

Abstract
It is over sixty years since the first administration of human growth hormone (GH) to children with GH deficiency, and over thirty years since recombinant human GH has been available for treatment of GH deficiency and a wider range of non-GH deficiency disorders. From a diagnostic perspective, genetic analysis, using single gene or Sanger sequencing and more recently next generation or whole exome sequencing, has brought advances in the diagnosis of specific causes of short stature, which has enabled therapy to be targeted more accurately. Genetic discoveries have ranged from defects of pituitary development and GH action to abnormalities in intracellular mechanisms, paracrine regulation and cartilage matrix formation. The strategy of GH therapy using standard doses has evolved to individualised GH dosing, depending on diagnosis and predictors of growth response. Evidence of efficacy of GH in GH deficiency, Turner syndrome and short children born small for gestational age is reviewed. The importance of critical assessment of growth response is discussed, together with the recognition and management of a poor or unsatisfactory growth response and the organisational issues related to prevention, detection and intervention regarding suboptimal adherence to GH therapy.
Keywords: Paediatrics, short stature, growth hormone therapy, growth hormone deficiency, Turner syndrome, small for gestational age
Introduction
Human pituitary-derived growth hormone (GH) has been in use to promote growth in short children for more than 25 years, until it was halted in 1985 due to recognition of the association with Creutzfeldt-Jakob disease (1). The first recombinant human GH received approval for paediatric clinical use for growth promotion in 1985, from both the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Initially, the approval was specifically for children with GH deficiency (Figure 1), but over time GH has been licensed for use in a number of non-GH deficiency growth disorders, including chronic renal insufficiency, Turner syndrome and short stature related to birth size in small for gestational age (SGA) children. The non-GH deficiency disorders of Noonan syndrome and idiopathic short stature have also received approval for GH use from the FDA, but not the EMA, and short stature due to SHOX gene haploinsufficiency and Prader-Willi syndrome (PWS) have received approval for some GH formulations in some countries (2,3,4).
Figure 1.
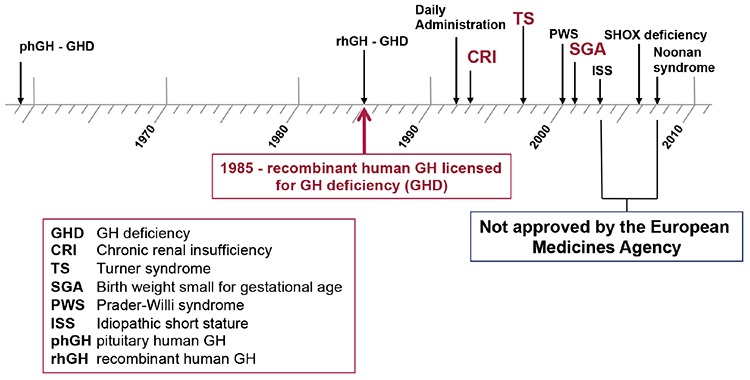
Growth disorders with approval for growth hormone therapy from the United States Food and Drug Administration and the European Medicines Agency
The capacity to secrete endogenous GH and the sensitivity to administered GH vary greatly, both within and among these disorders (5). Thus, there is a continuum whereby GH secretion is very low and responsiveness to treatment is high in patients with severe GH deficiency, in contrast to those with severe GH resistance (Laron syndrome) where GH secretion is high and response to administered GH is very low or non-existent (5,6,7). Conditions in-between include PWS and GH neurosecretory dysfunction, with good GH sensitivity but diminished secretion and idiopathic short stature, chronic renal insufficiency and SGA, with slightly reduced or normal GH secretion and variable GH responsiveness. When GH sensitivity is decreased, particularly in patients with SGA, Turner syndrome and idiopathic short stature, higher or pharmacological GH doses are generally required (5,8).
The differences in sensitivity to administered GH in children with growth failure arising from different conditions means that treatment management varies according to factors including the diagnosis, gender and age of the patient at GH initiation. An Advisory Board was convened in Dubai in December 2017, by Merck Serono Middle East FZ-LLC, Dubai, United Arab Emirates, with the aim of addressing the issues of the short- and long-term management of paediatric GH therapy for children with growth failure due to different conditions. This article reports the discussions and conclusions of the Advisory Board meeting.
The Genetic Basis of Short Stature
Human adult height is a polygenic trait in which the additive genetic contribution to normal variation is reported to be approximately 80% (9,10). While it is polygenic overall, multiple monogenic defects in genes coding for proteins with key functions in GH secretion and action have been identified to be associated with growth failure. At least eight genes have been identified in which individual abnormalities were associated with isolated GH deficiency and at least 19 genes where mutations resulted in combined pituitary hormone deficiency, with several related to specific syndromes (11,12). There were also 10 genes reported to be associated with GH resistance or insulin-like growth factor-1 (IGF-1) insensitivity. However, genome-wide association studies have indicated that only a minority of genes related to adult height are directly associated with the GH-IGF-1 axis and many genes act through other pathways (10).
Bone growth in the epiphyseal growth plate is influenced by many factors, including cytokines, nutritional status, other hormones such as thyroxine, glucocorticoids and gonadotropins, various paracrine factors within the extracellular matrix, and intracellular proteins (13,14). Next-generation sequencing has also shown that many genetic disorders that were previously thought to be only associated with skeletal dysplasia can present as dominant forms of apparent idiopathic short stature. These include, for example, abnormalities in the gene for the retinoic acid degrading enzyme CYP26C1 (15), coding and non-coding regions of the short-stature homeobox-containing gene SHOX (16,17), the ACAN gene coding for the growth plate extracellular matrix proteoglycan aggrecan (18,19), the natriuretic peptide receptor-B gene NPR2 (20,21,22) and the gene encoding Indian Hedgehog (IHH) (23).
These studies have led to a new paradigm (Opinion box) in which regulation of the epiphyseal plate is recognised to be pivotal to human linear growth, with disorders of the GH-IGF-1 axis making a less important contribution to height (13). The distinctions between idiopathic short or tall stature and skeletal dysplasias have become blurred and it is now understood that a genetic mutation can lead to a spectrum of phen. Gain of function mutations of various genes can lead to tall stature, whereas mild polymorphisms that modulate function and/or expression may result in low normal height. Mutations of genes that are not critical for growth, and mutations that are heterozygous or merely impair function of the gene product, can lead to isolated short stature, while mutations that cause severe loss of function affect critical genes causing skeletal dysplasias concomitant with growth failure (13,24). Therefore, testing for genetic defects in order to provide a diagnosis needs to be very specific and directed (25,26,27,28), although there are also arguments in favour of a hypothesis-free approach, using growth-specific whole exome sequencing-based gene panels (29,30,31).
Opinion Box. Consensus opinions of the advisory board.

GH Therapy for GH Deficiency and Non-GH Deficiency Causes of Short Stature
GH Therapy: Aims and Growth Response
For paediatric patients who receive exogenous GH for short stature, the therapy must be effective and safe. The induced catch-up growth should increase the height standard deviation score (SDS), with the aim of achieving an adult height SDS close to the genetic target height SDS, based on mid-parental height (2,32). Reports have suggested an association of excessive IGF-1 levels with adult morbidity, with the results that the aim of therapy for patients with initially low serum IGF-1 SDS is to treat with a GH dose that results in an IGF-1 increase to within the normal range (33,34). However, in patients with non-GH-deficient causes of growth failure, and initially normal IGF-1 SDS, elevated IGF-1 levels associated with higher GH doses may be required to achieve an acceptable height gain (34,35,36). GH treatment should also be as patient-friendly as possible, with appropriate injection devices and low injection volumes (37). Additionally, GH treatment should be cost-effective (34,38,39), providing sufficient efficacy with the lowest dose, which requires a personalised approach to therapy (40,41).
A number of different measurements have been used to assess growth response to GH treatment (42). These include the change in height SDS and height velocity at yearly intervals after starting GH therapy. Catch-up growth may also be determined from change in height over the first two years of GH treatment or using mathematical models of height SDS (43). However, there is no consensus on the definition of a good indicator of response and, if a relatively low cut-off level is taken, more than 50% of patients may be defined as poor responders (6,42,44). There may be multiple factors that affect response, such as concomitant disease, unanticipated GH insensitivity or poor adherence with the treatment (43). Nevertheless, this depends to a large extent on the correct diagnosis, because the aetiology has a major impact on the response to paediatric GH therapy (45).
Prediction of Response
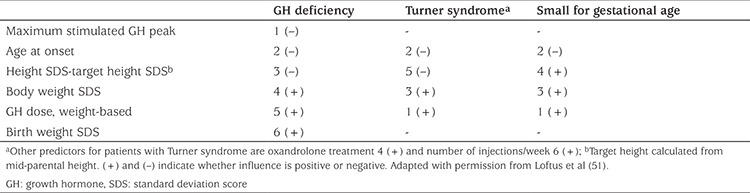
A number of models have been devised to predict the growth response to GH therapy for various aetiologies (41,46,47,48,49,50,51). Using such models, the mean predicted height velocity with a standard dose of 0.3 mg/kg/week is approximately 2 cm/year greater for children with GH deficiency, compared with children born SGA and girls with Turner syndrome. The factors that influence the prediction of the first-year growth response for patients with GH deficiency, Turner syndrome and SGA are shown in Table 1. The primary influence for children with GH deficiency is severity of the condition, whereas the primary influence for girls with Turner syndrome and children born SGA is the dose of GH per kg body weight per week (6,52,53).
Table 1. Predictive factors of the first year growth response to growth hormone treatment in patients with different causes of growth failure.
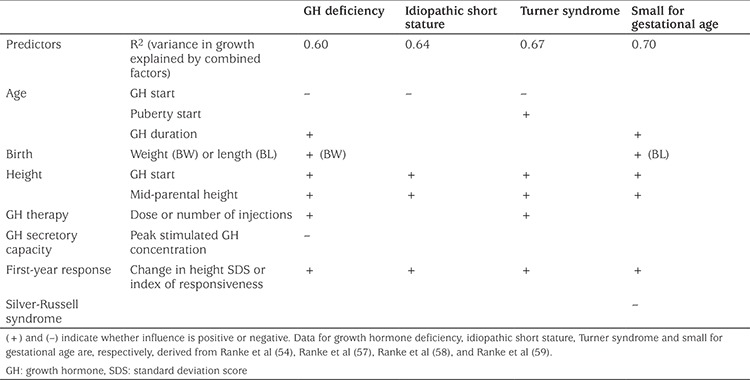
Predictive models can also be used for long-term growth response (54,55,56). Factors that influence adult height are shown in Table 2, including the calculated variation explained by all predictive factors combined for each diagnostic cause (54,57,58,59). Height at GH initiation and mid-parental target height has an impact on adult height for patients in each of the diagnostic categories. The first-year response to GH treatment was strongly correlated with gain to adult height and, therefore, change in height SDS should be formally assessed at the end of year one of GH therapy (58,59,60). These predictive factors can be used to tailor GH treatment even if a formal mathematical model is not used (Opinion Box). Alternatively, GH dose can be individualised by adjusting the dose according to serum IGF-1 level (41).
Table 2. Factors predictive of near adult height with growth hormone treatment in patients with short stature due to various causes.
GH Therapy in Specific Diagnoses
GH Deficiency
GH deficiency may either be isolated or occur together with deficiency of other pituitary hormones; patients with multiple pituitary hormone deficiencies will need replacement of additional hormones at appropriate levels over time, which could have a further impact on their growth rate. The severity of the GH deficiency, determined from the peak GH concentration seen on provocation testing, has a strong influence on the response rate. Children with more severe disease and very low stimulated GH peak have a greater response to GH therapy (44,46,61). Initiation of GH at as young an age as possible (Opinion Box) is also a key factor for a good response in GH-deficient patients and GH treatment should start when the patient is still pre-pubertal (45,57).
Turner Syndrome
Age at GH initiation is also strongly negatively correlated with growth response in girls with Turner syndrome (62,63). The genetic variability of girls with Turner syndrome results in marked differences among patients of different phenotypic characteristics (64). Dysmorphic features, such as webbed neck, cubitus valgus, shortening of the 4th/5th metacarpal and lymphoedema, may often be identified early, particularly if the chromosomal abnormalities include defects of the SHOX gene (17). While growth failure with decreased adult height is reported to occur in at least 95% of Turner syndrome cases, diagnosis and GH initiation is often delayed (64,65). Appropriate screening criteria, particularly using country-specific reference standards, can enable better and more efficient identification of short stature and earlier initiation of GH therapy (65). Dose of GH is also an important influence on growth in Turner syndrome and the recommended dose is generally around 50 µg/kg/day (64), which is higher than for patients with GH deficiency (3,35).
Short Stature Related to SGA
For children born SGA, approximately 90% will show catch-up growth within the first 2-3 years of life (66,67). However, GH therapy may be required for those children who do not show catch-up growth and can also improve body composition and metabolic health (67,68,69). Efficacy of GH treatment is greater if started at an early age (66) and height SDS was shown to be increased to a greater extent when patients at start of treatment were younger than four years compared with those over four years (70). However, treatment before four years of age is not recommended and not approved in Europe (3). The dose approved by the EMA is 33 µg/kg/day, although higher doses are approved by the FDA (3,71). While higher doses have been reported to provide better short-term efficacy, they are associated with supraphysiological IGF-1 levels and are, therefore, not recommended (71). Additionally, the increase in height gain with a higher dose was mainly in the short-term catch-up period and there was little additional benefit of a higher dose over the longer term at adult height (72).
Management of Poor Growth Response
Principles of Management
The recognition of a poor growth response to GH treatment is an important part of management of children with growth failure. However, a poor growth response is reported surprisingly frequently, particularly in children with Turner syndrome or born SGA, but the reported incidence depends to a large extent on the criteria used (42). The principles for management of a poor response are summarised in Figure 2A. To prevent occurrence of a poor response, the diagnosis of the cause of growth failure must be correct and the dose of GH administered should be based on that diagnosis, preferably with a prediction of the anticipated response (41). Growth must be determined accurately and the response at the end of the first year should be assessed; if the response is insufficient the primary diagnosis should be ascertained and the dose checked to ensure appropriateness (6). If the response is very low, then discontinuing GH treatment should be considered, particularly for non-GH deficiency diagnoses. However, there is no consensus on the cut-off of a low or very low response (6). Our preference is to use the change in height SDS in pre-pubertal children and consider a first-year change <0.3 SDS to be insufficient. A poor response following confirmation of the diagnosis may require an increase in GH dose, although this should be within the approved label.
Figure 2.
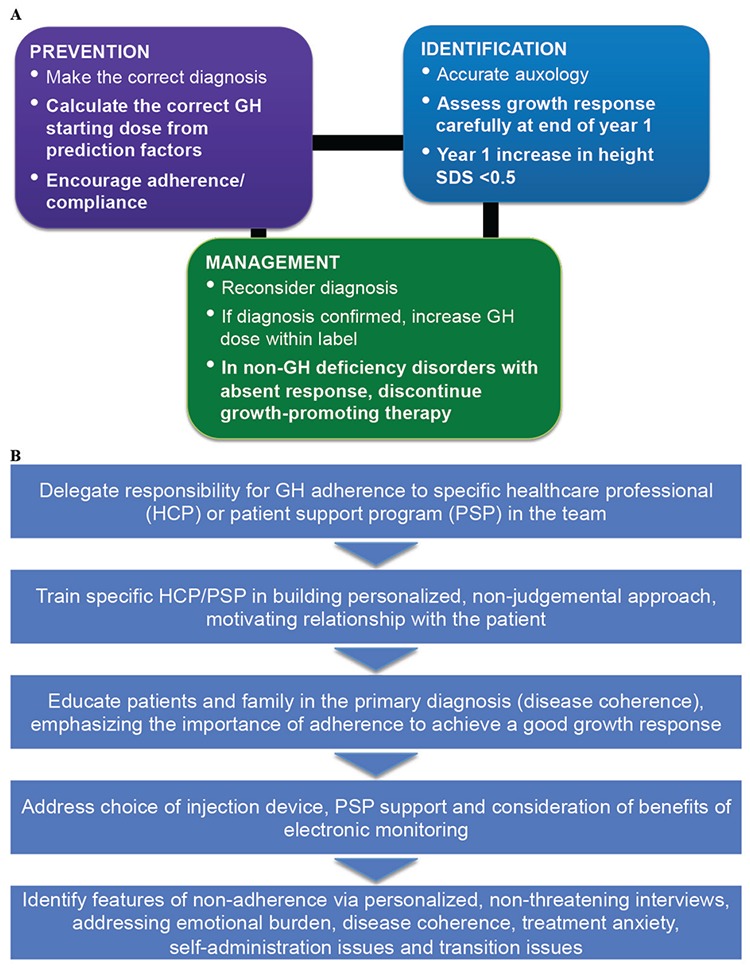
Management principles and strategy for patients with (A) a poor growth response and (B) non-adherence to GH therapy for short stature
Adherence to GH Therapy
The responsiveness of an individual patient to GH therapy can be determined from the difference between observed and predicted gain in height or height velocity (47). If responsiveness is reduced, while the diagnosis is deemed correct and presence of concomitant disease ruled out, then poor adherence to the treatment regimen should be considered (Opinion Box) (6,43,47,73). Monitoring of adherence should begin as soon as the treatment is initiated because some patients may not take the medication right from the start or renew their prescriptions, particularly if there is a lack of perceived need (74,75,76,77). While measurements of IGF-1 SDS may give an indication of adherence with GH, it is often not determined routinely and may not provide a definitive answer, because changes in concentration depend on multiple factors (77,78). Success of any therapy is dependent on good adherence and increased adherence may have a greater impact on health than improvements in specific medications. In the case of GH therapy, poor adherence has been shown to be associated with impaired clinical outcomes and reduced growth response (43,79,80,81).
Methods for assessing adherence have generally been poor, frequently relying on reporting by patients or carers, but have indicated that up to 82% of patients may miss at least some doses of GH (43,73,77). A strategy for prevention and management of adherence is outlined in Figure 2B. For effective management of poor adherence to GH, the paediatric endocrinologist or specialist nurse needs to learn techniques of non-judgemental motivational discussion. This requires time and organisation, knowledge of common issues affecting adherence at different treatment periods and ability to structure discussion with open questions, with emphasis on pre-GH treatment education. The same healthcare professional should discuss adherence at each outpatient visit. The strategy also involves addressing the choice of injection device and facility to identify adherence issues. New techniques of electronic monitoring are improving this process and provide important feedback data on evidence of sub-optimal adherence, which may not be available from self-reported data from patients and caregivers, clinical history or auxological measurements.
Electronic monitoring of GH injections is enabled through use of the easypod™ injection device and Easypod Connect© system, which is the only such solution currently approved and with published information (43,73). The device facilitates administration of a pre-set dose of GH, automatically records injection times and doses, and provides the patient with information such as number of doses remaining (82). The injection information can be downloaded at any time via Easypod Connect by healthcare personnel, which enables distance monitoring, with less need for frequent face-to-face visits. Thus, healthcare personnel are able to address issues of non-adherence with treatment at an early stage before any decrease in GH efficacy. Studies to date have indicated good acceptance of the device and high levels of adherence over several years (83,84,85). Studies with the device have also shown a significant correlation of high adherence with improved outcomes (43,85).
Manipulation of Puberty for Added Growth Advantage
Due to frequent delays in diagnosis of patients with short stature, GH treatment is often initiated close to, or even after, the start of the pubertal growth spurt. Studies have generally indicated that the growth response is greater if GH is started at a younger age, and particularly at the pre-pubertal stage, irrespective of the cause of short stature (45,57). While an increase in GH dose during puberty has been suggested, there are no clinical studies that have shown a convincing beneficial effect on adult height. Therefore, delaying puberty to allow exogenously administered GH to act for a longer period has been suggested as a strategy to improve overall linear growth (86). Oxandrolone administration has been examined in boys with constitutional delay of growth and idiopathic short stature, but had no significant effect on adult height (86,87). However, addition of oxandrolone to GH therapy has been studied in girls with Turner syndrome and provided approximately 3 cm of extra adult height gain (86,88).
Several small, off-label studies showed that delaying puberty with a gonadotropin-releasing hormone agonist (GnRHa) could increase adult height in children with idiopathic short stature or born SGA, but the effect was modest, not considered clinically significant and outweighed by adverse effects (89,90). When GH initiation is delayed and is close to or during puberty, adding a GnRHa may delay puberty and potentially prolong GH effectiveness. Although such combination treatment is not licensed, GnRHa added to GH has been examined in several studies in children with growth failure due to various different causes (86). The combination of a GnRHa with GH was reported to result in a significant increase in adult height in patients with GH deficiency (91,92) and also in those with idiopathic short stature (86,93) and born SGA (94). However, the GnRHa treatment has negative effects on body composition and, while these effects are reversible after GnRHa cessation, the effects on bone mineral density are of concern and could increase fracture risk (93,95,96).
Conclusions
GH therapy for growth failure due to causes with approval for use to promote growth in short children can induce clinically beneficial short-term and long-term gains in height. The sensitivity and responsiveness to GH treatment are increased in children with GH deficiency compared with children with non-GH deficiency disorders, such as Turner syndrome or SGA. GH therapy should be individualised for each patient, based on the diagnosis and factors predicting growth response, such as age, severity of GH deficiency and deficit from genetic target height. A formal assessment of the response after the first year of GH therapy is recommended, with calculation of the gain in height SDS. The gain should ideally be compared with the individualised prediction; alternatively, cut-off levels of first-year height SDS change of 0.3 or 0.5 have been suggested as lower limits of an acceptable capacity for catch-up growth.
Good adherence to GH therapy is essential to achieve optimal short- and long-term responses, although management of poor adherence generally requires time and organisation. Novel techniques of electronic monitoring are helpful and can provide data that demonstrate when adherence is reduced, which may not be detectable from the patient’s history or auxological observations. In some clinical situations, such as GH deficiency, SGA with short stature at onset of puberty and idiopathic short stature, addition of a GnRHa (for a minimum of two years) to GH therapy can increase adult height gain, but adverse effects should be carefully monitored and a positive benefit-risk ratio has not been formally assessed by regulatory authorities; these combinations were used in clinical trials only and are not included in any GH therapy label at present.
Acknowledgments
The authors acknowledge the contributions of the paediatric endocrinologists who attended the Advisory Board, ELITE III, organised by Akram Kharbat and Ayman Haridy of Merck Serono Middle East, Dubai, UAE, and Ekaterina Koledova of Merck KGaA, Darmstadt, held in Dubai, UAE, in December 2017. The contributing specialists were: Abdullah Al Herbish, Hesa Al Kandari, Hala Al Shaikh, Abdullah Alfaris, Afaf Alsagheir, Bassam Bin Abbass, Deepti Chaturvedi, Asma Deeb, Angham Mutair, Vidya Naryanan and Nandu Thalange. The authors also acknowledge the medical writing skills of Dr Peter Bates, Cambridge Medical Writing Services, UK, for assistance in preparation of this review.
Footnotes
Ethics
Peer-review: Internally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Concept: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Design: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Data Collection or Processing: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Analysis or Interpretation: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Literature Search: Jan M. Wit, Asma Deeb, Bassam Bin-Abbas, Angham Al Mutair, Ekaterina Koledova, Martin O. Savage, Writing: The authors also acknowledge the medical writing skills of Dr Peter Bates, Cambridge Medical Writing Services, UK, for assistance in preparation of this review. All authors contributed to the interpretation of the information from the advisory board and participated in the preparation of this manuscript, and approved the final manuscript for submission.
Conflict of Interest: JMW is member of several advisory boards (OPKO, Merck, Ammonett, Aeterna Zentaris, Agios, Biomarin) and has received speaker’s fees from Pfizer, Versartis, Sandoz, Lilly, Merck, JCR, and Novo Nordisk. EK is employed by Merck KGaA, Germany. MOS has consulting contracts with Merck KGaA, Germany, Sandoz, OPKO and Genexine and has received speaker’s fees from GeneScience and Sandoz, UK. The remaining authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Financial Disclosure: This work was supported by Merck Serono Middle East FZ-LLC, Dubai, UAE, an affiliate of Merck KGaA, Darmstadt, Germany.
References
- 1.Hintz RL. The prismatic case of Creutzfeldt-Jakob disease associated with pituitary growth hormone treatment. J Clin Endocrinol Metab. 1995;80:2298–2301. doi: 10.1210/jcem.80.8.7629222. [DOI] [PubMed] [Google Scholar]
- 2.Ranke MB, Wit JM. Growth hormone - past, present and future. Nat Rev Endocrinol. 2018;14:285–300. doi: 10.1038/nrendo.2018.22. [DOI] [PubMed] [Google Scholar]
- 3.Richmond E, Rogol AD. Current indications for growth hormone therapy for children and adolescents. Endocr Dev. 2010;18:92–108. doi: 10.1159/000316130. [DOI] [PubMed] [Google Scholar]
- 4.Navarro R, Dunn JD, Lee PA, Owens GM, Rapaport R. Translating clinical guidelines into practice: the effective and appropriate use of growth hormone. Am J Manag Care. 2013;19(15 Suppl):281–289. [PubMed] [Google Scholar]
- 5.Savage MO, Burren CP, Rosenfeld RG. The continuum of growth hormone-IGF-I axis defects causing short stature: diagnostic and therapeutic challenges. Clin Endocrinol (Oxf) 2010;72:721–728. doi: 10.1111/j.1365-2265.2009.03775.x. [DOI] [PubMed] [Google Scholar]
- 6.Bang P, Ahmed SF, Argente J, Backeljauw P, Bettendorf M, Bona G, Coutant R, Rosenfeld RG, Walenkamp MJ, Savage MO. Identification and management of poor response to growth-promoting therapy in children with short stature. Clin Endocrinol (Oxf) 2012;77:169–181. doi: 10.1111/j.1365-2265.2012.04420.x. [DOI] [PubMed] [Google Scholar]
- 7.David A, Hwa V, Metherell LA, Netchine I, Camacho-Hübner C, Clark AJ, Rosenfeld RG, Savage MO. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011;32:472–497. doi: 10.1210/er.2010-0023. [DOI] [PubMed] [Google Scholar]
- 8.Pfäffle R, Land C, Schönau E, Holterhus PM, Ross JL, Piras de Oliveira C, Child CJ, Benabbad I, Jia N, Jung H, Blum WF. Growth hormone treatment for short stature in the USA, Germany and France: 15 years of surveillance in the Genetics and Neuroendocrinology of Short-Stature International Study (GeNeSIS) Horm Res Paediatr. 2018;90:145–157. doi: 10.1159/000492397. [DOI] [PubMed] [Google Scholar]
- 9.Jelenkovic A, Sund R, Hur YM, Yokoyama Y, Hjelmborg JV, Möller S, Honda C, Magnusson PK, Pedersen NL, Ooki S, Aaltonen S, Stazi MA, Fagnani C, D’Ippolito C, Freitas DL, Maia JA, Ji F, Ning F, Pang Z, Rebato E, Busjahn A, Kandler C, Saudino KJ, Jang KL, Cozen W, Hwang AE, Mack TM, Gao W, Yu C, Li L, Corley RP, Huibregtse BM, Derom CA, Vlietinck RF, Loos RJ, Heikkilä K, Wardle J, Llewellyn CH, Fisher A, McAdams TA, Eley TC, Gregory AM, He M, Ding X, Bjerregaard-Andersen M, Beck-Nielsen H, Sodemann M, Tarnoki AD, Tarnoki DL, Knafo-Noam A, Mankuta D, Abramson L, Burt SA, Klump KL, Silberg JL, Eaves LJ, Maes HH, Krueger RF, McGue M, Pahlen S, Gatz M, Butler DA, Bartels M, van Beijsterveldt TC, Craig JM, Saffery R, Dubois L, Boivin M, Brendgen M, Dionne G, Vitaro F, Martin NG, Medland SE, Montgomery GW, Swan GE, Krasnow R, Tynelius P, Lichtenstein P, Haworth CM, Plomin R, Bayasgalan G, Narandalai D, Harden KP, Tucker-Drob EM, Spector T, Mangino M, Lachance G, Baker LA, Tuvblad C, Duncan GE, Buchwald D, Willemsen G, Skytthe A, Kyvik KO, Christensen K, Öncel SY, Aliev F, Rasmussen F, Goldberg JH, Sørensen TI, Boomsma DI, Kaprio J, Silventoinen K. Genetic and environmental influences on height from infancy to early adulthood: an individual-based pooled analysis of 45 twin cohorts. Sci Rep. 2016;6:28496. doi: 10.1038/srep28496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, Croteau-Chonka DC, Day FR, Duan Y, Fall T, Fehrmann R, Ferreira T, Jackson AU, Karjalainen J, Lo KS, Locke AE, Mägi R, Mihailov E, Porcu E, Randall JC, Scherag A, Vinkhuyzen AA, Westra HJ, Winkler TW, Workalemahu T, Zhao JH, Absher D, Albrecht E, Anderson D, Baron J, Beekman M, Demirkan A, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Fraser RM, Goel A, Gong J, Justice AE, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Lui JC, Mangino M, Mateo Leach I, Medina-Gomez C, Nalls MA, Nyholt DR, Palmer CD, Pasko D, Pechlivanis S, Prokopenko I, Ried JS, Ripke S, Shungin D, Stancáková A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, van der Laan SW, van Setten J, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Afzal U, Arnlöv J, Arscott GM, Bandinelli S, Barrett A, Bellis C, Bennett AJ, Berne C, Blüher M, Bolton JL, Böttcher Y, Boyd HA, Bruinenberg M, Buckley BM, Buyske S, Caspersen IH, Chines PS, Clarke R, Claudi-Boehm S, Cooper M, Daw EW, De Jong PA, Deelen J, Delgado G, Denny JC, Dhonukshe-Rutten R, Dimitriou M, Doney AS, Dörr M, Eklund N, Eury E, Folkersen L, Garcia ME, Geller F, Giedraitis V, Go AS, Grallert H, Grammer TB, Gräßler J, Grönberg H, de Groot LC, Groves CJ, Haessler J, Hall P, Haller T, Hallmans G, Hannemann A, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hemani G, Henders AK, Hillege HL, Hlatky MA, Hoffmann W, Hoffmann P, Holmen O, Houwing-Duistermaat JJ, Illig T, Isaacs A, James AL, Jeff J, Johansen B, Johansson Å, Jolley J, Juliusdottir T, Junttila J, Kho AN, Kinnunen L, Klopp N, Kocher T, Kratzer W, Lichtner P, Lind L, Lindström J, Lobbens S, Lorentzon M, Lu Y, Lyssenko V, Magnusson PK, Mahajan A, Maillard M, McArdle WL, McKenzie CA, McLachlan S, McLaren PJ, Menni C, Merger S, Milani L, Moayyeri A, Monda KL, Morken MA, Müller G, Müller-Nurasyid M, Musk AW, Narisu N, Nauck M, Nolte IM, Nöthen MM, Oozageer L, Pilz S, Rayner NW, Renstrom F, Robertson NR, Rose LM, Roussel R, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Schunkert H, Scott RA, Sehmi J, Seufferlein T, Shi J, Silventoinen K, Smit JH, Smith AV, Smolonska J, Stanton AV, Stirrups K, Stott DJ, Stringham HM, Sundström J, Swertz MA, Syvänen AC, Tayo BO, Thorleifsson G, Tyrer JP, van Dijk S, van Schoor NM, van der Velde N, van Heemst D, van Oort FV, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Waldenberger M, Wennauer R, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Arveiler D, Bakker SJ, Beilby J, Bergman RN, Bergmann S, Biffar R, Blangero J, Boomsma DI, Bornstein SR, Bovet P, Brambilla P, Brown MJ, Campbell H, Caulfield MJ, Chakravarti A, Collins R, Collins FS, Crawford DC, Cupples LA, Danesh J, de Faire U, den Ruijter HM, Erbel R, Erdmann J, Eriksson JG, Farrall M, Ferrannini E, Ferrières J, Ford I, Forouhi NG, Forrester T, Gansevoort RT, Gejman PV, Gieger C, Golay A, Gottesman O, Gudnason V, Gyllensten U, Haas DW, Hall AS, Harris TB, Hattersley AT, Heath AC, Hengstenberg C, Hicks AA, Hindorff LA, Hingorani AD, Hofman A, Hovingh GK, Humphries SE, Hunt SC, Hypponen E, Jacobs KB, Jarvelin MR, Jousilahti P, Jula AM, Kaprio J, Kastelein JJ, Kayser M, Kee F, Keinanen-Kiukaanniemi SM, Kiemeney LA, Kooner JS, Kooperberg C, Koskinen S, Kovacs P, Kraja AT, Kumari M, Kuusisto J, Lakka TA, Langenberg C, Le Marchand L, Lehtimäki T, Lupoli S, Madden PA, Männistö S, Manunta P, Marette A, Matise TC, McKnight B, Meitinger T, Moll FL, Montgomery GW, Morris AD, Morris AP, Murray JC, Nelis M, Ohlsson C, Oldehinkel AJ, Ong KK, Ouwehand WH, Pasterkamp G, Peters A, Pramstaller PP, Price JF, Qi L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Ritchie M, Rudan I, Salomaa V, Samani NJ, Saramies J, Sarzynski MA, Schwarz PE, Sebert S, Sever P, Shuldiner AR, Sinisalo J, Steinthorsdottir V, Stolk RP, Tardif JC, Tönjes A, Tremblay A, Tremoli E, Virtamo J, Vohl MC; Electronic Medical Records and Genomics (eMEMERGEGE) Consortium; MIGen Consortium; PAGEGE Consortium; LifeLines Cohort Study, Amouyel P, Asselbergs FW, Assimes TL, Bochud M, Boehm BO, Boerwinkle E, Bottinger EP, Bouchard C, Cauchi S, Chambers JC, Chanock SJ, Cooper RS, de Bakker PI, Dedoussis G, Ferrucci L, Franks PW, Froguel P, Groop LC, Haiman CA, Hamsten A, Hayes MG, Hui J, Hunter DJ, Hveem K, Jukema JW, Kaplan RC, Kivimaki M, Kuh D, Laakso M, Liu Y, Martin NG, März W, Melbye M, Moebus S, Munroe PB, Njølstad I, Oostra BA, Palmer CN, Pedersen NL, Perola M, Pérusse L, Peters U, Powell JE, Power C, Quertermous T, Rauramaa R, Reinmaa E, Ridker PM, Rivadeneira F, Rotter JI, Saaristo TE, Saleheen D, Schlessinger D, Slagboom PE, Snieder H, Spector TD, Strauch K, Stumvoll M, Tuomilehto J, Uusitupa M, van der Harst P, Völzke H, Walker M, Wareham NJ, Watkins H, Wichmann HE, Wilson JF, Zanen P, Deloukas P, Heid IM, Lindgren CM, Mohlke KL, Speliotes EK, Thorsteinsdottir U, Barroso I, Fox CS, North KE, Strachan DP, Beckmann JS, Berndt SI, Boehnke M, Borecki IB, McCarthy MI, Metspalu A, Stefansson K, Uitterlinden AG, van Duijn CM, Franke L, Willer CJ, Price AL, Lettre G, Loos RJ, Weedon MN, Ingelsson E, O’Connell JR, Abecasis GR, Chasman DI, Goddard ME, Visscher PM, Hirschhorn JN, Frayling TM. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016;174:145–173. doi: 10.1530/EJE-15-0937. [DOI] [PubMed] [Google Scholar]
- 12.Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, Daly AZ, Ajmal A, Pérez Millán MI, Ozel AB, Kitzman JO, Mills RE, Li JZ, Camper SA. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev. 2016;37:636–675. doi: 10.1210/er.2016-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, Nilsson O. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11:735–746. doi: 10.1038/nrendo.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005;64:157–165. doi: 10.1159/000088791. [DOI] [PubMed] [Google Scholar]
- 15.Montalbano A, Juergensen L, Fukami M, Thiel CT, Hauer NH, Roeth R, Weiss B, Naiki Y, Ogata T, Hassel D, Rappold GA. Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature. Eur J Hum Genet. 2018;26:1113–1120. doi: 10.1038/s41431-018-0148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alharthi AA, El-Hallous EI, Talaat IM, Alghamdi HA, Almalki MI, Gaber A. Screening of SHOX gene sequence variants in Saudi Arabian children with idiopathic short stature. Korean J Pediatr. 2017;60:327–332. doi: 10.3345/kjp.2017.60.10.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011;75:81–89. doi: 10.1159/000324105. [DOI] [PubMed] [Google Scholar]
- 18.Hauer NN, Sticht H, Boppudi S, Büttner C, Kraus C, Trautmann U, Zenker M, Zweier C, Wiesener A, Jamra RA, Wieczorek D, Kelkel J, Jung AM, Uebe S, Ekici AB, Rohrer T, Reis A, Dörr HG, Thiel CT. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. 2017;7:12225. doi: 10.1038/s41598-017-12465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J, Dauber A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. 2014;99:1510–1518. doi: 10.1210/jc.2014-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Belinchón A, Barreda-Bonis AC, Vasques GA, Ramirez J, Luzuriaga C, Carlone G, González-Casado I, Benito-Sanz S, Jorge AA, Campos-Barros A, Heath KE. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2015;100:1133–1142. doi: 10.1210/jc.2015-1612. [DOI] [PubMed] [Google Scholar]
- 21.Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, Miller TC, Moon JE, Mericq V, Potter LR, Warman ML, Hirschhorn JN, Dauber A. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. 2015;36:474–481. doi: 10.1002/humu.22773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasques GA, Amano N, Docko AJ, Funari MF, Quedas EP, Nishi MY, Arnhold IJ, Hasegawa T, Jorge AA. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab. 2013;98:1636–1644. doi: 10.1210/jc.2013-2142. [DOI] [PubMed] [Google Scholar]
- 23.Vasques GA, Funari MFA, Ferreira FM, Aza-Carmona M, Sentchordi-Montané L, Barraza-García J, Lerario AM, Yamamoto GL, Naslavsky MS, Duarte YAO, Bertola DR, Heath KE, Jorge AAL. IHH gene mutations causing short stature with nonspecific skeletal abnormalities and response to growth hormone therapy. J Clin Endocrinol Metab. 2018;103:604–614. doi: 10.1210/jc.2017-02026. [DOI] [PubMed] [Google Scholar]
- 24.Andrade AC, Jee YH, Nilsson O. New genetic diagnoses of short stature provide insights into local regulation of childhood growth. Horm Res Paediatr. 2017;88:22–37. doi: 10.1159/000455850. [DOI] [PubMed] [Google Scholar]
- 25.Murray PG, Clayton PE, Chernausek SD. A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol. 2018;6:564–574. doi: 10.1016/S2213-8587(18)30034-2. [DOI] [PubMed] [Google Scholar]
- 26.Sisley S, Trujillo MV, Khoury J, Backeljauw P. Low incidence of pathology detection and high cost of screening in the evaluation of asymptomatic short children. J Pediatr. 2013;163:1045–1051. doi: 10.1016/j.jpeds.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hattori A, Katoh-Fukui Y, Nakamura A, Matsubara K, Kamimaki T, Tanaka H, Dateki S, Adachi M, Muroya K, Yoshida S, Ida S, Mitani M, Nagasaki K, Ogata T, Suzuki E, Hata K, Nakabayashi K, Matsubara Y, Narumi S, Tanaka T, Fukami M. Next generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocr J. 2017;64:947–954. doi: 10.1507/endocrj.EJ17-0150. [DOI] [PubMed] [Google Scholar]
- 28.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167:2869–2892. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 29.Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, Klinger P, Kraus C, Trautmann U, Zenker M, Zweier C, Wiesener A, Abou Jamra R, Kunstmann E, Wieczorek D, Uebe S, Ferrazzi F, Büttner C, Ekici AB, Rauch A, Sticht H, Dörr HG, Reis A, Thiel CT. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet Med. 2018;20:630–638. doi: 10.1038/gim.2017.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams DR, Eng CM. Next-generation sequencing to diagnose suspected genetic disorders. New Engl J Med. 2018;379:1353–1362. doi: 10.1056/NEJMra1711801. [DOI] [PubMed] [Google Scholar]
- 31.Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99:3080–3092. doi: 10.1210/jc.2014-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deodati A, Cianfarani S. The rationale for growth hormone therapy in children with short stature. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):23–32. doi: 10.4274/jcrpe.2017.S003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen DB, Backeljauw P, Bidlingmaier M, Biller BM, Boguszewski M, Burman P, Butler G, Chihara K, Christiansen J, Cianfarani S, Clayton P, Clemmons D, Cohen P, Darendeliler F, Deal C, Dunger D, Erfurth EM, Fuqua JS, Grimberg A, Haymond M, Higham C, Ho K, Hoffman AR, Hokken-Koelega A, Johannsson G, Juul A, Kopchick J, Lee P, Pollak M, Radovick S, Robison L, Rosenfeld R, Ross RJ, Savendahl L, Saenger P, Sorensen HT, Stochholm K, Strasburger C, Swerdlow A, Thorner M. GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol. 2016;174:1–9. doi: 10.1530/EJE-15-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen P, Weng W, Rogol AD, Rosenfeld RG, Kappelgaard AM, Germak J. Dose-sparing and safety-enhancing effects of an IGF-I-based dosing regimen in short children treated with growth hormone in a 2-year randomized controlled trial: therapeutic and pharmacoeconomic considerations. Clin Endocrinol (Oxf) 2014;81:71–76. doi: 10.1111/cen.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfäffle R, Land C, Schönau E, Holterhus PM, Ross JL, Piras de Oliveira C, Child CJ, Benabbad I, Jia N, Jung H, Blum WF. Growth hormone treatment for short stature in the USA, Germany and France: 15 years of surveillance in the Genetics and Neuroendocrinology of Short-Stature International Study (GeNeSIS) Horm Res Paediatr. 2018;90:169–180. doi: 10.1159/000492397. [DOI] [PubMed] [Google Scholar]
- 36.Ballerini MG, Braslavsky D, Scaglia PA, Keselman A, Rodríguez ME, Martínez A, Freire AV, Domené HM, Jasper HG, Bergadá I, Ropelato MG. Circulating IGF-I, IGFBP-3 and the IGF-I/IGFBP-3 molar ratio concentration and height outcome in prepubertal short children on rhGH treatment over two years of therapy. Horm Res Paediatr. 2017;88:354–363. doi: 10.1159/000479691. [DOI] [PubMed] [Google Scholar]
- 37.Rohrer TR, Horikawa R, Kappelgaard AM. Growth hormone delivery devices: current features and potential for enhanced treatment adherence. Expert Opin Drug Deliv. 2017;14:1253–1264. doi: 10.1080/17425247.2017.1243526. [DOI] [PubMed] [Google Scholar]
- 38.Allen DB. Cost-conscious growth-promoting treatment: when discretion is the better part of value. Horm Res Paediatr. 2018;90:145–150. doi: 10.1159/000493397. [DOI] [PubMed] [Google Scholar]
- 39.Centonze C, Foo J, Vlachaki I, Novelli P. Cost-consequence analysis for recombinant human growth hormone treatment administered via different devices in children with growth hormone deficiency in Italy. Value Health. 2018;21(Suppl 3):128. doi: 10.2147/CEOR.S195265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koledova E, Stoyanov G, Ovbude L, Davies PSW. Adherence and long-term growth outcomes: results from the easypod™ connect observational study (ECOS) in paediatric patients with growth disorders. Endocr Connect. 2018;7:914–923. doi: 10.1530/EC-18-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wit JM, Ranke MB, Albertsson-Wikland K, Carrascosa A, Rosenfeld RG, Van Buuren S, Kristrom B, Schoenau E, Audi L, Hokken-Koelega AC, Bang P, Jung H, Blum WF, Silverman LA, Cohen P, Cianfarani S, Deal C, Clayton PE, de Graaff L, Dahlgren J, Kleintjens J, Roelants M. Personalized approach to growth hormone treatment: clinical use of growth prediction models. Horm Res Paediatr. 2013;79:257–270. doi: 10.1159/000351025. [DOI] [PubMed] [Google Scholar]
- 42.Bang P, Bjerknes R, Dahlgren J, Dunkel L, Gustafsson J, Juul A, Kriström B, Tapanainen P, Aberg V. A comparison of different definitions of growth response in short prepubertal children treated with growth hormone. Horm Res Paediatr. 2011;75:335–345. doi: 10.1159/000322878. [DOI] [PubMed] [Google Scholar]
- 43.van Dommelen P, Koledova E, Wit JM. Effect of adherence to growth hormone treatment on 0-2 year catch-up growth in children with growth hormone deficiency. PLoS One. 2018;13:e0206009. doi: 10.1371/journal.pone.0206009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Straetemans S, Thomas M, Craen M, Rooman R, De Schepper J; BESPEED. Poor growth response during the first year of growth hormone treatment in short prepubertal children with growth hormone deficiency and born small for gestational age: a comparison of different criteria. Int J Pediatr Endocrinol. 2018;2018:9. doi: 10.1186/s13633-018-0064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee PA, Sävendahl L, Oliver I, Tauber M, Blankenstein O, Ross J, Snajderova M, Rakov V, Pedersen BT, Christesen HT. Comparison of response to 2-years’ growth hormone treatment in children with isolated growth hormone deficiency, born small for gestational age, idiopathic short stature, or multiple pituitary hormone deficiency: combined results from two large observational studies. Int J Pediatr Endocrinol. 2012;2012:22. doi: 10.1186/1687-9856-2012-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ranke MB, Lindberg A; KIGS International Board. Observed and predicted growth responses in prepubertal children with growth disorders: guidance of growth hormone treatment by empirical variables. J Clin Endocrinol Metab. 2010;95:1229–1237. doi: 10.1210/jc.2009-1471. [DOI] [PubMed] [Google Scholar]
- 47.Kaspers S, Ranke MB, Han D, Loftus J, Wollmann H, Lindberg A, Roelants M, Kleintjens J. Implications of a data-driven approach to treatment with growth hormone in children with growth hormone deficiency and Turner syndrome. Appl Health Econ Health Policy. 2013;11:237–249. doi: 10.1007/s40258-013-0030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoyer-Kuhn H, Franklin J, Jones C, Blum WF, Schoenau E. Growth response to growth hormone treatment in patients with SHOX deficiency can be predicted by the Cologne prediction model. J Pediatr Endocrinol Metab. 2018;31:25–31. doi: 10.1515/jpem-2017-0282. [DOI] [PubMed] [Google Scholar]
- 49.Schönau E, Westermann F, Rauch F, Stabrey A, Wassmer G, Keller E, Brämswig J, Blum WF; German Lilly Growth Response Study Group. A new and accurate prediction model for growth response to growth hormone treatment in children with growth hormone deficiency. Eur J Endocrinol. 2001;144:13–20. doi: 10.1530/eje.0.1440013. [DOI] [PubMed] [Google Scholar]
- 50.Wikland KA, Kriström B, Rosberg S, Svensson B, Nierop AF. Validated multivariate models predicting the growth response to GH treatment in individual short children with a broad range in GH secretion capacities. Pediatr Res. 2000;48:475–484. doi: 10.1203/00006450-200010000-00010. [DOI] [PubMed] [Google Scholar]
- 51.Loftus J, Lindberg A, Aydin F, Gomez R, Maghnie M, Rooman R, Steinkamp H, Doerr H, Ranke M, Camacho-Hubner C. Individualised growth response optimisation (iGRO) tool: an accessible and easy-to-use growth prediction system to enable treatment optimisation for children treated with growth hormone. J Pediatr Endocrinol Metab. 2017;30:1019–1026. doi: 10.1515/jpem-2017-0120. [DOI] [PubMed] [Google Scholar]
- 52.Ranke MB, Lindberg A, Chatelain P, Wilton P, Cutfield W, Albertsson-Wikland K, Price DA. Derivation and validation of a mathematical model for predicting the response to exogenous recombinant human growth hormone (GH) in prepubertal children with idiopathic GH deficiency. KIGS International Board. J Clin Endocrinol Metab. 1999;84:1174–1183. doi: 10.1210/jcem.84.4.5634. [DOI] [PubMed] [Google Scholar]
- 53.Ranke MB, Lindberg A, Chatelain P, Wilton P, Cutfield W, Albertsson-Wikland K, Price DA; KIGS International Board. Kabi International Growth Study. Prediction of long-term response to recombinant human growth hormone in Turner syndrome: development and validation of mathematical models. Kabi International Growth Study. J Clin Endocrinol Metab. 2000;85:4212–4218. doi: 10.1210/jcem.85.11.6976. [DOI] [PubMed] [Google Scholar]
- 54.Ranke MB, Lindberg A, Mullis PE, Geffner ME, Tanaka T, Cutfield WS, Tauber M, Dunger D. Towards optimal treatment with growth hormone in short children and adolescents: evidence and theses. Horm Res Paediatr. 2013;79:51–67. doi: 10.1159/000347121. [DOI] [PubMed] [Google Scholar]
- 55.Straetemans S, De Schepper J, Thomas M, Verlinde F, Rooman R; BESPEED. Validation of prediction models for near adult height in children with idiopathic growth hormone deficiency treated with growth hormone: a Belgian registry study. Horm Res Paediatr. 2016;86:161–168. doi: 10.1159/000448553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Ridder MA, Stijnen T, Hokken-Koelega AC. Prediction model for adult height of small for gestational age children at the start of growth hormone treatment. J Clin Endocrinol Metab. 2008;93:477–483. doi: 10.1210/jc.2007-1381. [DOI] [PubMed] [Google Scholar]
- 57.Ranke MB, Lindberg A, Price DA, Darendeliler F, Albertsson-Wikland K, Wilton P, Reiter EO; KIGS International Board. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Horm Res. 2007;68:53–62. doi: 10.1159/000098707. [DOI] [PubMed] [Google Scholar]
- 58.Ranke MB, Lindberg A, Ferrández Longás A, Darendeliler F, Albertsson-Wikland K, Dunger D, Cutfield WS, Tauber M, Wilton P, Wollmann HA, Reiter EO; KIGS International Board. Major determinants of height development in Turner syndrome (TS) patients treated with GH: analysis of 987 patients from KIGS. Pediatr Res. 2007;61:105–110. doi: 10.1203/01.pdr.0000250039.42000.c9. [DOI] [PubMed] [Google Scholar]
- 59.Ranke MB, Lindberg A; KIGS International Board. Height at start, first-year growth response and cause of shortness at birth are major determinants of adult height outcomes of short children born small for gestational age and Silver-Russell syndrome treated with growth hormone: analysis of data from KIGS. Horm Res Paediatr. 2010;74:259–266. doi: 10.1159/000289570. [DOI] [PubMed] [Google Scholar]
- 60.Reiter EO, Price DA, Wilton P, Albertsson-Wikland K, Ranke MB. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J Clin Endocrinol Metab. 2006;91:2047–2054. doi: 10.1210/jc.2005-2284. [DOI] [PubMed] [Google Scholar]
- 61.Valsesia A, Chatelain P, Stevens A, Peterkova VA, Belgorosky A, Maghnie M, Antoniazzi F, Koledova E, Wojcik J, Farmer P, Destenaves B, Clayton P; PREDICT Investigator Group. GH deficiency status combined with GH receptor polymorphism affects response to GH in children. Eur J Endocrinol. 2015;173:777–789. doi: 10.1530/EJE-15-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ross J, Lee PA, Gut R, Germak J. Impact of age and duration of growth hormone therapy in children with Turner syndrome. Horm Res Paediatr. 2011;76:392–399. doi: 10.1159/000333073. [DOI] [PubMed] [Google Scholar]
- 63.Hughes IP, Choong CS, Harris M, Ambler GR, Cutfield WS, Hofman PL, Cowell CT, Werther G, Cotterill A, Davies PS; Australasian Paediatric Endocrine Group (APEG) Growth hormone treatment for Turner syndrome in Australia reveals that younger age and increased dose interact to improve response. Clin Endocrinol (Oxf) 2011;74:473–480. doi: 10.1111/j.1365-2265.2011.03937.x. [DOI] [PubMed] [Google Scholar]
- 64.Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, Lin AE, Mauras N, Quigley CA, Rubin K, Sandberg DE, Sas TCJ, Silberbach M, Söderström-Anttila V, Stochholm K, van Alfen-van derVelden JA, Woelfle J, Backeljauw PF; International Turner Syndrome Consensus Group. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177:1–70. doi: 10.1530/EJE-17-0430. [DOI] [PubMed] [Google Scholar]
- 65.Saari A, Sankilampi U, Hannila ML, Saha MT, Mäkitie O, Dunkel L. Screening of turner syndrome with novel auxological criteria facilitates early diagnosis. J Clin Endocrinol Metab. 2012;97:2125–2132. doi: 10.1210/jc.2012-1739. [DOI] [PubMed] [Google Scholar]
- 66.Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P; International Small for Gestational Age Advisory Board. International Small for Gestational Age Advisory Board consensus development conference statement: management of short children born small for gestational age, April 24-October 1, 2001. Pediatrics. 2003;111:1253–1261. doi: 10.1542/peds.111.6.1253. [DOI] [PubMed] [Google Scholar]
- 67.Finken MJJ, van der Steen M, Smeets CCJ, Walenkamp MJE, de Bruin C, Hokken-Koelega ACS, Wit JM. Children born small for gestational age: differential diagnosis, molecular genetic evaluation, and implications. Endocr Rev. 2018;39:851–894. doi: 10.1210/er.2018-00083. [DOI] [PubMed] [Google Scholar]
- 68.Breukhoven PE, Kerkhof GF, van Dijk M, Hokken-Koelega AC. Long-term impact of GH treatment during childhood on body composition and fat distribution in young adults born SGA. J Clin Endocrinol Metab. 2011;96:3710–3716. doi: 10.1210/jc.2011-1658. [DOI] [PubMed] [Google Scholar]
- 69.van der Steen M, Hokken-Koelega AC. Growth and metabolism in children born small for gestational age. Endocrinol Metab Clin North Am. 2016;45:283–294. doi: 10.1016/j.ecl.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 70.Argente J, Gracia R, Ibáñez L, Oliver A, Borrajo E, Vela A, López-Siguero JP, Moreno ML, Rodríguez-Hierro F; Spanish SGA Working Group. Improvement in growth after two years of growth hormone therapy in very young children born small for gestational age and without spontaneous catch-up growth: results of a multicenter, controlled, randomized, open clinical trial. J Clin Endocrinol Metab. 2007;92:3095–3101. doi: 10.1210/jc.2007-0078. [DOI] [PubMed] [Google Scholar]
- 71.van der Kaay D, Bakker B, van der Hulst F, Mul D, Mulder J, Schroor E, van Elswijk D, Rowaan I, Willeboer M, de Ridder M, Hokken-Koelega A. Randomized GH trial with two different dosages in combination with a GnRH analogue in short small for gestational age children: effects on metabolic profile and serum GH, IGF1, and IGFBP3 levels. Eur J Endocrinol. 2010;162:887–895. doi: 10.1530/EJE-09-1113. [DOI] [PubMed] [Google Scholar]
- 72.de Zegher F, Hokken-Koelega A. Growth hormone therapy for children born small for gestational age: height gain is less dose dependent over the long term than over the short term. Pediatrics. 2005;115:458–462. doi: 10.1542/peds.2004-1934. [DOI] [PubMed] [Google Scholar]
- 73.Acerini CL, Wac K, Bang P, Lehwalder D. Optimizing patient management and adherence for children receiving growth hormone. Front Endocrinol (Lausanne) 2017;8:313. doi: 10.3389/fendo.2017.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haverkamp F, Johansson L, Dumas H, Langham S, Tauber M, Veimo D, Chiarelli F. Observations of nonadherence to recombinant human growth hormone therapy in clinical practice. Clin Ther. 2008;30:307–316. doi: 10.1016/j.clinthera.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 75.Jackson C, Eliasson L, Barber N, Weinman J. Applying COM-B to medication adherence. Eur Health Psychologist. 2014;16:7–17. [Google Scholar]
- 76.Horne R. Compliance, adherence, and concordance: implications for asthma treatment. Chest. 2006;130(1 Suppl):65–72. doi: 10.1378/chest.130.1_suppl.65S. [DOI] [PubMed] [Google Scholar]
- 77.Fisher BG, Acerini CL. Understanding the growth hormone therapy adherence paradigm: a systematic review. Horm Res Paediatr. 2013;79:189–196. doi: 10.1159/000350251. [DOI] [PubMed] [Google Scholar]
- 78.Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, Savage MO. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect. 2018;7:212–222. doi: 10.1530/EC-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cutfield WS, Derraik JG, Gunn AJ, Reid K, Delany T, Robinson E, Hofman PL. Non-compliance with growth hormone treatment in children is common and impairs linear growth. PLoS One. 2011;6:e16223. doi: 10.1371/journal.pone.0016223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kapoor RR, Burke SA, Sparrow SE, Hughes IA, Dunger DB, Ong KK, Acerini CL. Monitoring of concordance in growth hormone therapy. Arch Dis Child. 2008;93:147–148. doi: 10.1136/adc.2006.114249. [DOI] [PubMed] [Google Scholar]
- 81.Bozzola M, Pagani S, Iughetti L, Maffeis C, Bozzola E, Meazza C. Adherence to growth hormone therapy: a practical approach. Horm Res Paediatr. 2014;81:331–335. doi: 10.1159/000357975. [DOI] [PubMed] [Google Scholar]
- 82.Dahlgren J. Easypod: a new electronic injection device for growth hormone. Expert Rev Med Devices. 2008;5:297–304. doi: 10.1586/17434440.5.3.297. [DOI] [PubMed] [Google Scholar]
- 83.Maggio MC, Vergara B, Porcelli P, Corsello G. Improvement of treatment adherence with growth hormone by easypod™ device: experience of an Italian centre. Ital J Pediatr. 2018;44:113. doi: 10.1186/s13052-018-0548-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bozzola M, Colle M, Halldin-Stenlid M, Larroque S, Zignani M; easypod™ survey study group. Treatment adherence with the easypod™ growth hormone electronic auto-injector and patient acceptance: survey results from 824 children and their parents. BMC Endocr Disord. 2011;11:4. doi: 10.1186/1472-6823-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koledova E, Stoyanov G, Ovbude L, Davies PSW. Adherence and long-term growth outcomes: results from the easypod™ connect observational study (ECOS) in paediatric patients with growth disorders. Endocr Connect. 2018;7:914–923. doi: 10.1530/EC-18-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wit JM, Oostdijk W. Novel approaches to short stature therapy. Best Pract Res Clin Endocrinol Metab. 2015;29:353–366. doi: 10.1016/j.beem.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 87.Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol. 2013;9:325–334. doi: 10.1038/nrendo.2013.71. [DOI] [PubMed] [Google Scholar]
- 88.Sas TC, Gault EJ, Bardsley MZ, Menke LA, Freriks K, Perry RJ, Otten BJ, de Muinck Keizer-Schrama SM, Timmers H, Wit JM, Ross JL, Donaldson MD. Safety and efficacy of oxandrolone in growth hormone-treated girls with Turner syndrome: evidence from recent studies and recommendations for use. Horm Res Paediatr. 2014;81:289–297. doi: 10.1159/000358195. [DOI] [PubMed] [Google Scholar]
- 89.Yanovski JA, Rose SR, Municchi G, Pescovitz OH, Hill SC, Cassorla FG, Cutler GB Jr. Treatment with a luteinizing hormone-releasing hormone agonist in adolescents with short stature. N Engl J Med. 2003;348:908–917. doi: 10.1056/NEJMoa013555. [DOI] [PubMed] [Google Scholar]
- 90.Dunkel L. Treatment of idiopathic short stature: effects of gonadotropin-releasing hormone analogs, aromatase inhibitors and anabolic steroids. Horm Res PaediatR. 2011;76(Suppl 3):27–29. doi: 10.1159/000330150. [DOI] [PubMed] [Google Scholar]
- 91.Mul D, Wit JM, Oostdijk W, Van den Broeck J; Dutch Advisory Group on Growth Hormone. The effect of pubertal delay by GnRH agonist in GH-deficient children on final height. J Clin Endocrinol Metab. 2001;86:4655–4656. doi: 10.1210/jcem.86.10.7910. [DOI] [PubMed] [Google Scholar]
- 92.Mericq MV, Eggers M, Avila A, Cutler GB Jr, Cassorla F. Near final height in pubertal growth hormone (GH)-deficient patients treated with GH alone or in combination with luteinizing hormone-releasing hormone analog: results of a prospective, randomized trial. J Clin Endocrinol Metab. 2000;85:569–573. doi: 10.1210/jcem.85.2.6343. [DOI] [PubMed] [Google Scholar]
- 93.van Gool SA, Kamp GA, Visser-van Balen H, Mul D, Waelkens JJ, Jansen M, Verhoeven-Wind L, Delemarre-van de Waal HA, de Muinck Keizer-Schrama SM, Leusink G, Roos JC, Wit JM. Final height outcome after three years of growth hormone and gonadotropin-releasing hormone agonist treatment in short adolescents with relatively early puberty. J Clin Endocrinol Metab. 2007;92:1402–1408. doi: 10.1210/jc.2006-2272. [DOI] [PubMed] [Google Scholar]
- 94.Lem AJ, van der Kaay DC, de Ridder MA, Bakker-van Waarde WM, van der Hulst FJ, Mulder JC, Noordam C, Odink RJ, Oostdijk W, Schroor EJ, Sulkers EJ, Westerlaken C, Hokken-Koelega AC. Adult height in short children born SGA treated with growth hormone and gonadotropin releasing hormone analog: results of a randomized, dose-response GH trial. J Clin Endocrinol Metab. 2012;97:4096–4105. doi: 10.1210/jc.2012-1987. [DOI] [PubMed] [Google Scholar]
- 95.Benabbad I, Rosilio M, Tauber M, Paris E, Paulsen A, Berggren L, Patel H, Carel JC; Phoenix Study Group. Growth hormone in combination with leuprorelin in pubertal children with idiopathic short stature. Endocr Connect. 2018;7:708–718. doi: 10.1530/EC-18-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mericq V, Gajardo H, Eggers M, Avila A, Cassorla F. Effects of treatment with GH alone or in combination with LHRH analog on bone mineral density in pubertal GH-deficient patients. J Clin Endocrinol Metab. 2002;87:84–89. doi: 10.1210/jcem.87.1.8148. [DOI] [PubMed] [Google Scholar]