

Summary
CHARGE syndrome is a congenital disorder with multiple malformations in the craniofacial structures, and cardiovascular and genital systems, which are mainly affected by neural crest defects caused by loss‐of‐function mutations within chromodomain helicase DNA‐binding protein 7 (CHD7). However, many patients with CHARGE syndrome test negative for CHD7. Semaphorin 3E (sema3E) is a gene reported to be mutated in patients with CHARGE syndrome. However, its role in the pathogenesis of CHARGE syndrome has not been verified experimentally. Here, we report that the knockdown of sema3E results in severe craniofacial malformations, including small eyes, defective cartilage and an abnormal number of otoliths in zebrafish embryos, which resemble the major features of CHARGE syndrome. Further analysis reveals that the migratory cranial neural crest cells are scattered in the region of the hindbrain, and the postmigratory neural crest cells are reduced in the pharyngeal arches upon sema3E knockdown. Notably, immunostaining and time‐lapse imaging analyses of a neural crest cell‐labelled transgenic fish line, sox10:EGFP, show that the migration of cranial neural crest cells is severely impaired, and many of these cells are misrouted upon sema3E knockdown. Furthermore, the sox10‐expressing cranial neural crest cells are scattered in chd7 homozygous mutants, which phenocopied the phenotype in sema3E morphants. Overexpression of sema3E rescues the phenotype of scattered cranial neural crest cells in chd7 homozygotes, indicating that chd7 may control the expression of sema3E to regulate cranial neural crest cell migration. Collectively, our data demonstrate that sema3E is involved in the pathogenesis of CHARGE syndrome by modulating cranial neural crest cell migration.
Keywords: CHARGE syndrome, Chd7, cranial neural crest cells, migration, semaphorin 3E
1. INTRODUCTION
CHARGE syndrome is a congenital disorder with multiple anomalies, including coloboma, heart defects, atresia choanae, retardation of growth, genital abnormalities and ear abnormalities.1 Craniofacial anomalies, including coloboma, choanal atresia/stenosis and hypoplasia/aplasia of the semicircular canals, are some of the main diagnostic criteria for this disease.2 Several anomalies, including craniofacial defects and heart defects, are life‐threatening, and about 30% of affected children die before their fifth birthday.3 Many, but not all, features of CHARGE syndrome can be attributed to the disruption of neural crest cells (NCCs), a transient population of multipotent cells that emerge from the dorsal neural tube and migrate dorsolaterally to populate diverse tissues and structures, including craniofacial bones and cartilages, the peripheral nervous system, pigmentation and cardiac structures.4, 5 Chromodomain helicase DNA‐binding protein 7 (CHD7) is the main known causative gene of this disease, and loss‐of‐function mutations or deletions within CHD7 are found in more than two‐thirds of CHARGE patients.6 A number of studies have shown that CHD7 is critical for the migration of neural crest (NC) cells, which is the so‐called neural crest hypothesis for CHARGE syndrome.7, 8, 9 However, many patients with CHARGE syndrome test negative for CHD7,10 and we currently know little about the pathogenic mechanisms induced by the other causative genes of this disease.
Semaphorins (SEMAs) are a family of secreted or transmembrane proteins, which were originally identified as guidance factors controlling axonal pathfinding during neural development.11 Class 3 semaphorins are a subfamily of secreted proteins, which have previously been identified as critical for NC development.12 For example, Yu et al. showed that sema3F and 3G were crucial for the separation of migratory cranial neural crest cell (CNCC) streams in zebrafish.13 Berndt showed that sema3D might regulate NC cell proliferation downstream of TCF in the zebrafish hindbrain.14 Importantly, it has been implicated previously that the role of sema3s in NC development is correlated with chd7. For instance, Payne et al. have suggested that Chd7 is localized at the promoter of sema3C and thus controls cardiovascular development.15 Ufartes et al16 reported that Chd7 was enriched at the promoter region of Sema3A in NCCs, and Chd7 loss of function inhibited the expression of sema3A. However, it was failure to find pathogenic mutations in sema3A for this disease in this study.16 SEMAPHORIN 3E (SEMA3E) is the fifth member of class three secreted semaphorins, and it is involved in axon guidance,17 vascular patterning,18 tumour metastasis 19 and immune regulation.20 Lalani et al21 reported that a SEMA3E missense mutation was found in a patient with CHARGE syndrome, and the location of SEMA3E was close to a chromosomal translocation breakpoint in an unrelated CHARGE patient, indicating that SEMA3E is likely to be another pathogenic gene responsible for CHARGE syndrome. However, the role of SEMA3E in NC development and the pathogenesis of CHARGE syndrome have not been studied.
To further clarify the role of SEMA3E in the pathogenesis of CHARGE syndrome, we examined the developmental role of sema3E in zebrafish models. We found that sema3E was expressed in the periphery of the hindbrain during the time of neural crest cell migration. Knockdown of sema3E resulted in small eyes and impaired cranial nerves and cartilage but relatively normal heart tube looping and expression of a T‐cell marker. Further analysis revealed that the crestin‐ and sox10‐positive migratory CNCCs were scattered in the hindbrain, and the expression of dlx2a‐ and hand2‐positive postmigratory CNCC markers was reduced in the pharyngeal region in sema3E morphants. Time‐lapse imaging revealed that the migration of sox10‐positive cranial NC cells was stalled near the dorsal crest, and some CNCCs were migrating in the dorsoposterior direction in sema3E morphants. Interestingly, sox10‐positive CNCCs were scattered, and the hand2‐positive postmigratory CNCCs were reduced in chd7 homozygotes, which phenocopied the phenotype in sema3E morphants. Finally, we found that overexpression of sema3E rescued the phenotype of scattered CNCCs and the reduction of postmigratory CNCCs in chd7 morphants, demonstrating that chd7 may control the expression of sema3E to regulate CNCC migration in zebrafish. Collectively, our data showed that sema3E is critical for the ventrolateral migration of CNCCs, and sema3E deficiency is involved in the pathogenesis of CHARGE syndrome.
2. MATERIALS AND METHODS
2.1. Zebrafish strains and embryos
Zebrafish strains, including wild‐type (AB strain), chd7 mutant,22 sox10:EGFP and islet:EGFP transgenic lines, were purchased from the China Zebrafish Resource Center. Embryos were obtained by natural spawning and raised in E3 buffer at 28°C. Adult zebrafish were raised in a zebrafish breeding system (HAISHENG Biotech) at 28°C.
2.2. Ethical approval
This study was approved by the Ethical Review Committee of Nanchang University, Nanchang, China.
2.3. Morpholinos, primers, mRNA synthesis and microinjection
Standard MO and antisense MO were purchased from GeneTools and prepared as 1 mM stock solutions using ddH2O (Millipore). The sequences of MOs and the primers used for amplification of RNA probes and coding sequences are listed in Table S1. Capped zebrafish full‐length sema3E mRNA for injection was synthesized in vitro using the mMessage mMachine SP6 transcription kit (Ambion, Thermo Fisher Scientific). MOs (6 ng for sema3E and standard MO) and capped mRNA (100 pg) were injected alone into the one‐cell‐stage zebrafish embryos at the yolk/blastomere boundary.
2.4. Polymerase chain reaction
All primers used for the preparation of WISH probes, reverse‐transcription PCR and quantitative real‐time PCR (qPCR) are listed in Table S1. Total RNA was extracted by grinding dechorionated 25‐30 embryos using RNAiso plus (Takara), followed by phenol‐chloroform extraction. Reverse‐transcription reaction was performed by using reverse transcriptase M‐MLV (Takara). Wild‐type and sema3E morphant transcripts were obtained using reverse transcription. qPCR was performed using the SYBR Premix Ex Taq II (Takara) on the Abi‐Step‐One plus Real‐Time PCR system (Applied Biosystems). All of the experiments were repeated three times in triplicates. The data were analysed using the △△C t method. Data are presented as mean ± SD, and Student's t test was performed for comparison between the control and injected groups. P‐value < 0.05 was considered to indicate a significant difference.
2.5. Whole‐mount in situ hybridization
Whole‐mount in situ hybridization (WISH) was performed as previously described using probes for hoxb2a, krox2a, hand2, crestin, dlx2a, sema3E and sox10. Briefly, staged embryos were fixed overnight in 4% paraformaldehyde, and then, they were dehydrated in a methanol gradient. When the embryos were ready for use, they were rehydrated in phosphate‐buffered saline containing 0.1% Tween‐20 (PBST). Embryos were permeabilized by proteinase K digestion, and then, they were hybridized with digoxin‐labelled probes overnight at 70°C. The next day, embryos were washed in a preheated mixture of 50% saline‐sodium citrate containing 0.1% Tween‐20 (SSCT) and 50% hybridization solution at 70°C. Embryos were washed again at room temperature and incubated in a staining solution in the dark until sufficient staining was visible in the embryos. Background staining was cleared by soaking the embryos in a benzylbenzoate‐benzylalcohol solution. Embryos were mounted in glycerol and were visualized using a Nikon AZ100 microscope. Images were captured using a Nikon DIGITAL SIGHT DS‐Fil1 digital camera (Nikon) and processed with NIS‐Elements F 3.0 (Nikon).
2.6. Whole‐mount immunostaining
For immunofluorescence microscopy, immunostaining was performed according to Chalasani et al.23 Larval zebrafish were fixed overnight with 4% paraformaldehyde in PBS, followed by methanol dehydration and rehydration. After permeabilization by ice‐cold acetone treatment and proteinase K digestion, embryos were incubated with primary and secondary antibodies at the following concentrations: 1:500 goat anti‐GFP, 1:500 anti‐goat IgG Alexa Fluor 488 (Invitrogen), 1:500 rabbit anti‐ph3, 1:500 rabbit anti‐active caspase 3, 1:500 anti‐rabbit IgG H&L (Cy3), 1:500 anti‐mouse 568 and 1:500 anti‐goat IgG Alexa Fluor 568.
2.7. Confocal microscopy
Live embryos were anesthetized with tricaine and then mounted on a coverslip with 1% low‐melting agarose. The agar layer was further covered with E3 buffer containing 0.0084 mg/mL tricaine to enable movies to be taken over a long time frame. For live imaging of the hindbrain, the recordings were started at 60 hpf and continued for 12 hours. Images were taken every 6 minutes or 5 minutes, and single frames were generated as projections of a stack of 46 planes with 2‐µm distance, covering a depth of 90 µm. For imaging immunostained samples, embryos were mounted on a coverslip with 1% low‐melting agarose. Confocal images were captured by an Olympus FV1000 confocal microscope, and 3D projections were generated with the FV100‐ASW 4.2 viewer (Olympus). Single frames were generated as projections of a stack of multiple planes. The number of immunofluorescence‐labelled cells was calculated using Imaris7 (Bitplane).
2.8. Alcian blue staining
Zebrafish larvae (5 dpf) were fixed in 4% paraformaldehyde at 4°C overnight. Then, the fixed larvae were digested in 10% trypsin for 1 hour. After washing three times with PBS, the larvae were stained with 0.1% (w/v) Alcian blue (Sigma) for 3 hours. Then, the staining reaction was terminated with the use of PBS and the larvae were washed again in PBS. The stained larvae were mounted in 90% glycerol and visualized using a Nikon AZ100 microscope. Images were captured using a Nikon DIGITAL SIGHT DS‐Fil1 digital camera and processed with NIS‐Elements F 3.0 software (Nikon).
2.9. Statistical analysis
Values are presented as the mean (SD). Student's t test was used to compare the means of different groups, and P‐values < 0.05 were considered significant.
3. RESULTS
3.1. sema3E is expressed in the hindbrain during the period of NC migration
To explore the developmental role of sema3E, we first examined the expression pattern of sema3E at various developmental stages using whole‐mount in situ hybridization (WISH). Minor expression of sema3E was detected at the shield stage and 15 hours postfertilization (hpf; Figure S1). At 18 hpf, the expression of sema3E emerged in the motor neurons and dorsal aorta (Figure 1A). At 22 hpf, sema3E was expressed more broadly, and the transcripts could be detected in the head, eyes, trunk, motor neurons and gut (Figure 1B). Notably, the expression of sema3E was found in the periphery of the rhombomeres at 22 hpf (Figure 1C) when NCCs were migrating in the ventrolateral direction, strongly indicating that sema3E was required for the migration of CNCCs. At 36 hpf, distinct expression of sema3E was detected at the midbrain‐hindbrain boundary, the otic vesicle and the jaw (Figure 1D), indicating that sema3E was also required for the development of the otic vesicle and the jaw.
Figure 1.
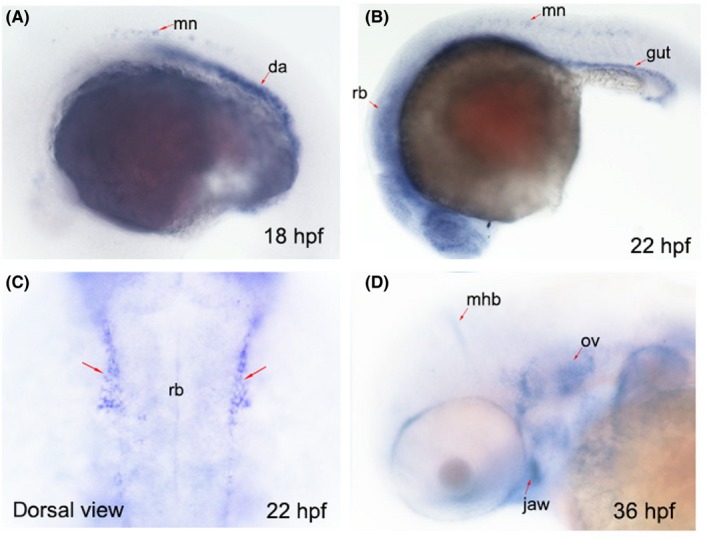
The expression patterns of sema3E at different developmental time points. A, The expression pattern of endogenous sema3E transcripts at 18 hpf. Red arrows indicate the expression in motor neurons and dorsal aorta; mn, motor neurons; da, dorsal aorta. B, The expression pattern of endogenous sema3E transcripts at 22 hpf. Red arrows indicate the expression of sema3E in motor neurons and gut; mn, motor neurons. C, The dorsal view of the expression pattern of endogenous sema3E transcripts at 22 hpf. Red arrows indicate the expression of sema3E at the periphery of the hindbrain; rb, rhombomere. D, The expression pattern of endogenous sema3E transcripts at 36 hpf. Red arrows indicate the expression of sema3E at the periphery of the hindbrain; rb, rhombomere; mhb, midbrain‐hindbrain boundary; ov, otic vesicle
3.2. Knockdown of sema3E results in severe craniofacial defects
To further analyse the role of sema3E in NC development, we used an antisense oligonucleotide morpholino (MO) targeting the junction of exon 5 and intron 5 of sema3E to block splicing and to knock down the expression.24 After injecting the MO into zebrafish zygotes, we observed several apparent phenotypes, including flattened head, small eyes and abnormal number of otoliths, which resembled the features of CHARGE syndrome (Figure 2A‐C). Craniofacial defects, including atresia choanae and cranial nerve defects, are the diagnostic criteria for CHARGE syndrome. We next checked the development of cranial cartilages by Alcian blue staining in sema3E morphants at 5 days postfertilization (dpf). As shown in Figure 2D, the development of cranial cartilages was severely impaired and the auditory ossicles and ceratobranchials were almost absent in sema3E morphants at 5 dpf. Moreover, we also injected the sema3E MO into a Isl1:EGFP transgenic fish line, which expressed EGFP under the control of a motor neuron–specific Isl1 promoter. As shown in Figure 2E, the vagal (nX) motor neurons exhibited shortened axonal branches and the trigeminal (nV) and facial (nVII) branchiomotor neurons exhibited aberrant medio‐lateral positioning and cell‐to‐cell spacing. To further verify the specificity of the sema3E MO, we performed rescue experiments and found that the CHARGE‐like phenotypes could be fully rescued by zebrafish sema3E overexpression in sema3E morphants (Figure 2A‐E). Since heart defects and T‐cell deficiency are the features of CHARGE syndrome, we examined the expression patterns of a heart tube marker, cmlc2, and a T‐cell marker, rag1, in sema3E morphants. The data showed that the development of the heart tube and T cells was unaffected at 48 hpf and 4 dpf respectively (Figure S2). Statistical analysis of these CHARGE‐like phenotypes showed that the percentages of flattened head, small eyes, abnormal otolith, cartilage defects and cranial nerve defects were 21.2 ± 1.2%, 22.7 ± 3.3%, 33.3 ± 2.8%, 77.4 ± 3.3% and 58.4 ± 3% (Figure 2F) respectively. Collectively, these data showed that knockdown of sema3E recapitulated the major craniofacial features, but not heart defects and T‐cell deficiency in CHARGE syndrome.
Figure 2.
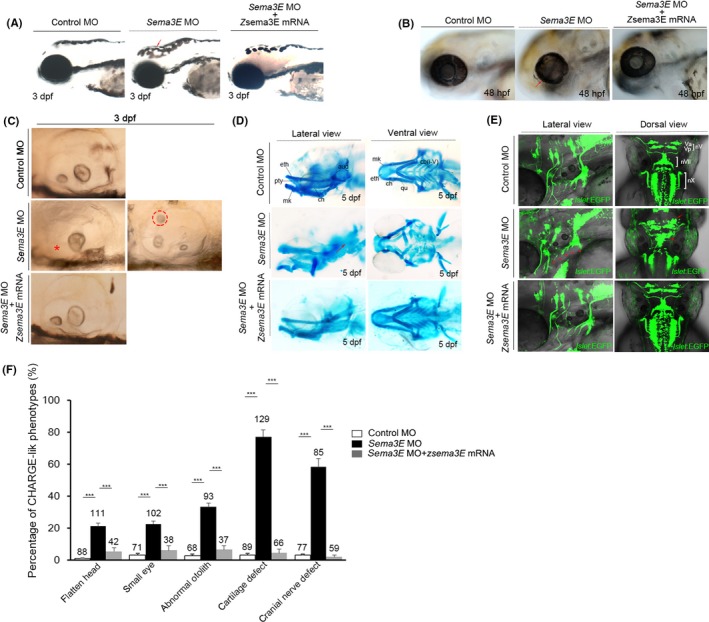
Knockdown of sema3E resulted in CHARGE‐like phenotypes. A, The head development in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA at 3 dpf. Red arrows indicate the flattened head in sema3E morphants. B, The eye development in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA at 48 hpf. Red arrows indicate the ocular tissue defect in sema3E morphants. C, The development of the otic vesicle in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA. A red asterisk indicates the absence of an otolith, and a red dashed circle denotes an abnormally positioned otolith. D, Cranial cartilage development in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA at 5 dpf. Red arrows indicate the malformations in the auditory ossicles and ceratobranchials. Eth, ethmoid plate; mk, Meckel's cartilage; pty, pterygoid process of the quadrate; aud, auditory ossicles; ch, ceratohyal; qu, quadrate; cb, ceratobranchials. E, Cranial nerve development in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA at 48 hpf. Red asterisks indicate the shortened branches of vagal (nX) motor neurons, and red arrows denote the aberrant medio‐lateral positioning and cell‐to‐cell spacing of the trigeminal (nV) and facial (nVII) branchiomotor neurons. F, Statistical analysis of the CHARGE‐like phenotypes in control, sema3E morphants and sema3E morphants co‐injected with sema3E mRNA. Numbers above the columns represent the total number of embryos in each experiment. ***P < 0.001, (n = 3)
3.3. Development of cranial neural crest cells is severely impaired in sema3E‐deficient embryos
CNCCs populate the face and pharyngeal arches to give rise to bones, cartilage, nerves and connective tissue. We questioned whether the craniofacial defects in sema3E morphants were attributed to the abnormal development of CNCCs. As the rhombomeres are the source of CNCCs, we first examined the expression of two rhombomere markers, krox20 and hoxa2b, by WISH. We found that, compared with controls, the location and expression of the two rhombomere markers appeared to be unaltered in sema3E morphants (Figure S3). To further address the development of CNCCs upon sema3E knockdown, we examined the expression patterns of two migratory NC cell markers, sox10 and crestin, by WISH. Interestingly, as shown in Figure 3A, the crestin‐ and sox10‐positive migratory CNCCs were scattered in the hindbrain in sema3E morphants, indicating that the migration of CNCCs was impaired. Then, we questioned whether scattered distribution of CNCCs was accompanied by a change in the amount of CNCCs in sema3E morphants. To answer this question, we quantified the expression level of sox10 and crestin transcripts by quantitative real‐time PCR. We found that there was no significant difference in the amount of the expression of these two genes between controls and sema3E morphants at 20 hpf (Figure 3B). Moreover, we examined the neural crest–specific proliferation and apoptosis by using a transgenic fish line, sox10:EGFP, which expressed EGFP under the control of an early neural crest‐specific sox10 promoter. As shown in Figure S4, there was no significant difference in the number of phosphohistone H3 and GFP double‐stained cells and GFP and activated caspase 3 double‐stained cells between control MO‐injected sox10:EGFP transgenic embryos and sema3E MO‐injected sox10:EGFP transgenic embryos, indicating that proliferation, survival and epithelial‐interstitial transformation of CNCCs were likely to be unaffected in sema3E morphants at 24 hpf. Since the migration of CNCCs is impaired, we questioned whether the postmigratory CNCCs were reduced in sema3E morphants. To address this question, we checked the development of postmigratory NC cells by examining the expression of two postmigratory NC markers, hand2 and dlx2a, by using WISH. As shown in Figure 3C, the staining for hand2 and dlx2a was moderately reduced in the pharyngeal arches in sema3E morphants on comparing with the control group at 23 hpf. The qPCR data confirmed the results of WISH (Figure 3D). Collectively, these data suggested that knockdown of sema3E caused migratory defects in CNCCs, which might underlie the craniofacial defects in sema3E morphants.
Figure 3.
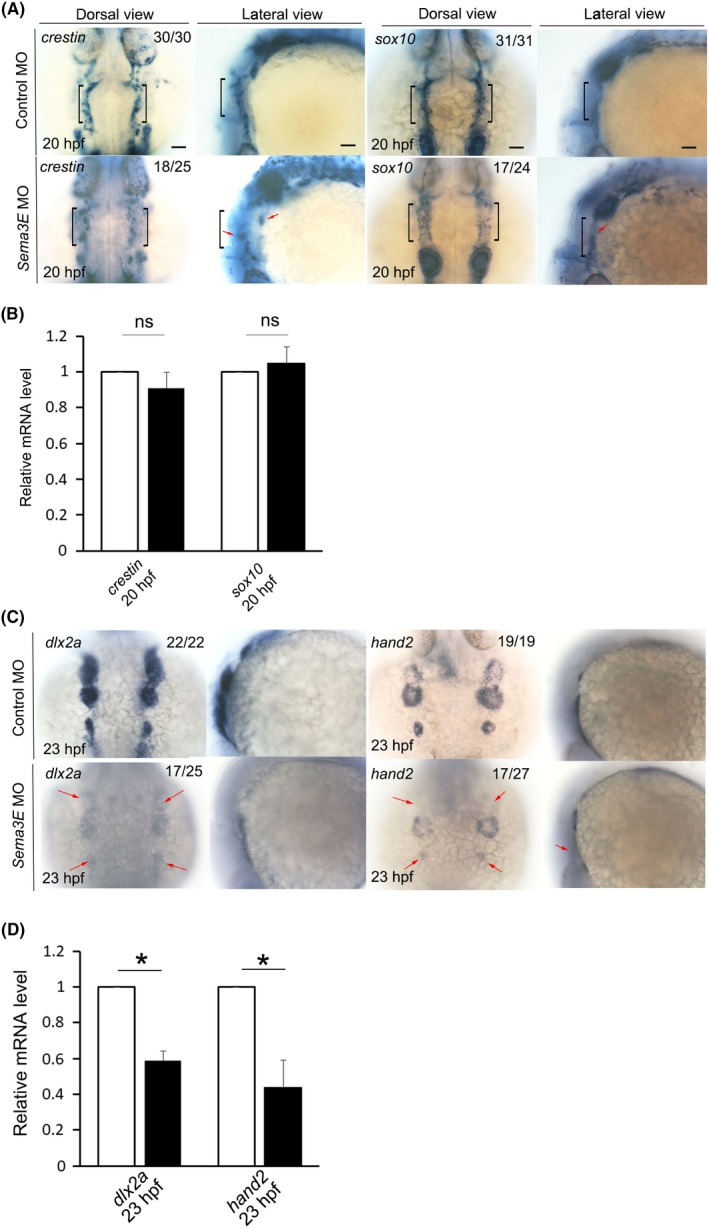
The expression of migratory cranial neural crest cell and postmigratory cranial neural crest cell markers in control and sema3E morphants. A, The expression of migratory cranial neural crest cell markers, crestin and sox10, in control and sema3E morphants at 20 hpf. Black brackets indicate the expression of crestin or sox10 in the rhombomeres. Red arrows denote the scattered cranial neural crest cells. B, Quantitative PCR to quantify the expression of crestin and sox10 in the hindbrain in control and sema3E morphants. C, The expression of postmigratory cranial neural crest cell markers, dlx2a and hand2, in control and sema3E morphants at 23 hpf. Red arrows denote the reduced postmigratory cranial neural crest cell markers in sema3E morphants. D, Quantitative PCR to quantify the expression of crestin and sox10 in the hindbrain in control and sema3E morphants (mean ± SD, Student's t test, *P < 0.05, n = 3)
3.4. Migration of sox10‐positive cranial neural crest cells is severely impaired upon sema3E knockdown
To further check the migration of CNCCs in sema3E morphants in detail, we injected the sema3E MO into the transgenic fish line, sox10:EGFP. After immunostaining the transgenic embryos with GFP antibody, we found that most of the sox10‐positive CNCCs were well patterned and distributed in the side of the hindbrain in the control MO‐injected transgenic embryos (Figure 4A). However, in the sema3E MO‐injected sox10:EGFP embryos, CNCCs were scattered and a number of them were misrouted and present within the hindbrain (Figure 4B). In the lateral view of these immunostained transgenic embryos, CNCCs were well patterned in the control MO‐injected transgenic embryos (Figure 4C). However, in the sema3E MO‐injected sox10:EGFP embryos, many of the CNCCs were scattered and present in the dorsal part of the hindbrain and the migration of many CNCCs seemed to have been halted (Figure 4D). To further analyse the dynamics of CNCCs in sema3E morphants, time‐lapse imaging was used from 22 to 23 hpf. We found that CNCCs with a round shape were migrating in the ventrolateral direction in the control MO‐injected group. However, in the sema3E MO‐injected transgenic embryos, the migration of a subset of CNCCs was halted and some CNCCs were even migrating in the dorsoposterior direction (Appendix S1), thus demonstrating that sema3E deficiency causes defective CNCC migration in zebrafish, which could be used to explain the pathogenic mechanism in sema3E‐positive patients with CHARGE syndrome.
Figure 4.
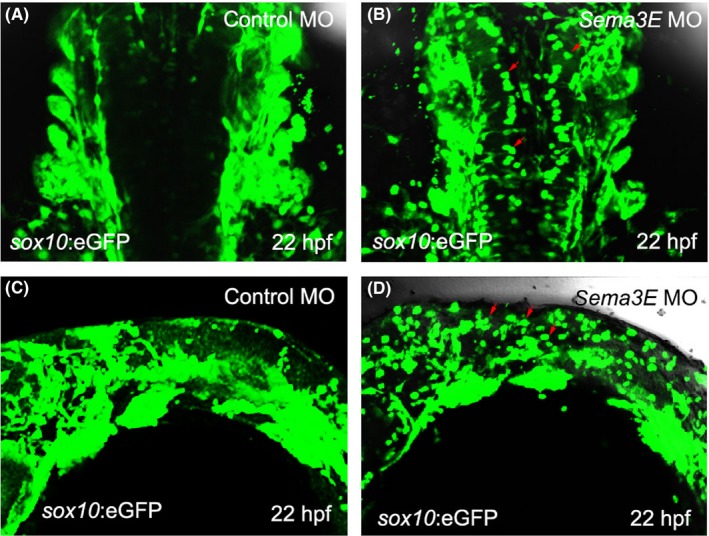
Knockdown of sema3E resulted in impaired migration of cranial neural crest cells in sox10:EGFP transgenic embryos at 22 hpf. A, Dorsal view of the immunostained control MO‐injected sox10:EGFP transgenic embryos. B, Dorsal view of the immunostained sema3E MO‐injected sox10:EGFP transgenic embryos. Red arrows denote the misrouted GFP‐expressing cranial neural crest cells. C, Lateral view of the immunostained control MO‐injected sox10:EGFP transgenic embryos. D, Lateral view of the immunostained sema3E MO‐injected sox10:EGFP transgenic embryos. Red arrows denote the error‐migrated GFP‐expressing cranial neural crest cells, which are present in the dorsal part of sema3E MO‐injected sox10:EGFP transgenic embryos
3.5. Overexpression of zebrafish sema3E rescues the phenotype of scattered cranial neural crest cells in chd7 homozygous mutants
To address whether ventrolateral migration errors of CNCCs are commonly found in the pathogenesis of CHARGE syndrome, we examined the development of CNCCs in chd7 zebrafish mutants.22 As shown in Figure 5A, sox10‐positive CNCCs were scattered in a quarter of (16/64) the offspring of chd7 heterozygous mutants at 22 hpf. Genotyping analysis showed that this quarter of embryos were chd7 homozygous mutants, indicating that the scattered distribution of CNCCs also existed in chd7 homozygous mutants and ventrolateral migration defects in CNCCs might be common among CHARGE patients, which is consistent with previous reports.7, 9 Moreover, we questioned whether chd7 controls the expression of sema3E to regulate CNCC migration. To address this question, we injected capped messenger RNA of zebrafish sema3E into the zygotes of chd7 heterozygous mutants at the single‐cell stage and we found that 41 out of the 56 sema3E‐overexpressing offspring of chd7 heterozygotes exhibited relatively normal distribution of sox10‐positive CNCCs. Genotyping analysis showed that 14 out of the 41 sema3E‐overexpressing offspring of chd7 heterozygotes were homozygous, and this proportion (14/56) is identical to Mendel's law of inheritance, thus demonstrating that overexpression of sema3E is sufficient to partially rescue the phenotype of scattered CNCCs in chd7 homozygous mutants. Moreover, we questioned whether overexpression of sema3E could also rescue the amount of postmigratory CNCCs in chd7 homozygous mutants. Indeed, as shown in Figure 5B, 33 out of the 49 sema3E‐overexpressing offspring of chd7 heterozygotes exhibited an increased number of hand2‐positive CNCCs on comparing with the un‐injected chd7 homozygotes. Genotyping analysis showed that 12 out of the 33 sema3E‐overexpressing offspring of chd7 heterozygotes were homozygous, and this proportion (12/49) is identical to Mendel's law of inheritance, thus demonstrating that overexpression of sema3E is sufficient to partially rescue the amount of postmigratory CNCCs in chd7 homozygous mutants. Collectively, these data suggest that chd7‐deficient embryos have migration defects in CNCCs. Sema3E deficiency contributed to the defective migratory CNCCs in chd7‐deficient embryos, and thus, it is involved in the pathogenesis of CHARGE syndrome.
Figure 5.
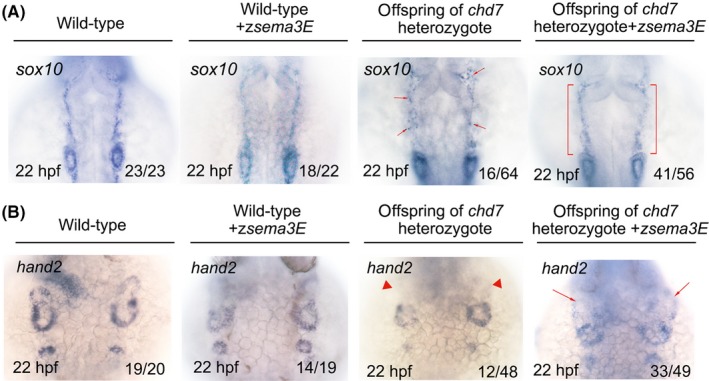
Overexpression of zebrafish sema3E partially rescued the phenotype of scattered cranial neural crest cells and the reduction of postmigratory neural crest cells in chd7 mutants. A, The expression of the migratory cranial neural crest marker, sox10, in wild‐type, sema3E‐overexpressing wild‐type, offspring of chd7 heterozygous mutants, sema3E‐overexpressing offspring of chd7 heterozygous mutants. Red arrows indicate the scattered migratory cranial neural crest cells in some offspring of chd7 heterozygous mutants. Red brackets denote the good‐patterned migratory cranial neural crest cells in sema3E‐overexpressing offspring of chd7 heterozygous mutants. B, The expression of the postmigratory cranial neural crest marker, hand2, in wild‐type, sema3E‐overexpressing wild‐type, offspring of chd7 heterozygous mutants, sema3E‐overexpressing offspring of chd7 heterozygous mutants. Red triangles indicate the reduced postmigratory cranial neural crest cells in some offspring of chd7 heterozygous mutants. Red arrows denote the amount of rescued postmigratory cranial neural crest cells in sema3E‐overexpressing offspring of chd7 heterozygous mutants
4. DISCUSSION
Haploinsufficiency of CHD7 is presumed to be the cause of CHARGE syndrome.25 Since it was found that more than two‐thirds of CHARGE patients carried heterozygous mutations of chd7, a number of studies were conducted to explore the pathogenic mechanism of CHARGE syndrome using chd7 knockdown or knockout models. These studies have given us some important insights into the understanding of the pathogenesis of CHARGE syndrome, and the contribution of defects in migration of CNCCs to the pathogenesis is widely accepted.26, 27 For example, the study by Bajpai et al. revealed that CHD7 cooperated with polybromo‐ and BRG1‐associated factor‐containing complex to regulate NC‐specific transcriptional factor expression and migration both in human neural crest‐like cells and in Xenopus.7 Okuno et al9 showed that induced pluripotent stem cell‐derived NC cells harboured CHD7 mutations from patients with CHARGE syndrome displayed defective migration. However, among the patients who meet the diagnostic criteria for CHARGE syndrome, up to ∼30% of the patients do not test positive for CHD7.3 To date, we know little about the pathogenic mechanism induced by the other causative genes in CHARGE patients. Recently, Bélanger et al28 reported that fam172a plays a key role in the regulation of co‐transcriptional alternative splicing, and mutations within this gene were found in CHD7 mutation‐negative CHARGE patients, indicating that dysregulation of co‐transcriptional alternative splicing caused by the deficiency of fam172a is likely to be a mechanism in CHD7 mutation‐negative CHARGE patients. Nevertheless, it is still uncertain whether migration defects in NC cells also exist in CHD7 mutation‐negative cases. In the previous report by Lalani et al., sequencing data from CHARGE patients suggested that sema3E mutation was likely to be a cause of CHARGE syndrome.21 Previous studies have revealed that class three semaphorins are involved in NC migration, and their expression is controlled by chd7.5, 15, 29 In the present study, our data showed that sema3E was expressed in the periphery of the hindbrain during the time window of NC migration in zebrafish. Knockdown of sema3E resulted in impaired migration of CNCCs, and it severely impaired the craniofacial structure, indicating that deficiency of sema3E alone is sufficient to induce CHARGE‐like features, further supporting the claim that sema3E is a causative gene of CHARGE syndrome. According to our data, heart looping and thymus development were seemingly unaffected upon sema3E knockdown (Figure S2), suggesting that deficiency of sema3E might not affect early cardiac and thymus development. However, the latter development of cardiovascular system is likely to be affected by sema3E deficiency, because, at least, sema3E is critical for vascular patterning.18 Moreover, our in situ hybridization data showed that the expression of sema3E was also found in the jaw and the otic vesicle at 48 hpf when the NC cells had finished their migration in zebrafish (data not shown), indicating that a defective craniofacial structure upon sema3E knockdown was attributed not only to defects in the migration of CNCCs but also to the expression of sema3E in the jaw and otic vesicle development. Further research should be conducted to explore the role of sema3E in jaw and otic vesicle development, especially with respect to the time window when NC cells have finished migration. Furthermore, we found that the sox10‐positive CNCCs were scattered in the hindbrain of chd7‐knockout zebrafish embryos, and overexpression of sema3E rescued this phenotype, indicating that the scattered distribution of CNCCs may be common in the pathogenesis of CHARGE syndrome, and chd7 controls the expression of sema3E during the migration of CNCCs.
Semaphorin signals have been reported to be crucial for the separation of NC migratory streams.13 It has been reported that Sema3 proteins, including Sema3A, Sema3F and Sema3G, are expressed in the NC‐free zone, while semaphorin receptors, neuropilins, are expressed on NC cells. Neuropilin‐expressing NC cells form separate streams when they are migrating dorsoventrally in response to the expression of semaphorins. Moreover, except for the role of sema3s in NC guidance, sema3D was found to be critical for the proliferation of CNCCs, and its expression is under the control of the wnt/△TCF signal.14 In the present study, our data showed that sema3E was expressed in the periphery of rhombomeres 3‐5, which include the NC zones (rhombomeres 3 and 5) and the NC‐free zone (rhombomere 4). The separation of dlx2a‐ and hand2‐positive NC cells is relatively normal in sema3E morphants, indicating that, unlike other sema3 proteins, sema3E may be not required for the formation of the NC migratory streams. Moreover, our live imaging data showed that the migration of CNCCs was stalled in the dorsal part of sema3E‐deficient sox10:EGFP transgenic embryos, thus indicating that sema3E may be required for the dorsoventral migration of CNCCs.
In conclusion, our data show that the knockdown of sema3E is sufficient to induce severe defects in craniofacial structures in zebrafish embryos, which phenocopy the major features of CHARGE syndrome. The craniofacial defects in sema3E‐deficient embryos may be attributed to the migration defects in CNCCs in sema3E morphants. Finally, overexpression of sema3E is sufficient to partially rescue the migration defects in chd7 homozygotes, which suggests that chd7 may regulate the expression of sema3E to modulate CNCC migration.
CONFLICT OF INTEREST
None.
Supporting information
ACKNOWLEDGEMENTS
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Liu Z‐Z, Guo J, Lu Y, et al. Sema3E is required for migration of cranial neural crest cells in zebrafish: Implications for the pathogenesis of CHARGE syndrome. Int. J. Exp. Path. 2019;100:234–243. 10.1111/iep.12331
Funding information
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81160144, 31400988, 81760216 and 31171044), the Natural Science Foundation of Jiangxi Province (Grant No. 20151BAB215015) and the Young Scientist of Jiangxi Province (20122BCB23007).
REFERENCES
- 1. Hudson A, Trider C‐L, Blake K. CHARGE syndrome. Pediatr Rev. 2017;38(1):56‐59. [DOI] [PubMed] [Google Scholar]
- 2. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A(3):306‐308. [DOI] [PubMed] [Google Scholar]
- 3. Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152A:674‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pauli S, Bajpai R, Borchers A. CHARGEd with neural crest defects. Am J Med Genet C Semin Med Genet. 2017;175:478‐486. [DOI] [PubMed] [Google Scholar]
- 5. Schulz Y, Wehner P, Opitz L, et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum Genet. 2014;133(8):997‐1009. [DOI] [PubMed] [Google Scholar]
- 6. Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955‐957. [DOI] [PubMed] [Google Scholar]
- 7. Bajpai R, Chen DA, Rada‐Iglesias A, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463(7283):958‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Nostrand JL, Brady CA, Jung H, et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature. 2014;514(7521):228‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okuno H, Renault Mihara F, Ohta S, et al. CHARGE syndrome modeling using patient‐iPSCs reveals defective migration of neural crest cells harboring CHD7 mutations. Elife. 2017;6:e21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hale CL, Niederriter AN, Green GE. Response to correspondence to Hale et al. atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170:3367‐3368. [DOI] [PubMed] [Google Scholar]
- 11. Jongbloets BC, Pasterkamp RJ. Semaphorin signalling during development. Development. 2014;141(17):3292‐3297. [DOI] [PubMed] [Google Scholar]
- 12. High F, Epstein JA. Signalling pathways regulating cardiac neural crest migration and differentiation. Novartis Found symp. 2007;283:152‐161:discussion 161‐154, 238‐141. [DOI] [PubMed] [Google Scholar]
- 13. Yu HH, Moens CB. Semaphorin signaling guides cranial neural crest cell migration in zebrafish. Dev Biol. 2005;280(2):373‐385. [DOI] [PubMed] [Google Scholar]
- 14. Berndt JD, Halloran MC. Semaphorin 3d promotes cell proliferation and neural crest cell development downstream of TCF in the zebrafish hindbrain. Development. 2006;133(20):3983‐3992. [DOI] [PubMed] [Google Scholar]
- 15. Payne S, Burney MJ, McCue K, et al. A critical role for the chromatin remodeller CHD7 in anterior mesoderm during cardiovascular development. Dev Biol. 2015;405(1):82‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ufartes R, Schwenty‐Lara J, Freese L, et al. Sema3A plays a role in the pathogenesis of CHARGE syndrome. Hum Mol Genet. 2018;27(8):1343‐1352. [DOI] [PubMed] [Google Scholar]
- 17. Xu H, Leinwand S g, Dell A l, Fried‐Cassorla E, Raper J a. The calmodulin‐stimulated adenylate cyclase ADCY8 sets the sensitivity of zebrafish retinal axons to midline repellents and is required for normal midline crossing. J Neurosci. 2010;30(21):7423‐7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu C, Yoshida Y, Livet J, et al. Semaphorin 3E and plexin‐D1 control vascular pattern independently of neuropilins. Science. 2005;307(5707):265‐268. [DOI] [PubMed] [Google Scholar]
- 19. Casazza A, Finisguerra V, Capparuccia L, et al. Sema3E‐plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J Clin Invest. 2010;120(8):2684‐2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi YI, Duke‐Cohan JS, Chen W, et al. Dynamic control of beta1 integrin adhesion by the plexinD1‐sema3E axis. Proc Natl Acad Sci U S A. 2014;11:379‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet. 2004;41(7):e94‐e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu ZZ, Wang ZL, Choi TI, et al. Chd7 is critical for early T‐cell development and thymus organogenesis in zebrafish. Am J Pathol. 2018;188(4):1043‐1058. [DOI] [PubMed] [Google Scholar]
- 23. Chalasani SH, Sabol A, Xu H, et al. Stromal cell‐derived factor‐1 antagonizes slit/robo signaling in vivo. J Neurosci. 2007;27(5):973‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dell AL, Fried‐Cassorla E, Xu H, Raper JA. cAMP‐induced expression of neuropilin1 promotes retinal axon crossing in the zebrafish optic chiasm. J Neurosci. 2013;33(27):11076‐11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Layman WS, Hurd EA, Martin DM. Chromodomain proteins in development: lessons from CHARGE syndrome. Clin Genet. 2010;78(1):11‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams MS. Speculations on the pathogenesis of CHARGE syndrome. Am J Med Genet A. 2005;133A(3):318‐325. [DOI] [PubMed] [Google Scholar]
- 27. Siebert JR, Graham JM Jr, MacDonald C. Pathologic features of the CHARGE association: support for involvement of the neural crest. Teratology. 1985;31(3):331‐336. [DOI] [PubMed] [Google Scholar]
- 28. Belanger C, Berube‐Simard FA, Leduc E, et al. Dysregulation of cotranscriptional alternative splicing underlies CHARGE syndrome. Proc Natl Acad Sci U S A. 2018;115(4):E620‐E629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li W, Xiong Y, Shang C, et al. Brg1 governs distinct pathways to direct multiple aspects of mammalian neural crest cell development. Proc Natl Acad Sci U S A. 2013;110(5):1738‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials