

Abstract
Structure-based drug design is critically dependent on accuracy of molecular docking scoring functions, and there is of significant interest to advance scoring functions with machine learning approaches. In this work, by judiciously expanding the training set, exploring new features related to explicit mediating water molecules as well as ligand conformation stability, and applying extreme gradient boosting (XGBoost) with Δ-Vina parametrization, we have improved robustness and applicability of machine-learning scoring functions. The new scoring function ΔvinaXGB can not only perform consistently among the top compared to classical scoring functions for the CASF-2016 benchmark but also achieves significantly better prediction accuracy in different types of structures that mimic real docking applications.
Graphical Abstract
1. Introduction
Molecular docking is a widely used computational approach that aims to predict binding pose and affinity for a small molecule to interact with a target protein and is playing an increasingly important role in structure-based drug design.1–3 A most critical component of molecular docking is its scoring function, and a robust scoring function should perform well across a variety of applications,4–13 including scoring (to estimate the binding affinity between a protein and a ligand given their complex structure), ranking (to rank the known ligands for a certain target protein when precise ligand binding poses provided), docking (to determine binding site and binding mode of a ligand on a protein), screening (to screen virtual small-molecule libraries and identify bioactive lead compound for further drug development). Extensive retrospective and comparative studies5–9, 14–25 have clearly demonstrated that those widely used classical scoring functions, such as Autodock Vina (Vina)26 and GlideScore,27–29 perform quite well in docking and screening tasks, but their scoring power remains to be less satisfactory. Meanwhile, most efforts aiming to improve scoring power, such as the development of X-Score30 and a variety of machine-learning based scoring functions,31–47 lead to significant under-performance in docking and screening tasks compared to classical scoring functions.48, 49 Although one can attempt to develop task-specific scoring functions using machine learning. i. e. different machine-learning scoring functions for distinct docking tasks,50, 51 such strategies can easily lead to overfitting due to the data limitation.52 In principle, a robust protein-ligand scoring function should perform well for a variety of different applications. Thus, this remains a challenge for the scoring function development to not only improve scoring accuracy, but also enhance robustness and applicability.
In order to tackle this challenge, recently we have introduced a Δ-Vina machine learning approach,53 in which machine learning is employed to parametrize corrections to the Vina scoring function.54 By taking advantage of the excellent docking power of Vina and the strength of random forest (RF) in efficiently utilizing large training data and feature sets, our previously developed scoring function ΔVinaRF2054 has improved scoring-docking-screening powers simultaneously and achieved superior performances in all power tests of CASF-2007,5 CASF-2013,6, 7 and CASF-20169 benchmarks compared to classical scoring functions.
Here we have explored the Δ-Vina machine-learning strategy via eXreme Gradient Boosting (XGBoost)55 in order to further enhance scoring accuracy, robustness and applicability of protein-ligand scoring functions. XGBoost is a scalable machine learning method for Gradient Boosting and has been demonstrated to achieve better performance than RF with much less computational cost.56, 57 Our main idea is to overcome two limitations which have not been addressed in the ΔVinaRF20 scoring function:
Explicit water molecules. Receptor-bound water (also known as ordered water networks) molecules play critical roles in protein-ligand recognition. Previous studies have indicated that more than 85% protein-ligand crystal structures have at least one water molecule in ligand binding site,58 and proper treatment of receptor-bound water molecules is beneficial for binding pose identification and binding affinity prediction.59–74 However, both Vina and ΔVinaRF20 scoring functions were parametrized with a training set where all explicit water molecules were removed from protein-ligand complex structures, and up to now this is also the case for all other machine-learning scoring functions. In this work, in order to properly treat explicit water molecules in our Δ-Vina machine-learning, we have judiciously expanded our training set to include protein-ligand complex structures with receptor-bound water molecules and explored mediating water related features. Our newly developed scoring function, ΔvinaXGB, has achieved superior and consistent performances in a series of tests using receptors with explicit water molecules in comparison with Vina and ΔVinaRF20, and hence it can take advantage of confident water predictions from water position analysis tools such as WaterMap73, 74 and WaterDock.62, 63
Ligand conformation stability. For a ligand molecule to adopt a complex-bound conformation, besides the change of intermolecular interactions and solvation, an internal free energy penalty may need to be paid, which originates from change of both ligand stability and flexibility. Previous studies have showed that the conformational energy changes of small molecules upon binding to target proteins can be in a range 0–25 kcal/mol75 and taking the ligand conformational energies into account can significantly improve the docking performance of classic scoring functions.59, 76, 77 The Vina scoring function employs one ligand-dependent term, the number of rotors, to approximate the free energy change due to the ligand flexibility, but does not explicitly consider ligand conformational energy change from the global minimum of the free ligand to the docked conformation. To our best knowledge, ligand conformation stability has not been considered in machine-learning scoring functions. In this work, we have explored new features to represent ligand conformation stability and demonstrated their importance in improving the accuracy and robustness of our machine-learning scoring function.
In this article, we describe the computational details for the development of our newly released model ΔvinaXGB, including data set preparation, feature generation, and algorithm application. ΔvinaXGB can not only perform consistently among the top compared to classical scoring functions in CASF-2016 benchmark, but also achieves significantly better prediction accuracy with Vina local optimized and flexible docked poses that mimic real docking applications. The ΔvinaXGB scoring function, code and data sets are freely available on the web at: http://www.nyu.edu/projects/yzhang/DeltaVina/
2. Methods
2.1. Data Preparation
Training Set.
As shown in Table 1, our training set is based on the PDBbind (v2016) database10 and CSAR data set,78, 79 and consists of three subsets: dry, water and decoy. For PDBbind (v2016) database, the high-quality PDBbind refined set (3<pKd<12) as well as weak binding complexes (0.4<pKd<3) selected from PDBbind general set (weak set) are used to construct our training set. To explore the effect of explicit water, we first analyzed the existence of receptor-bound water (RW) in these crystal structures. Water molecules in the crystal structure which are 2.0 to 3.5 Å away from protein polar atoms and possess theoretical binding affinities on receptor structure (Vina score < 0) are identified as RW. These requirements make sure that the RW are in proper positions and possess no clashes with protein. It should be mentioned that top-scored ligand poses usually adopt local minimum conformations, which might be slightly different from the conformation in crystal structure. Thus, we performed rigid local minimization of each selected complex using Vina local_search algorithm in the existence or absence of RW, which would better represent the real docking results. After removing complexes that possess distinct optimized ligand conformation compared with their native crystal structure (RMSD > 2 Å), dry subset and water subset have been built by 3264 optimized crystal structures and 3257 optimized crystal structures with RW , respectively. In addition, a total of 301 optimized protein-ligand complexes from the native pose of CSAR data set are also included in our dry set. The binding affinities of structures from water and dry are same as the experimental measured binding affinities (pKd(exp)) of their corresponding crystal structures. In terms of decoy set, which serves as a negative control of binding pose and binding affinity in the whole training set, a total of 7584 structures with estimated binding affinities (pKd(est)) are constructed using CSAR decoy set79 and PDBbind refined set. CSAR decoys are flexible docking poses generated by Zhou’s group79 using DOCK 4.0.180 based on crystal structures in CSAR-NRC HIQ benchmark release,78 and it includes up to 500 decoys for each complex. To balance the number of decoys and native poses in our training set, we calculated Vina scores for all CSAR decoys and only selected up to 21 decoys ranking at 0%, 5%, 10%, 25%, …, 95%, 100% for each protein-ligand complex based on Vina score. PDBbind decoys are rigid docking poses generated by us using Vina based on crystal strictures from PDBbind refined set. It includes up to 5 failed docking poses, which are docking poses with RMSD lager than and Vina scores lower than that of corresponding optimized crystal poses, for each of failed docking cases, where the top 1 docking pose is a failed docking pose. To determine the estimated binding affinity (pKd(est)), RMSD between decoys and crystal pose was first calculated, and if the RMSD is no larger than 1 Å, which means the decoy is similar as real bound pose, pKd(est) is assigned as the pKd(exp) of its corresponding native pose. For decoys with RMSD larger than 1 Å, pKd(Vina) was calculated and compared with the corresponding pKd(exp). If the pKd(Vina) is less than the pKd(exp), the pKd(est) of decoy is assigned as pKd(Vina); otherwise, pKd(est) is , and the pKd(est) is smaller when RMSD is larger. RMSD cutoff as 1 and 2 Å have been tested, and 1 Å can achieve better performance on Validation set. The complexes in the PDBbind (v2016) database released after 2015 as well as those overlap with the CASF (2013 and 2016) benchmark, which serve as a validation set and a test set, respectively, are excluded from our training set. With the addition of a receptor-bound water set and generation of a decoy set, our training set consists of 14406 complexes, which is significantly larger than the training set of ΔVinaRF20 (6658).
Table 1.
Data Sets.
| name | subset | source | year | size | typea | BA | note |
|---|---|---|---|---|---|---|---|
| Training | dry | PDBbind (v2016) +CSAR | before 2015 | 3565 | pKd(exp) | no water | |
| water | 3257 | with receptor-bound waters | |||||
| decoy | 7584 | pKd(est) | CSAR decoys and PDBbind decoys | ||||
| Validation | dry | PDBbind (v2016) | after 2015 | 315 | pKd(exp) | constructed by time-split and used to do early stop and model selection | |
| water | 315 | ||||||
| crystal | 316 | C | |||||
| LocalOpt | dry | CASF-2016 | 285 | pKd(exp) | constructed by Vina local minimization and used to evaluate scoring, ranking, screening power for optimized poses | ||
| water | 285 | ||||||
| decoy | 1624500 | ||||||
| FlexDock | dry | CASF-2016 | 5985 | pKd(exp) | constructed by Vina flexible docking and used to evaluate scoring and ranking power for docking poses | ||
| water | 5985 | ||||||
C: crystal structure, : optimized crystal structure, : optimized crystal structure with receptor-bound water, : docking poses, : docking poses with receptor-bound water.
Validation Set.
The validation set is constructed using structures from PDBbind (v2016) refined set and weak set that are released after 2015, which includes 316 complexes with three different structure types: C, , and . The reasons to build the time-split holdout validation set are (1) conducting the early stopping in model training to avoid the overfitting of XGBoost on training set; (2) selecting a model that can perform well on different structure types; (3) mimicking the potential lead-compound discovery process in docking-based drug design.
Test Set.
CASF-2016 benchmark9 has been used to evaluate the performances of our scoring function. CASF-2016 defines four different powers (scoring power, ranking power, docking power, and screening power) and provides corresponding test sets. To further test the postdocking rescoring performance and account for the explicit water effect, two subsets, noted as LocalOpt and FlexDock (Table 1), have been generated to mimic the poses needed to be rescored in real application. In the basis of crystal structures and computer-generated cross-docking poses from CASF-2016, optimized poses of LocalOpt are obtained by Vina local optimization with different environments: without water molecules and with explicit RW. Similarly, the FlexDock set is composed of docking poses produced by Vina flexible docking using crystal structures from CASF-2016. Each complex has one crystal structure and 20 docking poses from docking situations without or with explicit RW . Symmetry corrected RMSD between docking pose and crystal structure is calculated using Huangarian algorithm from Dock6.81–83
All structures from PDBbind(v2016) database are prepared using our data preparation schedule (Figure S1). The pKa has been determined via PROPKA 3.184, 85 in order to predict the protonation state of protein titratable residues.
2.2. Feature Generation
All features employed in our development of score function are summarized in Table S1. Same as ΔVinaRF20, 58 features from Vina source code are calculated, including protein-ligand interaction terms and a set of ligand property counts.86 A total of 30 buried solvent accessible surface area (bSASA) features are computed using three different structures (complex, ligand and protein), and 10 bSASA features for each structure consist of one total bSASA term and nine pharmacophore-based bSASA terms where pharmacophore types are characterized based on SYBYL87 atom types and DOCK88 neighboring atoms. MSMS89 program is used to calculate the atomic SASAs with a probe radius and the bSASA = SASAunbound − SASAcomplex.
To explore the water effect, certain RW that mediate protein-ligand interactions are considered as bridging water (BW) molecules according to the following criteria: (1) contact with both ligand and protein, the distances between polar atoms in ligand and BW are between 2.0 and ; (2) potential to form hydrogen bond networks, the angles between polar atoms in ligand, BW, and polar atoms in protein are no less than 60 degrees; (3) favorable for ligand binding, Vina score for BW are less than zero when using ligand as receptor. Finally, three BW features are calculated for the structures with RW, including the number of BW (NBW), the Vina score between BW and protein (Vina (P, BW)), and the Vina score between BW and ligand (Vina(L, BW)).
On the other hand, our feature set contains two features related to ligand stability in order to represent the ligand conformation change during the binding process. For each ligand, a maximum of 1000 conformers were generated using RDKit,90 and redundant conformers were removed on the basis of clustering analysis. The docked or crystal ligand conformer as well as newly generated conformers are minimized using MMFF91, 92 force filed with implicit water as solvent. The energy difference and RMSD between the conformer with lowest energy and the locally minimized docked/crystal conformer are designated as the feature ΔEconfs and RMSDconfs, which is assumed to reflect the stability of each docked/crystal ligand.
Furthermore, Nion, which is the number of binding site ions (distances between ions and polar atoms of ligand are less than ) is also included in our feature set.
2.3. Δ-Vina XGBoost Strategy
The Δ-Vina XGBoost strategy aims to correct the difference between Vina score and experimental binding affinity by XGBoost. The original Vina score with unit kcal/mol was converted to pKd using following formula: pKd(vina) = −0.73349E(Vina). Thus, the formula of our scoring function can be shown as the equation below:
Given a molecule i from the training set with an input feature vector , a XGBoost model uses K additive trees to predict the output and each tree corrects the difference between target and predictions made by all of the previous trees. Τhus
Here, is the space of all possible trees and λ is a shrinkage parameter, which used to reduce the overfitting and make the sequential training meaningful. The growing of each is rely on a regularized objective as follows:
where l is the loss function to compute the difference between the prediction and target , and the second regularized term Ω controls the complexity of the model via the number of leaves in and the scores on leaves . The feature, which can minimize this objective, will be chosen to split the terminal node of the tree into two child nodes, and only a random subset of features can be considered in each tree development to avoid the high correlation of trees. With the help of validation set, the early stopping with the patience of 50 training steps has been applied.
In our development of ΔvinaXGB, the output y for our training set is the ΔpKd and for the input feature vector x. The XGBoost package (version 0.80)55 in Python 3 is used to build the XGBoost model. The hyper-parameters utilized in current study are n_estimators (number of trees) = 500, learning_rate (shrinkage parameter) = 0.05, subsample (subsample ratio of the training instance when constructing each tree) = 0.8, colsample_bytree (subsample ratio of features when constructing each tree) = 0.8, min_child_weight (minimum sum of instance weight needed in a tree leaf node) = 6, max_depth (maximum depth of a tree) = 10, and objective (regression type) = “reg:linear”. Because of the high variance of each XGBoost model, the final prediction of our ΔvinaXGB model is the average of ten XGBoost models initialized with different random seed.
2.4. Evaluation Methods:
The comparative assessment of scoring functions (CASF) benchmark has been designed and updated by Wang’s group5–7, 9 and they defined four different powers (scoring power, ranking power, docking power and screening power) to evaluate the performance of scoring function in the CASF-2016, which is the latest version of CASF benchmark. Scoring power is aimed to evaluate the linear correlation between predicted binding affinity and experimental measured binding affinity by the classic Pearson’s correlation coefficient (R) between binding affinity and experimental measured binding affinity and the standard deviation (SD) in regression. Docking power is used to evaluate the ability of a scoring function to identify the native binding pose among computer-generated decoys and it calculates the success rate based on symmetry-corrected RMSDs between native binding pose and decoys. The top 1 docking success rate is computed as the percentage of complexes that have top-scored poses similar with native poses among all testing complexes. Ranking power is to check the ability of a scoring function to rank the activities of known ligands correctly using their predicted binding affinity. The CASF-2016 benchmark includes 57 targets and 5 known active ligands for each target. With the known active ligands for each target, ranking performances including Spearman’s rank correlation coefficient , Kendall’s rank correlation coefficient , and the predictive index (PI) are computed, and the overall performances for all targets (average) can indicate the ranking ability of a scoring function. Screening power is aimed to assess the ability of a scoring function to identify true binder for a given target among a pool of random molecules. The CASF-2016 provides true binders for each target and in screening power test, all ligands were cross docked to all target proteins and 100 docking poses were selected for each protein-ligand complex. Two indicators including enhancement factor and success rate are used for evaluating screening power. The enhancement factor is computed as flowing:
Here, is the number of true binders in top ranked candidates based on predicted binding scores. is the total number of true binders for the given target protein. Meanwhile, success rate at Top1% level is calculated as the percentage of tightest true binders in 1% of top-ranked molecules.
Besides standard test sets provided by CASF-2016 benchmark, we also evaluated the postdocking rescoring performance using two additional test sets: LocalOpt and FlexDock, as shown in Table 1. Scoring power, ranking power, and screening power defined by CASF-2016 have been reevaluated in LocalOpt for ΔvinaXGB, ΔVinaRF20, and Vina to estimate the scoring performance in local optimized poses with different water environments. Similarly, scoring correlation and ranking correlation were checked for best-scored pose from FlexDock for ΔvinaXGB, ΔVinaRF20, and Vina to show the scoring function’s performance on real docking poses.
3. Results and Discussion
Based on a judiciously enlarged training set which consists of 14406 protein-ligand complex structures and an enlarged feature set including new features accounting for explicit water effect and ligand stability, a new scoring function ΔvinaXGB was developed by incorporating XGBoost to Δ-Vina machine learning approach.
3.1. CASF-2016 Benchmark
The performances of ΔvinaXGB were tested on CASF-2016 benchmark to evaluate its scoring power, ranking power, docking power as well as screening power. In the original CASF-2016 paper,9 ΔvinaRF20 has been evaluated and their results show that ΔvinaRF20 can achieve superior performances on all tasks. However, the training set of the published ΔvinaRF20 that they tested consists of 140 molecules that have been included in the test set of CASF-2016. To avoid the bias due to the overlap between training set and test set, we retrained the ΔvinaRF20 using training set excluding CASF-2016 structures and checked the new ΔvinaRF20 performance on CASF-2016. All of the performances of ΔvinaRF20 shown here are from the retrained ΔvinaRF20.
As shown in Figure 1, ΔvinaXGB achieves Top 1 performances on scoring power, ranking power, docking power and the enhancement factor of screening power tasks. For the scoring power, the Pearson’s correlation coefficient (R) and standard deviation (SD) of ΔvinaXGB are 0.796 and 1.32, which are both better than ΔvinaRF20 and baseline model Vina, as shown in Figure 1A. Figure 2 also indicates that ΔvinaXGB has smallest number of cases with large errors (absolute error > 2). In ranking power, three quantitative indicators including Spearman’s rank correlation coefficient , Kendall’s rank correlation coefficient , and the predictive index (PI) are used to evaluate scoring functions. Similar as scoring power, ΔvinaXGB achieves highest values on , , and PI, which are 0.647, 0.568 and 0.674. In docking power, the success rate of best-scored pose with crystal structures has been shown in Figure 1C, and ΔvinaXGB’s performance is also ranked as top 1. Enhancement factor and success rate at top 1% level are calculated for screening power. As shown in Figure 1D, E, ΔvinaXGB achieves highest enhancement factor (13.14) and second highest success rate (36.8%, same as GlideScore-SP). Best scoring function in success rate of screening power is ΔvinaRF20 at top 1% level, and ΔvinaXGB can achieve better performance (ΔvinaXGB: 61.4% vs. ΔvinaRF20: 59.6%) when computing the success rate at the top 10% level.
Figure 1.

Performances of scoring functions on CASF-2016 benchmark. (A) Scoring power measured by Pearson correlation coefficient, (B) ranking power in terms of spearman correlation coefficient, (C) docking power measured by success rate for best-scored poses with crystal structures, and screening power measured by (D) enhancement factor and (E) success rate at top 1% level. All scoring functions are ranked in a descending order. ΔvinaXGB’s performances are colored as red and other scoring functions’ performances are colored as green.
Figure 2.

Scatter plots between experimental pKd and different scoring functions: (A) Vina predicted scores, (B) ΔvinaRF20 predicted scores, and (C) ΔvinaXGB predicted scores. The absolute error in pKd larger than 2 are colored as green, and in other cases are colored as orange. Mean absolute errors and root-mean-square errors between predicted scores and experimental pKd for three scoring functions have also been shown.
Based on crystal structures provided by CASF-2016, we also analyzed the performance of ΔvinaXGB and Vina with consideration of ligand stability and flexibility. As shown in Table 2 and Figure S3, Vina performs well for relatively rigid molecules but deteriorates significantly for highly flexible molecules. On the other hand, the performance of ΔvinaXGB is more stable which demonstrates its robustness. Furthermore, RMSDconfs and ΔEconfs, which are two features added to describe ligand stability, are ranked as top 1 and 2 among 94 features in feature importance analysis result (Figure S2). It should be noted that the accuracy of two ligand stability features is dependent on ligand conformation generation and ligand energy calculation, which may encounter difficulties for ligands with large number of rotors or large heterorings, as illustrated in Table S2, and can be improved by using deep learning methods in energy calculation.93
Table 2.
Scoring Performance of ΔvinaXGB and Vina on Different Ligand Stability and Flexibility Subsets of 285 Crystal Structures from CASF-2016.
| subset | ligand stabilitya | ligand flexibilitya | number | Vina | XGB |
|---|---|---|---|---|---|
| High | or | Nrot > 5 | 86 | 0.498 | 0.719 |
| Medium | Structures are not in low and high subset | 106 | 0.541 | 0.784 | |
| Low | and | 93 | 0.708 | 0.816 | |
All complexes in each subset should satisfy both ligand stability and flexibility requirements.
3.2. Rescoring Performance
ΔvinaXGB is mainly developed for rescoring, which needs docked poses as input. Vina is a widely used docking software and its good docking power has been validated in CASF benchmarks. To evaluate the performance of ΔvinaXGB as a rescoring tool for complex structures that mimic real docking applications, we employed Vina local optimization and flexible docking to generate potential docked ligand poses. Here, both docked poses with and without receptor-bound water molecules are considered, and results are compared with both Vina and ΔvinaRF20.
3.2.1. Local Optimized Poses (LocalOpt)
LocalOpt data set has been built by Vina rigid local optimization based on crystal structures and computer-generated decoys from CASF-2016. Scoring power, ranking power, and screening power defined by CASF-2016 have been re-evaluated on LocalOpt for ΔvinaXGB, ΔvinaRF20 and Vina. In order to accounting for explicit water effect, we also considered protein-ligand complexes which include receptor-bound water molecules.
Figure 3A,B illustrates the scoring and ranking performance on LocalOpt ( and ), together with performance on crystal structures without local optimization . For locally optimized protein-ligand structures, ΔvinaXGB’s performance has been improved from 0.796 to 0.809 in Pearson’s R. In Spearman’s R, the performances of ΔvinaXGB on C (0.647) and are similar. When keeping explicit water molecules, ΔvinaXGB can achieve 0.757 scoring correlation and 0.614 ranking correlation, which are close to the performances of crystal structures. However, ΔvinaRF20’s performance decreases significantly when it comes to locally optimized poses, especially for structures with water molecules, where Pearson’s R decreases from 0.732 to 0.626 and Spearman’s R decreases from 0.626 to 0.547 for . Both ΔvinaXGB and ΔvinaRF20 have improved over Vina, whose performance is relatively stable on different types of structures.
Figure 3.

Performances of ΔvinaXGB, ΔvinaRF20 and Vina on LocalOpt. (A) Pearson correlation coefficient for scoring power. (B) Spearman correlation coefficient for ranking power. (C) Enhancement factor and (D) success rate for screening power at top 1% level. Performances of ΔvinaXGB, ΔvinaRF20, and Vina are colored as orange, green, and blue. For each scoring function, performance on crystal structures (C, shown as reference), local optimized poses , and local optimized poses with receptor-bound water molecules are displayed from left to right with gradually changed color.
In terms of screening power, local optimizations for computer generated cross-docking poses have been conducted with/without receptor water molecules. Similar as scoring power, ΔvinaXGB achieves much higher enhancement factor and success rate at top 1% level after local optimization compared with ΔvinaRF20 and Vina, as shown in Figure 3C,D. One interesting thing is that all scoring functions reach the best performance on , except success rate of ΔvinaRF20, which is slightly worse than performance on C. Here ΔvinaXGB achieves highest enhancement factor as 16.74 and success rate as 58%. These results suggest the importance of considering receptor-bound water molecules in virtual screening.
3.2.2. Flexible Docking Poses (FlexDock)
To further test rescoring performances on real docking poses, we conducted flexible docking using Vina for CASF-2016 test set, and 20 poses were generated for each complex. Similar as in LocalOpt, we also considered the docking environment with receptor-bound water molecules. In the evaluation process, all docking poses as well as crystal structure poses are rescored by ΔvinaRF20 and ΔvinaXGB.
The flexible docking success rates of best-scored pose in different docking situations and RMSD cutoff have been illustrated in Figure S4. Only the best-scored pose with RMSD less than RMSD cutoff is considered as a successful docking case. It is clear that Vina always achieves best docking performance among three scoring functions when RMSD cutoff is ; however, the performances of three scoring functions are very close and the largest difference is only 2% in dry docking and 3% in docking with receptor-bound water molecules. All scoring function’s performances have been improved significantly after keeping RW, which demonstrates the importance of explicit water molecules in molecular docking. In addition, ΔvinaXGB can achieve better docking performance than Vina and ΔvinaRF20 when RMSD cutoff is less than .
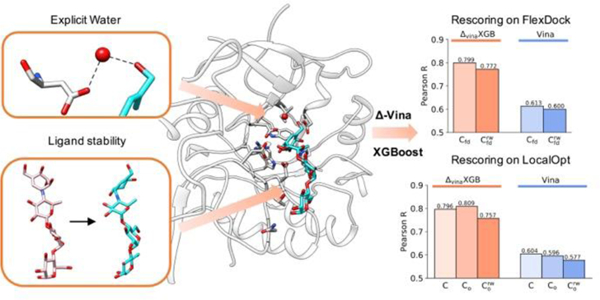
To evaluate the rescoring performance of ΔvinaRF20 and ΔvinaXGB on flexible docking poses, we ranked docking poses based on predicted score and calculated the scoring and ranking correlation coefficient for best-scored poses. Figure 4A shows the scoring correlation, and both ΔvinaRF20 and ΔvinaXGB improve the initial Vina performance significantly in dry and water docking situations. ΔvinaXGB achieves the best performances, which are 0. 799 on and 0.772 on in Pearson’s R, and are close to the scoring performance on crystal structures. Rescoring using only Vina best-scored pose for each structure has also been conducted. Different from choosing best-scored poses from 20 poses generated by Vina, selecting Vina best-scored pose to rescore is an efficient rescoring method and is more convenient for Vina users. As expected, the performances of scoring have been improved a lot by ΔvinaXGB on and (Figure 4(B)). Similar performance improvements by ΔvinaXGB have also been found in ranking correlations (Figure S5). These good performances indicate the potential application of ΔvinaXGB in Vina-based rescoring, where using Vina to generate docking poses, and rescoring all docking poses or even top 1 pose to get more accurate binding affinity prediction using ΔvinaXGB. Based on the script we provide, Vina docking poses can be directly used as input structures to be rescored using ΔvinaXGB.
Figure 4.

Scoring performances of ΔvinaXGB, ΔvinaRF20, and Vina on FlexDock. Pearson correlation coefficient for (A) best-scored docking poses and (B) Vina best-scored docking pose in dry environment and water environment . Here, best-scored poses are determined based on predicted scores. Performances of Vina, ΔvinaRF20, and ΔvinaXGB are colored as blue, green, and orange.
4. Conclusion
Explicit water molecules and ligand conformation stability are two issues that have not been considered in our previous scoring function ΔvinaRF20. In this work, we have further explored the Δ-Vina machine-learning strategy via XGBoost and addressed these two limitations by judiciously expanding our training set with receptor-bound water molecules and enlarging the feature set with bridging water and ligand stability features. Our newly developed scoring function ΔvinaXGB achieves superior performance compared with ΔvinaRF20 and Vina in two different level evaluations, including four power tests in CASF-2016 benchmark and rescoring performance for Vina optimized poses and flexible docking poses with or without explicit water molecules. This work suggests that ΔvinaXGB significantly improved both robustness and applicability of machine-learning scoring functions and could serve as an effective rescoring tool in structure-based inhibitor design.
Supplementary Material
Figure S1: Data preparation schedule for PDBbind(v2016) database.
Figure S2: Feature importance.
Figure S3: MAE of Vina and ΔvinaXGB for structures with different number of rotors.
Figure S4: Success rates of best-scored pose in different RMSD values.
Figure S5. Ranking performances of ΔvinaXGB, ΔvinaRF20 and Vina on FlexDock
Table S1: Feature set of ΔvinaXGB.
Table S2: Failed conformation generation cases in CASF-2016.
Acknowledgements
We would like to acknowledge the support by NIH (R35-GM127040) and computational resources provided by NYU-ITS.
Reference
- 1.Forli S; Huey R; Pique ME; Sanner MF; Goodsell DS; Olson AJ, Computational Protein-Ligand Docking and Virtual Drug Screening with the Autodock Suite. Nat. Protoc 2016, 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irwin JJ; Shoichet BK, Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem 2016, 59, 4103–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ, Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guedes IA; Pereira FSS; Dardenne LE, Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol 2018, 9, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng TJ; Li X; Li Y; Liu ZH; Wang RX, Comparative Assessment of Scoring Functions on a Diverse Test Set. J. Chem. Inf. Model 2009, 49, 1079–1093. [DOI] [PubMed] [Google Scholar]
- 6.Li Y; Han L; Liu ZH; Wang RX, Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model 2014, 54, 1717–1736. [DOI] [PubMed] [Google Scholar]
- 7.Li Y; Liu ZH; Li J; Han L; Liu J; Zhao ZX; Wang RX, Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model 2014, 54, 1700–1716. [DOI] [PubMed] [Google Scholar]
- 8.Li Y; Su MY; Liu ZH; Li J; Liu J; Han L; Wang RX, Assessing Protein-Ligand Interaction Scoring Functions with the Casf-2013 Benchmark. Nat. Protoc 2018, 13, 666–680. [DOI] [PubMed] [Google Scholar]
- 9.Su MY; Yang QF; Du Y; Feng GQ; Liu ZH; Li Y; Wang RX, Comparative Assessment of Scoring Functions: The Casf-2016 Update. J. Chem. Inf. Model 2019, 59, 895–913. [DOI] [PubMed] [Google Scholar]
- 10.Liu ZH; Su MY; Han L; Liu J; Yang QF; Li Y; Wang RX, Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions. Acc. Chem. Res 2017, 50, 302–309. [DOI] [PubMed] [Google Scholar]
- 11.Gohlke H; Hendlich M; Klebe G, Knowledge-Based Scoring Function to Predict Protein-Ligand Interactions. J. Mol. Biol 2000, 295, 337–356. [DOI] [PubMed] [Google Scholar]
- 12.Leach AR; Shoichet BK; Peishoff CE, Prediction of Protein-Ligand Interactions. Docking and Scoring: Successes and Gaps. J. Med. Chem 2006, 49, 5851–5855. [DOI] [PubMed] [Google Scholar]
- 13.Huang SY; Grinter SZ; Zou XQ, Scoring Functions and Their Evaluation Methods for Protein-Ligand Docking: Recent Advances and Future Directions. Phys. Chem. Chem. Phys 2010, 12, 12899–12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J; Wang RX, Classification of Current Scoring Functions. J. Chem. Inf. Model 2015, 55, 475–482. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara P; Gohlke H; Price DJ; Klebe G; Brooks CL, Assessing Scoring Functions for Protein-Ligand Interactions. J. Med. Chem 2004, 47, 3032–3047. [DOI] [PubMed] [Google Scholar]
- 16.Gaieb Z; Liu S; Gathiaka S; Chiu M; Yang HW; Shao CH; Feher VA; Walters WP; Kuhn B; Rudolph MG; Burley SK; Gilson MK; Amaro RE, D3r Grand Challenge 2: Blind Prediction of Protein-Ligand Poses, Affinity Rankings, and Relative Binding Free Energies. J. Comput. Aided Mol. Des 2018, 32, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gathiaka S; Liu S; Chiu M; Yang HW; Stuckey JA; Kang YN; Delproposto J; Kubish G; Dunbar JB; Carlson HA; Burley SK; Walters WP; Amaro RE; Feher VA; Gilson MK, D3r Grand Challenge 2015: Evaluation of Protein-Ligand Pose and Affinity Predictions. J. Comput. Aided Mol. Des 2016, 30, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halperin I; Ma BY; Wolfson H; Nussinov R, Principles of Docking: An Overview of Search Algorithms and a Guide to Scoring Functions. Proteins 2002, 47, 409–443. [DOI] [PubMed] [Google Scholar]
- 19.Kim R; Skolnick J, Assessment of Programs for Ligand Binding Affinity Prediction. J. Comput. Chem 2008, 29, 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsden PM; Puvanendrampillai D; Mitchell JBO; Glen RC, Predicting Protein-Ligand Binding Affinities: A Low Scoring Game? Org. Biomol. Chem 2004, 2, 3267–3273. [DOI] [PubMed] [Google Scholar]
- 21.Perola E; Walters WP; Charifson PS, A Detailed Comparison of Current Docking and Scoring Methods on Systems of Pharmaceutical Relevance. Proteins 2004, 56, 235–249. [DOI] [PubMed] [Google Scholar]
- 22.Plewczynski D; Lazniewski M; Augustyniak R; Ginalski K, Can We Trust Docking Results? Evaluation of Seven Commonly Used Programs on Pdbbind Database. J. Comput. Chem 2011, 32, 742–755. [DOI] [PubMed] [Google Scholar]
- 23.Warren GL; Andrews CW; Capelli AM; Clarke B; LaLonde J; Lambert MH; Lindvall M; Nevins N; Semus SF; Senger S; Tedesco G; Wall ID; Woolven JM; Peishoff CE; Head MS, A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem 2006, 49, 5912–5931. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z; Sun HY; Yao XJ; Li D; Xu L; Li YY; Tian S; Hou TJ, Comprehensive Evaluation of Ten Docking Programs on a Diverse Set of Protein-Ligand Complexes: The Prediction Accuracy of Sampling Power and Scoring Power. Phys. Chem. Chem. Phys 2016, 18, 12964–12975. [DOI] [PubMed] [Google Scholar]
- 25.Gaillard T, Evaluation of Autodock and Autodock Vina on the Casf-2013 Benchmark. J. Chem. Inf. Model 2018, 58, 1697–1706. [DOI] [PubMed] [Google Scholar]
- 26.Trott O; Olson AJ, Software News and Update Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 28.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL, Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 30.Wang RX; Lai LH; Wang SM, Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J. Comput. Aided Mol. Des 2002, 16, 11–26. [DOI] [PubMed] [Google Scholar]
- 31.Zilian D; Sotriffer CA, Sfcscore(Rf): A Random Forest-Based Scoring Function for Improved Affinity Prediction of Protein-Ligand Complexes. J. Chem. Inf. Model 2013, 53, 1923–1933. [DOI] [PubMed] [Google Scholar]
- 32.Li GB; Yang LL; Wang WJ; Li LL; Yang SY, Id-Score: A New Empirical Scoring Function Based on a Comprehensive Set of Descriptors Related to Protein-Ligand Interactions. J. Chem. Inf. Model 2013, 53, 592–600. [DOI] [PubMed] [Google Scholar]
- 33.Ragoza M; Hochuli J; Idrobo E; Sunseri J; Koes DR, Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jimenez J; Skalic M; Martinez-Rosell G; De Fabritiis G, K-Deep: Protein-Ligand Absolute Binding Affinity Prediction Via 3d-Convolutional Neural Networks. J. Chem. Inf. Model 2018, 58, 287–296. [DOI] [PubMed] [Google Scholar]
- 35.Ain QU; Aleksandrova A; Roessler FD; Ballester PJ, Machine-Learning Scoring Functions to Improve Structure-Based Binding Affinity Prediction and Virtual Screening. WIREs Comput Mol Sci 2015, 5, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wójcikowski M; Ballester PJ; Siedlecki P, Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep 2017, 7, 46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ballester PJ; Mitchell JBO, A Machine Learning Approach to Predicting Protein-Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li HJ; Leung KS; Wong MH; Ballester PJ, Substituting Random Forest for Multiple Linear Regression Improves Binding Affinity Prediction of Scoring Functions: Cyscore as a Case Study. BMC Bioinformatics 2014, 15, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durrant JD; McCammon JA, Nnscore: A Neural-Network-Based Scoring Function for the Characterization of Protein-Ligand Complexes. J. Chem. Inf. Model 2010, 50, 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durrant JD; McCammon JA, Nnscore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf. Model 2011, 51, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feinberg EN; Sur D; Wu ZQ; Husic BE; Mai HH; Li Y; Sun SS; Yang JY; Ramsundar B; Pande VS, Potentialnet for Molecular Property Prediction. ACS Cent. Sci 2018, 4, 1520–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavecchia A, Machine-Learning Approaches in Drug Discovery: Methods and Applications. Drug Discov. Today 2015, 20, 318–331. [DOI] [PubMed] [Google Scholar]
- 43.Li HJ; Leung KS; Wong MH; Ballester PJ, Improving Autodock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inform 2015, 34, 115–126. [DOI] [PubMed] [Google Scholar]
- 44.Sunseri J; Ragoza M; Collins J; Koes DR, A D3r Prospective Evaluation of Machine Learning for Protein-Ligand Scoring. J. Comput. Aided Mol. Des 2016, 30, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunseri J; King JE; Francoeur PG; Koes DR, Convolutional Neural Network Scoring and Minimization in the D3r 2017 Community Challenge. J. Comput. Aided Mol. Des 2019, 33, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen DD; Cang ZX; Wu KD; Wang ML; Cao Y; Wei GW, Mathematical Deep Learning for Pose and Binding Affinity Prediction and Ranking in D3r Grand Challenges. J. Comput. Aided Mol. Des 2019, 33, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomes J; Ramsundar B; Feinberg EN; Pande VS Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity. arXiv.org e-Print archive2017; https://ui.adsabs.harvard.edu/abs/2017arXiv170310603G (accessed March 01, 2017).
- 48.Gabel J; Desaphy J; Rognan D, Beware of Machine Learning-Based Scoring Functions-on the Danger of Developing Black Boxes. J. Chem. Inf. Model 2014, 54, 2807–2815. [DOI] [PubMed] [Google Scholar]
- 49.Ballester PJ; Schreyer A; Blundell TL, Does a More Precise Chemical Description of Protein-Ligand Complexes Lead to More Accurate Prediction of Binding Affinity? J. Chem. Inf. Model 2014, 54, 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashtawy HM; Mahapatra NR, Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model 2018, 58, 119–133. [DOI] [PubMed] [Google Scholar]
- 51.Kinnings SL; Liu NN; Tonge PJ; Jackson RM; Xie L; Bourne PE, A Machine Learning-Based Method to Improve Docking Scoring Functions and Its Application to Drug Repurposing. J. Chem. Inf. Model 2011, 51, 1195–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y; Yang JY, Structural and Sequence Similarity Makes a Significant Impact on Machine-Learning-Based Scoring Functions for Protein-Ligand Interactions. J. Chem. Inf. Model 2017, 57, 1007–1012. [DOI] [PubMed] [Google Scholar]
- 53.Ramakrishnan R; Dral PO; Rupp M; von Lilienfeld OA, Big Data Meets Quantum Chemistry Approximations: The Delta-Machine Learning Approach. J. Chem. Theory Comput 2015, 11, 2087–2096. [DOI] [PubMed] [Google Scholar]
- 54.Wang C; Zhang YK, Improving Scoring-Docking-Screening Powers of Protein-Ligand Scoring Functions Using Random Forest. J. Comput. Chem 2017, 38, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen TQ; Carlson G Xgboost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, August 13–17, 2016; Krishnapuram B; Shah A; Aggarwal C; Shen D; Rastogi R, Eds. ACM: San Francisco, CA, USA, 2016; pp 785–794. [Google Scholar]
- 56.Sheridan RP; Wang WM; Liaw A; Ma JS; Gifford EM, Extreme Gradient Boosting as a Method for Quantitative Structure-Activity Relationships. J. Chem. Inf. Model 2016, 56, 2353–2360. [DOI] [PubMed] [Google Scholar]
- 57.Ericksen SS; Wu HZ; Zhang HK; Michael LA; Newton MA; Hoffmann FM; Wildman SA, Machine Learning Consensus Scoring Improves Performance across Targets in Structure-Based Virtual Screening. J. Chem. Inf. Model 2017, 57, 1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu YP; Wang RX; Yang CY; Wang SM, Analysis of Ligand-Bound Water Molecules in High-Resolution Crystal Structures of Protein-Ligand Complexes. J. Chem. Inf. Model 2007, 47, 668–675. [DOI] [PubMed] [Google Scholar]
- 59.Murphy RB; Repasky MP; Greenwood JR; Tubert-Brohman I; Jerome S; Annabhimoju R; Boyles NA; Schmitz CD; Abel R; Farid R; Friesner RA, Wscore: A Flexible and Accurate Treatment of Explicit Water Molecules in Ligand-Receptor Docking. J. Med. Chem 2016, 59, 4364–4384. [DOI] [PubMed] [Google Scholar]
- 60.Breiten B; Lockett MR; Sherman W; Fujita S; Al-Sayah M; Lange H; Bowers CM; Heroux A; Krilov G; Whitesides GM, Water Networks Contribute to Enthalpy/Entropy Compensation in Protein-Ligand Binding. J. Am. Chem. Soc 2013, 135, 15579–15584. [DOI] [PubMed] [Google Scholar]
- 61.Bortolato A; Tehan BG; Bodnarchuk MS; Essex JW; Mason JS, Water Network Perturbation in Ligand Binding: Adenosine a(2a) Antagonists as a Case Study. J. Chem. Inf. Model 2013, 53, 1700–1713. [DOI] [PubMed] [Google Scholar]
- 62.Ross GA; Morris GM; Biggin PC, Rapid and Accurate Prediction and Scoring of Water Molecules in Protein Binding Sites. PLoS ONE 2012, 7, e32036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sridhar A; Ross GA; Biggin PC, Waterdock 2.0: Water Placement Prediction for Holo-Structures with a Pymol Plugin. PLoS ONE 2017, 12, e0172743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lie MA; Thomsen R; Pedersen CNS; Schiott B; Christensen MH, Molecular Docking with Ligand Attached Water Molecules. J. Chem. Inf. Model 2011, 51, 909–917. [DOI] [PubMed] [Google Scholar]
- 65.Roberts BC; Mancera RL, Ligand-Protein Docking with Water Molecules. J. Chem. Inf. Model 2008, 48, 397–408. [DOI] [PubMed] [Google Scholar]
- 66.Thilagavathi R; Mancera RL, Ligand-Protein Cross-Docking with Water Molecules. J. Chem. Inf. Model 2010, 50, 415–421. [DOI] [PubMed] [Google Scholar]
- 67.Huggins DJ; Tidor B, Systematic Placement of Structural Water Molecules for Improved Scoring of Proteinligand Interactions. Protein Eng Des Sel 2011, 24, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verdonk ML; Chessari G; Cole JC; Hartshorn MJ; Murray CW; Nissink JWM; Taylor RD; Taylor R, Modeling Water Molecules in Protein-Ligand Docking Using Gold. J. Med. Chem 2005, 48, 6504–6515. [DOI] [PubMed] [Google Scholar]
- 69.Ladbury JE, Just Add Water! The Effect of Water on the Specificity of Protein-Ligand Binding Sites and Its Potential Application to Drug Design. Chem Biol 1996, 3, 973–980. [DOI] [PubMed] [Google Scholar]
- 70.Liu JF; He X; Zhang JZH, Improving the Scoring of Protein-Ligand Binding Affinity by Including the Effects of Structural Water and Electronic Polarization. J. Chem. Inf. Model 2013, 53, 1306–1314. [DOI] [PubMed] [Google Scholar]
- 71.Garcia-Sosa AT, Hydration Properties of Ligands and Drugs in Protein Binding Sites: Tightly-Bound, Bridging Water Molecules and Their Effects and Consequences on Molecular Design Strategies. J. Chem. Inf. Model 2013, 53, 1388–1405. [DOI] [PubMed] [Google Scholar]
- 72.de Beer SBA; Vermeulen NPE; Oostenbrink C, The Role of Water Molecules in Computational Drug Design. Curr. Top. Med. Chem 2010, 10, 55–66. [DOI] [PubMed] [Google Scholar]
- 73.Abel R; Young T; Farid R; Berne BJ; Friesner RA, Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc 2008, 130, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Young T; Abel R; Kim B; Berne BJ; Friesner RA, Motifs for Molecular Recognition Exploiting Hydrophobic Enclosure in Protein-Ligand Binding. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sitzmann M; Weidlich IE; Filippov IV; Liao C; Peach ML; Ihlenfeldt WD; Karki RG; Borodina YV; Cachau RE; Nicklaus MC, Pdb Ligand Conformational Energies Calculated Quantum-Mechanically. J. Chem. Inf. Model 2012, 52, 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nivedha AK; Makeneni S; Foley BL; Tessier MB; Woods RJ, Importance of Ligand Conformational Energies in Carbohydrate Docking: Sorting the Wheat from the Chaff. J. Comput. Chem 2014, 35, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hou X; Rooklin D; Yang D; Liang X; Li K; Lu J; Wang C; Xiao P; Zhang Y; Sun JP; Fang H, Computational Strategy for Bound State Structure Prediction in Structure-Based Virtual Screening: A Case Study of Protein Tyrosine Phosphatase Receptor Type O Inhibitors. J Chem Inf Model 2018, 58, 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dunbar JB; Smith RD; Yang CY; Ung PM; Lexa KW; Khazanov NA; Stuckey JA; Wang S; Carlson HA, Csar Benchmark Exercise of 2010: Selection of the Protein-Ligand Complexes. J. Chem. Inf. Model 2011, 51, 2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang SY; Zou X, Scoring and Lessons Learned with the Csar Benchmark Using an Improved Iterative Knowledge-Based Scoring Function. J. Chem. Inf. Model 2011, 51, 2097–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ewing TJA; Kuntz ID, Critical Evaluation of Search Algorithms for Automated Molecular Docking and Database Screening. J. Comput. Chem 1997, 18, 1175–1189. [Google Scholar]
- 81.Allen WJ; Rizzo RC, Implementation of the Hungarian Algorithm to Account for Ligand Symmetry and Similarity in Structure-Based Design. J. Chem. Inf. Model 2014, 54, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lang PT; Brozell SR; Mukherjee S; Pettersen EF; Meng EC; Thomas V; Rizzo RC; Case DA; James TL; Kuntz ID, Dock 6: Combining Techniques to Model Rna-Small Molecule Complexes. Rna 2009, 15, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mukherjee S; Balius TE; Rizzo RC, Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J. Chem. Inf. Model 2010, 50, 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sondergaard CR; Olsson MHM; Rostkowski M; Jensen JH, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of Pk(a) Values. J. Chem. Theory Comput 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- 85.Olsson MHM; Sondergaard CR; Rostkowski M; Jensen JH, Propka3: Consistent Treatment of Internal and Surface Residues in Empirical Pk(a) Predictions. J. Chem. Theory Comput 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- 86.Koes DR; Baumgartner MP; Camacho CJ, Lessons Learned in Empirical Scoring with Smina from the Csar 2011 Benchmarking Exercise. J. Chem. Inf. Model 2013, 53, 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Clark M; Cramer RD; Vanopdenbosch N, Validation of the General-Purpose Tripos 5.2 Force-Field. J. Comput. Chem 1989, 10, 982–1012. [Google Scholar]
- 88.Jiang LL; Rizzo RC, Pharmacophore-Based Similarity Scoring for Dock. J. Phys. Chem. B 2015, 119, 1083–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanner MF; Olson AJ; Spehner JC, Reduced Surface: An Efficient Way to Compute Molecular Surfaces. Biopolymers 1996, 38, 305–320. [DOI] [PubMed] [Google Scholar]
- 90.RDKit: Open-source cheminformatics; http://www.rdkit.org/. (accessed March 2, 2018).
- 91.Tosco P; Stiefl N; Landrum G, Bringing the Mmff Force Field to the Rdkit: Implementation and Validation. J. Cheminformatics 2014, 6, 37. [Google Scholar]
- 92.Halgren TA, Merck Molecular Force Field .2. Mmff94 Van Der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem 1996, 17, 520–552. [Google Scholar]
- 93.Lu JN; Wang C; Zhang YK, Predicting Molecular Energy Using Force-Field Optimized Geometries and Atomic Vector Representations Learned from an Improved Deep Tensor Neural Network. J. Chem. Theory Comput 2019, 15, 4113–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Data preparation schedule for PDBbind(v2016) database.
Figure S2: Feature importance.
Figure S3: MAE of Vina and ΔvinaXGB for structures with different number of rotors.
Figure S4: Success rates of best-scored pose in different RMSD values.
Figure S5. Ranking performances of ΔvinaXGB, ΔvinaRF20 and Vina on FlexDock
Table S1: Feature set of ΔvinaXGB.
Table S2: Failed conformation generation cases in CASF-2016.