovery at this time point.
Statistical difference between measurements at different time points calculated by 1‐way ANOVA with Tukey's post hoc test: * p < .05, ** p < .01.
(F) Similar average cross‐sectional areas of individual insulin‐ (INS) and glucagon‐ (GCG) positive cells at PT‐week 20.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.