

Abstract
The ovarian surface epithelium (OSE) is a monolayer of cells surrounding the ovary that is ruptured during ovulation. After ovulation, the wound is repaired, however, this process is poorly understood. In epithelial tissues, wound repair is mediated by an epithelial-to-mesenchymal transition (EMT). Transforming Growth Factor Beta-1 (TGFβ1) is a cytokine commonly known to induce an EMT and is present throughout the ovarian microenvironment. We, therefore, hypothesized that TGFβ1 induces an EMT in OSE cells and activates signaling pathways important for wound repair.
Treating primary cultures of mouse OSE cells with TGFβ1 induced an EMT mediated by TGFβRI signaling. The transcription factor Snail was the only EMT-associated transcription factor increased by TGFβ1 and, when overexpressed, was shown to increase OSE cell migration. A polymerase chain reaction array of TGFβ signaling targets determined Cyclooxygenase-2 (Cox2) to be most highly induced by TGFβ1. Constitutive Cox2 expression modestly increased migration and robustly enhanced cell survival, under stress conditions similar to those observed during wound repair. The increase in Snail and Cox2 expression with TGFβ1 was reproduced in human OSE cultures, suggesting these responses are conserved between mouse and human. Finally, the induction of Cox2 expression in OSE cells during ovulatory wound repair was shown in vivo, suggesting TGFβ1 increases Cox2 to promote wound repair by enhancing cell survival. These data support that TGFβ1 promotes ovulatory wound repair by induction of an EMT and activation of a COX2-mediated pro-survival pathway. Understanding ovulatory wound repair may give insight into why ovulation is the primary non-hereditary risk factor for ovarian cancer.
Keywords: ovarian surface epithelium, wound repair, epithelial-to-mesenchymal transition, transforming growth factor beta 1, Cyclooxygenase 2
Summary sentence: COX2 increases epithelial cell survival under the stress conditions observed during ovulatory wound repair, therefore promoting survival in cells that might otherwise undergo apoptosis due to the accumulation of DNA damage present at ovulation.
Introduction
Although the events leading up to ovulation have been extensively studied, the processes mediating wound repair after ovulation have not. This is likely because genetic manipulation in the ovaries of animal models frequently result in infertility due to improper follicle development and anovulation, rendering the study of ovulatory wound repair more challenging. Ovulation is triggered by a rapid increase in luteinizing hormone (LH) from the pituitary gland. This LH surge activates a cascade of events such as the production of proteases that work to degrade the follicle wall and allow ovulation to occur. Specifically, activation of Progesterone Receptor (PR) by the LH surge induces the expression of genes encoding proteases such as ADAM Metallopeptidase With Thrombospondin Type 1 Motif 1 (ADAMTS1) and Cathepsin L (CTSL) [1]. LH also induces the expression of Cyclooxygenase 2 (Cox2) in cumulus cells to facilitate cAMP production, allowing for the cumulus expansion required for ovulation [2,3]. Previous studies have demonstrated that by inhibiting PR, or the induction of either ADAMTS1 or COX2, ovulation does not occur, resulting in infertility [1,2]. Although these consequences highlight the importance of these factors for the process of ovulation, the factors that contribute to the repair of the ovulatory wound are less well defined.
During ovulation in mice and sheep, the basement membrane at the apex of the ovulating follicle degrades and the ovarian surface epithelium (OSE) overlying the ovulatory follicle sloughs off, thereby creating a wound site [4,5]. Electron microscopy of the rabbit ovary revealed swelling under the OSE, degenerated fibroblasts and disintegrated collagen just prior to follicular rupture. This disintegration was observed in the outer part of the tunica albuginea and gradually spread down to the follicle wall [6]. This has also been reported in the macaque, where a reduction in cadherin-mediated OSE adhesion was observed at the time of ovulation [7]. Little has been confirmed in the human OSE, because of the inaccessibility of ovulating human ovaries.
Once ovulation has occurred, the ovulatory wound is repaired in as little as 12 h in mice [8]. Dysregulated wound repair has been associated with fibrosis and chronic inflammation in tissues such as the liver, kidney, and lung and can lead to oncogenic transformation [9]. Fibrosis is a condition where there is an excessive accumulation of extracellular matrix (ECM) proteins and is observed in aged mouse ovaries [10]. Briley et al. [10] found that aged fibrotic ovaries also displayed markers of chronic inflammation such as multinucleated macrophage giant cells. Ovulation represents a time with extensive ECM remodeling, requiring a balance of ECM production and degradation. Dysregulation of this process could lead to the fibrosis and chronic inflammation seen in aged ovaries.
One key mechanism that has been implicated in both wound repair and fibrosis is the epithelial-to-mesenchymal transition (EMT) [9,11]. EMT is a transient process where cells lose their epithelial characteristics, such as apical–basal polarity and cell–cell junctions, and acquire mesenchymal characteristics, such as front-rear polarity and cytoskeletal reorganization [12–14]. Epithelial cells undergoing an EMT lose their epithelial barrier function and become migratory and invasive, as well as resistant to apoptosis [13,14]. EMT is required for wound repair, allowing cells to migrate and secrete ECM proteins into the wounded area [11]. However, in tissues such as kidney, liver, skin, and lungs, induction of an EMT in the epithelium has been associated with the development and progression of fibrosis [15–20]. OSE undergoing a morphological EMT during wound repair has been studied in the macaque and the rat where the OSE layer assumes a flattened morphology over the developing corpus luteum [7, 21].
In this study, we evaluate signaling pathways that contribute to the EMT occurring during ovulatory wound repair. During a previous assessment of cytokine levels in preovulatory follicular fluid, we identified an abundance of Transforming Growth Factor Beta 1 (TGFβ1) [22], a cytokine well established for its capacity to induce an EMT. We also showed that TGFβ1 could induce similar responses to follicular fluid, strengthening its potential role in inducing EMT in the OSE cells. At ovulation, TGFβ1 that is found within the follicular fluid bathes the surrounding OSE cells after follicular rupture. TGFβ1 is also secreted by immune cells present at the wound site, such as macrophages [11], and is produced by other ovarian cells (granulosa cells) [23] and can, therefore, be found throughout the ovarian microenvironment. In this study, we demonstrate that TGFβ1 induces an EMT in OSE cells through the canonical Smad2/3 signaling pathway to induce the expression of Snai1 (Snail) and Ptgs2 (Cox2). Forced Snail expression induces a partial EMT with enhanced migration. Forced Cox2 expression modestly increases cell migration, but also strongly activates the protein kinase B (AKT) pathway and confers resistance to cell stress from hypoxia and reactive oxygen species (ROS)—common stressors in tissues undergoing repair [24]. Immunohistochemical analysis of superovulated mice shows Cox2 expression is induced in OSE cells around ovulatory wound sites, confirming this protein is induced in vivo. Furthermore, human OSE cells treated with TGFβ1 also exhibit increased SNAIL and COX2 expression, suggesting this signaling pathway is conserved in humans. This study suggests that the OSE surrounding the ovulatory wound induce a canonical EMT and the expression of Cox2 to promote wound repair through cell migration into the wound site and by activating survival pathways. Cox2 may promote survival of the repairing OSE cells, but may also promote survival of OSE cells harboring DNA damage and mutations. This work provides novel insights into mechanisms that may promote ovulatory wound repair, but that also could promote the age-associated fibrosis and shed light onto why ovulation is a primary risk factor for ovarian cancer.
Materials and methods
For complete materials and methods, please refer to the supplemental information.
OSE cell isolation and culture
Mouse ovarian surface epithelial (mOSE) cells were isolated from mouse ovaries and cultured in mOSE medium as previously described [22]. Briefly, ovaries were isolated from randomly cycling, 6-week-old FVB/N mice and were incubated in 0.25% Trypsin/phosphate buffered saline (PBS; Invitrogen) at 37°C, 5% CO2 for 30 min to remove the OSE layer. mOSE cells were isolated via centrifugation and plated onto tissue culture plates (Corning). The purity of epithelial cell isolations was assessed using immunofluorescence. In addition to Cadherin-1 (CDH1) staining, as previously reported [25], mOSE cells show appropriate localization of the tight and adherens junction proteins Zonula occludens-1 (ZO1) and Catenin beta-1 (CTNNB1; Supplementary Figure S1).
Human OSE cells used in this study were previously isolated and cultured in human OSE medium as previously described in Tonary et al. [26]. They were accessed under protocol #1999540-01H approved by the Ottawa Health Science Network Research Ethics Board. Briefly, ovaries were obtained by the Ottawa Ovarian Cancer Tissue Bank with informed consent from women undergoing surgery for reasons other than ovarian pathology. OSE cells were scraped from the ovarian surface using a scalpel blade and transferred into human OSE media. OSE cells were isolated via centrifugation and plated onto tissue culture plates (Corning). All mouse and human OSE cells were passaged 2–3 times prior to experimental use and experiments were conducted with cells of a passage number less than 25. In all experiments, control and treated samples were assessed at the same passage number at the same cell density.
TGFβ1 signaling targets PCR array
mOSE cells (1 × 106 cells) were plated 24 h prior to treatment with TGFβ1 (10 ng/mL). RNA was collected 4 days after TGFβ1 treatment (RNAeasy Kit, Qiagen). Complementary DNA (cDNA) synthesis was performed using RT2 First Strand Kit (Qiagen). cDNA was run on a RT2 Profiler Array—TGFβ (Qiagen). The array was run in triplicate (N = 3) and analyzed using the DataAnalysis Excel platform provided with the array kit.
Prostaglandin E2 enzyme-linked immunosorbent assay
Media was collected from mOSE cell cultures in TGFβ1-treated or control conditions and diluted 1:500 using fresh mOSE media. Prostaglandin E2 (PGE2) concentrations were determined using the Prostaglandin E2 enzyme-linked immunosorbent assay (PGE2 ELISA) Kit, Monoclonal (Cayman Chemical) as per manufacturer’s directions. The analysis was performed using the excel platform provided with the kit and values were expressed as the concentration of PGE2 in media per cell number.
Cox2 overexpressing mOSE cells
The vector pWpi (referred to as WPI) was given as a gift from Didier Trono (Addgene plasmid # 12254). WPI encoded enhanced GFP (eGFP) regulated by the EF1-α promoter. Murine Cox2 cDNA was cloned from mOSE cells using the In-Fusion polymerase chain reaction (PCR) Cloning Kit (Clontech) following the manufacturer’s protocol and then used to generate the lentivirus expression vector WPI-Cox2-IRES-eGFP (WPI-Cox2). Lentiviral vectors were prepared by transfecting human embryonic kidney 293 (HEK293) cells (American Type Culture Collection). Viruses were harvested by collecting the media from the transfected cells 48 h after transfection and filtered using a 45 μm filter. mOSE cells were infected with 200 μL of either the WPI-Cox2 or WPI virus. Infected mOSE cells were isolated using fluorescence-activated cell sorting for GFP expression.
Inducible Snail expressing mOSE cells
mOSE cells were transduced with the lentiviral vector pEF1-tet and selected with G418 to generate the cell line mOSE-tet. pEF1-tet is a derivative of pLVX-Tet-On Advanced (Clontech) utilizing the human EF1-α promoter to drive the expression of reverse tetracycline-controlled transactivator (rtTA) and neomycin resistance. The mOSE-tet cell line was subsequently transduced with the lentiviral vector pLVX-imSnail and selected in hygromycin B. pLVX-imSnail is a derivative of pLVX-Tight-Puro (Clontech) designed to express the murine Snai1 under the control of the doxycycline-inducible promoter (TRE) and hygromycin resistance under the control of the PGK promoter. Snail expression was induced by treating cells with doxycycline (Sigma-Aldrich) for 48 h prior to an experimental readout.
Statistics
All experiments were conducted a minimum of three times and plotted as an average of replicates with error bars showing the standard error of the mean. All statistical analyses were carried out using GraphPad Prism 6 software unless otherwise stated. An unpaired Student t-test was used when comparing two groups. A one-way analysis of variance (ANOVA) was used with a Dunnett’s post-test to compare three or more groups. For comparisons of more than one variable, a two-way ANOVA was used with a Bonferroni post-test. An ANCOVA analysis was used to compare migration, proliferation and survival assays. Significance was determined using a P value < 0.05.
Mouse superovulation
Adult female FVB/N mice were injected intraperitoneally with 5 IU of equine chorionic gonadotropin (eCG; Calbiochem) followed by 5 IU of human chorionic gonadotropin (hCG; Sigma) 46 h later. Ovaries were collected at 6 h after eCG and at 14–18 h post-hCG, fixed overnight in 10% formalin and paraffin embedded. Investigations using experimental animals were conducted in accordance with the SSR’s specific guidelines and standards. They were carried out using the Guidelines for the Care and Use of Animals established by the Canadian Council on Animal Care, with protocols approved by the University of Ottawa Animal Care Committee.
RNA sequencing library preparation and sequencing
mOSE cells (1 × 106 cells) were plated 24 h prior to treatment with TGFβ1 (10 ng/mL). RNA was collected 4 days after TGFβ1 treatment (RNAeasy Kit, Qiagen).
Total RNA was quantified using a NanoDrop Spectrophotometer ND-1000 (NanoDrop Technologies, Inc.) and its integrity was assessed on a 2100 Bioanalyzer (Agilent Technologies). Libraries were generated from 250 ng of total RNA as follows: messenger RNA enrichment was performed using the NEBNext Poly(A) Magnetic Isolation Module (New England BioLabs). cDNA synthesis was achieved with the NEBNext RNA First-Strand Synthesis and NEBNext Ultra Directional RNA Second Strand Synthesis Modules (New England BioLabs). The remaining steps of library preparation were done using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs). Adapters and PCR primers were purchased from New England BioLabs. Libraries were quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies) and the Kapa Illumina GA with Revised Primers-SYBR Fast Universal kit (Kapa Biosystems). Average size fragment was determined using a LabChip GX (PerkinElmer) instrument.
The libraries were normalized, denatured in 0.05 N NaOH and then were diluted to 200 pM and neutralized using HT1 buffer. ExAMP was added to the mix and the clustering was carried out on a Illumina cBot and the flowcell was run on a HiSeq 4000 for 2 × 100 cycles (paired-end mode) following the manufacturer’s instructions. A phiX library was used as a control and mixed with libraries at 1% level. The Illumina control software was HCS HD 3.4.0.38, the real-time analysis program was RTA v. 2.7.7. Program bcl2fastq2 v2.18 was then used to demultiplex samples and generate fastq reads.
RNA sequencing analysis
Transcript quantification for each sample was performed using Kallisto [27] with the GRCm38 transcriptome reference and the -b 50 bootstrap option. The R package Sleuth [28] was then used for determining differentially expressed genes between control and TGFβ1-treated cells. Significant genes were defined as genes with a q value < 0.05 (Wald test) and a beta coefficient > 0.5 or <−0.5.
Data availability
Raw sequencing files have been deposited and are available along with processed transcript quantifications at GSE121936.
Results
TGFβ1 induces an EMT by upregulating Snail expression in vitro
To study the effects of TGFβ1 on OSE cells in vitro, mouse OSE (mOSE) primary cell cultures were exposed to recombinant TGFβ1 protein and assessed for classical EMT characteristics such as a spindle-like morphology, enhanced migration and the loss in cell–cell junctions [9]. Previous studies from our lab have shown that bovine follicular fluid contains 10 ng/mL TGFβ1 at the time of ovulation, and, therefore, these studies were carried out using this dose [22]. TGFβ1 treatment-induced actin cytoskeleton rearrangement observed as elongated cell shape with cellular projections—characteristic of mesenchymal cells (Figure 1A) and increased cellular migration (Figure 1B), compared to untreated cells. Gene expression changes resulting from TGFβ1 treatment were consistent with an EMT, including decreased Cdh1 and Keratin 19 (Krt19) and increased Alcam (Figure 1C, Supplementary Figure S2). The morphological EMT observed in Figure 1A was visualized after 4 days in culture. To assess the transcriptional EMT response more thoroughly, a TGFβ1 time course was used and included earlier time points. Unlike most epithelial cells, OSE cells have been reported to express the mesenchymal markers Vim and Cdh2 and therefore were not included in this characterization [29]. To determine how TGFβ1 elicits this response, the expression of traditional EMT transcription factors was assessed during TGFβ1 treatment. Snail was the only transcription factor whose expression was increased by TGFβ1 (3.5-fold), whereas Twist was decreased approximately 50% and Zeb1 and Slug remained unchanged (Figure 1D, Supplementary Figure S2). These data demonstrate that TGFβ1 induces an EMT in mOSE cultures.
Figure 1.
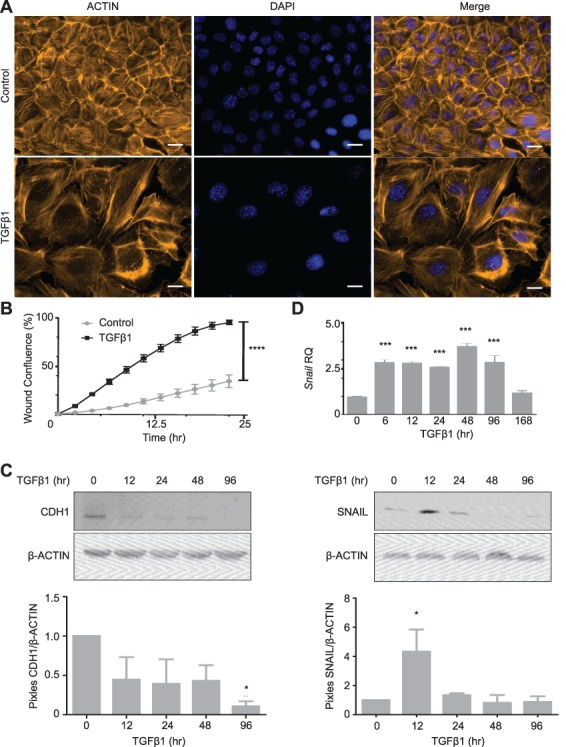
Epithelial-to-mesenchymal transition in mOSE cells treated with TGFβ1 (10 ng/mL). A: mOSE cells display actin cytoskeletal rearrangement upon TGFβ1 treatment (4 d), as determined by ACTIN immunofluorescence (representative image, N = 6). B: TGFβ1 enhances mOSE cell migration for 24 h of treatment compared to untreated controls (N = 3, ANCOVA, R2 = 0.82 and 0.97 for control and TGFβ1 treatment respectively). C: TGFβ1 treatment of mOSE cells decreases CDH1 (91% at 96 h of treatment) compared to untreated controls, as determined by western blot. β-ACTIN was used as a loading control (representative western blot, N = 3, One-way ANOVA with Dunnett’s post-test). D: mOSE cells treated with TGFβ1 have increased Snail expression compared to untreated controls, as determined by Q-PCR (top) and western blot (bottom). β-ACTIN was used as a loading control. mOSE cells were treated with TGFβ1 for up to 168 h for Q-PCR and up to 96 h for western blot (N = 3, One-way ANOVA with Dunnett’s post-test). *** and **** indicate a significant difference from the untreated control group, P < 0.001 and P < 0.0001 respectively. Scale bar = 15 μm. RQ = relative quantity. Experiments were performed using cells under passage number 25.
We next sought to confirm that the transcriptional targets of TGFβ1 treatment in mOSE cells were dependent on the activity of TGFβRI, the receptor that is responsible for phosphorylating SMADs 2 and 3, members of the canonical TGFβ1 signaling pathway. In the presence of a TGFβRI inhibitor (SB431542), SMAD2 and SMAD3 phosphorylation were decreased and the downstream increase in Snail was inhibited (Supplementary Figure S3). These data show that the TGFβ1-mediated EMT in mOSE cells involves the activity of TGFβRI, signaling through the canonical Smad2/3 pathway.
Forced Snail expression induces a partial EMT in vitro
To determine the transcriptional targets of SNAIL, its expression was activated using a doxycycline induction system (Figure 2A). Snail induction did not alter cytoskeletal organization or genes normally associated with an EMT (Krt19, Cdh1, and Alcam; Figure 2B–D), but did increase the migratory capacity of mOSE cells (Figure 2C). In the presence of doxycycline, inducible Snail mOSE cells migrate significantly faster than the vector control (mOSE cells with induced eGFP expression). In the absence of doxycycline, these mOSE cells migrate at the same rate. Doxycycline treatment of the vector control also did not alter EMT characteristics (Supplementary Figure S4). These data suggest Snail induction causes a partial EMT in mOSE cells, contributing to the increased migration observed with the TGFβ1-induced EMT, but does not affect the expression of several genes known to be associated with an EMT.
Figure 2.
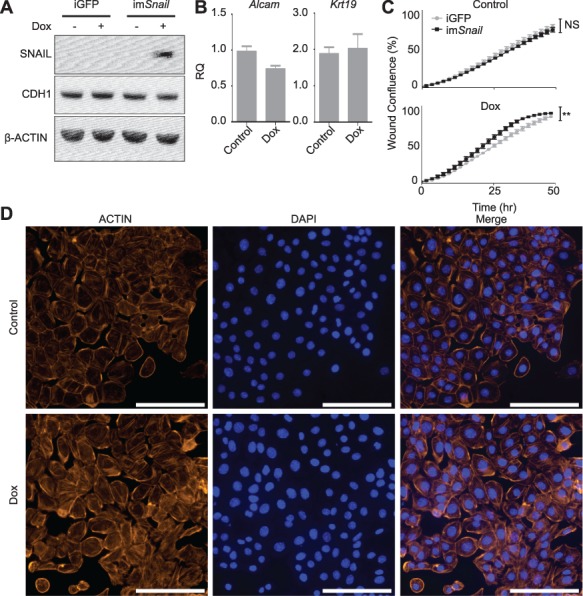
Forced Snail expression induces a partial EMT in vitro. A: Inducible Snail (imSnail) in mOSE cells show increased SNAIL in the presence of doxycycline (200 ng/mL, 48 h) compared to the control mOSE cells bearing inducible GFP (iGFP), as determined by western blot. CDH1 levels remain unaffected by doxycycline treatment. β-ACTIN was used as a loading control (representative western blot, N = 3). B: imSnail mOSE cells do not show altered expression of EMT markers (Alcam, Krt19) in the presence of doxycycline for 48 h (N = 3, Student t-test). C: imSnail cells have a modestly increased migration in response to doxycycline treatment for up to 48 h (100 ng/mL), compared to iGFP cells, whereas their migration is not significantly different in the absence of doxycycline (control), as determined using a migration assay (N = 3, ANCOVA, R2 = 0.93 and 0.99 for untreated iGFP and imSnail cells respectively and 0.98 and 0.95 for Dox treated iGFP and imSnail cells respectively-). ** indicates a significant difference between treatment groups, P < 0.005. D: imSnail mOSE cells do not alter actin cytoskeletal organization in the presence of doxycycline, as determined by ACTIN immunofluorescence (representative image, N = 3). Scale bar = 100 μm. RQ = relative quantity. Experiments were performed using cells under passage number 25 and Snail expression was induced by treating cells with dox for 48 h prior to an experimental readout.
TGFβ1 increases Cox2 expression and PGE2 secretion in vitro
A TGFβ Signaling Targets PCR Array was used to identify additional targets of TGFβ1 treatment that may promote an EMT and wound repair during ovulation. The gene most upregulated following TGFβ1 exposure was Cox2 (8-fold) (Figure 3A). Using a TGFβ1 treatment time course, Cox2 expression was shown to be increased as early as 12 h after treatment (Figure 3B and C) and was unchanged by forced Snail expression (Figure 3D). COX2 is the rate-limiting enzyme in prostaglandin production, and PGE2 is the most widely produced prostaglandin in the body, with the most versatile actions [30]. Therefore, a Prostaglandin E2 enzyme-linked immunosorbent assay (PGE2 ELISA) was used to confirm that the increase in COX2 leads to increased prostaglandin production and showed that, after 48 h of TGFβ1 treatment, PGE2 levels were significantly increased compared to the control OSE cells (Figure 3E). Using a TGFβRI inhibitor, we confirmed that the TGFβ1-induced increase in Cox2 was dependent on this receptor’s activity, suggesting this target is downstream of the canonical Smad2/3 pathway (Supplementary Figure S3).
Figure 3.
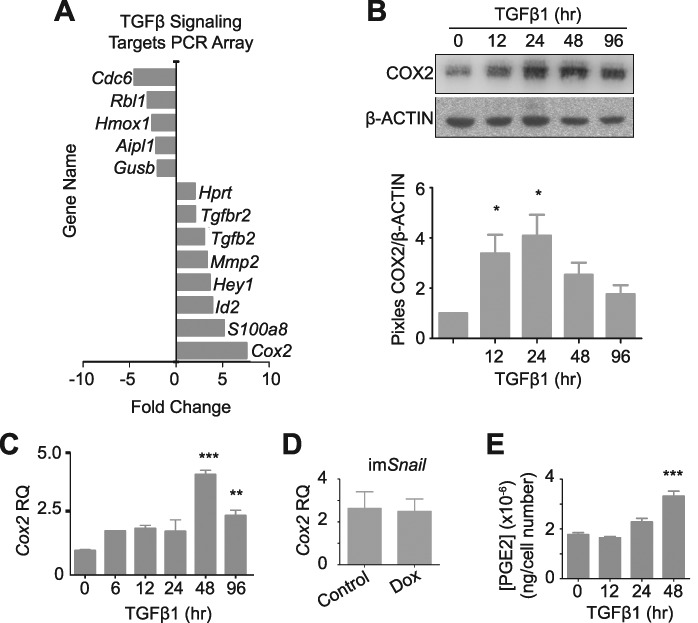
mOSE cells treated with TGFβ1 (10 ng/mL) increase Cox2 expression and PGE2 secretion. A–C: mOSE cells treated with TGFβ1 (4 days) have increased Cox2 expression as determined by a TGFβ1 Signaling Targets PCR array (A, N = 3, Student t-test), and validated by western blot (B, representative western blot, N = 3, One-way ANOVA with Dunnett’s post-test) and Q-PCR (C, N = 3, One-way ANOVA with Dunnett’s post-test). β-ACTIN was used as a loading control for the western blot. mOSE cells were treated with TGFβ1 up to 96 h. D: Cox2 expression is unaffected in imSnail mOSE cells treated with doxycycline (200 ng/mL, 48 h) as determined by Q-PCR (N = 3, Student t-test). E: mOSE cells treated with TGFβ1 have increased PGE2 production at 48 h after treatment, compared to untreated mOSE cells, determined by a PGE2-ELISA (N = 3, One-way ANOVA with Dunnett’s post-test). ** and *** indicate significant differences from the untreated control, P < 0.01 and P < 0.001 respectively. RQ = relative quantity. Experiments were performed using cells under passage number 25.
Forced Cox2 expression increases OSE cell survival
To determine if COX2 plays a role in inducing an EMT, we transduced mOSE cells with a lentiviral Cox2 expression vector. Overexpression modestly increased cell migration but did not alter the expression of Snail or Krt19, suggesting it plays a small role in inducing a partial EMT (Supplementary Figure S5). Further analysis revealed that Cox2 overexpression and PGE2 treatment can increase phosphorylation of AKT at serine 473 (Ser473), an indicator of AKT activation, which is known to regulate cell proliferation and survival (Figure 4A–C). Proliferation was however not affected by Cox2 overexpression in mOSE cells (data not shown). Treating mOSE cells with Celecoxib (a COX2 inhibitor) decreased their viability compared to control cells (Figure 4D). Furthermore, mOSE cells overexpressing Cox2 (WPI-Cox2) had increased resistance to wound repair-related cell stress, showing increased survival in the presence of hydrogen peroxide-induced ROS compared to control cells (WPI). In a hydrogen peroxide dose response, Cox2 overexpression increased the LD50 of mOSE cells from 85 μM to 150 μM (Figure 4E). When cultured in hypoxic conditions, vector control-infected mOSE cells showed decreased viability, whereas Cox2-overexpressing mOSE cells were able to maintain viability (Figure 4F). This was supported by a growth curve showing Cox2-overexpressing cells maintained their proliferation until day 6 in low oxygen conditions, whereas mOSE infected with the vector control have decreased proliferation within 4 days (Figure 4G). Together, these data demonstrate that COX2 plays a role in promoting mOSE cell survival when exposed to ovulation-associated stressors.
Figure 4.
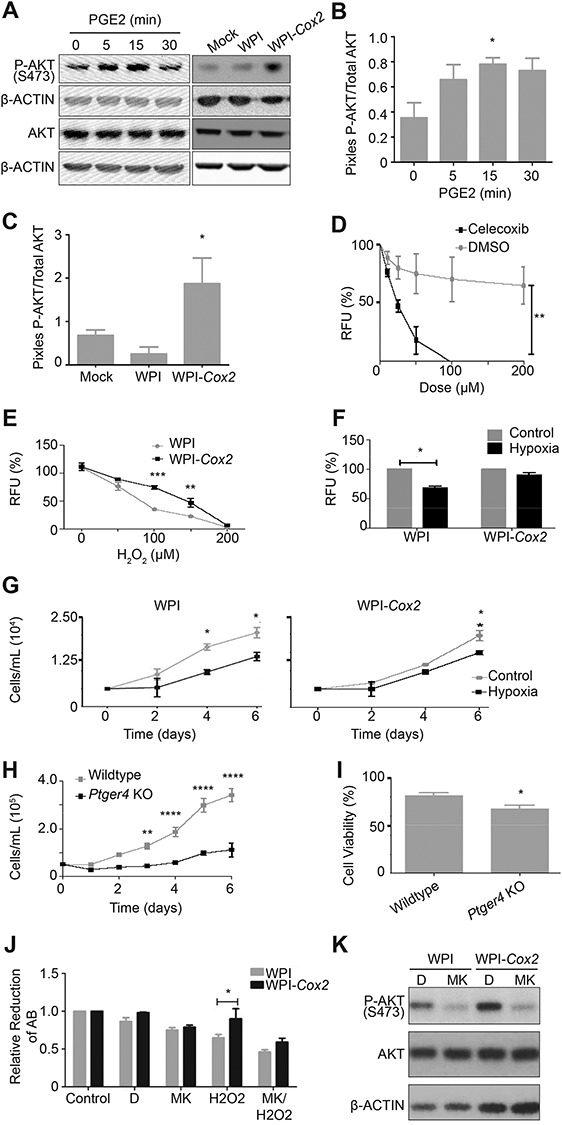
Constitutive Cox2 overexpression and PGE2 treatment promote cell survival through the PTGER4 receptor and activation of AKT signaling. A–C: mOSE cells either treated with PGE2 (8 μg/mL, up to 30 min), or overexpressing Cox2 (WPI-Cox2) have increased levels of P-AKT compared to control cells or mOSE cells infected with the vector control (WPI) as determined by western blot. β-ACTIN was used as a loading control (representative western blot, N = 3, One-way ANOVA with Dunnett’s post-test). D: mOSE cells treated with celecoxib (2 days) have reduced viability compared to control-treated mOSE cells (DMSO), as determined by an Alamar Blue assay (N = 3, Two-way ANOVA with Bonferroni’s post-test). E: WPI-Cox2 mOSE cells are more resistant to H2O2 treatment than WPI mOSE cells, as determined using an Alamar Blue assay after 2 days of treatment (N = 3, Two-way ANOVA with Bonferroni’s post-test). F: WPI mOSE cells cultured in low oxygen (5% O2, 6 days) have reduced viability compared to WPI-Cox2 mOSE cells which maintain their viability, as determined using an Alamar Blue assay (N = 3, Student t-test). G: WPI mOSE cells cultured under low oxygen have decreased proliferation at 4 days in culture whereas WPI-Cox2 mOSE cells maintain their proliferation rate at least until 6 days in culture (N = 3, Two-way ANOVA with Bonferroni’s post-test). H-I: Ptger4 knockout mOSE cells (Ptger4 KO) have reduced proliferation and viability compared to Ptger4 wildtype mOSE cells (wildtype), as determined using a growth curve (H, N = 3, Two-way ANOVA with Bonferroni’s post-test) and trypan blue exclusion (I, N = 3, Student t-test). J-K: The enhanced viability of WPI-Cox2 mOSE cells treated with H2O2 (100 μM, 48 h) compared to WPI mOSE cells is no longer present when pre-treated for 30 min with the P-AKT inhibitor MK-2206 (MK, 2 μM), as determined by an Alamar Blue assay (J, N = 3, Two-way ANOVA with Bonferroni’s post-test, AB = Alamar Blue). Western blot (K) was used to show the efficiency of P-AKT inhibition of the vehicle-treated (DMSO, D) and MK-treated (2 μM, 5 h) mOSE cells. β-ACTIN was used as a loading control for the western blot (representative western blot, N = 3). *, **, ***, **** indicate a significant difference from the untreated control or wildtype group, P < 0.05, 0.01, 0.001, and 0.0001, respectively. RFU = relative fluorescence units. Experiments were performed using cells under passage number 25.
Prostaglandin E2 receptors (PTGER2 and PTGER4) are the receptors known to mediate PGE2 signaling through the AKT pathway in epithelial cells [31]. Analysis of the expression levels of these receptors showed that Ptger4 is expressed in mOSE cells whereas Ptger2 is not (data not shown). To determine if PTGER4 is required for maintaining cell proliferation and survival, mOSE cells were isolated from Ptger4-floxed mice [32] and infected with adenovirus expressing Cre-recombinase (Ad-cre) to achieve Ptger4 deletion (Supplementary Figure S6A). Following Ad-cre infection, mOSE cells had a much-reduced proliferation rate and had reduced viability (Figure 4H and I). These data suggest PTGER4 is important to maintain cell survival and proliferation in these cells. Treating the Ptger4 knockout cells with TGFβ1 still enhanced migration, suggesting PTGER4 is not required for TGFβ1-induced migration (Supplementary Figure S6B).
To assess whether the survival phenotype associated with Cox2 expression is dependent on the activation of the AKT pathway, WPI and WPI-Cox2 mOSE cells were treated with H2O2 in the presence or absence of MK-2206, an AKT inhibitor. MK-2206 was able to decrease the levels of P-AKT-Ser473, and prevent the survival phenotype associated with Cox2 expression in the presence of H2O2 (Figure 4J and K). These data suggest that COX2 promotes cell survival in mOSE cells by activating the AKT pathway through PTGER4.
TGFβ1 induces an EMT in human OSE cells
Primary human OSE cells were isolated to assess whether the cell morphology and transcriptional changes in Snail and Cox2 observed with TGFβ1 treatment in mice were conserved in human. Human OSE cultures treated with TGFβ1 acquire a mesenchymal morphology and have 3- and 4-fold increases in Snail and Cox2 transcripts respectively (Supplementary Figure S7). These data suggest the signaling pathway observed in mouse OSE cells is conserved in human OSE cells.
Cox2 is expressed during ovulatory wound repair in mouse ovaries
The involvement of COX2 in ovulatory wound repair is not known; however, its promotion of cell survival may contribute to efficient repair. Given Cox2 expression increases as cells undergo an EMT, we predicted that its expression would increase during ovulatory wound repair. To explore this in vivo, ovaries from superovulated mice were assessed before and after ovulation for Cox2 expression. In preovulatory follicles examined 6 h after hCG treatment and in non-ovulating areas of ovaries collected after ovulation (14–18 h post hCG), Cox2 was expressed in the granulosa cells, as expected, but remained undetectable in the OSE cells (Figure 5). However, after ovulation occurred, the OSE cells surrounding the ovulatory wound sites expressed Cox2 (Figure 5). Cox2 expression is therefore induced after ovulation, in vivo, and our data suggests it contributes to maintaining cell viability during wound repair.
Figure 5.
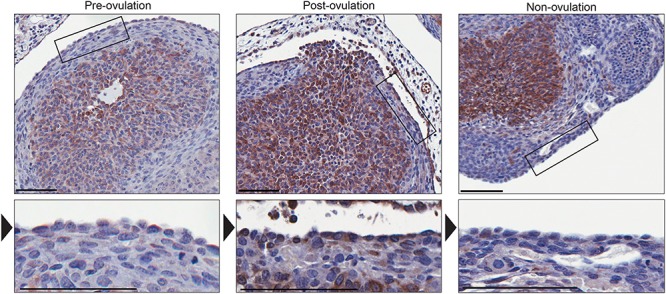
Immunohistochemistry detecting COX2 during post-ovulatory wound repair. The granulosa cells in ovary sections collected prior to ovulation (6 h post-hCG) express COX2 whereas the OSE layer surrounding the antral follicles (indicated by the box and arrowhead) is negative for Cox2 expression. After ovulation (14–16 h post-hCG), the OSE layer surrounding the ovulatory wound site (indicated by the box and arrowhead) expresses COX2 whereas OSE surrounding non-ovulating sites remain negative for COX2 expression. Representative sections, N = 5. Scale bar = 100 μm.
RNA sequencing of mOSE cells treated with TGFβ1 reveals upregulation of ECM deposition
These experiments have identified TGFβ1 targets that promote some aspects of the EMT. However, activation of these targets alone does not promote the full EMT response that is observed with TGFβ1 treatment. To broaden our understanding of the effects of TGFβ1 on OSE cells, RNA sequencing was used to assess mOSE cells with and without TGFβ1 treatment. RNA sequencing revealed 372 significantly upregulated and 220 downregulated genes between control and TGFβ1-treated mOSE cells. Cox2 and Snail were among the significantly upregulated genes and Krt19 was downregulated with TGFβ1 treatment, in agreement with our quantitative PCR (Q-PCR) and western blot findings (Figure 6A). Gene ontology (GO) term enrichment analysis revealed many significantly enriched terms associated with TGFβ1 treatment, including system development, regulation of multicellular organismal process and movement of a cell or subcellular component (Figure 6B). The most significant terms enriched in the downregulated genes included cellular response to cytokine stimulus, flavonoid metabolic process and organic hydroxy compound catabolic process (Figure 6B). When examining the entirety of the enriched terms, it is clear that EMT-associated terms are abundant with TGFβ1 treatment. Locomotion, cell migration, and cell adhesion were all upregulated in TGFβ1 treated mOSE cells (Figure 6B). Many collagens, integrins, matrix metalloproteinases, and ADAMTS proteins were increased in TGFβ1 treated mOSE cells, further characterizing the ECM remodeling that occurs with TGFβ1 treatment (Figure 6A). These data give a broad view of the TGFβ1-induced EMT in mOSE cells and highlight the ECM restructuring that occurs with this process. A full list of significantly upregulated and downregulated genes seen with TGFβ1 can be found in Supplementry Table S1.
Figure 6.
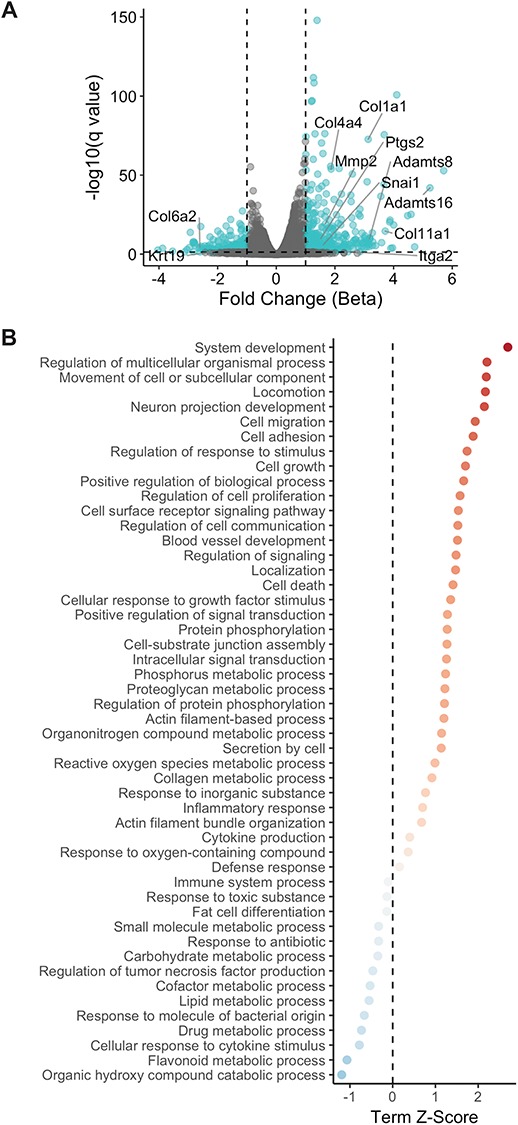
RNA sequencing results of TGFβ1-treated mOSE cells (10 ng/mL, 96 h) shows increased ECM deposition with treatment. A: Volcano plot demonstrating increased expression of ECM deposition and remodeling genes observed with TGFβ1 treatment. Ptgs2 (Cox2) and Snail are also increased with TGFβ1 treatment whereas Krt19 is decreased with treatment. Fold change was determined by assessing the beta coefficient of the general linear models used for differential expression analysis B: Plot of significantly enriched terms upregulated and downregulated by TGFβ1 treatment. Experiments were performed using cells under passage number 25.
Discussion
It is becoming increasingly apparent that the EMT is best described as a spectrum, where cells can transiently lie at intermediate locations along the continuum, exhibiting simultaneously epithelial and mesenchymal characteristics [9, 12, 13, 33, 34]. This “partial EMT” occurs during wound repair, where cells at the edge of the wound site migrate to close the injury, but retain their adherens junctions (epithelial characteristic), migrating as a sheet [13]. This has been reported previously in OSE cells, which maintain expression of Keratin 8 and E-cadherin throughout wound repair [4]. Once migration into the wound is complete, the edge cells re-form the epithelial barrier, completing the re-epithelialization process.
This study demonstrates that TGFβ1 induces an EMT in OSE cells, through the activity of TGFβRI. The sole traditional transcription factor induced by TGFβ1 treatment in the mOSE is Snail, but interestingly, and contrary to the mammary epithelium [35], Snail induction independent of TGFβ1 treatment only induced a partial EMT. This finding has not been previously reported in the ovary; however, a partial EMT following Snail expression has been observed in renal epithelial cells [15]. These results suggest the additional characteristics driven by TGFβ1 are mediated by other genes either alone, or in conjunction with Snail. This concept supports the theory proposed by Jolly et al. [36] where EMT characteristics are best observed as separate vectors on the EMT continuum and are controlled by different factors.
In the OSE cells, we have found that SNAIL mediates their migratory capacity and COX2 promotes their survival, but other factors downstream of TGFβ1 are required for altered morphology or loss of additional epithelial characteristics. The TGFβ Signaling Targets array identified genes such as Hey1 and Mmp2 as being significantly increased by TGFβ1 treatment. In addition, RNA sequencing of TGFβ1-treated mOSE cells identified a large number of collagens and ADAMTS genes increased with treatment. These genes are also known to induce an EMT in other tissues [14,37–39], although none have yet been reported to play a role in the ovulatory wound repair process in mammals. Perhaps these genes contribute to the additional EMT characteristics seen with TGFβ1 treatment, not observed with Snail expression alone. Furthermore, both the TGFβ Signaling Targets array and RNA sequencing were used to determine the effects of TGFβ1 treatment after 96 h, when OSE cells have morphologically undergone an EMT. A more thorough investigation of the transcriptional targets of short term TGFβ1 treatment might shed light on additional TGFβ1 targets that would lead to these mesenchymal characteristics.
Remodeling of the ECM is essential for wound repair, but in the case of dysregulation, it can result in fibrosis or scarring. Fibrosis, which is a feature of aged mouse ovaries [10], occurs when the amount of ECM deposited is greater than the amount being degraded and induces a chronically inflamed environment which can lead to transformation [9]. Our RNA sequencing data revealed a large number of ECM proteins upregulated by TGFβ1 treatment, many of which were collagens. Dysregulation of ovulatory wound repair could lead to excessive collagen deposition and formation of a fibrotic lesion. Interestingly, wound healing in the fetus is scarless, whereas wound healing in adult tissue is not [40]. This is suggested to be due, in the fetus, to a decreased inflammatory response to wounding, leading to a lack of COX2 induction and PGE2 secretion [40]. Introducing exogenous PGE2 to a fetal model of wound repair induces scar formation [41] and blocking COX2 using Celecoxib in adult wound repair reduces scarring [42]. This suggests that in the adult ovary, the increase in COX2 following ovulation promotes scarring and may lead to the accumulation of fibrotic lesions.
Cell survival is another important component of wound repair, as OSE cells mediating this repair are surrounded by harsh environmental conditions. In the hours preceding ovulation, vasoconstriction at the apex of the ovulating follicle occurs, which decreases blood flow to the follicle and its surrounding ovarian tissue [43]. Additionally, ovulatory wound sites are in contact with the immune cells that populate the forming corpus luteum after ovulation to promote repair [44–46]. Because of the increase in OSE proliferation around the wound site and to the influx of immune cells and transient loss of local vasculature, the ovulating environment is transiently hypoxic [44–46]. There is an elevation of ROS in the OSE at the time of ovulation, which also contributes to this harsh environment that is necessary for the OSE to survive and for efficient wound repair to occur [47]. We found that Cox2 induction was associated with enhanced survival when mOSE cells were exposed to these stressors in vitro. In vivo, Cox2 expression in the OSE follows ovulation and is, therefore, only present during ovulatory wound repair. Therefore, the induction of Cox2 by TGFβ1 may facilitate wound repair by improving OSE cell survival after ovulation. Alternatively, the high COX2 levels in granulosa cells, resulting in PGE2 in the follicular fluid [48], could promote OSE cell survival directly. It is notable that the timing of Cox2 induction by TGFβ1 treatment in vitro does not coincide with the timing of Cox2 induction during ovulatory wound repair in vivo. This is likely due to the limitations of mimicking the complex ovarian microenvironment composed of multiple cytokines and cell types in an in vitro setting where cells have been stimulated by a single agent.
The observation that PGE2 promotes survival through the activation of AKT is supported by research in endothelial cells showing small interfering RNA-mediated knockdown of mTORC2 impairs PGE2-enhanced AKT activation and cell survival [49]. These findings shed light on potential issues of using COX2 inhibitors for oral contraceptive use—a promising contraceptive option as reviewed by Duffy [50]. These data show that inhibition of COX2 decreases OSE cell viability and suggests that use of COX2 inhibitors could inhibit efficient wound repair, should ovulation occur. This is supported by studies in the skin showing a decreased rate of wound closure with the application of a topical COX2 inhibitor [51]. The data presented here are in vitro, however, so the potential consequences of decreased OSE viability with COX2 inhibition in vivo should be evaluated. Unfortunately, this study cannot be carried out using the mouse model because COX2 knockout mice are infertile due to anovulation [2]. Treatment of rats with indomethacin, a non-selective COX inhibitor, did not affect OSE cell proliferation; while not specifically assessed, there was no effect on cell survival noted [21]. It is also notable that, in macaque ovaries, removal of the OSE cell layer did not appear to have lasting consequences on ovarian function [52].
Ovulation is the primary non-hereditary risk factor for ovarian cancer, with ovarian cancer risk increasing with the number of lifetime ovulation cycles [53]. The incessant ovulation hypothesis suggests that the high rate of cell turnover during ovulation, in conditions with high ROS, can lead to DNA damage and mutations in the OSE [54]. Murdoch and Martinchick showed an accumulation of mutagenic 8-oxoguanine modifications in the surviving OSE cells after ovulation [47]. If the induction of Cox2 promotes the survival of cells that have acquired ovulation-associated DNA mutations, this could increase the potential for the transformation of these cells. Furthermore, COX2 may promote the formation of fibrotic lesions over time. These data are supported by a recent review showing the benefits of aspirin, a nonsteroidal anti-inflammatory drug (NSAID) which acts as a COX inhibitor, as a preventative agent for ovarian cancer [55]. The authors state a more thorough study of non-aspirin NSAIDs, selective for COX2 inhibition is warranted due to the present lack of comprehensive data available for these NSAIDs and ovarian cancer risk [55].
This study is the first to characterize a signaling pathway, summarized in Supplementary Figure S8, that may drive EMT-mediated ovulatory wound repair. The EMT is widely accepted as a mediator for wound repair and can be induced by many cytokines found in the ovarian environment or follicular fluid. A limitation of this study is that we have focused on only one of these cytokines in isolation, and so have likely just scratched the surface of the complexity of this process. In this study, we assessed TGFβ1 signaling and found that it signals through the canonical Smad2/3 pathway to induce the expression of Snail and Cox2. SNAIL acts to increase the migratory capacity of OSE cells along with COX2; however, COX2 primarily induces PGE2 secretion which activates the AKT pathway to maintain cell proliferation and survival. The induction of an EMT and maintenance of cell survival may facilitate ovulatory wound repair but may also promote fibrosis and its associated inflammation and render cells more susceptible to transformation. Future studies examining how other cytokines present at the wound site, such as Insulin-like growth factor 1 (IGF1) or Dickkopf 3 (DKK3; [56]), act independently, or in conjunction with TGFβ1, are warranted. Defining the mechanisms of ovulatory wound repair is imperative to understanding age-associated fibrosis as well as ovulation as a risk factor for ovarian cancer.
Supplemental methods
Quantitative RT-PCR
RNA extraction, cDNA synthesis, and quantitative reverse transcription PCR (RT-PCR) were performed as previously described [57]. Briefly, RNA was extracted using the RNeasy Mini Kit (Qiagen) and cDNA was synthesized using the OneStep RT-PCR Kit (Qiagen). The ABI 7500 FAST qRT-PCR machine (Applied Biosystems) was used for Quantitative PCR using the Taqman gene expression (Life Technologies) and SsoFast gene expression (Bio-Rad) assays. Tbp was used as an endogenous control for the assays. Primer sequences are listed in Supplementary Table S2.
Western blot
Protein was extracted and used for western blot analysis as previously described [57]. Briefly, protein from mOSE cells was extracted using M-PER mammalian protein extraction reagent (GE Healthcare) as per the manufacturer’s directions and run on NuPAGE 4–12% Bis-Tris gradient gels (Life Technologies). Protein samples were transferred to a polyvinylidene difluoride membrane and were then blocked in 5% non-fat milk or 5% bovine serum albumin prior to antibody incubation. Antibody conditions are described in Supplemental Table S3. Western blots were developed using Clarity Western ECL Substrate (Bio-Rad) and the FluorChem FC2 imaging system (Alpha Innotech). β-ACTIN was used as a loading control for western blots [58,59]. Densitometry was done using ImageJ software and protein expression was plotted as pixels of the gene of interest/pixels of β-ACTIN or GAPDH and normalized to the control/untreated sample. For phosphorylated proteins, protein expression was plotted as pixels of phosphorylated proteins/pixels of total protein.
Immunohistochemistry
Tissue sections from superovulated mouse ovaries were used for immunohistochemical analysis as previously described [57]. Briefly, 5 μm sections were deparaffinized in xylenes and rehydrated in an ethanol gradient. Endogenous peroxidase activity was blocked using 3% H2O2 and endogenous avidin and biotin were blocked using an Avidin/Biotin Blocking kit (Dako). Tissue sections were then blocked in a Serum-Free Blocking Reagent (Dako) and incubated overnight at 4°C in mouse anti-COX2 (Abcam, ab15191, RRID AB_2085144) diluted 1:1500 using Antibody Diluent (Dako). Tissue sections were incubated in anti-rabbit horseradish peroxidase-labeled polymer (Dako) for 30 min, room temperature and developed using diaminobenzidine. Sections were counterstained with hematoxylin, dehydrated using an ethanol gradient and mounted using Permount (Fisher Scientific). The Scanscope CS2 (Aperio) was used to acquire images.
PCR
OSE cells were isolated from Ptger4fl/fl mice [32] as described above and DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen). DNA was used in PCR analysis using the CloneAmp Hifi PCR Premix (Takara) to detect the unrecombined Ptger4 gene and the recombined Ptger4 gene using the following primer pairs: unrecombined: Forward—5′ GGA GTC ACT TTT CCC TTG AGA AG 3′ and Reverse—5′AAC GAG CCA TTT ACC ACT TGC AA 3′, recombined: Forward—5′ GGA GTC ACT TTT CCC TTG AGA AG 3′ and Reverse—5′CGA GTC CTT AGG CTT TTA AGT GGA 3′. The PCR reaction was run under the following conditions: 94°C 2 min initial denaturation, 94°C 10 s, 62°C 15 s, 72°C 2 min for 35 cycles, 7°C 5 min.
The proliferation assay
mOSE cells (50 000) were seeded into 24-well tissue culture plates (Corning) in mOSE media. Proliferation was assessed by counting viable cells using the Vi-CELL XR cell viability analyzer (Beckman Coulter) for each day of the experiment.
The migration assay
mOSE cells (70 000) were plated into 96-well tissue culture plates (Essen ImageLock, Essen Instruments) with or without TGFβ1 (10 ng/mL), 24 h prior to scratch assay. The scratch assay was performed at 100% cell confluence and carried out using the Wound Maker (Essen Instruments). Wound confluence was monitored every 2 h with the Incucyte Live-Cell Imaging System using the 10× objective and wound confluence was determined using the instrument software (Essen Instruments). At time 0 the wounded area was imaged and recorded as a wound confluence of 0%. The images collected every 2 h were then analyzed using the same software for the percentage of wounded space now occupied by cells. This data was recorded throughout the experiment and plotted as Wound Confluence (%)/time (hour). An ANCOVA was used to statistically assess the rate of migration within the linear range of the experiment.
Alamar Blue
mOSE cells (10 000) were seeded into 96-well tissue culture plates (Corning) 24 h prior to administration of treatment. 1× AlamarBlue (Life Technologies) was added to each well and incubated for 8 h. Fluorescence was measured using excitation and emission values of 530 and 590 nm, respectively or absorbance was measured at A570 and A600 using the Fluoroskan Ascent FL (Thermo Fisher Scientific) and used to calculate relative fluorescence units or relative reduction of Alamar Blue.
Immunofluorescence
ACTIN
mOSE cells were plated on glass coverslips in mOSE media 24 h prior to TGFβ1 treatment. Cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 (10 min). Cells were probed for Actin using ActinRed 555 ReadyProbes Reagent (Life Technologies) following the manufacturer’s directions. After staining, coverslips were mounted onto slides using ProLong Gold antifade mountant with DAPI and maintained in the dark at room temperature for 24 h to allow mounting media to cure. Wide-field fluorescence images were acquired using a Zeiss Axioskop 2 microscope using Plan Apochromat 63x/1.40NA Oil (DIC III) objective and images were processed using Image J.
OSE Cell Purity Validation
Cells were seeded onto glass coverslips and grown to 90% confluence. Cells were washed twice with PBS then fixed with 4% paraformaldehyde (w/v, in PBS) for 15 min, permeabilized with 0.2% Triton X-100 (v/v, in PBS) for 10 min, and then blocked with 5% normal goat serum (v/v, in PBS) (Sigma) for 1 h at room temperature. Cells were probed with anti-CTNNB1 (1:500; Abcam, ab6302) or anti-ZO1 (1:100; Abcam, ab216880) diluted in blocking buffer overnight at 4°C. The next day, cells were probed with AlexaFluor488 goat anti-rabbit secondary antibody (1,500; Invitrogen, A-11034) diluted in blocking buffer for 1 h at room temperature in the dark. Cells were mounted onto slides using ProLong Gold antifade mountant with DAPI and maintained in the dark at room temperature for 24 h to allow mounting media to cure. Confocal images were acquired using a Zeiss LSM800 AxioObserver Z1 system using Plan-Apochromat 40x/1.3NA oil objective and were processed using Image J. Combined assessment of CDH1, ZO1, and CTNNB1 indicated >95% purity of epithelial cells in the primary cultures.
Supplementary Material
Acknowledgments
We thank the tissue donors for making this research possible. We are grateful to Dr. Christopher Kennedy (University of Ottawa) for kindly providing the conditional Ptger4-floxed mice [32]. We wish to acknowledge the contribution of staff of the McGill University and Génome Québec Innovation Centre, Montreal, Canada, for performing library preparation and sequencing associated with RNA-seq experiments.
Conference Presentation: This work was presented in part at the 49th Annual Meeting of the Society for the Study of Reproduction, 19 July 2016, San Diego, California.
Author contributions
Experiments were designed by LEC and BCV. LEC and DPC performed the Transforming Growth Factor β1 (TGFβ1)-treatment time course, Transforming Growth Factor Receptor 1 (TGFβR1) inhibition and Prostaglandin E2 enzyme-linked immunosorbent assay (PGE2-ELISA) experiments on mouse ovarian surface epithelial (mOSE) cells. DPC analyzed the RNA sequencing results. OC derived the mOSE primary cultures. LEC and OC performed the TGFβ1-treatment human OSE experiments. LFG produced the preliminary data and contributed to preliminary experimental designs for this project with BCV. KG made the mOSE cells with inducible Snail and inducible enhanced green fluorescent protein (eGFP) expression and LEC performed all experiments with these cells. LEC derived the Cox2-overexpressing mOSE cells and was assisted by HAD in assessing the epithelial-to-mesenchymal characteristics and by HWW in characterizing the pro-survival phenotype in these cells. LEC and KG performed the MK-2206 experiments. CWM performed the TGFβ Signaling Targets the PCR array. NHV performed the immunofluorescence for mOSE cell purity and TGFβ1-treatment ACTIN staining. LEC performed the mouse superovulation experiments and the immunohistochemistry on collected ovary samples. LEC and BCV wrote the manuscript with input from all authors.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
- [1]. Robker RL, Russell DL, Espey LL, Lydon JP, O’Malley BW, Richards JS. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. PNAS 2000; 97:4689–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2–deficient mice. Cell 1997; 91:197–208. [DOI] [PubMed] [Google Scholar]
- [3]. Richards JS. Ovulation: New factors that prepare the oocyte for fertilization. Mol Cell Endocrinol 2005; 234:75–79. [DOI] [PubMed] [Google Scholar]
- [4]. Singavarapu R, Buchinsky N, Cheon D-J, Orsulic S. Whole ovary immunohistochemistry for monitoring cell proliferation and ovulatory wound repair in the mouse. Reprod Biol Endocrinol 2010; 8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Murdoch WJ. Ovarian surface epithelium during ovulatory and anovulatory ovine estrous cycles. Anat Rec 1994; 240:322–326. [DOI] [PubMed] [Google Scholar]
- [6]. Bjersing L, Cajander S. Ovulation and the role of the ovarian surface epithelium. Experientia 1975; 31:605–608. [DOI] [PubMed] [Google Scholar]
- [7]. Wright JW, Jurevic L, Stouffer RL. Dynamics of the primate ovarian surface epithelium during the ovulatory menstrual cycle. Hum Reprod 2011; 26:1408–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Burdette JE, Kurley SJ, Kilen SM, Mayo KE, Woodruff TK. Gonadotropin-induced superovulation drives ovarian surface epithelia proliferation in CD1 mice. Endocrinology 2006; 147:2338–2345. [DOI] [PubMed] [Google Scholar]
- [9]. Nieto MA, Huang RYJ, Jackson RA, Thiery JP. EMT: 2016. Cell 2016; 166:21–45. [DOI] [PubMed] [Google Scholar]
- [10]. Briley SM, Jasti S, McCracken JM, Hornick JE, Fegley B, Pritchard MT, Duncan FE. Reproductive age-associated fibrosis in the stroma of the mammalian ovary. Reproduction 2016; 152:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]. Shaw TJ, Martin P. Wound repair: a showcase for cell plasticity and migration. Curr Opin Cell Biol 2016; 42:29–37. [DOI] [PubMed] [Google Scholar]
- [12]. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Savagner P. Epithelial-mesenchymal transitions: from cell plasticity to concept elasticity. Curr Top Dev Biol 2015; 112:273–300. [DOI] [PubMed] [Google Scholar]
- [14]. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 2015; 21:989–997. [DOI] [PubMed] [Google Scholar]
- [16]. Morizane R, Fujii S, Monkawa T, Hiratsuka K, Yamaguchi S, Homma K, Itoh H. miR-34c attenuates epithelial-mesenchymal transition and kidney fibrosis with ureteral obstruction. Sci Rep 2014; 4:4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. PNAS 2006; 103:13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Hosoki K, Yasukawa A, Toda M, Miyake Y, Ruiz DB, Bernabe PG, Takagi T, Morooka R, Nagao M, DAlessandro-Gabazza CN, Taguchi O, Gabazza EC et al. . Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. J Allergy Clin Immunol 2012; 129:AB117. [Google Scholar]
- [19]. Nikitorowicz-Buniak J, Denton CP, Abraham D, Stratton R. Partially evoked epithelial-Mesenchymal transition (EMT) is associated with increased TGFβ Signaling within Lesional scleroderma skin. PLoS One 2015; 10:e0134092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, Han Y-P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol 2010; 176:2247–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Gaytán M, Sánchez MA, Morales C, Bellido C, Millán Y, Martín de Las Mulas J, Sánchez-Criado JE, Gaytán F. Cyclic changes of the ovarian surface epithelium in the rat. Reproduction 2005; 129:311–321. [DOI] [PubMed] [Google Scholar]
- [22]. Gamwell LF, Collins O, Vanderhyden BC. The mouse ovarian surface epithelium contains a population of LY6A (SCA-1) expressing progenitor cells that are regulated by ovulation-associated factors. Biol Reprod 2012; 87:80, 1–10. [DOI] [PubMed] [Google Scholar]
- [23]. Knight PG, Glister C. TGF-β superfamily members and ovarian follicle development. Reproduction 2006; 132:191–206. [DOI] [PubMed] [Google Scholar]
- [24]. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res 2010; 89:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Vuong NH, Salah Salah O, Vanderhyden BC. 17β-Estradiol sensitizes ovarian surface epithelium to transformation by suppressing Disabled-2 expression. Sci Rep 2017; 7:16702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Tonary AM, Macdonald EA, Faught W, Senterman MK, Vanderhyden BC. Lack of expression of c-KIT in ovarian cancers is associated with poor prognosis. Int J Cancer 2000; 89:242–250. [DOI] [PubMed] [Google Scholar]
- [27]. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- [28]. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 2017; 14:687–690. [DOI] [PubMed] [Google Scholar]
- [29]. Auersperg N, Wong AST, Choi KC, Kang SK, Leung PCK. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocrine Reviews 2001; 22:255–288. [DOI] [PubMed] [Google Scholar]
- [30]. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem 2007; 282:11613–11617. [DOI] [PubMed] [Google Scholar]
- [31]. George RJ, Sturmoski MA, Anant S, Houchen CW. EP4 mediates PGE dependent cell survival through the PI3 kinase/AKT pathway. Prostaglandins Other Lipid Mediat 2007; 83:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Schneider A, Guan YF, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM. Generation of a conditional allele of the mouse prostaglandin EP receptor. Genesis 2004; 40:7–14. [DOI] [PubMed] [Google Scholar]
- [33]. Jolly MK, Jia D, Boareto M, Mani SA, Pienta KJ, Ben-Jacob E, Levine H. Coupling the modules of EMT and stemness: a tunable ‘stemness window’ model. Oncotarget 2015; 6:25161–25174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Carter LE, Cook DP, Vanderhyden BC. Phenotypic plasticity and the origins and progression of ovarian cancer In: Leung PCK, Adashi EY (eds.), The Ovary, 3rd ed. San Diego, California, United States: Elsevier (Academic Press); 2018: Chapter 33. [Google Scholar]
- [35]. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K et al. . The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol 2017; 11:755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7:131–142. [DOI] [PubMed] [Google Scholar]
- [38]. Ruff M, Leyme A, Le Cann F, Bonnier D, Le Seyec J, Chesnel F, Fattet L, Rimokh R, Baffet G, Théret N. The disintegrin and metalloprotease ADAM12 is associated with TGF-β-induced epithelial to mesenchymal transition. PLoS One 2015; 10:e0139179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am J Respir Cell Mol Biol 2008; 38:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Rolfe KJ, Grobbelaar AO. A review of fetal scarless healing. ISRN Dermatol 2012; 698034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Wilgus TA, Bergdall VK, Tober KL, Hill KJ, Mitra S, Flavahan NA, Oberyszyn TM. The impact of cyclooxygenase-2 mediated inflammation on scarless fetal wound healing. Am J Pathol 2004; 165:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Wilgus TA, Vodovotz Y, Vittadini E, Clubbs EA, Oberyszyn TM. Reduction of scar formation in full-thickness wounds with topical celecoxib treatment. Wound Repair Regen 2003; 11:25–34. [DOI] [PubMed] [Google Scholar]
- [43]. Migone FF, Cowan RG, Williams RM, Gorse KJ, Zipfel WR, Quirk SM. In vivo imaging reveals an essential role of vasoconstriction in rupture of the ovarian follicle at ovulation. PNAS 2016; 113:2294–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Brannstrom M, Giesecke L, Moore IC, Van Den Heuvel CJ. Leukocyte subpopulations in the rat corpus luteum during pregnancy and pseudopregnancy’. Biol Reprod 1994; 50:1161–1167. [DOI] [PubMed] [Google Scholar]
- [45]. Brannstrom M, Pascoe V, Norman RJ, McClure N. Localization of leukocyte subsets in the follicle wall and in the corpus luteum throughout the human menstrual cycle. Fertil Steril 1994; 61:488–495. [PubMed] [Google Scholar]
- [46]. Cohen PE, Zhu L, Pollard JW. Absence of colony stimulating factor-1 in osteopetrotic (csfmop/csfmop) mice disrupts estrous cycles and ovulation. Biol Reprod 1997; 56:110–118. [DOI] [PubMed] [Google Scholar]
- [47]. Murdoch WJ, Martinchick JF. Oxidative damage to DNA of ovarian surface epithelial cells affected by ovulation: carcinogenic implication and chemoprevention. Exp Biol Med 2004; 229:546–552. [DOI] [PubMed] [Google Scholar]
- [48]. Pier B, Edmonds JW, Wilson L, Arabshahi A, Moore R, Bates GW, Prasain JK, Miller MA. Comprehensive profiling of prostaglandins in human ovarian follicular fluid using mass spectrometry. Prostaglandins Other Lipid Mediat 2018; 134:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Dada S, Demartines N and Dormond O. mTORC2 regulates PGE2-mediated endothelial cell survival and migration. Biochem. Biophys. Res. Commun 2008; 4(8):857–879. [DOI] [PubMed] [Google Scholar]
- [50]. Duffy DM. Novel contraceptive targets to inhibit ovulation: The prostaglandin E2 pathway. Hum Reprod Update 2015; 21:652–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Futagami A, Ishizaki M, Fukuda Y, Kawaba S, Yamanaka N. Wound healing involves induction of cyclooxygenase-2 expression in rat skin. Lab Invest 2002; 82(11):1503–13. [DOI] [PubMed] [Google Scholar]
- [52]. Wright JW, Pejovic T, Lawson M, Jurevic L, Hobbs T, Stouffer RL. Ovulation in the absence of the ovarian surface epithelium in the primate. Biol. Reprod 2010; 82(3):599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53]. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet 2014; 384:1376–1388. [DOI] [PubMed] [Google Scholar]
- [54]. Fathalla MF. Incesssant ovulation – a factor in ovarian neoplasia? Lancet 1971; 2:163. [DOI] [PubMed] [Google Scholar]
- [55]. Verdoodt F, Kjaer SK, Friis S. Influence of aspirin and non-aspirin NSAID use on ovarian and endometrial cancer: summary of epidemiologic evidence of cancer risk and prognosis. Maturitas 2017; 100:1–7. [DOI] [PubMed] [Google Scholar]
- [56]. Zamah AM, Hassis ME, Albertolle ME, Williams KE. Proteomic analysis of human follicular fluid from fertile women. Clin Proteomics 2015; 12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing files have been deposited and are available along with processed transcript quantifications at GSE121936.