

Abstract
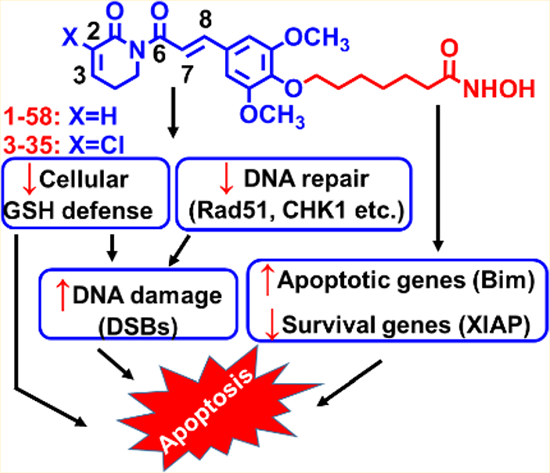
Synergistic-to-additive antileukemic interactions of piperlongumine (PL) and HDAC inhibitor (HDACi) SAHA (Vorinostat) provide a compelling rationale to construct PL-HDACi hybrids, such as 1–58, which recapitulated the synergism between the parental compounds in high-risk and chemoresistant AML cells. Both PL and HDACi components, either in combination or in hybrid molecules, are essential for inducing significant DNA damage and apoptosis. Introducing C2-chloro substituent to 1–58 yielded 3–35 with increased cytotoxicity but decreased selectivity in noncancerous MCF-10A cells; eliminating C7–C8 olefin of PL obtained 3–31/3–98 scaffolds which were still more active than PL or SAHA in AML and were well-tolerated by MCF-10A cells. The HDACi function was crucial for modulating expression of DNA repair and apoptosis-related proteins. Collectively, PL and SAHA hybrids are potent, multifunctional anti-AML agents, acting in part, by interfering cellular GSH defense, suppressing expression of DNA repair and pro-survival proteins, and inducing expression of pro-apoptotic proteins.
Graphical Abstract
INTRODUCTION
Acute myeloid leukemia (AML) remains a devastating disease with poor outcomes, especially in elderly patients.1 In 2015, the disease was projected to affect 20 830 new cases and to cause 10 460 deaths in the U.S.2 Although the prognosis of AML in younger patients has been steadily improved, long-term disease-free survival of elderly patients remains poor, only 10–15%. Except acute promyelocytic leukemia, which can be treated with all-trans retinoic acid and arsenic trioxide, the mainstay of initial treatment for all other subtypes of AML using a combination of cytosine arabinoside (Ara-C, also known as cytarabine) and an anthracycline (e.g., daunorubicin) has not been changed for more than three decades. Treatment failure is mainly due to resistance to chemotherapy and disease relapse arising from leukemic stem cells (LSCs).3,4 LSCs are defined as CD34+CD38−CD123+ cells and are the primary origin of AML growth and relapse.5 The discovery of innovative therapeutic agents for AML treatment represents an urgent and essential medical need.
Histone deacetylase inhibitors (HDACis), such as suberoylanilide hydroxamic acid (SAHA, Scheme 1), display diverse mechanisms of action that are attractive for AML therapy in preclinical models.6 DNA damage is one of the mechanisms contributing to HDACi-induced growth arrest and apoptosis in both AML cell lines and patient blasts.7–9 HDACis may cause DNA damage and subsequent leukemia cell death through induction of reactive oxygen species (ROS),7,10 down-regulation and/or impairment of the function of DNA repair enzymes (e.g., Ku70, RAD51, CHK1, etc.),10–14 modulation of apoptotic pathways (such as upregulation of pro-apoptotic genes and down-regulation of pro-survival genes),14 and disruption of chaperone function of Hsp90, resulting in degradation of pro-growth/pro-survival client proteins (Bcr-Abl, mutant FLT-3, c-Raf, AKT, etc.).15 Recent evidence also supports the role of HDACi in targeting LSCs (e.g., using drug combinations of HDACi and CHK116 or Wee117 inhibitors to suppress primitive leukemia cell populations). On the other hand, clinical efficacy of single-agent HDACis, such as SAHA, in AML patients remains modest,18,19 indicating that the ultimate role of HDACi for AML treatment may lie in combinatorial approaches.20
Scheme 1.

Chemical Structures of (A) Piperlongumine (PL), SAHA, and Hybrid Molecules 1–58, 3–35, 3–31, 3–98 and (B) 1–58/3–35 Analogues 27, 28, 35, and 36
Piperlongumine (PL, Scheme 1) is a natural product reported to selectively kill cancer cells over nonmalignant cells in vitro and in vivo.21 PL induces primary myeloid leukemia cell death22 and causes severe cytotoxicity against CD34+ AML cells from patient specimens, which may be attributed to its electrophilic GSH-depleting properties.23 Its selective toxicity to LSCs relative to hematopoietic stem and progenitor cells (EC50: 380 nM vs. 4900 nM) was also reported.24 By using a DNA-repair-deficient DT40 cell line as genotoxic model, PL was found to induce DNA double strand breaks (DSBs) and to suppress a homologous recombination (HR) DNA repair mechanism.25 PL-induced DNA damage (e.g., increased levels of 8-OHdG and DNA strand breaks) was also observed in pancreatic cancer cell lines and in the corresponding in vivo mouse model.26
Genetic alterations resulting in intrinsic DNA DSBs and defective DNA repair mechanisms have been reported in AML cells,27 which makes targeting genomic integrity and DNA damage response an attractive anti-AML strategy that can be therapeutically exploited to induce synthetic lethality in AML cells.28 Given the observations that PL is able to induce DNA DSBs and to inhibit the HR DNA repair mechanism25 and that HDACis directly cause DNA damage while interfering with DNA repair processes at multiple levels, such as down-regulating DNA repair proteins, inhibiting both HR and NHEJ repair,29 and disrupting cell cycle check points,9 we envisioned that HDACi and PL can cooperatively cause DNA damage in AML cells and synergistically induce AML cell death.
Clinical resistance to SAHA in leukemia patients has been correlated with overexpression of antioxidant defense enzymes,18 and cellular GSH-depleting agent phenethyl isothiocyanate (PEITC) was able to significantly enhance cytotoxicity of HDACi in drug-resistant AML cells and in primary leukemia cells.30 Because of the α,β-unsaturated imide pharmacophore, electrophilic PL could compromise GSH-related antioxidant capacity by depleting the cellular GSH pool via direct conjugation and/or inhibiting critical GSH-detoxification pathway enzymes, such as GSTP1.21,23 PL treatment has been shown to significantly reduce total GSH level and to subsequently induce cell death in CD34+ primitive AML cells.23 Therefore, besides the aforementioned DNA damage, electrophilic PL and PL analogues could also act synergistically with HDACis by interfering with cellular GSH levels and GSH regulatory pathways. It is noteworthy that attenuated GSH defense could also lead to elevated DNA damage,31,32 contributing to the suppression of AML cells.
In addition to coadministration, combined PL and HDACi activities could also be realized by constructing hybrid molecules that contain both pharmacophores (Scheme 1). This single-molecule “hybrid drug” approach more efficiently colocalizes PL and HDACi activities in the same cell and could greatly simplify the optimization of drug-like properties, PK, and dose-toxicity profiles33 at a later drug discovery and development stage. Because of complementary and overlapped anti-AML properties, we hypothesized that when combined together, either in a drug combination or hybridized in a single molecule, PL could potentiate HDACi activities leading to potent antileukemic effects. In the present study, we investigated in vitro anti-AML properties of PL and SAHA either in combination or in hybrid molecules (PL-HDACis). PL and SAHA combinatorial treatment synergistically induced apoptosis in phenotypically distinct AML cells, and this synergy is maintained by PL-HDACis, such as compounds 1–58 and 3–35 (Scheme 1). Similar to drug combination, prototype PL-HDACis displayed potent anti-AML activities, in part, by interfering with cellular GSH defense, inducing DNA damage (e.g., DSBs), suppressing DNA repair and pro-survival genes, and up-regulating the expression of pro-apoptotic genes in AML cells.
RESULTS AND DISCUSSION
1. Chemistry.
In order to incorporate both PL and HDACi activities into a single chemical entity, the prototype hybrid molecule 1–58 was designed by introducing a seven-carbon linker at the 4′ position of PL. The linker was further connected to the hydroxamic acid moiety, a zinc-binding group (ZBG), to mimic the partial structure of SAHA (Scheme 1). The modified PL structure served as a “cap” that could bind at the surface of HDAC enzymes. Two electrophilic olefins are present in the PL structure; published PL structure–activity relationship (SAR) studies suggested that the C2–C3 olefin was responsible for GSH conjugation via Michael addition and was vital for ROS induction. The C2 substituent could potentially affect the chemical reactivity of C2–C3 olefin and impact anticancer potency of PL. For instance, electron-withdrawing C2-chloride increases, while C2-methyl group decreases cytotoxicity of PL to cancer cells.34,35 After conjugation with cellular GSH, close interactions of PL-GSH conjugate with GSH-binding proteins may increase the availability of electrophilic C7–C8 olefin to protein thiol groups and therefore promote C7–C8 olefin-caused covalent protein modification, this process was also referred as cellular glutathionylation because of the attachment of PL-GSH conjugate to a cellular protein.35 Elimination of the C7–C8 double bond has been shown to reduce cytotoxicity,35 which might be a consequence of decreased PL covalent protein binding. To investigate how the C2 substituent and the C7–C8 olefin affect the overall anti-AML properties of 1–58, compounds 3–35 with a C2–Cl substituent and 3–31/3–98 (Scheme 1) of which the internal C7–C8 double bond was eliminated, were also synthesized and tested in cell culture models. Although just containing partial PL structure, we hypothesized that the C2–C3 olefin of 3–31 or 3–98 may still work cooperatively with the HDACi functional group to achieve improved anti-AML activities compared to parental drug, either SAHA or PL. Instead of having both HDACi and PL pharmacophores, compounds 27, 28, 35, and 36 are close structural analogues of 1–58 or 3–35 with just one active pharmacophore (Scheme 1), for example, ZBG of 1–58 or 3–35 was replaced by a methyl ester in 27 and 28, respectively, and the electrophilic C2–C3 olefin of 1–58 was blocked by either a C3-methyl substituent (compound 35) or by hydrogen saturation (compound 36). These compounds were used to make comparisons with 1–58 and 3–35 to demonstrate both PL and HDACi pharmacophores contribute to the observed superior anti-AML properties of PL-HDACis.
The synthesis of 1–58/3–35 and 3–31/3–98 scaffold were started from commercially available sinapinic acid 3 and syringic acid 5, respectively (Scheme 2). Carboxylic acids 3 and 5 were first converted to corresponding methyl esters 4 and 6 followed by the alkylation of phenol hydroxyl groups with compound 2, a t-butyl ester of 7-bromoheptanoic acid, to afford 7 and 8, respectively. Free carboxylic acids 9 and 10 were obtained via basic hydrolysis of methyl esters 7 and 8, while the t-butyl esters of these molecules were intact. In order to conduct SAR studies of PL-like pharmacophores, α002Cβ-unsaturated lactams 11,36 1234 which have a C2–Cl substituent, and 1337 with a C3-methyl group were synthesized according to published procedures. Compound 14, δ-valerolactam, was purchased. Different lactams 11–14 were deprotonated by n-BuLi and then coupled with pivaloyl chloride or oxalyl chloride-activated 9 or 10, respectively, to afford 15–20. In following steps, t-butyl esters were cleaved using TFA, and the obtained acids 21–26 were coupled to THP-protected hydroxyl amine. Final acidic deprotection using TFA in MeOH gave hydroxamic acid-containing 1–58, 3–35, 3–31, 3–98, 35, and 36. Methyl esters 27 and 28 were prepared from 21 and 22 by using diazomethane as esterification reagent, respectively. Synthetic intermediates and final products were characterized using 1H, 13C NMR and mass spectrometry; for the biological studies, purity of the compounds was further confirmed using HPLC analysis.
Scheme 2. Synthesis of PL-HDACi Hybrid Molecules 1–58, 3–35, 3–31, 3–98 and Related Analogues 27, 28, 35, 36a.

aReagents and conditions: (a) (BOC)2O, DMAP, t-BuOH, THF; (b) concentrated sulfuric acid, MeOH, reflux; (c) K2CO3, DMF, 70 °C; (d) LiOH, THF, H2O; (e) (1) pivaloyl chloride, TEA, THF, −20 °C; (2) lactam, n-BuLi, THF, −78 °C; (f) (1) oxalyl chloride, THF, −20 °C; (2) lactam, n-BuLi, THF, −78 °C (g) 20% TFA, DCM; (h) diazomethane, DCM; (i) HBTU, NH2OTHP, DIPEA, DMF; (j)10% TFA, MeOH.
2. Antileukemic Interactions between SAHA and PL.
To investigate if PL could enhance the anti-AML activity of SAHA, U937 (p53 null) and MOLM-13 (p53 wt, FLT3-ITD) cells were treated with either SAHA or PL, alone or in combination, at the indicated concentrations for 24 h. The presence of FLT3/ITD in MOLM-13 cells represents an aggressive AML disease phenotype.38 After drug treatment, cell death was assessed by Annexin V–FITC/PI (propidium iodide) staining and flow cytometry analysis. In U937 cells, the combination of SAHA with PL at 2 μM and 4 μM (1:1 ratio), respectively, resulted in significant induction of cell death compared to individual drug treatment (Figure 1A). Furthermore, for U937 cells the interaction of PL and SAHA was synergistic, as determined by calculating CI (combination Index) values (CI < 0.9), while the interaction observed in MOLM-13 cells was additive (0.9 ≤ CI ≤ 1.1, Figure 1A). Drug-induced cell death was accompanied by cleavage of PARP-1 and caspase-3, demonstrating that the cells underwent apoptosis (Figure 1B). The synergistic to additive antileukemic interactions between PL and SAHA provide a compelling rationale to construct PL-HDACi hybrid molecules against AML cells.
Figure 1.

Antileukemic activity of PL-HDACi 1–58 is similar to that of combined SAHA and PL. (A) U937 and MOLM-13 cells were treated with SAHA or PL, alone or in combination, for 24 h and then subjected to Annexin V/PI staining and flow cytometry analyses. *indicates p < 0.05, **indicates p < 0.005, and ***indicates p < 0.0005. CI values were calculated using CompuSyn software. (B) U937 cells were treated with SAHA and PL alone and in combination for 24 h. Whole cell lysates were subjected to Western blotting and probed with the indicated antibody. (C) U937 and MOLM-13 cells were treated with 1–58 for 24 h and then subjected to Annexin V/PI staining and flow cytometry analyses. ***indicates p < 0.0005. (D) U937 cells were treated with 1–58 for 24 h. Whole cell lysates were subjected to Western blotting and probed with the indicated antibody. (E) EC50s, calculated from the flow data presented in panels A and C, are graphed as median ± SEM for 1–58, SAHA, PL, and the combination of SAHA and PL (1:1) in U937 and MOLM-13 cells. EC50 values were calculated as drug concentrations necessary to induce 50% Annexin V positive cells compared to vehicle control treated cells. ***indicates p < 0.0005.
3. Antileukemic Activity of the PL-HDACi Hybrid 1–58 in AML Cell Lines.
On the basis of favorable anti-AML interactions between PL and SAHA, we explored a “hybrid drug” approach to discover a single chemical entity possessing both HDACi and PL-related activities. In both U937 and MOLM-13 cell lines, the prototype hybrid compound 1–58 triggered concentration-dependent apoptotic cell death, as indicated by a significant increase of Annexin V-positive cells (Figure 1C) and cleavage of PARP and caspase-3 (Figure 1D). As shown in Table 1, apoptosis-inducing activity of 1–58 recapitulated those of the PL and SAHA combinations (1:1 ratio) at all tested concentrations (1, 2, 4 μM). EC50s of 1–58 and the combination of PL and SAHA were also very similar, significantly lower than those of single agents, indicating improved anti-AML potency (Figure 1E). 1–58 was further tested in other AML cell lines representing various disease subtype and status, for instance, in pediatric AML cell lines CMS, CMK, and CMY and in Ara-C-resistant HL60/ARA-C and CMK/ARA-C cells (Ara-C IC50s > 8 μM in CMY, HL60/ARA-C, and CMK/ARA-C cells, unpublished data). The MTT assay was conducted (72h treatment), and IC50s of 1–58 were submicromolar for the tested cell lines and were equivalent or substantially lower than combined PL and SAHA treatment (Table 2), even in the Ara-C-resistant cell lines (Table 2). Collectively, 1–58 was effective against Ara-C-resistant AML cells and showed a broad spectrum of anti-AML activities regardless of many disease-specific molecular characteristics, such as p53 and FLT3-ITD mutational status.
Table 1.
Similar Levels of Apoptosis Induced by 1–58 and PL+SAHA in AML Cell Linesa
| 1–58 | SAHA plus PL (1:1, concn of each drug) | |||||
|---|---|---|---|---|---|---|
| cells | 1 μM | 2 μM | 4 μM | 1 μM | 2 μM | 4 μM |
| U937 | 13.7% | 56.9% | 88.5% | 6.2% | 62.5% | 81.1% |
| MOLM-13 | 24.6% | 65.9% | 80.3% | 8.0% | 53.6% | 77.5% |
Apoptosis levels are presented as mean % Annexin V+ cells of triplicates from one representative experiment.
Table 2.
Antileukemic Activities of SAHA, PL, SAHA+PL, and 1–58, a PL-HDACi Hybrid Drug, in AML Cell Linesa
| cell line | SAHA | PL | SAHA+PL (1:1) | 1–58 |
|---|---|---|---|---|
| OCI-AML3 | 1365.0 ± 56.3 | 1868.0 ± 118.7 | 1109.0 ± 40.7 | 615.0 ± 67.8 |
| CMS | 1021.0 ± 86.9 | 3097.0 ± 176.3 | 1037.0 ± 24.1 | 384.6 ± 44.3 |
| CMK | 439.3 ± 53.8 | 2815.0 ± 198.9 | 612.7 ± 34.0 | 255.8 ± 23.9 |
| CMY | 553.3 ± 39.8 | 1620.0 ± 143.6 | 442.4 ± 31.3 | 184.7 ± 15.9 |
| U937 | 907.5 ± 120.8 | 2174.0 ± 384.6 | 767.0 ± 61.2 | 498.2 ± 106.9 |
| MV4–11 | 496.1 ± 23.9 | 1439.0 ± 114.7 | 289.1 ± 5.3 | 229.8 ± 40.72 |
| MOLM-13 | 984.9 ± 25.8 | 1838.0 ± 194.7 | 934.5 ± 37.1 | 454.2 ± 77.8 |
| HL60 | 570.5 ± 27.3 | 1778.0 ± 140.5 | 529.2 ± 29.0 | 357.2 ± 44.0 |
| HL60/ARA-C | 1331.0 ± 144.9 | 2250.0 ± 109.8 | 1128.0 ± 119.6 | 692.1 ± 51.1 |
| CMK/ARA-C | 490.2 ± 44.3 | 1911.0 ± 206.4 | 421.8 ± 13.7 | 214.9 ± 15.1 |
The numbers represent IC50 values (nM) ± SEM. IC50s were measured using the MTT assay after 72 h treatment.
4. Structure–Activity Relationships (SAR) of 1–58.
4.1. Cellular HDACi Activities.
Designed as multifunctional anticancer agents, hybrid molecules were expected to retain HDACi activities. Since HDACs remove acetyl groups from lysine residues of histone and nonhistone proteins, the acetylation status of nuclear histone H4 and cytosolic α-tubulin have been frequently used as markers of cellular HDACi activity on nuclear HDACs and on cytosolic HDAC6 respectively. In our experiments, U937 cells were treated with 1 μM PL-HDACis and related analogues for 4 h and cell lysates were analyzed by Western blotting. Like SAHA (the positive control), hybrid molecules 1–58, 3–35, 3–31, and 3–98 induced hyper-acetylation of histone H4 and α-tubulin (Figure 2), indicating strong pan-HDACi activity. Comparisons between 1–58/3–35 and 3–31/3–98 demonstrated that the absence of C7–C8 olefin in 3–31 and 3–98 did not change cellular HDACi efficacy in U937 cells. As expected, PL itself did not provoke an increase and had no effect on SAHA-induced acetylation of H4 and α-tubulin. Likewise, 27 and 28, which lack the hydroxamic acid functional group, did not affect acetylation of H4 and α-tubulin. The cellular HDACi activity of compound 35 with C3-methyl group was better than that of compound 36 in which C2–C3 olefin was saturated. Compared to 1–58, structural modification of the 5,6-dihydro-2(1H)-pyridinone ring of PL, by introducing a C3-methyl group (compound 35) or saturation of C2–C3 olefin (compound 36), decreased cellular HDACi activities. On the contrary, C2–Cl substitution and elimination of C7–C8 olefin did not. These results suggested that the PL or certain partial PL scaffold was an acceptable “cap” group to render effective HDACi activities to the hybrid molecules.
Figure 2.

PL-HDACis 1–58, 3–35, 3–31, and 3–98 demonstrate HDACi activity. U937 cells were treated with 1 μM of SAHA, PL, PL +SAHA, 1–58, 3–35, 3–31, 3–98, 35, 36, 27, or 28 for 4 h. Whole cell lysates were subjected to Western blotting. Western blots were probed with anti–Ac-H4 (acetyl-histone H4), -H4, -Ac-Tubulin, or β-actin antibody.
HDACs are critical regulators of gene expression by changing acetylation status of histones and modifying chromatin structure. In the following studies, we further demonstrated that the HDACi function in drug combination or in the chimeric molecules was responsible for the modulation of DNA repair and apoptotic pathways in AML cells and resulted in improved anti-AML effects.
4.2. Impact of HDACi and PL Moieties.
First, to clarify the contributions of HDACi and PL moieties to the overall anti-AML effects of 1–58, close structural analogues, i.e. compounds 27, 35 and 36, were tested in U937 cells. Hydroxamic acid, the ZBG of 1–58, was replaced by a methyl ester in 27 to eliminate HDACi activity. On the other hand, compounds 35 and 36 retained the ZBG but the electrophilic C2–C3 olefin of PL was blocked by using C3-methyl substituent in compound 35 or via hydrogen saturation in compound 36. In contrast to 1–58, 27 did not induce apoptosis in U937 cells (Figure 3A). Without an active C2–C3 olefin, compounds 35 and 36 also failed to induce significant apoptosis (Figure 3A), which was likely due to reduced cellular HDACi activity compared to 1–58 (Figure 2) and the lack of C2–C3 olefin-provoked PL-like cellular activities, such as GSH conjugation and/or protein alkylation. The impact of functional C2–C3 olefin can also be emphasized by the comparisons of cellular HDACi potency and the apoptotic EC50s of 1–58 and SAHA: although the two compounds caused similar histone H4 and α-tubulin hyperacetylation, a reflection of close cellular HDACi activity (Figure 2), the apoptotic EC50s of 1–58 in AML cells are significantly lower than those of SAHA (Figure 1E). Nevertheless, these results suggest that the integration of intact HDACi and PL moieties into one chemical entity is necessary for the observed superior anti-AML effects, blocking either pharmacophore in close structural analogues significantly decreased their apoptosis induction activity. Once they have entered AML cells, 1–58 may have a dual action mode: some drug molecules act as HDAC inhibitors, and others may induce PL-like effects. Synergism of HDACi and PL structural components is reflected by the dramatically enhanced apoptotic effects of 1–58 compared to PL or SAHA alone (Figure 1A).
Figure 3.

Structure–activity relationship: effects of C2–Cl substitution and elimination of the internal C7–C8 olefin on the anti-AML potency of PL-HDACis. (A) U937 cells were treated with 1–58, 3–35, 3–31, 3–98, 28, 27, 35, or 36 for 24 h and then subjected to Annexin V/PI staining and flow cytometry analyses. (B) U937 cells were treated with 3–35 for 24 h. Whole cell lysates were subjected to Western blotting. (C) AML patient samples AML#91 and AML#92 were treated with 3–35 for 24 h and then subjected to Annexin V/PI staining and flow cytometry analyses. (D) AML#91 and AML#92 were treated with 3–35 for 24 h and then whole cell lysates were subjected to Western blotting.
Next we examined if the chemical reactivity of the C2–C3 olefin plays a role in the biological properties of hybrid molecules. C2 chloro-substituted PL was reported to be more cytotoxic to cancer cells and a C2-chloro PL analogue was well-tolerated in an in vivo lung cancer mouse model.34 To test if the introduction of an electron-withdrawing group at C2 position could increase electrophilicity of the C2–C3 olefin and therefore results in more potent PL-HDACis, 3–35 was designed as an analogue of 1–58 with a C2-chloro substituent. We found that 3–35 was much more potent than 1–58 in inducing apoptosis in U937 AML cells. Although 29.9% and 49.2% apoptosis were detected with 0.25 and 0.5 μM 3–35 treatments, respectively, 1–58 was not active at these concentrations (Figure 3A). The cooperative actions of PL and HDACi moieties was also observed by comparing compounds 3–35 and 28: 28 is a 3–35 analogue with a methyl ester instead of a ZBG which is critical for HDAC inhibition. At 1 μM, 28 induced 29.7% apoptosis, whereas 3–35, having both C2-chloro PL and HDACi functionalities, induced 88.0% apoptosis. Although 28 is a potent apoptosis inducer at 2 μM, its effect was still inferior to that of 3–35, which caused almost 100% apoptotic cell death (Figure 3A). Furthermore, 3–35 treatment resulted in concentration-dependent apoptosis in two primary AML patient samples (Figure 3C). Again, 3–35-induced apoptosis in U937 cells and in primary AML patient samples was further verified by the detections of cleavage of PARP-1 and caspase-3 via Western blots (Figure 3B,D). These results demonstrate that introducing an electron-withdrawing group at the C2 position of PL enhanced the potency of hybrid molecules, which could be attributed to the electron-deficiency of C2–C3 olefin that facilitates the conjugation with cellular nucleophiles.
We then looked at the effects of eliminating the C7–C8 olefin of PL. 3–31 and 3–98 were synthesized as 1–58 and 3–35 analogues without the C7–C8 double bond. It has been reported that disrupting the electrophilicity/reactivity of the C7–C8 olefin by saturation, steric blockade, or cyclization into a heterocycle could substantially diminish PL’s cytotoxicity and covalent modification of GSH-binding proteins.35 Interestingly, these modifications did not interfere with PL’s ROS production and GSH conjugation properties as long as C2–C3 olefin was intact. Indeed, although 3–31/3–98 had similar cellular HDACi activities (Figure 2) as 1–58/3–35, they induced less apoptotic cell death than 1–58/3–35 at 2 μM, likely due to the lack of the C7–C8 olefin (Figure 3A). It is noteworthy that 3–31 and 3–98 are still more active than SAHA or PL at inducing U937 cell death. After 24 h drug treatment at 2 μM, SAHA induced 25% apoptosis (Figure 1A), whereas 3–31 and 3–98 induced approximately 40% (Figure 3A). Because 3–31 and 3–98 showed similar cellular HDAC inhibition effects as SAHA (Figure 2), the increased cytotoxic effects are due to the cellular actions of the remaining PL-related structures (i.e., the imide bond and the dihydropyridinone ring). Interestingly, 28 consistently outperformed 3–98 at 1 and 2 μM, demonstrating that the presence of C7–C8 olefin, which is related to covalent protein glutathionylation (i.e., cross-linking of drug-GSH conjugate with cellular proteins35), contributed more to the overall cytotoxicity of 3–35 than HDAC inhibition. In contrast, without the C2-chloro substituent, the HDACi functional group became more critical than the internal C7–C8 olefin: removal of ZBG from 1–58 resulted in a complete loss of apoptotic effect, as demonstrated by compound 27. The C2-chloro substituent seems to rely on C7–C8 olefin to enhance cytotoxicity of PL-HDACis: without C7–C8 olefin, cellular apoptotic effects of 3–31 and 3–98 were comparable; on the contrary, in the presence of C7–C8 olefin, 3–35 with C2-chloro substituent was much more potent than 1–58 (Scheme 3A).
4.3. Cytotoxicity vs Selectivity.
Because both cytotoxicity and selectivity (i.e., sparing noncancer cells) are important for an anticancer compound, we further evaluated PL, SAHA, PL plus SAHA, and hybrid compounds 1–58, 3–31, 3–35, and 3–98 in noncancer cells. PL has been shown to be nontoxic to MCF-10A, a noncancerous breast epithelial cell line, at concentrations up to 15 μM in 24 h treatment,21 which makes it an interesting model for comparison. Although normal human blood cells would be more relevant to leukemia treatment, because of limited availability of clinical samples and the sensitivities of a few triple negative breast cancer cell lines to 1–58, for example, 1–58 showed IC50 of 382.4 ± 12.9 nM in MDA-MB-231 cells (MTT assay, activities of PL-HDACis in solid tumor cell lines will be described separately), we started our selectivity evaluation using MCF-10A cells to rank order drug candidates for further studies. Both MCF-10A and U937 cells were treated with escalating concentrations (0.5–5 μM) of PL, SAHA, PL plus SAHA (1:1), 1–58, 3–31, 3–35, and 3–98 for 24 h and viable cells were measured using MTT assays. As expected, SAHA and PL were well-tolerated in MCF-10A cells in 24 h treatments (Figure S1). Interestingly, although 1–58 and PL+SAHA (1:1) had similar effects in U937 cells, 1–58 had little to no effect on MCF-10A cells, suggesting that 1–58 was more selective than PL+SAHA (Figure 4A). Similar to 1–58, 3–31 and 3–98 also showed selectivity between the two cell lines (Figure 4B). Strikingly different than the previous compounds, 3–35 caused a significant reduction of viable cells in both MCF-10A and U937 cells (Figure 4C).
Figure 4.

Selectivity of PL-HDACis in U937 and MCF-10A cells. (A–C) U937 (AML cell line) or MCF-10A (normal breast cell line) cells were treated with PL+SAHA, 1–58, 3–98, 3–31, or 3–35 at indicated concentrations (0.5–5 μM) for 24 h. Viable cells were determined using MTT reagent. Cell viabilities were graphed as mean ± SD of four to six independent experiments. Comparisons were made between the two cell lines, the unpaired t test (one-tailed) was conducted using GraphPad Prism 5.0. The significant level was set as 0.05, * represents P < 0.05, ** represents P < 0.005, *** represents P < 0.0005. (D) U937 cells were pretreated with NAC (3 mM) or vehicle for 3h, washed with PBS and followed by the treatment of SAHA, PL, SAHA+PL, 1–58 or 3–35 for 24 h. The cells were then subjected to Annexin V/PI staining and flow cytometry analyses. Vehicle treatment (DMSO < 0.1%, v/v) was used as control in each experiment.
Distinct from 1–58, the C2–C3 olefin of 3–35 is more electron deficient and we envisioned that the increased electrophilicity of 3–35 was the major reason leading to the nonselective cytotoxicity. To test this hypothesis, we incubated 3–35 and 1–58 (50 μM) with and without GSH (5 mM) in phosphate buffer (pH 7.4) at 37 °C up to 4h, and analyzed the incubation mixture by HPLC. Both compounds remained intact in the absence of GSH; however, whereas 1–58 was still stable in the presence of GSH, 3–35 was consumed and gave new peaks (Figure S2A). Interestingly, we also observed reaction of 1–58 and GSH in the presence of U937 cell lysate (Figure S2B). These results suggest that 3–35 may efficiently cause cellular GSH conjugation and trigger the subsequent massive protein glutathionylation which is responsible for the non-selective toxicity. In support of this assumption, we observed that 3–98 was still able to react with GSH in the absence of cell lysate (Figure S2A), however, due to the lack of the glutathionylation-required C7–C8 olefin, it was tolerable in MCF-10A cells (Figure 4C). On the other hand, 1–58-caused GSH conjugation/glutathionylation may occur at a lower extent compared to 3–35, either due to the relatively low intrinsic chemical reactivity of C2–C3 olefin, or because the GSH conjugation caused by 1–58 might be an enzymatic process and therefore could be affected by the distinct expression levels of the responsible enzyme(s) (such as certain GST enzyme) in U937 and in MCF-10A cells. Further studies (such as comparing drug-induced cellular glutathionylations etc.) are needed to understand the more detailed mechanisms of the selectivity for PL-HDACi hybrid molecules. Nonetheless, the comparison of 1–58 and 3–35 in U937 and in MCF-10A cells suggest that 1–58 is more suitable than 3–35 for further research.
To build a connection between cellular GSH consumption and apoptosis induction, we conducted an N-acetyl cysteine (NAC) rescue experiment. Preincubation of NAC (3 mM), a well-known GSH precursor and antioxidant, with U937 cells was conducted (3 h, followed by PBS wash to remove excess NAC in culture media) to increase intracellular free thiol/GSH levels. We found that NAC rescued U937 cells from SAHA, PL, SAHA plus PL, 1–58 or 3–35-induced apoptosis at indicated concentrations (Figure 4D). The result emphasized the role of GSH consumption and/or ROS induction played in the overall apoptotic effects. It is interesting to note that compared to the partial rescue observed in SAHA-related treatments (SAHA or SAHA+PL), NAC completely rescued U937 cells from 1–58 and 3–35 treatments. This observation could be explained by the conjugation between PL-HDACis and cellular GSH/NAC: once the PL moiety is conjugated, the resulted bulky and highly polar conjugate cannot serve as an effective “cap” group binding at the surface of HDAC enzymes, which leads to a complete inactivation of both PL and HDACi activities. In the case of SAHA, because of the pleiotropic anticancer characteristics,6 although its ROS induction/GSH consumption effects could be neutralized by NAC treatment, SAHA can still elevate apoptosis levels through other pathways.
5. DNA Damage Caused by PL and HDACi Treatments in U937 Cells.
Because both PL25,26 and HDACis, such as SAHA,7 are able to induce DNA damage and inhibit DNA repair mechanisms, we investigated their impact on the integrity of DNA in U937 cells. We first treated U937 cells with PL and SAHA, alone or in combination, for 20 h and then assessed DNA damage using alkaline comet assay. The alkaline comet assay is a single cell gel electrophoresis assay to analyze DNA damage ranging from DNA single strand breaks, DSBs, and alkali-labile sites in the DNA.39 This assay allows the direct observation of the “comet tail” which is correlated with the damaged DNA content. In our experiment, PL (2 μM) and SAHA (2 μM) combination caused more prominent DNA damage than each drug alone (Figure 5A). At 2 μM, although the damaged DNA via SAHA or PL treatments were undetectable, their combinatorial effect was significant; moreover, the DNA content in the tail induced by 1–58 (2 μM) treatment was comparable to that of combined SAHA and PL (Figure 5B).
Figure 5.

Combination of SAHA+PL and the hybrid compound 1–58 induce DNA strand breaks. (A) U937 cells were treated with SAHA, PL, SAHA+PL, or 1–58 for 20 h and then subjected to alkaline comet assay analysis. Representative images are shown. (B) Comet assay results are graphed as median percent DNA in the tail from 4 replicate gels ± s.e.m. *indicates p < 0.05 and **indicates p < 0.005. (C) U937 cells were treated with SAHA, PL, SAHA+PL, or 1–58 for 24 h and then whole cell lysates were subjected to Western blotting. The fold changes for the γH2AX, CHK1, and RAD51 densitometry measurements, normalized to β-actin, and then compared to no drug treatment control, are indicated. (D) U937 cells were treated with SAHA, PL, SAHA+PL, or 1–58 for 24 h, and then whole cell lysates were subjected to Western blotting. The fold changes for the Bim, and XIAP densitometry measurements, normalized to β-actin, and then compared to no drug treatment control, are indicated.
To further confirm drug-induced DNA damage and to exploit the mechanisms behind the increased DNA-damaging effects, protein levels of γH2AX and a few DNA repair-related proteins, CHK1 and Rad51, were analyzed by Western blots. As shown in Figure 5C, SAHA (2 μM) treatment resulted in increased expression of γH2AX, an established biomarker of DNA double-strand breaks.40 Although PL itself did not cause increased γH2AX, it substantially potentiated SAHA’s effects (2.5-fold) and resulted in a 4.1-fold increase of γH2AX in combinatorial treatment. Compound 1–58, which has both HDACi and PL-related properties, acted comparably to the drug combination and induced a 4.5-fold increase of γH2AX. Furthermore, Rad51, an essential protein in the HR DNA repair pathway,41 was up-regulated 1.4-fold by PL (2 μM), which could be a cellular adaptive reaction to the PL-induced DNA damage. Strikingly, this increase was abolished by combination with SAHA or via the hybrid compound 1–58 (Figure 5C). Meanwhile, the protein levels of CHK1, a checkpoint regulator that participates in multiple pathways to protect cells from DNA damages,42 were also reduced by HDACi treatments to 0.6, 0.8 and 0.7-fold in SAHA, SAHA plus PL and 1–58 treatments, respectively. Our experiments suggest that 1–58 treatment-associated CHK1 and/or Rad51 reduction was related to its HDACi activities. Current literature gave a few possibilities to understand the observed CHK1/Rad 51 downregulation, such as HDACi-induced downregulation of E2F1 transcriptional factor14 and HDAC inhibition-mediated induction of microRNA miR-182 which was linked to time- and dose-dependent decreases in the levels of Rad51.43 Nonetheless, the reduction of Rad51 and CHK1 contribute to the observed significant DNA damage induced by SAHA and PL combination and their hybrid molecule 1–58.
6. Apoptotic Proteins Affected by PL-HDACi Hybrid Molecule.
The balance between the expression/activity of the pro- and antiapoptotic pathways could ultimately determine the fate of a given cell. It is well-established that HDACis can induce apoptosis by up-regulating pro-apoptotic proteins (e.g., Bim, Bax, Puma, Noxa, etc.) and down-regulating pro-survival proteins (e.g., survivin, XIAP).44 Among the Bcl-2 and IAP (inhibitors of apoptosis) families, our Western blotting analysis revealed up-regulation of pro-apoptotic Bim (EL isoform, 1.8 fold) and down-regulation of antiapoptotic protein XIAP (to 0.6 fold) by 1–58 (Figure 5D). Because PL itself did not change the expression of Bim and XIAP (Figure 5D), the observed modulations were mainly attributed to the retained HDACi activity of 1–58. PL and SAHA combination, as well as SAHA alone, also showed similar effects.
BH3-only protein Bim plays a role in initiating apoptosis and has been shown to be up-regulated by different HDACis, for instance, by belinostat in AML and acute lymphoblastic leukemia cells;45 HDACi-mediated transcriptional activation of Bim may occur through histone H4 hyperacetylation within the gene promoter region.46 Similarly, the expression of short-lived antiapoptotic proteins XIAP can also be modulated by HDACis; our results are consistent with a previous report which demonstrated that XIAP proteins were reduced by SAHA in U937 cells.47
In our experiments, HDACi activities of SAHA, in the PL plus SAHA drug combination, or PL-HDACi hybrid modulated the expression of proteins involved in DNA repair and apoptotic pathways, which collectively favored enhanced DNA damage and apoptotic cell death. The ultimate contribution of each protein can be further evaluated using ectopic overexpression or shRNA knockdown methodologies in future studies.
CONCLUSIONS
We found that PL and HDACi SAHA, either used in combination or hybridized into a hybrid molecule, synergistically caused apoptotic cell death of AML cells. The IC50s of 1–58 (a prototype PL-HDACi) in multiple AML cell lines, including aggressive and chemotherapy-resistant cell models, are significantly lower than those of PL, SAHA, and their combination (1:1 ratio). SAR study of 1–58 suggested that both PL and HDACi pharmacophores are required for superior anti-AML activities. Introducing electron-withdrawing C2–Cl substituent to 1–58 yielded 3–35 with increased cytotoxicity but decreased selectivity in a MCF-10A noncancerous cell culture model; eliminating C7–C8 olefin of PL obtained 3–31/3–98 scaffold, which are still more active than PL or SAHA alone against U937 cells. On the contrary to 3–35, compounds 1–58, 3–31 and 3–98 were well-tolerated by MCF-10A cells, suggesting better selectivity of these compounds. The difference of 3–35 and 1–58 in AML cells and in MCF-10A cells could be partially attributed to their distinct reactivity to cellular thiol components, such as GSH. Pretreatment with NAC, the precursor of GSH biosynthesis and an antioxidant/thiol-based nucleophile, rescued U937 cells from drug-induced cytotoxicity, which emphasized the critical role of GSH conjugation in the overall antiproliferative effects of drug combinations and hybrid molecules. Revealed by comet assays and Western blots, PL and SAHA demonstrated their ability to cooperatively induce DNA damage in U937 cells, either in combination or in hybrid. The hyperacetylation patterns of histone H4 and cytosolic α-tubulin suggested that PL-HDACis 1–58, 3–35, 3–31, and 3–98 retained SAHA comparable pan-HDACi activities. The HDACi functions of drug combination or 1–58 were responsible for the modulated expression of proteins in DNA repair and apoptotic pathways (e.g., downregulation of DNA repair-related CHK1 and Rad51, upregulation of pro-apoptotic Bim, and suppression of pro-survival XIAP). These effects collectively favor DNA damage and apoptotic cell death. Our current results demonstrate that PL and HDACi combination or PL-HDACi hybrids are potent antileukemic agents against AML, they act in part, by interfering cellular GSH defense, suppressing expression of DNA repair and pro-survival genes, and inducing expression of pro-apoptotic genes. Prototype hybrid compounds in this study could be a promising starting point to discover more potent and cancer-cell-selective PL-HDACis against hematological malignancies.
EXPERIMENTAL SECTION
General Methods for Chemistry.
1H and 13C NMR spectra were obtained using Varian Mercury 600 MHz and Advance 400 MHz spectrometers. Chemical shifts are reported as δ values in parts per million (ppm) relative to tetramethylsilane (TMS) for all recorded NMR spectra. All reagents and solvents were obtained commercially from Acros, Aldrich, or Fisher and were used without purification. Flash column chromatography was performed over 200–300 mesh silica gel. High-resolution mass spectral data were collected using a LCT Premier XE KD128 instrument. All compounds submitted for biological testing were found to be at least 95% pure by analytical HPLC. Purity check and HPLC analysis of incubations were conducted using a Phenomenex (LunaR) C18, 3.5 μm, 100 mm × 3.0 mm column on a Shimadzu UFLC instrument. HPLC parameters were: solvent A, water containing 0.1% formic acid (FA) and 10% (v/v) CH3OH; solvent B, 0.1% FA in CH3CN; flow rate 0.5 mL/min; gradient, t = 0 min, 10% B; t = 1 min, 10% B; t = 18 min, 80% B; t = 20 min, 80% B; t = 21 min, 10% B.
tert-Butyl 7-Bromoheptanoate (2).
7-Bromoheptanoic acid (1.11 g, 5.3 mmol, 1 equiv) and Boc anhydride (1.38g, 6.3 mmol, 1.2 equiv) were mixed in tert-butyl alcohol (5 mL). After the mixture was stirred at room temperature for 10 min, DMAP (195 mg, 1.6 mmol, 0.3 equiv) was added, and the reaction mixture was stirred for another 2 h. Upon completion, the solvent was removed under reduced pressure, and the residue was redissolved in ethyl acetate, washed with water and brine, and dried over anhydrous Na2SO4. Purification by flash chromatography (Hex/EA, 20/1) afforded product (colorless oil, 570 mg, 40%). 1H NMR (600 MHz, CDCl3): δ 3.38 (t, J = 6.6 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 1.82–1.86 (m, 2H), 1.55–1.60 (m, 2H), 1.40–1.45 (m, 2H), 1.42 (s, 9H), 1.29–1.34 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 173.06, 80.01, 35.39, 33.81, 32.55, 28.16, 28.09, 27.81, 24.83.
(E)-Methyl 3-(4-Hydroxy-3,5-dimethoxyphenyl)acrylate (4).
Sinapinic acid (1.03 g, 4.6 mmol, 1 equiv) was dissolved in methanol (20 mL) followed by addition of several drops of concd H2SO4. The reaction mixture was refluxed for 6 h. Upon completion, the reaction mixture was cooled to ambient temperature, and solvent was removed under vacuum. The resulting residue was diluted with ethyl acetate and washed using sat. aq NaHCO3, water, and brine, and dried over anhydrous Na2SO4. Purification by flash chromatography (Hex/EA, 2:1) afforded product (yellow oil, 1.11g, quantitative yield). 1H NMR (600 MHz, CDCl3): δ 7.55 (d, J = 15.6 Hz, 1H), 6.72 (s, 2H), 6.26 (d, J = 15.6 Hz, 1H), 5.79 (s, 1H), 3.89 (s, 6H), 3.76(s, 3H). 13C NMR (125 MHz, CDCl3): δ 167.61, 147.18, 145.15, 137.13, 125.77, 115.41, 105.00, 56.27, 51.62.
Methyl 3,5-Dimethoxy-4-hydroxybenzoate (6).
Prepared from syringic acid using a similar procedure described for compound 4 (quantitative yield). 1H NMR (600 MHz, CDCl3): δ 7.32 (s, 2H), 6.04 (s, 1H), 3.93 (s, 6H), 3.90 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 166.85, 146.59, 139.16, 120.98, 106.57, 56.36, 52.07.
(E)-tert-Butyl 7-(2,6-Dimethoxy-4-(3-methoxy-3-oxoprop-1-enyl)-phenoxy)heptanoate (7).
Compound 2 (570 mg, 2.1 mmol, 1.1 equiv), compound 4 (476 mg, 2 mmol, 1 equiv), and K2CO3 (838 mg, 6 mmol, 3 equiv) were mixed in DMF (4 mL). The reaction mixture was stirred at 60 °C overnight. The reaction mixture was cooled to room temperature and diluted with water. After extraction using ethyl acetate three times, the combined organic phase was washed with water and brine, and dried over anhydrous Na2SO4. Purification by flash chromatography (Hex/EA, 6:1) afforded product compound 7 (colorless oil, 630 mg, 75%). 1H NMR (600 MHz, CDCl3): δ 7.55 (d, J = 15.6 Hz, 1H), 6.69 (s, 2H), 6.28 (d, J = 15.6 Hz, 1H), 3.94 (t, J = 6.6 Hz, 2H), 3.81(s, 6H), 3.75 (s, 3H), 2.16 (t, J = 7.2 Hz, 2H), 1.67–1.72 (m, 2H), 1.53–1.68(m, 2H), 1.41–1.46(m, 2H), 1.38 (s, 9H), 1.28–1.33(m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.16, 167.36, 153.60, 144.89, 139.40, 129.57, 116.76, 105.23, 79.84, 73.41, 56.07, 51.61, 35.49, 29.86, 28.80, 28.04, 25.47, 25.01. HRMS(ESI): calcd for [C23H34O7+H]+, 423.2383; found, 423.2404.
Methyl 4-(7-tert-Butoxy-7-oxoheptyloxy)-3,5-dimethoxybenzoate (8).
A flask was charged with compound 6 (1.76 g, 8.23 mmol) and compound 2 (2.4 g, 9.06 mmol). DMF (10 mL) and potassium carbonate (3.41 g, 24.70 mmol) were added at room temperature. The reaction was allowed to undergo at 80 °C overnight. Once completed, the resulting mixture was washed with water and extracted with DCM three times, and then the organic phase was dried over Na2SO4 for 30 min. Purification done by flash chromatography (Hex/EA, 8:1) produces compound 8 (colorless oil, 3.7 g, 94%). 1H NMR (600 MHz, CDCl3): δ 7.26 (s, 2H), 3.99 (t, J = 7.2 Hz, 2H), 3.87 (s, 3H), 3.86 (s, 6H), 2.18 (t, 7.2 Hz, 2H), 1.70–1.75 (m, 2H), 1.55–1.60 (m, 2H), 1.42–1.47 (m, 2H), 1.41 (s, 9H), 1.31–1.36 (m, 2H).13C NMR (125 MHz, CDCl3): δ 173.20, 166.75, 153.12, 141.43, 124.88, 106.74, 79.88, 73.37, 56.16, 52.16, 35.52, 29.89, 28.82, 28.08, 25.48, 25.03. HRMS (ESI): calcd. for [C21H32O7+Na]+, 419.2046; found, 419.2046.
(E)-3-(4-(7-tert-Butoxy-7-oxoheptyloxy)-3,5-dimethoxyphenyl)-acrylic Acid (9).
Compound 7 (588 mg, 1.4 mmol, 1 equiv) was dissolved in THF/H2O (5 mL/5 mL) followed by the addition of LiOH (40 mg, 1.7 mmol, 1.2 equiv). The reaction mixture was stirred at room temperature overnight. Upon completion, 2 N HCl was added to acidify the reaction, and the solution was extracted with ethyl acetate three times. The collected organic phase was washed with water and brine, and dried over anhydrous Na2SO4. Evaporation of solvent afforded product compound 9 (yellow oil, 600 mg, 100%), which was used in the next step without further purification. 1H NMR (600 MHz, CDCl3): δ 7.68 (d, J = 15.6 Hz, 1H), 6.75 (s, 2H), 6.33 (d, J = 15.6 Hz, 1H), 3.98 (t, J = 6.6 Hz, 2H), 3.85 (s, 6H), 2.19 (t, J = 7.2 Hz, 2H), 1.71–1.75 (m, 2H), 1.56–1.61 (m, 2H), 1.43–1.48 (m, 2H), 1.42 (s, 9H), 1.33–1.37 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.31, 172.21, 153.66, 147.04, 139.82, 129.26, 116.31, 105.57, 79.97, 73.50, 56.13, 35.54, 29.89, 28.83, 28.09, 25.49, 25.04.
4-(7-tert-Butoxy-7-oxoheptyloxy)-3,5-dimethoxybenzoic Acid (10).
Compound 10 was made from 8 (3.7 g, 7.7 mmol) as described for compound 9. Product was obtained as yellow oil (1.7 g, 47%). 1H NMR (600 MHz, CDCl3): δ 7.37 (s, 2H), 4.05(t, J = 7.2 Hz, 2H), 3.90(s, 6H), 2.23(t, J = 7.8 Hz, 2H), 1.74–1.79(m, 2H), 1.59–1.64(m, 2H), 1.46–1.51(m, 2H), 1.44 (s, 9H), 1.35–1.39(m, 2H); 13C NMR (125 MHz, CDCl3): δ 173.33, 171.37, 153.17, 142.18, 124.04, 107.34, 79.99, 73.44, 56.18, 35.53, 29.90, 28.82, 28.08, 25.47, 25.04.
(E)-tert-Butyl 7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxo-5,6-dihydropyridin-1(2H)-yl)prop-1-enyl)phenoxy)heptanoate (15).
Step 1: compound 9 (540 mg, 1.3 mmol, 1 equiv) was dissolved in DCM (12 mL) followed by addition of oxalyl chloride (0.56 mL, 6.5 mmol, 5 equiv) and a catalytic amount of DMF (two drops). The reaction mixture was stirred at room temperature for 1 h after which the solvent was removed under reduced pressure. The residue was dissolved in THF (3 mL) and directly used in the next step without purification. Step 2: compound 11 (150 mg, 1.56 mmol, 1.2 equiv) was dissolved in anhydrous THF (5 mL), and n-BuLi (2.5 M in hexane, 0.68 mL, 1.7 mmol, 1.3 equiv) was added dropwise via syringe at −78 °C. The reaction mixture was stirred for 15 min at the same temperature followed by the dropwise addition of the THF solution in the first step. TLC showed the completion of the reaction within about 30 min at −78 °C. Saturated aq NH4Cl was added to quench this reaction, and the resulting solution was extracted with ethyl acetate three times. The combined organic phase was washed with water and brine, and dried over anhydrous Na2SO4. Flash chromatography (Hex/EA, 4:1) purification afforded product as yellow oil (358 mg, 56%). 1H NMR (600 MHz, CDCl3): δ 7.66 (d, J = 15.6 Hz, 1H), 7.39 (d, J = 15.6 Hz, 1H), 6.91–6.94 (m, 1H), 6.77(s, 2H), 6.02 (m, 1H), 4.02 (t, J = 6.6 Hz, 2H), 3.96 (t, J = 6.6 Hz, 2H), 3.85 (s, 6H), 2.45–2.47 (m, 2H), 2.19 (t, J = 7.8 Hz, 2H), 1.70–1.75 (m, 2H), 1.56–1.61(m, 2H), 1.48–1.44 (m, 2H), 1.42 (s, 9H), 1.32–1.37 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.24, 168.90, 165.84, 153.55, 145.49, 143.94, 139.31, 130.37, 125.83, 120.83, 105.55, 79.90, 73.46, 56.15, 41.63, 35.56, 29.91, 28.86, 28.10, 25.52, 25.07, 24.79. HRMS (ESI): calcd. for [C27H37NO7+Na]+, 510.2468; found, 510.2455.
(E)-tert-Butyl 7-(4-(3-(3-Chloro-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxoprop-1-enyl)-2,6-dimethoxyphenoxy)heptanoate (16).
Compound 16 was synthesized from compounds 9 (360 mg, 0.88 mmol) and 12 (139 mg, 1.1 mmol) following the procedure described for compound 15, except 9 was activated using pivaloyl chloride (0.18 mL, 0.97 mmol) in the presence of TEA (0.24 mL, 1.8 mmol) in THF (5 mL) at −10 °C. Flash chromatography (Hex/EA, 4:1) purification afforded product as colorless oil (150 mg, oil, 33%). 1H NMR (600 MHz, CDCl3): δ 7.68 (d, J = 15.0 Hz, 1H), 7.38 (d, J = 15.6 Hz, 1H), 7.07 (t, J = 4.8 Hz, 1H), 6.77 (s, 2H), 4.06 (t, J = 6.6 Hz, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.85 (s, 6H), 2.53–2.56 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 1.71–1.74 (m, 2H), 1.57–1.59 (m, 2H), 1.43–1.63 (m, 2H), 1.41 (s, 9H), 1.33–1.36 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.21, 168.50, 161.37, 153.59, 145.12, 141.10, 139.58, 130.09, 128.20, 119.99, 105.69, 79.89, 73.46, 56.20, 41.77, 35.55, 29.90, 28.85, 28.09, 25.51, 25.28, 25.05. HRMS (ESI): calcd for [C27H36ClNO7+Na]+, 544.2078; found, 544.2054.
(E)-tert-Butyl 7-(2,6-Dimethoxy-4-(3-(4-methyl-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxoprop-1-enyl)phenoxy)heptanoate (17).
Compound 17 was synthesized from compounds 9 and 13 following the procedure described for compound 16. 9 (141 mg, 0.34 mmol) was activated using pivaloyl chloride (0.068 mL, 0.37 mmol, 1.1 equiv) in the presence of TEA (0.09 mL, 2 equiv) in THF at −10 °C. Compound 13 (44 mg, 0.4 mmol) was deprotonated by using n-BuLi (2 equiv) in THF and then reacted with activated 9 to afford product 17 (24 mg, 23%). 1H NMR (600 MHz, CDCl3): δ7.63 (d, J = 15.6 Hz, 1H), 7.40 (d, J = 15.6 Hz, 1H), 6.76 (s, 2H), 5.81 (d, J = 1.2 Hz, 1H), 4.00 (t, J = 6.6 Hz, 2H), 3.96 (t, J = 6.6 Hz, 2H), 3.84(s, 6H), 2.37 (d, J = 6.6 Hz, 2H), 2.19 (t, J = 7.2 Hz, 2H), 1.99 (s, 3H), 1.72 (m, 2H),1.58 (m, 2H), 1.43–1.48 (m, 2H), 1.41 (s, 9H), 1.33–1.36 (m, 2H). 13C NMR (125 MHz, CDCl3): δ173.25, 168.87, 166.09, 157.85, 153.54, 143.61, 139.21, 130.46, 121.29, 120.97, 105.50, 79.89, 73.44, 56.13, 41.53, 35.55, 29.90, 29.89, 28.84, 28.09, 25.50, 25.05, 22.97. HRMS (ESI): calcd for [C28H39NO7+H]+, 502.2805; found, 502.2807.
(E)-tert-Butyl 7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxopiperidin-1-yl)-prop-1-enyl)phenoxy)heptanoate (18).
Compound 18 was synthesized from compounds 9 and 14 following the procedure described for compound 16. 9 (900 mg, 2.21 mmol) was activated using pivaloyl chloride (0.44 mL, 2.43 mmol, 1.1 equiv) in the presence of TEA (0.6 mL, 2 equiv) in THF at −10 °C. Compound 14 (263 mg, 2.65 mmol) was deprotonated by using n-BuLi (2 equiv) in THF and then reacted with activated 9 to afford product 18 (760 mg, 75%). 1H NMR (600 MHz, CDCl3): δ 7.59 (d, J = 15.0 Hz, 1H), 7.31 (d, J = 15.6 Hz, 1H), 6.74 (s, 2H), 3.95 (t, J = 6.6 Hz, 2H), 3.83(s, 6H), 3.76 (t, J = 4.8 Hz, 2H), 2.57(t, J = 6.6 Hz, 2H), 2.18 (t, J = 7.2 Hz, 2H), 1.84–1.86 (m, 4H), 1.69–1.72 (m, 2H), 1.55–1.58 (m, 2H), 1.42–1.46 (m, 2H), 1.40 (s, 9H), 1.31–1.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ173.86, 173.21, 169.59, 153.53, 143.53, 139.23, 130.39, 121.07, 105.48, 79.87, 73.42, 56.12, 44.60, 35.53, 34.92, 29.88, 28.84, 28.08, 25.50, 25.04, 22.54, 20.60. HRMS (ESI): calcd for [C27H39NO7+Na]+, 512.2624; found, 512.2625.
tert-Butyl 7-(2,6-Dimethoxy-4-(6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)phenoxy)heptanoate (19).
Compound 19 was synthesized from compounds 10 and 11 following the procedure described for compound 16. 10 (1.0 g, 2.62 mmol) was activated using pivaloyl chloride (0.52 mL, 2.88 mmol, 1.1 equiv) in the presence of TEA (0.72 mL, 2 equiv) in THF at −10 °C. Compound 11 (305 mg, 3.14 mmol) was deprotonated by using n-BuLi (2 equiv) in THF and then reacted with activated 10 to afford product 19 (340 mg, 28%).1H NMR (600 MHz, CDCl3): δ 6.96–6.99 (m, 1H), 6.85 (s, 2H), 5.98–6.00 (m, 1H), 4.01 (t, J = 6.6 Hz, 2H), 3.96 (t, J = 6.6 Hz, 2H), 3.84 (s, 6H), 2.59–2.61 (m, 2H), 2.22 (t, J = 7.8 Hz, 2H), 1.73–1.77 (m, 2H), 1.58–1.63 (m, 2H), 1.45–1.49 (m, 2H), 1.44 (s, 9H), 1.33–1.39 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.64, 173.22, 165.56, 152.96, 145.48, 140.82, 130.66, 125.71, 106.24, 79.87, 73.39, 56.18, 43.72, 35.54, 29.91, 28.84, 28.08, 25.49, 25.05, 24.88. HRMS (ESI): calcd for [C25H35NO7+H]+, 462.2492; found, 462.2491.
tert-Butyl 7-(4-(5-Chloro-6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)-2,6-dimethoxy phenoxy)heptanoate (20).
Compound 20 was synthesized from compounds 10 and 12 following the procedure described for compound 16. 10 (1.0 g, 2.62 mmol) was activated using pivaloyl chloride (0.52 mL, 2.88 mmol, 1.1 equiv) in the presence of TEA (0.72 mL, 2 equiv) in THF at −10 °C. Compound 12 (413 mg, 3.14 mmol) was deprotonated by using n-BuLi (1.5 equiv) in THF and then reacted with activated 10 to afford product 20 (350 mg, 27%). 1H NMR (600 MHz, CDCl3): δ 7.11 (t, J = 4.2 Hz, 1H), 6.83 (s, 2H), 3.99–4.02 (m, 4H), 3.84 (s, 6H), 2.67–2.70 (m, 2H), 2.22 (t, J = 7.8 Hz, 2H), 1.73–1.77 (m, 2H), 1.58–1.63 (m, 2H), 1.46–1.48 (m, 2H), 1.44 (s, 9H), 1.34–1.39 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.25, 173.15, 161.18, 153.07, 141.19, 140.88, 130.02, 127.75, 106.28, 79.90, 73.47, 56.26, 43.90, 35.55, 29.92, 28.84, 28.09, 25.49, 25.42, 25.06. HRMS (ESI): calcd for [C25H34ClNO7+Na]+, 518.1922; found, 518.1922.
(E)-7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxo-5,6-dihydropyridin-1(2H)-yl)prop-1-enyl) phenoxy)heptanoic Acid (21).
Compound 15 (350 mg, 0.72 mmol) was dissolved in a mixture of DCM/TFA (10 mL/1 mL), and the reaction mixture was stirred at room temperature overnight. Upon completion shown by TLC, the reaction mixture was diluted with DCM and washed with water and brine, and dried over anhydrous Na2SO4. Evaporation of solvent afforded product as yellow oil (311 mg, quantitative yield), which was used in the next step without purification. 1H NMR (600 MHz, CDCl3): δ 7.67 (d, J = 15.6 Hz, 1H), 7.41 (d, J = 15.0, 1H), 6.93–6.95 (m, 1H), 6.79 (s, 2H), 6.04–6.06 (m, 1H), 4.04 (t, J = 6.6 Hz, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.87 (s, 6H), 2.47–2.49 (m, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.73–1.76 (m, 2H), 1.64–1.67 (m, 2H), 1.47–1.51 (m, 2H), 1.39–1.43 (m, 2H).
(E)-7-(4-(3-(3-Chloro-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxo-prop-1-enyl)-2,6-dimethoxyphenoxy)heptanoic Acid (22).
Compound 22 was synthesized from 16 following the procedure described for compound 21. Compound 16 (130 mg, 0.25 mmol) was dissolved in DCM (5 mL) followed by the addition of TFA (1 mL) to afford compound 22 (110 mg, 95%). 1H NMR (600 MHz, CDCl3): δ 7.69 (d, J = 15.6 Hz, 1H), 7.39 (d, J = 15.0 Hz, 1H), 7.08 (t, J = 4.8 Hz, 1H), 6.78 (s, 2H), 4.07 (t, J = 6.6 Hz, 2H), 3.98 (t, J = 6.6 Hz, 2H), 3.85 (s, 6H), 2.54–2.57 (m, 2H), 2.35 (t, J = 7.2 Hz, 2H), 1.71–1.76 (m, 2H), 1.62–1.67 (m, 2H), 1.45–1.50 (m, 2H), 1.36–1.41 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 179.33, 168.59, 161.41, 153.57, 145.19, 141.15, 139.51, 130.12, 128.19, 119.98, 105.70, 73.38, 56.20, 41.79, 35.85, 29.84, 28.78, 25.44, 25.29, 24.61.
7-(2,6-Dimethoxy-4-(6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)phenoxy)heptanoic Acid (23).
Compound 23 was synthesized from 19 following the procedure described for compound 21. Compound 19 (340 mg, 0.74 mmol) was dissolved in DCM (10 mL) followed by the addition of TFA (2 mL) to afford compound 23 (242 mg, 81%). 1H NMR (600 MHz, CDCl3): δ 6.99–7.02 (m, 1H), 6.84 (s, 2H), 6.00–6.03 (m, 1H), 4.03 (t, J = 6.6 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.84 (s, 6H), 2.60–2.63 (m, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.73–1.77 (m, 2H), 1.63–1.68 (m, 2H), 1.46–1.51 (m, 2H), 1.36–1.41 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 179.71, 173.77, 166.01, 152.94, 145.99, 140.88, 130.44, 124.97, 106.29, 73.35, 56.19, 43.83, 33.89, 29.80, 28.69, 25.36, 24.85, 24.59.
7-(4-(5-Chloro-6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)-2,6-dimethoxyphenoxy)heptanoic Acid (24).
Compound 24 was synthesized from 20 following the procedure described for compound 21. Compound 20 (310 mg, 0.63 mmol) was dissolved in DCM (10 mL) followed by the addition of TFA (1.5 mL) to afford compound 24 (275 mg, quantitative yield). 1H NMR (600 MHz, CDCl3): δ 7.09 (t, J = 4.2 Hz, 1H), 6.80 (s, 2H), 4.00 (t, J = 6.6 Hz, 2H), 3.98 (t, J = 6.6 Hz, 2H), 3.81 (s, 6H), 2.65–2.68 (m, 2H), 2.35 (t, J = 7.2 Hz, 2H), 1.70–1.75 (m, 2H), 1.61–1.66 (m, 2H), 1.43–1.48 (m, 2H), 1.34–1.39 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 180.10, 173.35, 161.43, 153.01, 141.28, 141.04, 129.97, 127.59, 106.29, 73.47, 56.26, 43.98, 33.89, 29.76, 28.67, 25.37, 25.34, 24.55.
(E)-7-(2,6-Dimethoxy-4-(3-(4-methyl-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxoprop-1-enyl)phenoxy)heptanoic Acid (25).
Compound 25 was synthesized from 17 following the procedure described for compound 21. Compound 17 (136 mg, 0.27 mmol) was dissolved in DCM (10 mL) followed by the addition of TFA (1.5 mL) to afford compound 25 (120 mg, quantitative yield). 1H NMR (600 MHz, CDCl3): δ 7.63 (d, J = 15.6 Hz, 1H), 7.37 (d, J = 15.6 Hz, 1H), 6.77(s, 2H), 5.84 (s, 1H), 3.96–4.00(m, 4H), 3.83(s, 6H), 2.39(t, J = 6 Hz, 2H), 2.33(t, J = 7.2 Hz, 2H), 2.00(s, 3H), 1.69–1.74(m, 2H), 1.61–1.65(m, 2H), 1.43–1.48(m, 2H), 1.34–1.39(m, 2H). 13C NMR (125 MHz, CDCl3): δ 179.72, 169.28, 166.85, 159.21, 153.49, 144.23, 139.15, 130.37, 120.78, 120.71, 105.58, 73.41, 56.09, 41.72, 33.90, 29.85, 29.76, 28.74, 25.40, 24.58, 23.03.
(E)-7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxopiperidin-1-yl)prop-1-enyl)phenoxy)heptanoic Acid (26).
Compound 26 was synthesized from 18 following the procedure described for compound 21. Compound 18 (400 mg, 0.82 mmol) was dissolved in DCM (10 mL) followed by the addition of TFA (2.0 mL) to afford compound 26 (354 mg, quantitative yield). 1H NMR (600 MHz, CDCl3): δ 7.61 (d, J = 15.6 Hz, 1H), 7.31 (d, J = 15.6 Hz, 1H), 6.76 (s, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.84 (s, 6H), 3.76 (t, J = 6.0 Hz, 2H), 2.59 (t, J = 6.6 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.84–1.87 (m, 4H), 1.70–1.75 (m, 2H), 1.61–1.66 (m, 2H), 1.44–1.49 (m, 2H), 1.35–1.40 (m, 2H). 13C NMR (125 MHz, CDCl3): δ179.54, 174.14, 169.71, 153.52, 143.71, 139.20, 130.39, 121.07, 105.51, 73.34, 56.13, 44.68, 34.88, 33.90, 29.82, 28.77, 25.44, 24.60, 22.52, 20.57.
(E)-Methyl 7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxo-5,6-dihydropyridin-1(2H)-yl)prop-1-enyl)phenoxy)heptanoate (27).
Compound 21 was dissolved in diethyl ether/MeOH (6 mL/0.6 mL), and trimethylsilyldiazomethane (2 M in diethyl ether, 0.18 mL, 1.2 equiv) was added at 0 °C. The reaction mixture was stirred at room temperature overnight. Solvent was removed under reduced pressure, and the residue was redissolved in ethyl acetate, washed with water and brine, and dried over anhydrous Na2SO4. Crude product was purified by flash chromatography (Hex/EA, 2:1) to afford 27 (90 mg, yellow oil, 64%). 1H NMR (600 MHz, CDCl3): δ 7.62 (d, J = 15.6 Hz, 1H), 7.36 (d, J = 15.6 Hz, 1H), 6.88–6.90 (m, 1H), 6.75 (s, 2H), 5.98 (m,1H), 3.98 (t, J = 6.6 Hz, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.82 (s, 6H), 3.61 (s, 3H), 2.41–2.44 (m, 2H), 2.27 (t, J = 7.2 Hz, 2H), 1.67–1.72 (m, 2H), 1.58–1.63 (m, 2H), 1.41–1.46 (m, 2H), 1.30–1.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 174.20, 168.85, 165.82, 153.53, 145.53, 143.82, 139.25, 130.38, 125.76, 120.87, 105.51, 73.35, 56.12, 51.42, 41.61, 33.99, 29.86, 28.86, 25.46, 24.88, 24.77. HRMS (ESI): calcd for [C24H31NO7+H]+, 446.2179; found, 446. 2188.
(E)-Methyl 7-(4-(3-(3-chloro-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxoprop-1-enyl)-2,6-dimethoxyphenoxy)heptanoate (28).
Compound 28 was synthesized from 22 following the procedure described for compound 27. Compound 22 (70 mg, 0.15 mmol) was reacted with trimethylsilyldiazomethane (2 M in diethyl ether, 0.13 mL, 1.2 equiv) in a mixture of diethyl ether and MeOH (5 mL:0.5 mL) to afford compound 28 (22 mg, colorless oil, 31%). 1H NMR (600 MHz, CDCl3): δ 7.70 (d, J = 15.6 Hz, 1H), 7.41 (d, J = 15.6 Hz, 1H), 7.09 (t, J = 4.2 Hz, 1H), 6.80 (s, 2H), 4.09 (t, J = 6.0 Hz, 2H), 3.99 (t, J = 6.0 Hz, 2H), 3.88 (s, 6H), 3.67 (s, 3H), 2.57 (q, J = 6.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.73–1.78 (m, 2H), 1.63–1.68 (m, 2H), 1.46–1.51 (m, 2H), 1.35–1.40 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 174.25, 168.51, 161.38, 153.58, 145.11, 141.11, 139.55, 130.11, 128.20, 120.00, 105.69, 73.40, 56.20, 51.45, 41.78, 34.02, 29.88, 28.88, 25.47, 25.29, 24.90.
HRMS (ESI): calcd for [C24H30ClNO7+H]+, 480.1789; found, 480.1795.
General procedure for compounds 29–34.
To a flask were added carboxylic acids 21–26 (1 equiv), THP protected hydroxyl amine (2.5 equiv) and condensing reagent HBTU (2 equiv) and filled with argon. DMF and DIPEA (2 equiv) were added via syringe. The reaction mixture was stirred at room temperature overnight and was diluted with water, extracted with ethyl acetate three times. The combined organic phase was washed with brine and dried over anhydrous Na2SO4. Crude products were briefly purified by flash column chromatography (EA/DCM, 1:1) to afford compounds 29–34 with yields from 50% to 80%.
(E)-7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxo-5,6-dihydropyridin-1(2H)-yl)prop-1-enyl) phenoxy)-N-hydroxyheptanamide (1–58).
Compound 29 (100 mg, 0.19 mmol, 1 equiv) was dissolved in MeOH (10 mL) and then TFA (2 mL) was added. The reaction mixture was continued to stir at room temperature for 1 h. Solvent was removed under reduced pressure. Crude product was purified by flash column chromatography (DCM/MeOH, 20:1) to afford product 1–58 (white solid, 57 mg, 67%). 1H NMR (600 MHz, CDCl3): δ 8.77 (br, s 1H), 7.64 (d, J = 15.0 Hz, 1H), 7.37 (d, J = 15.6 Hz, 1H), 6.92–6.95 (m, 1H), 6.77 (s, 2H), 6.02 (d, J = 9.6 Hz, 1H), 4.02 (t, J = 6.6 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.84 (s, 6H), 2.46 (m, 2H), 2.12 (m, 2H), 1.67–1.70 (m, 2H), 1.60–1.63 (m, 2H), 1.43–1.45 (m, 2H), 1.31–1.34 (m, 2H). 13C NMR (125 MHz, CDCl3): δ171.45, 168.95, 165.92, 153.48, 145.64, 143.85, 139.15, 130.42, 125.75, 120.93, 105.58, 73.27, 56.17, 41.66, 32.71, 29.72, 28.59, 25.27, 25.16, 24.78. HRMS (ESI): calcd for [C23H30N2O7+H]+, 447.2131; found, 447.2129.
(E)-7-(4-(3-(3-Chloro-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxo-prop-1-enyl)-2,6-dimethoxyphenoxy)-N-hydroxyheptanamide (3–35).
Compound 30 (100 mg, 0.18 mmol) was reacted with 0.5 mL TFA in 10 mL MeOH to afford compound 3–35 (50 mg, colorless oil, 67%). 1H NMR (600 MHz, CDCl3): δ 7.69 (d, J = 15.6, 1H), 7.39 (d, J = 15.6, 1H), 7.09 (t, J = 4.8 Hz, 1H), 6.79 (s, 2H), 4.08 (t, J = 6.6 Hz, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.86(s, 6H), 2.57 (q, J = 6.0 Hz, 2H), 2.14 (t, J = 7.8 Hz, 2H), 1.69–1.73 (m, 2H), 1.62–1.67 (m, 2H), 1.43–1.48 (m, 2H),1.34–1.37 (m, 2H). 13C NMR (125 MHz, CDCl3): δ171.55, 168.55, 161.43, 153.50, 145.04, 141.19, 139.39, 130.17, 128.16, 120.09, 105.73, 73.30, 56.23, 41.81, 32.70, 29.72, 28.57, 25.29, 25.25, 25.15. HRMS (ESI): calcd for [C23H29N2O7Cl+H]+, 481.1742; found, 481.1734.
7-(2,6-Dimethoxy-4-(6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)phenoxy)-N-hydroxyheptanamide (3–31).
Compound 31 (184 mg, 0.35 mmol) was dissolved in MeOH (10 mL) followed by the addition of TFA (1 mL) at room temperature. After stirring for 1 h, solvent was removed under reduced pressure. Crude product was purified by flash column chromatography (DCM/MeOH, 20:1) to afford product 3–31 (59 mg, colorless oil, 40%). 1H NMR (600 MHz, CDCl3): δ 6.96–6.99 (m, 1H), 6.83 (s, 2H), 5.99(d, J = 9.6 Hz, 1H), 4.04 (t, J = 6.6 Hz, 2H), 3.95 (t, J = 6.6 Hz, 2H), 3.82 (s, 6H), 2.58–2.60 (m, 2H), 2.06 (t, J = 7.2 Hz, 2H), 1.67–1.71 (m, 2H), 1.57–1.62 (m, 2H), 1.39–1.44 (m, 2H), 1.28–1.31 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.61, 171.22, 165.93, 152.84, 145.85, 140.73, 130.58, 125.09, 106.28, 73.02, 56.25, 43.76, 32.53, 29.69, 28.36, 25.10, 25.03, 24.88. HRMS (ESI): calcd for [C21H28N2O7+H]+, 421.1975; found, 421.1975.
7-(4-(5-Chloro-6-oxo-1,2,3,6-tetrahydropyridine-1-carbonyl)-2,6-dimethoxyphenoxy)-N-hydroxyheptanamide (3–98).
Compound 3–98 was synthesized by treating 32 (80 mg, 0.15 mmol) with TFA(0.4 mL) in MeOH (5 mL) at room temperature. Crude product was purified by flash column chromatography (DCM/MeOH, 20:1) (light yellow solid, 38 mg, 56%). 1H NMR (600 MHz, CDCl3): δ 7.09 (t, J = 4.8 Hz, 1H), 6.78 (s, 2H), 3.96–4.00 (m, 4H), 3.80 (s, 6H), 2.65–2.68 (m, 2H), 2.06 (t, J = 6.6 Hz, 2H), 1.66–1.70 (m, 2H), 1.57–1.62 (m, 2H), 1.38–1.42 (m, 2H), 1.26–1.32 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 173.13, 171.51, 161.38, 152.99, 141.19, 141.04, 130.02, 127.61, 106.28, 73.24, 56.31, 43.94, 32.59, 29.71, 28.46, 25.39, 25.21, 25.16. HRMS (ESI): calcd for [C21H27N2O7Cl+H]+, 455.1590; found, 455.1585.
(E)-7-(2,6-Dimethoxy-4-(3-(4-methyl-2-oxo-5,6-dihydropyridin-1(2H)-yl)-3-oxoprop-1-enyl)phenoxy)-N-hydroxyheptanamide (35).
Compound 35 was synthesized by treating 33 (67 mg, 0.12 mmol) with TFA (0.5 mL) in MeOH (6.0 mL) at room temperature. Crude product was purified by flash column chromatography (DCM/MeOH, 30:1) (lightly yellow solid, 20 mg, 36%). 1H NMR (600 MHz, CDCl3): δ 8.77(br s, 1H), 7.63 (d, J = 15.6 Hz, 1H), 7.39(d, J = 15.0 Hz, 1H), 6.77(s, 2H), 5.82(s, 1H), 3.99(t, J = 6.0 Hz, 2H), 3.97(t, J =6.0 Hz, 2H), 3.84 (s, 6H), 2.38(t, J = 5.4 Hz, 2H), 2.12(m, 2H), 2.00 (s, 3H), 1.68–1.70(m, 2H), 1.61–1.64(m, 2H), 1.40–1.46(m, 2H),1.30–1.36 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 171.37, 168.92, 166.18, 158.01, 153.46, 143.55, 139.06, 130.51, 121.24, 121.05, 105.55, 73.25, 56.16, 41.57, 32.72, 29.91, 29.71, 28.58, 25.27, 25.16, 22.99. HRMS (ESI): calcd for [C24H32N2O7+H]+, 461.2288; found, 461.2299.
(E)-7-(2,6-Dimethoxy-4-(3-oxo-3-(2-oxopiperidin-1-yl)prop-1-enyl)phenoxy)-N-hydroxyheptanamide (36).
Compound 36 was synthesized by treating 34 (70 mg, 0.13 mmol) with TFA (0.4 mL) in MeOH (10 mL) at room temperature. Crude product was purified by flash column chromatography (DCM/MeOH, 20:1) (15 mg, 26%). 1H NMR (400 MHz, DMSO-d6): δ 10.32(s,1H), 7.32 (d, J = 16.0 Hz, 1H), 6.85 (s, 2H), 6.54 (d, J = 15.6 Hz, 1H), 3.82 (t, J = 6.4 Hz, 2H), 3.77 (s, 6H), 3.12–3.17 (m, 2H), 2.31 (t, J = 7.6 Hz, 2H), 1.93 (t, J = 7.6 Hz, 2H), 1.36–1.61 (m, 10H), 1.20–1.28 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ173.73, 169.57, 165.39, 153.68, 139.06, 138.21, 130.80, 121.97, 105.33, 72.80, 56.33, 38.63, 33.36, 32.70, 29.94, 28.99, 28.78, 25.60, 25.54, 22.38. HRMS (ESI): calcd for [C23H32N2O7+Na]+, 471.2107; found, 471.2100.
Cell Culture.
U937, MV4–11, HL60, MCF-10A were purchased from the American Type Culture Collection (Manassas, VA, U.S.A.). MOLM-13 cells were purchased from AddexBio (San Diego, CA). CMY and CMS cells were gifts from Dr. A Fuse from the National Institute of Infectious Diseases, Tokyo, Japan. The OCI-AML3 and CMK cell lines were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). HL-60 and CMK cytarabine resistant cells (designated HL-60/ARA-C and CMK/ARA-C, respectively) were generated by stepwise selection of HL-60 or CMK cells in the presence of cytarabine, until they could be maintained in the presence of 600 nM or 300 nM cytarabine, respectively. The cell lines were cultured in RPMI 1640 media with 10% fetal bovine serum (Life Technologies, Carlsbad, CA, U.S.A.; 20% FBS for MOLM-13 cells) and 2 mM L-glutamine, plus 100 U/mL penicillin and 100 μg/mL streptomycin, in a 37 °C humidified atmosphere containing 5% CO2/95% air. MCF-10A cells were maintained in Dulbecco’s modified Eagle’s medium-F12 (DMEM/F12) (Invitrogen) supplemented with 5% horse serum (Invitrogen), 0.5 μg/mL hydrocortisone (Sigma), 100 ng/mL cholera toxin (Sigma), 10 μg/mL insulin (Sigma), and 20 ng/mL recombinant human EGF (Peprotech). SAHA and piperlongumine were purchased from Cayman (item no.: 10009929; Ann Arbor, MI) and Indofine (catalog no.: P-004; Hillsborough, NJ) respectively.
Clinical Samples.
Diagnostic blast samples were obtained from the First Hospital of Jilin University, Changchun, China. Written informed consent was provided according to the Declaration of Helsinki. This study was approved by the Human Ethics Committee of The First Hospital of Jilin University. The samples were purified by standard Ficoll-Hypaque density centrifugation, then cultured in RPMI 1640 with 20% fetal bovine serum, ITS solution (Sigma-Aldrich) and 20% supernatant of the 5637 bladder cancer cell line (as a source of granulocyte-macrophage colony-stimulating factor).48
MTT Assay.
AML cell lines or MCF-10A cell line were treated with the indicated compounds, alone or in combination, for 24 or 72 h. After drug treatment, MTT assay was performed according to the standard protocol (0.05 mg/mL MTT incubated with cell at 37 °C for 3h, supernatant was removed, 100 μL of DMSO was added, incubated for 30 min at 37 °C, absorbance was read at 570 nM using a microplate reader).
Western Blot Analysis.
Cells were lysed in the presence of protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN, U.S.A.). Whole-cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidenedifluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, U.S.A.), and immunoblotted with anti-Bcl-2, -Bcl-xL, -Mcl-1, -XIAP, -PARP, -Bim, -Bak, -Bax, -survivin, -γH2AX, -cleaved caspase-3 (designated-cf caspase-3, Cell Signaling Technology, Danvers, MA, U.S.A.), −CHK1, -RAD51 (Santa Cruz), -acetyl-histone 4 (ac-H4), -H4 (Upstate Biotechnology, Lake Placid, NY), -acetyl-tubulin (actubulin), or -β-actin (Sigma-Aldrich) antibody, as previously described. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, U.S.A.), as described by the manufacturer. Western blots were repeated at least three times, and one representative blot is shown. Densitometry measurements were made using Odyssey V3.0 (Li-Cor), normalized to β-actin, and then compared to the corresponding no drug treatment control.
Apoptosis Assay (Annexin V/PI Staining).
AML cells were treated with the indicated concentration of SAHA, piperlongumine, 1–58, 3–35, 3–31, 3–98, 27, 28, 35, or 36, alone or in combination, for 24 h and subjected to flow cytometry analysis using the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide Apoptosis Kit (Beckman Coulter; Brea, CA, U.S.A.), as previously described. Results are expressed as percent Annexin V+. Experiments were performed three independent times in triplicate, for the AML cell lines, data presented are from one representative experiment, while patient sample experiments were performed once in triplicate due to limited sample. Patient samples were chosen solely on the basis of the availability of adequate sample for the assay. The extent and direction of antileukemic interaction was determined by calculating the combination index (CI) values using CompuSyn software (Combosyn Inc., Paramus, NJ, U.S.A.). CI < 1, CI = 1, and CI > 1 indicate synergistic, additive, and antagonistic effects, respectively.
Alkaline Comet Assay.
U937 cells were treated for 20 h with SAHA, PL alone or in combination, 16 h for compounds 1–58 and 3–35, and subjected to alkaline comet assay as previously described.14,49,50 Slides were stained with SYBR Gold (Life Technologies) and then imaged on an Olympus BX-40 microscope equipped with a DP72 microscope camera and Olympus cellSens Dimension software (Olympus America Inc., Center Valley, PA). Approximately 50 comets per gel were scored using CometScore (TriTek Corp, Sumerduck, VA, U.S.A.). The median percent DNA in the tail was calculated and graphed ± SEM.
Statistical Analysis.
Differences were compared using the two-sample t test. Statistical analyses were performed with GraphPad Prism 5.0. Error bars represent ± SEM. The level of significance was set at P <0.05.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a Wayne State University startup fund (Z.Q.) and Barbara Ann Karmanos Cancer Institute McLaren Program Funding Support (Z.Q., 2014); Children’s Hospital of Michigan Foundation (J.W.T., Y.G., Z.Q.); Jilin University, Changchun, China (H.L.), Decerchio/Guisewite Family (J.W.T and Y.G.), and the Barbara Ann Karmanos Cancer Institute (Y.G.), and grants from the National Natural Science Foundation of China, NSFC 31271477 (Y.G.) and NSFC 31471295 (Y.G.), Hyundai Hope On Wheels (J.W.T. and Y.G.), and the Ring Screw Textron Endowed Chair for Pediatric Cancer Research (J.W.T.). Ginopolos/Karmanos Endowment (J.W.T.) and Levy Family Cancer Research Fund (J.W.T). The funders had no role in study design, data collection, analysis and interpretation of data, decision to publish, or preparation of the manuscript.
ABBREVIATIONS USED
- AML
acute myeloid leukemia
- PL
piperlongumine
- HDACi
histone acetylase inhibitor
- ROS
reactive oxygen species
- Ara-C
cytarabine
- LSCs
leukemic stem cells
- DSBs
double strand breaks
- HR
homologous recombination
- NHEJ
nonhomologous end joining
- cf-caspase-3
cleaved caspase-3
- CI
combination index
- TFA
trifluoroacetic acid
- TEA
triethyl amine
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00772.
Comparison of cell viability of MCF-10A and U937 cell after PL or SAHA treatment, chemical stability and reactivity of PL-HDACis to GSH, and related experimental details (PDF)
Selected data for compounds (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Roboz GJ Novel approaches to the treatment of acute myeloid leukemia. Hematology Am. Soc. Hematol Educ Program 2011, 2011, 43–50. [DOI] [PubMed] [Google Scholar]
- (2).Siegel RL; Miller KD; Jemal A Cancer statistics, 2015. Ca-Cancer J. Clin 2015, 65, 5–29. [DOI] [PubMed] [Google Scholar]
- (3).Pollyea DA; Gutman JA; Gore L; Smith CA; Jordan CT Targeting acute myeloid leukemia stem cells: a review and principles for the development of clinical trials. Haematologica 2014, 99, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Barabe F; Kennedy JA; Hope KJ; Dick JE Modeling the initiation and progression of human acute leukemia in mice. Science 2007, 316, 600–604. [DOI] [PubMed] [Google Scholar]
- (5).Hope KJ; Jin L; Dick JE Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol 2004, 5, 738–743. [DOI] [PubMed] [Google Scholar]
- (6).Quintas-Cardama A; Santos FP; Garcia-Manero G Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [DOI] [PubMed] [Google Scholar]
- (7).Petruccelli LA; Dupere-Richer D; Pettersson F; Retrouvey H; Skoulikas S; Miller WH Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 2011, 6, e20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Robert C; Rassool FV HDAC inhibitors: roles of DNA damage and repair. Adv. Cancer Res 2012, 116, 87–129. [DOI] [PubMed] [Google Scholar]
- (9).Bose P; Dai Y; Grant S Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol. Ther 2014, 143, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rosato RR; Almenara JA; Maggio SC; Coe S; Atadja P; Dent P; Grant S Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther 2008, 7, 3285–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen CS; Wang YC; Yang HC; Huang PH; Kulp SK; Yang CC; Lu YS; Matsuyama S; Chen CY; Chen CS Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007, 67, 5318–5327. [DOI] [PubMed] [Google Scholar]
- (12).Munshi A; Kurland JF; Nishikawa T; Tanaka T; Hobbs ML; Tucker SL; Ismail S; Stevens C; Meyn RE Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res 2005, 11, 4912–4922. [DOI] [PubMed] [Google Scholar]
- (13).Shubassi G; Robert T; Vanoli F; Minucci S; Foiani M Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res. 2012, 72, 1332–1335. [DOI] [PubMed] [Google Scholar]
- (14).Xie C; Drenberg C; Edwards H; Caldwell JT; Chen W; Inaba H; Xu X; Buck SA; Taub JW; Baker SD; Ge Y Panobinostat Enhances Cytarabine and Daunorubicin Sensitivities in AML Cells through Suppressing the Expression of BRCA1, CHK1, and Rad51. PLoS One 2013, 8, e79106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bali P; Pranpat M; Bradner J; Balasis M; Fiskus W; Guo F; Rocha K; Kumaraswamy S; Boyapalle S; Atadja P; Seto E; Bhalla K Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem 2005, 280, 26729–26734. [DOI] [PubMed] [Google Scholar]
- (16).Dai Y; Chen S; Kmieciak M; Zhou L; Lin H; Pei XY; Grant S The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Mol. Cancer Ther 2013, 12, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Grant S Targeting leukemia stem cells with HDAC inhibitors and modulators of the DNA damage response. Leuk. Suppl 2014, 3, S14–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Garcia-Manero G; Yang H; Bueso-Ramos C; Ferrajoli A; Cortes J; Wierda WG; Faderl S; Koller C; Morris G; Rosner G; Loboda A; Fantin VR; Randolph SS; Hardwick JS; Reilly JF; Chen C; Ricker JL; Secrist JP; Richon VM; Frankel SR; Kantarjian HM Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [DOI] [PubMed] [Google Scholar]
- (19).Schaefer EW; Loaiza-Bonilla A; Juckett M; DiPersio JF; Roy V; Slack J; Wu W; Laumann K; Espinoza-Delgado I; Gore SD A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica 2009, 94, 1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bose P; Grant S Rational Combinations of Targeted Agents in AML. J. Clin. Med 2015, 4, 634–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Raj L; Ide T; Gurkar AU; Foley M; Schenone M; Li X; Tolliday NJ; Golub TR; Carr SA; Shamji AF; Stern AM; Mandinova A; Schreiber SL; Lee SW Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (22).Xiong XX; Liu JM; Qiu XY; Pan F; Yu SB; Chen XQ Piperlongumine induces apoptotic and autophagic death of the primary myeloid leukemia cells from patients via activation of ROS-p38/JNK pathways. Acta Pharmacol. Sin 2015, 36, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pei S; Minhajuddin M; Callahan KP; Balys M; Ashton JM; Neering SJ; Lagadinou ED; Corbett C; Ye H; Liesveld JL; O’Dwyer KM; Li Z; Shi L; Greninger P; Settleman J; Benes C; Hagen FK; Munger J; Crooks PA; Becker MW; Jordan CT Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem 2013, 288, 33542–33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hartwell KA; Miller PG; Mukherjee S; Kahn AR; Stewart AL; Logan DJ; Negri JM; Duvet M; Jaras M; Puram R; Dancik V; Al-Shahrour F; Kindler T; Tothova Z; Chattopadhyay S; Hasaka T; Narayan R; Dai M; Huang C; Shterental S; Chu LP; Haydu JE; Shieh JH; Steensma DP; Munoz B; Bittker JA; Shamji AF; Clemons PA; Tolliday NJ; Carpenter AE; Gilliland DG; Stern AM; Moore MA; Scadden DT; Schreiber SL; Ebert BL; Golub TR Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat. Chem. Biol 2013, 9, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Okamoto S; Narita T; Sasanuma H; Takeda S; Masunaga S; Bessho T; Tano K Impact of DNA repair pathways on the cytotoxicity of piperlongumine in chicken DT40 cell-lines. Genes Cancer 2014, 5, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dhillon H; Chikara S; Reindl KM Piperlongumine induces pancreatic cancer cell death by enhancing reactive oxygen species and DNA damage. Toxicol Rep 2014, 1, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sallmyr A; Fan J; Rassool FV Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [DOI] [PubMed] [Google Scholar]
- (28).Bouwman P; Jonkers J The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [DOI] [PubMed] [Google Scholar]
- (29).Koprinarova M; Botev P; Russev G Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair 2011, 10, 970–977. [DOI] [PubMed] [Google Scholar]
- (30).Hu Y; Lu W; Chen G; Zhang H; Jia Y; Wei Y; Yang H; Zhang W; Fiskus W; Bhalla K; Keating M; Huang P; Garcia-Manero G Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound beta-phenylethyl isothiocyanate. Blood 2010, 116, 2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Crook TR; Souhami RL; Whyman GD; McLean AE Glutathione depletion as a determinant of sensitivity of human leukemia cells to cyclophosphamide. Cancer Res. 1986, 46, 5035–5038. [PubMed] [Google Scholar]
- (32).Hedley DW; McCulloch EA; Minden MD; Chow S; Curtis J Antileukemic action of buthionine sulfoximine: evidence for an intrinsic death mechanism based on oxidative stress. Leukemia 1998, 12, 1545–1552. [DOI] [PubMed] [Google Scholar]
- (33).Morphy R; Rankovic Z Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem 2005, 48, 6523–6543. [DOI] [PubMed] [Google Scholar]
- (34).Wu Y; Min X; Zhuang C; Li J; Yu Z; Dong G; Yao J; Wang S; Liu Y; Wu S; Zhu S; Sheng C; Wei Y; Zhang H; Zhang W; Miao Z Design, synthesis and biological activity of piperlongumine derivatives as selective anticancer agents. Eur. J. Med. Chem 2014, 82, 545–551. [DOI] [PubMed] [Google Scholar]
- (35).Adams DJ; Dai M; Pellegrino G; Wagner BK; Stern AM; Shamji AF; Schreiber SL Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci.U. S. A 2012, 109, 15115–15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Winter DK; Drouin A; Lessard J; Spino C Photochemical rearrangement of N-chlorolactams: a route to N-heterocycles through concerted ring contraction. J. Org. Chem 2010, 75, 2610–2618. [DOI] [PubMed] [Google Scholar]
- (37).Kawanaka Y; Kobayashi K; Kusuda S; Tatsumi T; Murota M; Nishiyama T; Hisaichi K; Fujii A; Hirai K; Nishizaki M; Naka M; Komeno M; Nakai H; Toda M Design and synthesis of orally bioavailable inhibitors of inducible nitric oxide synthase. synthesis and biological evaluation of dihydropyridin-2(1H)-imines and 1,5,6,7-tetrahydro-2H-azepin-2-imines. Bioorg. Med. Chem 2003, 11, 689–702. [DOI] [PubMed] [Google Scholar]
- (38).Sallmyr A; Fan J; Datta K; Kim KT; Grosu D; Shapiro P; Small D; Rassool F Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [DOI] [PubMed] [Google Scholar]
- (39).Olive PL; Banath JP The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc 2006, 1, 23–29. [DOI] [PubMed] [Google Scholar]
- (40).Redon CE; Nakamura AJ; Zhang YW; Ji JJ; Bonner WM; Kinders RJ; Parchment RE; Doroshow JH; Pommier Y Histone δH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res 2010, 16, 4532–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sung P; Robberson DL DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 1995, 82, 453–461. [DOI] [PubMed] [Google Scholar]
- (42).Sakurikar N; Eastman A Will targeting Chk1 have a role in the future of cancer therapy? J. Clin. Oncol 2015, 33, 1075–1077. [DOI] [PubMed] [Google Scholar]
- (43).Lai TH; Ewald B; Zecevic A; Liu C; Sulda M; Papaioannou D; Garzon R; Blachly JS; Plunkett W; Sampath D HDAC inhibition induces microRNA-182 which targets Rad51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin. Cancer Res 2016, 22, 3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Carew JS; Giles FJ; Nawrocki ST Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [DOI] [PubMed] [Google Scholar]
- (45).Dai Y; Chen S; Wang L; Pei XY; Kramer LB; Dent P; Grant S Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-κB and Bim. Br. J. Haematol 2011, 153, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Xargay-Torrent S; Lopez-Guerra M; Saborit-Villarroya I; Rosich L; Campo E; Roue G; Colomer D Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin. Cancer Res 2011, 17, 3956–3968. [DOI] [PubMed] [Google Scholar]
- (47).Rosato RR; Almenara JA; Kolla SS; Maggio SC; Coe S; Gimenez MS; Dent P; Grant S Mechanism and functional role of XIAP and Mcl-1 down-regulation in flavopiridol/vorinostat antileukemic interactions. Mol. Cancer Ther 2007, 6, 692–702. [DOI] [PubMed] [Google Scholar]
- (48).Quentmeier H; Zaborski M; Drexler HG The human bladder carcinoma cell line 5637 constitutively secretes functional cytokines. Leuk. Res 1997, 21, 343–350. [DOI] [PubMed] [Google Scholar]
- (49).Niu X; Zhao J; Ma J; Xie C; Edwards H; Wang G; Caldwell JT; Xiang S; Zhang X; Chu R; Wang J; Lin H; Taub JW; Ge Y Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin. Cancer Res 2016, DOI: 10.1158/1078-0432.CCR-15-3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhao J; Niu X; Li X; Edwards H; Wang G; Wang Y; Taub JW; Lin H; Ge Y Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget 2016, 7, 34785–34799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.