

Abstract
Alzheimer’s disease (AD) is the most common form of dementia. The accumulation of β-amyloid plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau protein are two hallmarks of AD. The β-amyloid and tau proteins have been at the center of AD research and drug development for decades. However, most of the clinical trials targeting β-amyloid have failed. Whereas the safety and efficacy of most tau-targeting drugs have not yet been completely assessed, the first tau aggregation inhibitor, LMTX, failed in a late-stage trial, leading to further recognition of the complexities of AD and reconsideration of the amyloid hypothesis and perhaps the tau hypothesis as well. Multilevel complex interactions between genetic, epigenetic, and environmental factors contribute to the occurrence and progression of AD. Formaldehyde (FA) is a widespread environmental organic pollutant. It is also an endogenous metabolite in the human body. Recent studies suggest that elevation of FA in the body by endogenous and/or exogenous exposure may play important roles in AD development. We have demonstrated that FA reduces lysine acetylation of cytosolic histones, thereby compromising chromatin assembly and resulting in the loss of histone content in chromatin, a conserved feature of aging from yeast to humans. Aging is an important factor for AD progression. Therefore, FA-induced inhibition of chromatin assembly and the loss of histones may contribute to AD initiation and/or development. This review will briefly summarize current knowledge on mechanistic insights into AD, focusing on epigenetic alterations and the involvement of FA in AD development. The exploration of chemical exposures as contributing factors to AD may provide new insights into AD mechanisms and could identify potential novel therapeutic targets.
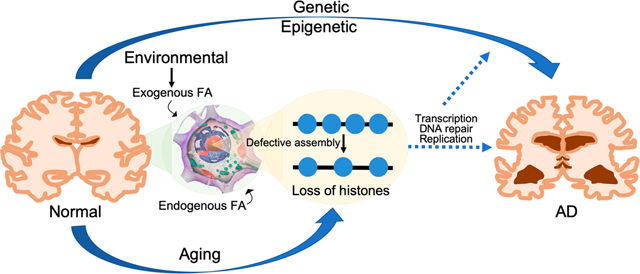
Graphical Abstract
1. INTRODUCTION
In 2017, the World Health Organization (WHO) reported that worldwide there are around 50 million people with dementia and nearly 10 million new cases every year. Alzheimer’s disease (AD) is the most common form of dementia and may contribute to 60–70% of cases.1 In the United States, it was estimated in 2010 that there were 4.7 million individuals aged 65 years or older with AD dementia, and the total number of AD patients was projected to rise to 13.8 million by 2050.2 Numerous factors contribute to the risk of developing AD, including aging, genetic factors, family history, a history of head trauma, midlife hypertension, obesity, diabetes, hypercholesterolemia, and certain dietary and lifestyle factors.3 AD is a progressive disease that begins with mild memory loss and possibly leads to loss of the ability for conversation and responses to the environment. AD dysfunction involves brain regions that control thought, memory, and language.4 AD can be classified into two main types on the basis of the age of onset: early-onset AD (EOAD) and late-onset AD (LOAD). EOAD exhibits cognitive symptoms roughly from 30 to 60 or 65 years of age and accounts for approximately 5% of all cases.5 LOAD, which is defined as AD with onset later than age 60 or 65, accounts for more than 90% of diagnosed AD patients. Both EOAD and LOAD may occur in individuals with a positive family history of AD, but the primary etiology in most AD patients is likely due to multiple susceptibility genes and environmental factors.6 The failures of most clinical trials targeting β-amyloid and tau proteins led to further recognition of the complexities of AD. In this review, we briefly summarize the mechanism(s) of AD focusing on epigenetic regulation and the effects of formaldehyde (FA), a ubiquitous environmental contaminant, in AD occurrence and development.
2. HYPOTHESIS OF AD PATHOGENESIS
The “accumulation of amyloid-β” hypothesis is the most common explanation for the etiology of AD. Amyloid-β (Aβ) peptides result from the sequential cleavage of amyloid precursor protein (APP), which is a large transmembrane protein found in many tissues.7 The proteolytic processing of APP to produce amyloidogenic fragments results from sequential proteolysis by the γ-secretase complex, composed of presenilin 1 (PSEN1) and presenilin 2 (PSEN2), which themselves are subject to mutations that can alter APP cleavage to form longer, toxic Aβ peptides.8 The neurotoxic Aβ peptides can cause neuronal dysfunction and induce neuronal apoptosis through interactions with cell-surface receptors. Long-term accumulation of toxic Aβ species in the brain parenchyma (composed of neurons and glial cells) leads to the formation of aggregates that can result in oxidative damage of DNA and proteins9 and progressively contributes to tau pathology, synaptic dysfunction, inflammation, neuronal loss, and ultimately dementia.10 Although tau pathology is not a result of Aβ accumulation, it plays an important role in the appearance of Aβ-induced toxicity.11 When tau is hyperphosphorylated, it can break away from microtubules and produce neurofibrillary tangles that cause neuronal dysfunction in AD.
Currently, there are several other hypotheses to explain the neurobiological dysfunctions underlying AD. These include but are not limited to the cholinergic hypothesis that includes glutamatergic neurotransmission alterations, the involvement of oxidative stress, disruptions of calcium channels,12 persistent inflammatory responses,13 microRNA activity,14 loss of DNA integrity, faulty cell-cycle regulation, regression of myelination,14 and autophagy.15,16
3. GENETIC STUDIES IN AD
The first case of AD was reported in 1906, but the precise pathogenesis leading to the development of AD is still unknown. With regard to genetic risk considerations, mutations of APP, PSEN1, and PSEN2 genes are well-established genetic risk factors for EOAD and familial AD.17–19 In LOAD cases, apolipoprotein E (APOE) plays an important role. APOE4 fragments increase tau phosphorylation, leading to GABAergic (GABA = γ-aminobutyric acid) interneuron impairment.20 The levels of butyrylcholinesterase (BCHE) in cortical regions have been found to be increased and to be enriched within Aβ plaques and in the neurofibrillary tangles in AD brains.21 Genetic variations of BCHE can increase the cortical butyrylcholinesterase activity, leading to decreased acetylcholine levels and inducing synaptic functioning disruption, glial cell activation, Aβ plaque formation, and neurodegeneration.22 APOE4- and BCHE-positive cases are suggested to be associated with EOAD diagnosis and accelerated cognitive decline.23 In addition, other genes have also been found to be involved in AD. With the application of genome-wide association studies (GWAS), some newly described putative risk genes for LOAD include ATP-binding cassette subfamily A member 7 (ABCA7), bridging integrator 1 gene (BIN1), triggering receptor expressed on myeloid cells (TREM), cluster of differentiation 33 (CD33), clusterin (CLU), complement receptor 1 (CR1), ephrin type-A receptor 1 (EPHA1), CD2-associated protein (CD2AP), membrane-spanning 4-domains subfamily A (MS4A) gene cluster and phosphatidylinositol-binding clathrin assembly protein (PIC-ALM). The proteins coded by these various potential AD candidate genes participate in a variety of cellular processes such as oxidative homeostasis, protein metabolism, inflammatory responses, cholesterol metabolism, and synaptic function.24
4. EPIGENETIC STUDIES IN AD
Whereas genomic studies have identified several genes potentially associated with AD, the vast majority of AD cases do not show strong genetic underpinnings. Some recent evidence suggests that epigenetics may also contribute to influence the risk of AD development. Epigenetics is the study of modifications in gene expression and/or chromatin structure, RNA editing, and RNA interference without changes in the underlying DNA sequence.25,26 These modifications can occur on DNA molecules (mainly methylation on cytosine bases at cytosine–guanine dinucleotide [CpG] sites) or on the histone proteins that form the fundamental structure of chromatin, primarily via histone methylation, acetylation, phosphorylation, ubiquitination, and sumoylation. Furthermore, noncoding RNAs (ncRNAs) can regulate gene expression by recruiting DNA methyltransferases and chromatin modifiers to their targets, inhibiting translation of mRNA and aiding in the degradation or stability of mRNA by sequence complementarity with their targets.27
4.1. DNA Methylation.
DNA methylation refers to the addition of a methyl group to the cytosine nucleotide at CpG sites via methyl group donation from S-adenosylmethionine (SAM), which is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, −2, −3a/b, and −4. DNA methylation of a gene promoter usually represses transcription. Hyper- or hypomethylation of CpG sites can change over time as a function of genes interacting with their environments, and the changes in DNA methylation levels have implications for gene expression changes via alterations in chromatin structure. CpG sites are the most important methylation sites associated with aging, which is also an important risk factor of AD, while loci not associated with CpG islands were most often hypomethylated.28
DNA-methylated cytosine bases (5mC) can be further modified sequentially to form 5-hydroxymethylcytosine (5hmC) and then 5-formylcytosine, which is modified further to 5-carboxymethylcytosine by the family of Tet enzymes.29 Thus, studies on changes in global DNA modification have focused on measurement of both 5mC or 5hmC. Some of these studies found lower levels of 5mC and 5hmC associated with AD in the glioblastoma cell line H4,30 in the neuron-like cell line SH-SY5Y,31 and in the entorhinal cortex in postmortem brain tissue.32 In contrast, other studies found increased 5mC and 5hmC levels, or no change, in different cell lines or brain tissues. These inconsistent results may be due in part to different techniques, heterogeneous tissue, and limited sample sizes. Ellison et al.33 used gas chromatography/mass spectrometry (GC/MS) to examine global levels of 5mC and 5hmC in different brain regions affected by AD pathology across various disease stages. They found that changes in DNA modification are progression-related, as they occurred in the early stages of AD but reversed to levels observed in controls in later AD stages. These results may partly explain the conflict between previous studies.
Gene-specific DNA methylation changes have also been examined in AD. Strong associations were found between decreased DNA methylation and amyloid load, neuritic plaques, and diffuse plaques.34,35 De Jager et al.36 reported that the level of methylation at 71 of the 415 848 analyzed CpG sites was associated with AD pathology, including CpGs in the ABCA7 and BIN1 gene regions, which are susceptible variants in AD. Other CpG sites nearby genes whose DNA methylation and RNA expression levels were altered in AD included those associated with ANK1, CDH23, DIP2A, RHBDF2, RPL13, SERPINF1, and SERPINF2. Changes in DNA methylation of AD susceptibility genes could occur in the presymptomatic stage of AD and may be an early feature of the disease.36 In several studies, APP and APOE promoter hypomethylation has been found in AD patients.37–40 Among female monkeys who sustained infantile exposure to lead (Pb), DNMT1 activity was found to be decreased, resulting in increases in APP, β-site APP cleaving enzyme-1 (BACE1), and specificity protein 1 (SP1) mRNA and protein levels.41 PSEN1 promoter hypomethylation has been found to be associated with increased Aβ.42 Interestingly, APP and PSEN1 were found to be hypomethylated after S-adenosylhomocysteine (SAH) administration,43 and PSEN1 expression and Aβ production decrease with increasing SAM levels.44 In fact, high homocysteine, low vitamin B12, and low folate levels are often found in AD, suggesting a dysregulation in the SAM methyl donor cycle that is required for epigenetic regulation through DNA methylation.45,46 However, there also exist conflicting results.47 DNA methylation changes have also been found to be involved in the progression of AD. Neprilysin (NEP), an important enzyme for the degradation of Aβ, is decreased in the brains of AD patients.48 It has been verified that Aβ increased NEP DNA methylation, resulting in suppressing the NEP mRNA and protein expression in vitro.49 That may partially explain why accumulating Aβ cannot be removed effectively in AD progression. Although other critical AD-related genes such as the APP, PSEN1, and TAU genes, along with a vast array of other candidate genes, were investigated for changes in their DNA methylation, coincident conclusions have been ambiguous and difficult to solidify.50
Recently, genome-wide screening and epigenome-wide association studies (EWAS) were performed. The transmembrane protein 59 gene (TMEM59) was found to be hypomethylated in frontal cortex tissue of LOAD patients by Illumina Infinium Human Methylation 27K array analysis.51 In other studies, Watson et al.52 used the Illumina Infinium 450 K array to analyze neighboring AD-associated CpG sites of superior temporal gyrus tissue from AD patients. In this study, 479 differentially methylated regions (DMRs) were detected, and most of them were found to be hypermethylated. Another recently published study concluded that cell-type composition and aging were the major sources of data noise.53 Gasparoni et al.53 identified numerous genes with cell-type-specific methylation signatures that showed differential methylation dynamics associated with aging, specifically neuronal genes including CLU, SYNJ2, and NCOR2 or glail cell genes including RAI1, CXXC5, and INPP5A. Differentially methylated CpGs (DMCGs) identified in neuronal genes MCF2L, MAP2, STK32C, and S100B, in LRRC8B in both neurons and glia, and in ANK1 in glia were found to be associated with AD Braak stage progression. Although there are some limitations of this research, it has provided clues for validating the results of cell-specific studies. Additional research is needed to continue to enlighten us about the roles of DNA methylation related to AD disease etiology and progression.
4.2. Histone Modification.
In contrast to DNA methylation, histone modifications have been less studied in AD to date. As “writers” and “erasers” of acetylation, histone acetyltransferases (HATs) catalyze the acetylation of lysine residues (mostly in histone tails) and loosen chromatin, thereby facilitating gene transcription, whereas histone deacetylases (HDACs) catalyze removal of acetyl groups from lysine residues and condense chromatin, thereby repressing gene expression. Many studies have demonstrated significant roles for HATs and HDACs in learning and memory formation.54 For example, the stabilization of short-term memory into long-term memory was impaired in transgenic mice that express a mutant HAT, i.e., cAMP response element binding protein (CREB) binding protein (CBP), while the long-term memory and long-term potentiation (LTP) were impaired in a mouse model of the haploinsufficiency form of Rubinstein–Taybi syndrome (RTS), a disorder caused by CBP mutations. By contrast, improved memory formation and synaptic plasticity were observed in mice lacking HDAC2 or HDAC3. HDACs are assigned to classes I, II, III, and IV according to their sequence homology.55 HDAC2 (class I), HDAC6 (class IIb), and sirtuins1 (SIRTs1; class III) have primarily been linked to the pathobiology of AD. For example, neuron-specific over-expression of HDAC2 was shown to be associated with decreased synaptic plasticity and memory formation in mice.55 Conversely, HDAC2 deficiency increased synapse number and memory facilitation in mice.56 Moreover, HDAC6 has been found to be overexpressed in the brains of AD patients.57 Indeed, targeting HDACs is considered as a potential promsing therapy for AD, and several HDAC inhibitor therapeutics have been shown to play a protective role in AD.50
Given the observed changes in expression of HDACs and/or HATs in AD, it is not surprising to detect perturbations of histone lysine acetylations in AD patients.54 Using LC–MS/MS selected reaction monitoring (SRM) spectrometry, Zhang et al.58 found a significant decrease in global levels of H3K18 and H3K23 acetylations, two activating histone marks, in the temporal lobes of AD subjects compared with an age-matched control group. The results were further validated by LC–MS/MS–TMT (tandem-mass-tagging) and Western blot analysis. In another study, Hernandez-Ortega et al. examined global levels of H3K9 dimethylation (H3k9me2), a repressive histone mark, and H4K12 acetylation (H4K12ac), an active mark, in the hippocampi of 47 AD cases by immunohistochemistry and found that both H3K9me2 and H4K12ac were decreased in the cornu ammonis 1 (CA1) brain region in AD.59 This is consistent with a report showing that the level of histone H4 acetylation is 50% lower in APP/PS1 mice than in wild-type littermates.60 Interestingly, restoration of H4ac by HDAC inhibitors enabled improved learning in mice, suggesting a possible role for the loss of H4Kac in cognitive impairment. Increases in global histone modifications in post-mortem AD brain were also reported in several studies. Narayan et al. reported significant increase of H3 acetylation (K9/K14) and H4 acetylation (K5/K8/K12/K16), active histone marks, in free-floating AD interior temporal gyrus sections and in tissue microarrays of middle temporal gyrus compared with controls.61 Each marker correlated significantly with tau and amyloid load. Using immunohistochemistry, Western blot analysis, and ELISA, Mastroeni et al. showed an increase in H3K4 trimethylation (H3K4me3), an active mark, in cytoplasmic labeling and a decrease in nuclear labeling in AD brains compared with controls.62 Interestingly, cytoplasmic localization of H3K4me3 occurred before observation of tau hyperphosphorylation, suggesting that intracellular localization of H3K4me3 may represent an early epigenetic change in AD. Aβ peptide misfolding is considered to be critical for the progression of AD. Lithner et al. demonstrated hyper-acetylation of H3K14 in response to secreted Aβ or by direct exposure to soluble Aβ in cell and mouse model systems.63 They further found by Western blot analysis that H3K14 acetylation as well as H3K9 dimethylation were significantly increased in the occipital cortex in post-mortem AD compared to nondemented and age-matched control subjects. Overall, these findings reveal the complicated features of global histone modification alterations in AD, and it is likely that the observed inconsistencies and variations might be due to the different brain regions studied or to the various methodologies used.
Using chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-seq), Gjoneska et al.64 profiled epigenome landscapes and correlated them with transcriptional changes during neurodegeneration in the hippocampus of the CK-p25 mouse model of AD and CK littermate controls. Among several chromatin marks tested, relative differences in H3K4me3 levels (associated with active promoters) resulted in 3667 increased-level and 5056 decreased-level peaks, while relative levels of H3K27ac (associated with enhancers) resulted in 2456 increased-level and 2154 decreased-level peaks. Differences in polycomb-repressed (H3K27me3) and hetero-chromatin (H3K9me3) regions were seen in only a small number of peaks. They found that genes with increased-level peaks for H3Kme3 (promoters) or H3K27ac (enhancers) in AD are associated with immune and stimulus-response functions and that genes with decreased-level peaks for H3Kme3 and H3K27ac are related to synapse- and learning-associated functions. Interestingly, genetic variants associated with AD from GWAS were enriched in increased-level enhancer (H3K27ac) orthologues but not in decreased-level enhancer orthologues, suggesting that the immune basis of AD is likely mediated by both genetic and epigenetic mechanisms while cognitive impairment is more likely influenced by environmentally driven epigenomic alterations in neuronal cells. Recently, the genome-wide profile of H4K16ac, a histone mark implicated in aging, showed that normal aging leads to gain of H4K16ac while dramatic loss of H4K16ac is observed in the proximity of genes linked to aging and AD in the lateral temporal lobe of AD individuals compared with younger, cognitively normal controls.65 In addition, this study found a strong association between AD GWAS single-nucleotide polymorphisms and AD expression quantitative trait loci with the regions of significant H4K16ac changes.65 In conclusion, these studies implicate histone modification alterations in neurodegeneration and AD development.
4.3. MicroRNAs.
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression at the post-translational level. There are more than 5000 known miRNAs, and each miRNA can regulate over 1000 specific targets.66 MicroRNAs exist in various body tissues, including brain and cerebrospinal fluid (CSF), and up to 70% of recognized miRNAs are known to be crucial regulators of neuronal and glial functions,67 as they constitute a regulatory network for stringent spatial and temporal gene expression.68 To date, there have been many studies focused on miRNAs and AD. Reduced expression of miR-29a, −29b-1,69 −107,70 −9,71 −298, and −32872 have been identified, targeting genes such as APP and BACE. MiR-9, which is expressed specifically in neuro-genic regions of the brain during neural development and in adulthood, had candidate binding sites within the 3′ untranslated regions of BACE1 and PSEN1.69 MiR-125b is one of the highly abundant miRNAs in the human brain and CSF, and it was found to induce tau phosphorylation at the AT 180 epitope and correlated best with AD progression.73 MiR-134 was reported to be increased as a result of the loss of SIRT1, resulting in the downregulated expression of CREB and brain-derived neurotrophic factor (BDNF), thereby impairing synaptic plasticity. MiR-34c was noted to be upregulated in AD brains and caused synaptic dysfunction as well as memory impairment through SIRT1.74 Overall, this brief survey substantiates the roles to which miRNAs contribute in most aspects of AD pathogenesis.75 However, the results of studies of archived human brain tissues were highly variable, with some miRNAs upregulated and others downregulated, and the results were not found to be consistent with AD progression.76
5. FORMALDEHYDE AND ALZHEIMER’S DISEASE
In addition to genetics and epigenetics, environmental factors play important roles in AD. A large body of literature shows the positive associations between AD and air pollutants (PM2.5),77 heat waves,78 heavy metals (notably cadmium79 and zinc80), type 2 diabetes, smoking,81,82 and others. Formaldehyde (FA), a highly reactive single-carbon aldehyde, is widely distributed in living organisms and environments.83 Exposure to FA is known to cause acute health problems, such as upper respiratory illness, allergies, and possible death.84 Recently, many studies have focused on evaluating the association between chronic exposure to FA and its neuro-toxicity. Epidemiological studies have shown excess FA accumulated in AD patients,85 associated with impaired performance in learning and memory.86 Moreover, excessive exposure to FA was reported to induce amyloid aggregation87,88 and tau protein aggregation and hyperphosphorylation in vitro and in vivo.89,90 Rodent studies have shown that excess endogenous FA can lead to memory impairments, tau protein hyperphosphorylation, and neuronal loss.91,92 Mean-while, Yang et al. observed that FA, induced by chronic exposure to methanol, also formed β-amyloid plaques and caused memory impairments in monkeys.93 Moreover, Zhai et al. reported that FA induces AD-like pathologies and cognitive impairments in rhesus monkeys.94 Monkeys without AD-related mutations in the APP, PSEN1, and PSEN2 genes were chronically exposed to FA by intracerebroventricular injections over a 1 year period. The spatial working memory ability of the FA group was found to be remarkably decreased after 2 months of treatment, and the notable presence of AD pathological markers was observed in memory-related brain regions of FA-treated monkeys after 12 months of administration.94 FA-induced AD-like features in primate brain suggest that FA may play an important role in AD pathogenesis.
The FA burden in the human body originates from both endogenous and exogenous sources. Endogenous FA is the byproduct of aldehyde metabolism inside the body. There are three primary sources: oxidation of methanol, histone demethylation, and methylamine deamination. Exogenous FA exists ubiquitously in the environment in construction materials, agricultural fertilizers, fumigants, paints, cosmetics, polish, and cleaning agents.95 Typically, the level of FA is about 0.1–0.4 mM in the normal physiological brain,95 specifically 0.25–0.32 mM in the hippocampus.96 Recent data from clinical studies show that the elevation of endogenous FA concentrations in urine from dementia patients is positively correlated with cognitive impairment.97 Similarly, increased FA levels have also been observed in the hippocampi of AD patients.98 Taken together, these findings suggest that FA toxicity can be closely related to the critical hallmarks of AD pathology. The roles of FA in each of the specific AD pathology mechanisms reviewed herein are discussed below topically.
5.1. FA in Amyloid Peptide Aggregation.
Extracellular amyloid-β plaque is a major pathological feature of AD.99 Amyloid fibrils fuse to and emanate from the cerebral vascular basement membrane. Semicarbazide-sensitive amine oxidase (SSAO) is an enzyme that facilitates methylamine deamination and generates FA.95 SSAO is colocalized with cerebral vascular amyloid-β deposits in Alzheimer’s brains.100 FA, derived from SSAO-mediated deamination of methylamine,101 is an electrophilic compound; therefore, it can form protein adducts by reacting with the nucleophilic side chains of amino acids, such as lysine and arginine. Aβ-amyloid molecules are composed of two lysines and one arginine, which are vulnerable to react with FA and other aldehydes.101 The reaction between lysine residues and FA generates the intermediate N6-hydroxymethyllysine, from which a labile Schiff base or the more stable N6-formyllysine can be produced by a subsequent dehydration or oxidation reaction, respectively. Two adjacent Schiff bases would cross-link to each other and form a stable methylene bridge.102 These formaldehyde–protein adducts may alter protein structure, protein deposition, and subsequent plaque formation in the compartment adjacent to the cerebrovessels.101 Meanwhile, FA has the capacity to increase the rate of formation of β-amyloid β-sheets, oligomers, and protofibrils and also may alter the size of the aggregates.87,102 Moreover, FA is a potential inflammatory agent,103 and inflammation is well-known to play an important role in AD.104 The cytotoxicity of FA stimulates inflammation and releases more SSAO, which generates a cascading toxic cycle.105
Genetic variation of Apolipoprotein E (APOE) and aging are two major risk factors for AD. Among three isoforms, i.e., apolipoprotein E allele 2 (APOE2), APOE3, and APOE4, APOE4 is considered as the major genetic risk factor associated with AD, whereas APOE3 is neutral and APOE2 is protective.106,107 Recently, Huang et al. demonstrated that APOE4 secreted by glia enhanced APP transcription and Aβ synthesis the most compared with APOE3 and APOE2 in ES-cell-derived human neurons.107 Interestingly, in the presence of 10 mM FA in vitro, APOE4 formed aggregates with more Aβ and APOE protein content than aggregates formed with either APOE3 or APOE2.88 The three APOE isoforms differ by only two residues, with APOE4 having arginine residues at 112 and 158. FA may preferentially target APOE4 more than APOE2 or APOE3 since FA forms cross-links with larger lysine, tryptophan, and arginine residues. FA has been found to be increased in AD patients. Taken together, these results suggest that FA may play a role in the differential pattern of amyloid plaque formation in AD patients with differing APOE genetic backgrounds.
5.2. FA in Tau Protein Phosphorylation and Aggregation.
In the AD brain, tau is about 3–4-fold more phosphorylated than in the normal adult brain.108 There is evidence that FA can induce tau hyperphosphorylation and aggregation in vivo and in vitro.91,109,110 Yang et al.111 have found that FA, as a methanol metabolite, contributed to amyloid peptide aggregation and tau protein phosphorylation and aggregation in chronic feeding studies of young male monkeys given 3% methanol. The enhanced tau phosphorylation observed in the brain tissue was noted to be persistent six months after the methanol feeding had stopped. Moreover, studies showed that compared with equal concentrations of methanol, FA induced tau protein to form spherical cytotoxic aggregates and led to significant increases in tau phosphorylation at both the T181 and S396 sites, indicating that FA readily leads to cellular impairments.89,92 These findings suggest the pathological changes related to AD can be long-lasting and persistent. Glycogen synthase kinase-3 (GSK-3) is a proline-directed serine/threonine kinase that plays essential roles in physiological processes. Phosphorylated tau, which is catalyzed by GSK-3β, interferes with the bindings between tau protein and DNA. Thus, hyperphosphorylation disturbs the interaction between tau protein and DNA, which may subsequently lead to DNA damage and even cell death.112 It has been reported that GSK-3β, tau oligomers, and phosphorylated and truncated forms of tau are elevated in a mouse model of AD.113 To summarize, the neurotoxicity of FA has been associated with neuronal tau aggregation.102
5.3. FA and Microtubule Disintegration.
Tau protein promotes the assembly and maintenance of microtubules.108 Abnormally, tau phosphorylation detaches tau protein from microtubules, causing microtubules to be diminished in number and density.114 This loss of microtubules disturbs the capacity of neurons to maintain axonal transport, synaptic connections, and neuronal porphogenesis.115 The loss of microtubules from axons and dendrites is a key contributor to nervous system degeneration during AD.116 FA was found to induce morphological changes of microtubules in mouse embryonic cerebral cortical neurons. Moreover, FA-induced neuronal microtubule deterioration is observed at lower concentrations than with methanol and formic acid.92
5.4. FA and DNA Methylation.
DNA methylation, which is regulated by enzymes with DNA methyltransferase (DNMT) activity, is critically required for memory formation in the brain.117 DNMT1 and DNMT3a knockout mice exhibit reduced global DNA methylation in the brain and show evidence of resulting deficits of memory acquisition.118,119 A decline in global DNA methylation was found in the autopsied hippocampi of patients with AD.34,120 Tong et al.121 reported that a marked increase in endogenous FA levels is associated with a decline of global DNA methylation in the autopsied hippocampi from AD patients. Thus, excess FA could reduce global DNA methylation by interfering with the function of DNMTs in vitro and in vivo. This is an area of study that is ripe for further research.
5.5. FA and Histone Modification.
As an electrophile, FA can potentially react with nucleophilic side chains of amino acids, such as lysine, arginine, histidine, and cysteine. However, FA formed adducts only with lysine residues on histone H4 in vitro.122 In vivo studies have also confirmed the formation of FA–histone lysine adducts (Figure 1A). Since the formation of FA–histone lysine adducts is refractory to lysine acetylation by histone acetyltransferase(s), we examined whether the levels of histone lysine modifications, in particular lysine acetylations, are changed following FA exposure.122 We found that total levels of H3K18ac, H3K9ac, and H3K14ac were decreased in BEAS-2B cells treated with FA, whereas total levels of lysine methylations, including H3K4me3, H3K9me2, and H3K27me3, were not changed by FA exposure. Cell fractionation analysis further demonstrated that in addition to H3K9 and H3K14 acetylations, the level of H4K12ac in cytosolic and chromatin fractions was also dramatically decreased upon FA treatment. The downregulation of these modifications was likely due to the formation of FA–histone lysine adducts, as the expression of histone acetyltransferase(s) was not changed by FA exposure. These experimental results are partially consistent with reported observations of a significant decrease in H3K18ac in the temporal lobe of AD subjects58 and a decrease in H4K12ac in the CA1 region in AD.
Figure 1.

FA induces loss of histones in chromatin. (A) Formation of FA–histone adducts. The reaction between lysine residues and FA generates the intermediate N6-hydroxymethyllysine, from which a labile Schiff base (primary FA–lysine adduct) or the more stable N6-formyllysine can be produced by a subsequent dehydration or oxidation reaction, respectively. Adapted from ref 122. (B) FA forms adducts with lysines on histone proteins, preventing the sites from being physiologically modified. FA exposure reduces acetylations of lysines on newly synthesized histones H3 and H4, thereby inhibiting nucleosome assembly, which contributes to the loss of histones in chromatin, a conserved feature of aging.
Since histone H4K5 and H4K12 acetylations as well as lysine acetylations of the N-terminal tail of H3 are considered to be important for histone nuclear translocation and assembly into chromatin, we also tested whether FA exposure regulates chromatin assembly pathways that can alter chromatin structure.122 The amount of histone H3 in chromatin fractions isolated from BEAS-2B cells treated with FA was lower than that in chromatin from untreated cells. Moreover, the enrichment of histone H3 in certain genomic locations was also decreased following FA exposure, suggesting that FA might be able to compromise chromatin assembly, resulting in the loss of histones in chromatin (Figure 1B). Aberrant chromatin assembly can induce defects in DNA repair, replication, and transcription. Thus, RNA-seq was first used to characterize FA-regulated genes in BEAS-2B cells exposed to 100 μM FA for 48 h. A total of 654 genes were identified as FA-inducible genes (see Table S1 in ref 122). Interestingly, ingenuity pathway analysis (IPA) showed that neurological disease was one of top five diseases and biological functions related to these FA-response genes.122 A growing number of studies have demonstrated that the loss of histone proteins is a conserved feature of aging. Age-related histone losses have been observed in Caenorhabditis elegans, mice, and humans. In yeast, aging is accompanied by a profound loss of nucleosomal histones, and overexpression of core histones enhances yeast longevity, suggesting that histone losses may be critical for aging. Because AD is an aging disease, establishing the correlations of FA to histone loss, aging, and AD has important implications for our understanding of the mechanisms of AD and for identifying potential preventative or therapeutic interventions.
5.6. FA and MicroRNAs.
MicroRNAs are well-known to influence diseases caused by various environmental exposures, yet miRNAs have been severely understudied in relation to formaldehyde exposure. One study123 found that FA significantly disrupts miRNA expression profiles within the nasal epithelium of cynomolgus macaques induced by inhaled formaldehyde exposure in vivo. Among 13 miRNAs profiled, miR-125b, miR-152, miR-219–5p, and miR-532–5p were increased and miR-22, miR-26b, miR-29a, miR-140–5p, miR-142–3p, miR-203, and miR-374a were decreased, and these alterations likely influenced apoptosis signaling. MicroRNAs are largely represented in the nervous system, playing an essential role in nerve cell differentiation and neuronal function.124–126 Changes in expression of specific miRNAs have been found in neurodegenerative diseases, including spinal motor neuron disease, Huntington’s disease, and AD.102,127,128 For example, the activity of miR-134 is upregulated in the brains of animals with status epilepticus, and when it was silenced, hippocampal dendritic spine density was reduced and mice became refractory to hippocampal damage and seizures.129 In addition, miR-17–5p, miR-20a, and miR-106b were associated with downregulation of the expression of endogenous APP in vitro.130 Thus, it is possible that dysregulation of miRNAs might be involved in FA-induced AD pathogenesis. It is imperative to fully understand the mechanisms underlying FA-induced toxicity in the brain through modulation of miRNA pathways.
5.7. FA and Inflammation and Oxidative Stress.
The levels of pro-inflammatory markers, including NF-κB, TNF-α, IL-1β, and COX-2, have been shown to be increased significantly in mice after 15.5 mg kg−1 day−1 FA exposure.109 16s rRNA next-generation sequencing analysis showed an increase in bacterial populations in AD brain tissues compared with normal controls.131 Inflammatory reactions included activated microglia and astrocytes, which are important for blood–brain barrier integrity, formation of synapses, and homeostasis of neurotransmitters, among other things. C-reactive protein (CRP) has been demonstrated not only as a marker of inflammation but also to act as a direct mediator of inflammatory reactions and the innate immune response. Research has found that CRP is highly expressed in neuron tissue of AD patients and that monomeric CRP (mCRP) directly contributes to AD pathogenesis.132 In an epidemiology investigation, serum CRP levels were lower in FA-exposed workers than in unexposed controls.133 Thus, the relationship between CRP and FA exposure related to AD needs further exploration. In vivo, significant increases in reactive oxygen species and malondialdehyde levels and decreases in glutathione and superoxide dismutase levels have been reported after FA exposure in mouse brain. Increased 8-OH-dG levels suggest that there is DNA damage induced by oxidative stress in mouse brain.109 A dramatic decrease in melatonin levels in post-mortem cerebrospinal fluid of AD patients has been reported.134 FA can directly inactivate melatonin and induce intensive oxidative stress by reducing glutathione levels,135 linking FA to AD through oxidative stress.
6. CONCLUSIONS AND FUTURE PERSPECTIVES
Alzheimer’s disease is the consequence of a complex combination of effects related to genetic, epigenetic, and environmental factors. The elevation of FA in brain tissue may play an important role in AD development involving multifarious mechanisms. However, direct causal links and interconnected mechanisms involving FA and amyloid-β, tau protein, histone modifications, chromatin assembly, and histone loss in particular, remain to be fully demonstrated. Since most of the amyloid-targeting and some of the tau-targeting AD clinical trials have ended in failure, identifying possible novel mechanisms underlying AD is a major challenge that we are facing. Continuous study of the involvement of chemical exposures in AD development and progress may provide us with new insight into AD mechanisms and potential therapeutic targets.
Funding
This work was supported in part by National Institutes of Health Grants ES026138 and ES029359 to C.J. and NIEHS Center of Excellence Pilot Project Program ES000260 to C.J.
Biography
Fei Wang is an Assistant Professor in the School of Public Health at China Medical University. She obtained her Ph.D. in Public Health from China Medical University in 2015. She joined Dr. Jin’s lab at New York University School of Medicine as a visiting scholar in 2018 and worked on the mechanisms underlying formaldehyde-induced Alzheimer’s disease. Meanwhile, she also studied the effect of chromium exposure on post-translational histone modifications.
Danqi Chen is a Research Scientist in the Department of Environmental Medicine at New York University School of Medicine. She obtained her B.S. from China Medical University in 2008 and her Ph.D. in Environmental Health Science from New York University in 2018. Her research has focused on arsenic-induced polyadenylation of canonical histone mRNAs and its roles in arsenic carcinogenesis as well as aldehyde-induced chromatin dysregulation.
Peipei Wu obtained her M.D. in Clinical Medicine from Zhengzhou University in China. She is currently in the Environmental Health Sciences Master’s Program at New York University and is a graduate assistant in the Department of Environmental Medicine at NYU School of Medicine. Her research concentrates on epigenetics and human disease.
Catherine Klein is an Assistant Professor in the Department of Environmental Medicine at New York University School of Medicine. She obtained an M.S. in Human Genetics from George Washington University and her Ph.D. from NYU. Her work has focused on understanding the mutagenic and epigenetic mechanisms involved in the toxic and adverse health effects of metals, including nickel, chromium, and arsenic.
Chunyuan Jin is an Assistant Professor in the Department of Environmental Medicine at New York University School of Medicine. His laboratory is interested in understanding the mechanistic insight into dysregulation of chromatin assembly following environmental exposures and its roles in environmental diseases. Before joining NYU, he obtained his Ph.D. from the University of Tokyo and finished his postdoctoral training with Dr. Gary Felsenfeld at NIH.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).World Health Organization (2017) Dementia Factsheet, https://www.who.int/news-room/fact-sheets/detail/dementia.
- (2).Hebert LE, Weuve J, Scherr PA, and Evans DA (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80, 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barnard ND, Bush AI, Ceccarelli A, Cooper J, de Jager CA, Erickson KI, Fraser G, Kesler S, Levin SM, Lucey B, Morris MC, and Squitti R (2014) Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol. Aging 35, S74–S78. [DOI] [PubMed] [Google Scholar]
- (4).Centers for Disease Control and Prevention (2018) Alzheimer’s Disease and Healthy Aging, https://www.cdc.gov/aging/aginginfo/alzheimers.htm.
- (5).Freudenberg-Hua Y, Li W, and Davies P (2018) The Role of Genetics in Advancing Precision Medicine for Alzheimer’s Disease—A Narrative Review. Front. Med 5, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bekris LM, Yu CE, Bird TD, and Tsuang DW (2010) Genetics of Alzheimer disease. J. Geriatr Psychiatry Neurol 23, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang H, Ma Q, Zhang Y-W, and Xu H (2012) Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem 120 (s1), 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hardy JA, and Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- (9).Canter RG, Penney J, and Tsai LH (2016) The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539, 187–196. [DOI] [PubMed] [Google Scholar]
- (10).De Strooper B, and Karran E (2016) The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- (11).Mandelkow EM, and Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harbor Perspect. Med 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sanabria-Castro A, Alvarado-Echeverria I, and Monge-Bonilla C (2017) Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci 24, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, and Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schonrock N, Matamales M, Ittner LM, and Gotz J (2012) MicroRNA networks surrounding APP and amyloid-beta metabolism–implications for Alzheimer’s disease. Exp. Neurol 235, 447–454. [DOI] [PubMed] [Google Scholar]
- (15).Chen Y, Wei G, Nie H, Lin Y, Tian H, Liu Y, Yu X, Cheng S, Yan R, Wang Q, Liu DH, Deng W, Lai Y, Zhou JH, Zhang SX, Lin WW, and Chen DF (2014) Beta-Asarone prevents autophagy and synaptic loss by reducing ROCK expression in asenescence-accelerated prone 8 mice. Brain Res 1552, 41–54. [DOI] [PubMed] [Google Scholar]
- (16).Zhang Y, Chen X, Zhao Y, Ponnusamy M, and Liu Y (2017) The role of ubiquitin proteasomal system and autophagy–lysosome pathway in Alzheimer’s disease. Rev. Neurosci 28, 861–868. [DOI] [PubMed] [Google Scholar]
- (17).Goate A (2006) Segregation of a missense mutation in the amyloid beta-protein precursor gene with familial Alzheimer’s disease. J. Alzheimer’s Dis 9, 341–347. [DOI] [PubMed] [Google Scholar]
- (18).Brickell KL, Leverenz JB, Steinbart EJ, Rumbaugh M, Schellenberg GD, Nochlin D, Lampe TH, Holm IE, Van Deerlin V, Yuan W, and Bird TD (2007) Clinicopathological concordance and discordance in three monozygotic twin pairs with familial Alzheimer’s disease. J. Neurol., Neurosurg. Psychiatry 78, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tanzi RE, and Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- (20).Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, Halabisky B, Deng C, Mahley RW, and Huang Y (2009) GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Darvesh S, Hopkins DA, and Geula C (2003) Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci 4, 131–138. [DOI] [PubMed] [Google Scholar]
- (22).Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, Foroud TM, Hakonarson H, Huentelman MJ, Aisen PS, Petersen RC, Green RC, Jack CR, Koeppe RA, Jagust WJ, Weiner MW, and Saykin AJ (2014) APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol. Psychiatry 19, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).De Beaumont L, Pelleieux S, Lamarre-Theroux L, Dea D, and Poirier J (2016) Butyrylcholinesterase K and Apolipoprotein E-varepsilon4 Reduce the Age of Onset of Alzheimer’s Disease, Accelerate Cognitive Decline, and Modulate Donepezil Response in Mild Cognitively Impaired Subjects. J. Alzheimer’s Dis 54, 913–922. [DOI] [PubMed] [Google Scholar]
- (24).Misra A, Chakrabarti SS, and Gambhir IS (2018) New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med. Res 148, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Holliday R (2006) Epigenetics: a historical overview. Epigenetics 1, 76–80. [DOI] [PubMed] [Google Scholar]
- (26).Dupont C, Armant DR, and Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin. Reprod. Med 27, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Veerappan CS, Sleiman S, and Coppola G (2013) Epigenetics of Alzheimer’s disease and frontotemporal dementia. Neurotherapeutics 10, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, Sugarbaker DJ, Yeh RF, Wiencke JK, and Kelsey KT (2009) Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 5, No. e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Roubroeks JAY, Smith RG, van den Hove DLA, and Lunnon K (2017) Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neuro-chem 143, 158–170. [DOI] [PubMed] [Google Scholar]
- (30).Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, and Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360, 672–674. [DOI] [PubMed] [Google Scholar]
- (31).Murrell J, Farlow M, Ghetti B, and Benson MD (1991) A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99. [DOI] [PubMed] [Google Scholar]
- (32).Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, and Rogers J (2010) Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol. Aging 31, 2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ellison EM, Abner EL, and Lovell MA (2017) Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J. Neurochem 140, 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, and van den Hove DL (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 34, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Levine ME, Lu AT, Bennett DA, and Horvath S (2015) Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 7, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, Tang A, Raj T, Replogle J, Brodeur W, Gabriel S, Chai HS, Younkin C, Younkin SG, Zou F, Szyf M, Epstein CB, Schneider JA, Bernstein BE, Meissner A, Ertekin-Taner N, Chibnik LB, Kellis M, Mill J, and Bennett DA (2014) Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci 17, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).West RL, Lee JM, and Maroun LE (1995) Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci 6, 141–146. [DOI] [PubMed] [Google Scholar]
- (38).Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, and Ukitsu M (1999) Reduction with age in methylcytosine in the promoter region –224 approximately –101 of the amyloid precursor protein gene in autopsy human cortex. Mol. Brain Res 70, 288–292. [DOI] [PubMed] [Google Scholar]
- (39).Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Ukitsu M, and Genda Y (1999) The methylation status of cytosines in a tau gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett 275, 89–92. [DOI] [PubMed] [Google Scholar]
- (40).Wang SC, Oelze B, and Schumacher A (2008) Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One 3, No. e2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, and Zawia NH (2008) Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J. Neurosci 28, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, and Scarpa S (2011) Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 32, 187–199. [DOI] [PubMed] [Google Scholar]
- (43).Lin HC, Hsieh HM, Chen YH, and Hu ML (2009) S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. NeuroToxicology 30, 622–627. [DOI] [PubMed] [Google Scholar]
- (44).Scarpa S, Fuso A, D’Anselmi F, and Cavallaro RA (2003) Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Lett 541, 145–148. [DOI] [PubMed] [Google Scholar]
- (45).Choi SW, and Mason JB (2002) Folate status: effects on pathways of colorectal carcinogenesis. J. Nutr 132, 2413S–2418S. [DOI] [PubMed] [Google Scholar]
- (46).Smith AD (2008) The worldwide challenge of the dementias: a role for B vitamins and homocysteine? Food Nutr Bull 29, S143–172. [DOI] [PubMed] [Google Scholar]
- (47).Barrachina M, and Ferrer I (2009) DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol 68, 880–891. [DOI] [PubMed] [Google Scholar]
- (48).Park MH, Lee JK, Choi S, Ahn J, Jin HK, Park JS, and Bae JS (2013) Recombinant soluble neprilysin reduces amyloid-beta accumulation and improves memory impairment in Alzheimer’s disease mice. Brain Res 1529, 113–124. [DOI] [PubMed] [Google Scholar]
- (49).Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, and Hu CJ (2009) The epigenetic effects of amyloid-beta(1–40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem. Biophys. Res. Commun 378, 57–61. [DOI] [PubMed] [Google Scholar]
- (50).Sanchez-Mut JV, and Graff J (2015) Epigenetic Alterations in Alzheimer’s Disease. Front. Behav. Neurosci 9, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, and Rozek LS (2012) Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimer’s Dis 29, 571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Watson CT, Roussos P, Garg P, Ho DJ, Azam N, Katsel PL, Haroutunian V, and Sharp AJ (2016) Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Gasparoni G, Bultmann S, Lutsik P, Kraus TFJ, Sordon S, Vlcek J, Dietinger V, Steinmaurer M, Haider M, Mulholland CB, Arzberger T, Roeber S, Riemenschneider M, Kretzschmar HA, Giese A, Leonhardt H, and Walter J (2018) DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenet. Chromatin 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Narayan P, and Dragunow M (2017) Alzheimer’s Disease and Histone Code Alterations. Adv. Exp. Med. Biol 978, 321–336. [DOI] [PubMed] [Google Scholar]
- (55).Xu WS, Parmigiani RB, and Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552. [DOI] [PubMed] [Google Scholar]
- (56).Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, and Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ding H, Dolan PJ, and Johnson GV (2008) Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem 106, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, and Kirsch W (2012) Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 12, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hernandez-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, and Ferrer I (2016) Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol 26, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, and Arancio O (2009) Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimer’s Dis 18, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Narayan PJ, Lill C, Faull R, Curtis MA, and Dragunow M (2015) Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol. Dis 74, 281–294. [DOI] [PubMed] [Google Scholar]
- (62).Mastroeni D, Delvaux E, Nolz J, Tan Y, Grover A, Oddo S, and Coleman PD (2015) Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol. Aging 36, 3121–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lithner CU, Lacor PN, Zhao WQ, Mustafiz T, Klein WL, Sweatt JD, and Hernandez CM (2013) Disruption of neocortical histone H3 homeostasis by soluble Abeta: implications for Alzheimer’s disease. Neurobiol. Aging 34, 2081–2090. [DOI] [PubMed] [Google Scholar]
- (64).Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, and Kellis M (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F, Toledo JB, Gosai SJ, Gregory BD, Torres C, Trojanowski JQ, Wang LS, Johnson FB, Bonini NM, and Berger SL (2018) Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci 21, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Londin E, Loher P, Telonis AG, Quann K, Clark P, Jing Y, Hatzimichael E, Kirino Y, Honda S, Lally M, Ramratnam B, Comstock CE, Knudsen KE, Gomella L, Spaeth GL, Hark L, Katz LJ, Witkiewicz A, Rostami A, Jimenez SA, Hollingsworth MA, Yeh JJ, Shaw CA, McKenzie SE, Bray P, Nelson PT, Zupo S, Van Roosbroeck K, Keating MJ, Calin GA, Yeo C, Jimbo M, Cozzitorto J, Brody JR, Delgrosso K, Mattick JS, Fortina P, and Rigoutsos I (2015) Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. U. S. A 112, E1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Boudreau RL, Jiang P, Gilmore BL, Spengler RM, Tirabassi R, Nelson JA, Ross CA, Xing Y, and Davidson BL (2014) Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 81, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Cheng C, Bhardwaj N, and Gerstein M (2009) The relationship between the evolution of microRNA targets and the length of their UTRs. BMC Genomics 10, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, and De Strooper B (2008) Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U. S. A 105, 6415–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, and Nelson PT (2008) The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci 28, 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schonrock N, Humphreys DT, Preiss T, and Gotz J (2012) Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-beta. J. Mol. Neurosci 46, 324–335. [DOI] [PubMed] [Google Scholar]
- (72).Boissonneault V, Plante I, Rivest S, and Provost P (2009) MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J. Biol. Chem 284, 1971–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, and Terro F (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev 12, 289–309. [DOI] [PubMed] [Google Scholar]
- (74).Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, Mak G, Kim D, Su SC, and Tsai LH (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Dehghani R, Rahmani F, and Rezaei N (2018) MicroRNA in Alzheimer’s disease revisited: implications for major neuro-pathological mechanisms. Rev. Neurosci 29, 161–182. [DOI] [PubMed] [Google Scholar]
- (76).Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, and De Jager PL (2015) Epigenomics of Alzheimer’s disease. Transl. Res 165, 200–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Liu XD, Zhang YC, Luo C, Kang J, Li JQ, Wang K, Ma P, and Yang X (2017) At seeming safe concentrations, synergistic effects of PM2.5 and formaldehyde co-exposure induces Alzheimer-like changes in mouse brain. Oncotarget 8, 98567–98579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Culqui DR, Linares C, Ortiz C, Carmona R, and Diaz J (2017) Association between environmental factors and emergency hospital admissions due to Alzheimer’s disease in Madrid. Sci. Total Environ 592, 451–457. [DOI] [PubMed] [Google Scholar]
- (79).Popugaeva E, and Bezprozvanny I (2014) Can calcium hypothesis explain synaptic loss in Alzheimer’s disease? Neurodegener. Dis 13, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lui E, Fisman M, Wong C, and Diaz F (1990) Metals and the liver in Alzheimer’s disease. An investigation of hepatic zinc, copper, cadmium, and metallothionein. J. Am. Geriatr. Soc 38, 633–639. [DOI] [PubMed] [Google Scholar]
- (81).Ampuero I, Bermejo-Pareja F, García Ribas G, and García de Yébenes J (2009) Environmental and Genetic Risk Factors for Alzheimer’s Disease in Spain. Eur. Neurol. Rev 4 (2), 125–128. [Google Scholar]
- (82).Reitz C, and Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol 88, 640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).He R, Lu J, and Miao J (2010) Formaldehyde stress. Sci. China: Life Sci 53, 1399–1404. [DOI] [PubMed] [Google Scholar]
- (84).Tang X, Bai Y, Duong A, Smith MT, Li L, and Zhang L (2009) Formaldehyde in China: production, consumption, exposure levels, and health effects. Environ. Int 35, 1210–1224. [DOI] [PubMed] [Google Scholar]
- (85).Tong Z, Wang W, Luo W, Lv J, Li H, Luo H, Jia J, and He R (2016) Urine Formaldehyde Predicts Cognitive Impairment in Post-Stroke Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis 55, 1031–1038. [DOI] [PubMed] [Google Scholar]
- (86).Perna RB, Bordini EJ, and Deinzer-Lifrak M (2001) A case of claimed persistent neuropsychological sequelae of chronic formaldehyde exposure: clinical, psychometric, and functional findings. Arch. Clin. Neuropsychol 16, 33–44. [PubMed] [Google Scholar]
- (87).Chen K, Kazachkov M, and Yu PH (2007) Effect of aldehydes derived from oxidative deamination and oxidative stress on beta-amyloid aggregation; pathological implications to Alzheimer’s disease. J. Neural Transm (Vienna) 114, 835–839. [DOI] [PubMed] [Google Scholar]
- (88).Rizak JD, Ma Y, and Hu X (2014) Is formaldehyde the missing link in AD pathology? The differential aggregation of amyloid-beta with APOE isoforms in vitro. Curr. Alzheimer Res 11, 461–468. [DOI] [PubMed] [Google Scholar]
- (89).Nie CL, Wei Y, Chen X, Liu YY, Dui W, Liu Y, Davies MC, Tendler SJ, and He RG (2007) Formaldehyde at low concentration induces protein tau into globular amyloid-like aggregates in vitro and in vivo. PLoS One 2, No. e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Lu J, Miao J, Su T, Liu Y, and He R (2013) Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim. Biophys. Acta, Gen. Subj 1830, 4102–4116. [DOI] [PubMed] [Google Scholar]
- (91).Tong Z, Han C, Luo W, Wang X, Li H, Luo H, Zhou J, Qi J, and He R (2013) Accumulated hippocampal formaldehyde induces age-dependent memory decline. Age (Dordr) 35, 583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Yang M, Lu J, Miao J, Rizak J, Yang J, Zhai R, Zhou J, Qu J, Wang J, Yang S, Ma Y, Hu X, and He R (2014) Alzheimer’s disease and methanol toxicity (part 1): chronic methanol feeding led to memory impairments and tau hyperphosphorylation in mice. J. Alzheimer’s Dis 41, 1117–1129. [DOI] [PubMed] [Google Scholar]
- (93).Yang M, Miao J, Rizak J, Zhai R, Wang Z, Huma T, Li T, Zheng N, Wu S, Zheng Y, Fan X, Yang J, Wang J, Yang S, Ma Y, Lu L, He R, and Hu X (2014) Alzheimer’s disease and methanol toxicity (part 2): lessons from four rhesus macaques (Macaca mulatta) chronically fed methanol. J. Alzheimer’s Dis 41, 1131–1147. [DOI] [PubMed] [Google Scholar]
- (94).Zhai R, Rizak J, Zheng N, He X, Li Z, Yin Y, Su T, He Y, He R, Ma Y, Yang M, Wang Z, and Hu X (2018) Alzheimer’s Disease-Like Pathologies and Cognitive Impairments Induced by Formaldehyde in Non-Human Primates. Curr. Alzheimer Res 15, 1304–1321. [DOI] [PubMed] [Google Scholar]
- (95).Tulpule K, and Dringen R (2013) Formaldehyde in brain: an overlooked player in neurodegeneration? J. Neurochem 127, 7–21. [DOI] [PubMed] [Google Scholar]
- (96).Tong Z, Han C, Luo W, Li H, Luo H, Qiang M, Su T, Wu B, Liu Y, Yang X, Wan Y, Cui D, and He R (2013) Aging-associated excess formaldehyde leads to spatial memory deficits. Sci. Rep 3, 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Tong ZQ, Zhang JL, Luo WH, Wang WS, Li FX, Li H, Luo HJ, Lu J, Zhou JN, Wan Y, and He RQ (2011) Urine formaldehyde level is inversely correlated to mini mental state examination scores in senile dementia. Neurobiol. Aging 32, 31–41. [DOI] [PubMed] [Google Scholar]
- (98).Tong Z, Zhang J, Luo W, Wang W, Li F, Li H, Luo H, Lu J, Zhou J, Wan Y, and He R (2011) Urine formaldehyde level is inversely correlated to mini mental state examination scores in senile dementia. Neurobiol. Aging 32, 31–41. [DOI] [PubMed] [Google Scholar]
- (99).Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev 81, 741–766. [DOI] [PubMed] [Google Scholar]
- (100).Ferrer I, Lizcano JM, Hernandez M, and Unzeta M (2002) Overexpression of semicarbazide sensitive amine oxidase in the cerebral blood vessels in patients with Alzheimer’s disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurosci. Lett 321, 21–24. [DOI] [PubMed] [Google Scholar]
- (101).Gubisne-Haberle D, Hill W, Kazachkov M, Richardson JS, and Yu PH (2004) Protein cross-linkage induced by formaldehyde derived from semicarbazide-sensitive amine oxidase-mediated deamination of methylamine. J. Pharmacol. Exp. Ther 310, 1125–1132. [DOI] [PubMed] [Google Scholar]
- (102).Chen K, Maley J, and Yu PH (2006) Potential inplications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem 99, 1413–1424. [DOI] [PubMed] [Google Scholar]
- (103).Tao YX, and Johns RA (2002) Activation and up-regulation of spinal cord nitric oxide receptor, soluble guanylate cyclase, after formalin injection into the rat hind paw. Neuroscience 112, 439–446. [DOI] [PubMed] [Google Scholar]
- (104).McGeer PL, and McGeer EG (2001) Inflammation, autotoxicity and Alzheimer disease. Neurobiol. Aging 22, 799–809. [DOI] [PubMed] [Google Scholar]
- (105).Yu PH (2001) Involvement of cerebrovascular semi-carbazide-sensitive amine oxidase in the pathogenesis of Alzheimer’s disease and vascular dementia. Med. Hypotheses 57, 175–179. [DOI] [PubMed] [Google Scholar]
- (106).Roses AD, Strittmatter WJ, Pericak-Vance MA, Corder EH, Saunders AM, and Schmechel DE (1994) Clinical application of apolipoprotein E genotyping to Alzheimer’s disease. Lancet 343, 1564–1565. [DOI] [PubMed] [Google Scholar]
- (107).Huang YA, Zhou B, Wernig M, and Sudhof TC (2017) ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 168, 427–441e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, and Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U. S. A 83, 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Liu X, Zhang Y, Wu R, Ye M, Zhao Y, Kang J, Ma P, Li J, and Yang X (2018) Acute formaldehyde exposure induced early Alzheimer-like changes in mouse brain. Toxicol. Mech. Methods 28, 95–104. [DOI] [PubMed] [Google Scholar]
- (110).He X, Li Z, Rizak JD, Wu S, Wang Z, He R, Su M, Qin D, Wang J, and Hu X (2017) Resveratrol Attenuates Formaldehyde Induced Hyperphosphorylation of Tau Protein and Cytotoxicity in N2a Cells. Front. Neurosci 10, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Yang MF, Miao JY, Rizak J, Zhai RW, Wang ZB, Huma T, Li T, Zheng N, Wu SH, Zheng YW, Fan XN, Yang JZ, Wang JH, Yang SC, Ma YY, Lu LB, He RQ, and Hu XT (2014) Alzheimer’s Disease and Methanol Toxicity (Part 2): Lessons from Four Rhesus Macaques (Macaca mulatta) Chronically Fed Methanol. J. Alzheimer’s Dis 41, 1131–1147. [DOI] [PubMed] [Google Scholar]
- (112).Lu J, Miao JY, Pan R, and He RQ (2011) Formaldehyde-mediated Hyperphosphorylation Disturbs The Interaction Between Tau Protein and DNA. Shengwu Huaxue Yu Shengwu Wuli Jinzhan 38, 1113–1120. [Google Scholar]
- (113).Kisby GE, Ryan A, Beam M, and Woltjer R (2011) The cycad genotoxin methylazoxymethanol (MAM) induces brain tissue DNA damage and accelerates tau pathology in htau mice. Society of Neuroscience Abstracts [Google Scholar]
- (114).Iqbal K, Alonso A. d. C., Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, and Grundke-Iqbal I (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta, Mol. Basis Dis 1739, 198–210. [DOI] [PubMed] [Google Scholar]
- (115).Feijoo C, Campbell DG, Jakes R, Goedert M, and Cuenda A (2005) Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci 118, 397–408. [DOI] [PubMed] [Google Scholar]
- (116).Jean DC, and Baas PW (2013) It cuts two ways: microtubule loss during Alzheimer disease. EMBO J 32, 2900–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Day JJ, and Sweatt JD (2010) DNA methylation and memory formation. Nat. Neurosci 13, 1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, and Fan G (2010) Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci 13, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, and Sweatt JD (2010) Cortical DNA methylation maintains remote memory. Nat. Neurosci 13, 664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, and Rogers J (2011) Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 32, 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Tong Z, Han C, Qiang M, Wang W, Lv J, Zhang S, Luo W, Li H, Luo H, Zhou J, Wu B, Su T, Yang X, Wang X, Liu Y, and He R (2015) Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer’s disease. Neurobiol. Aging 36, 100–110. [DOI] [PubMed] [Google Scholar]
- (122).Chen D, Fang L, Mei S, Li H, Xu X, Des Marais TL, Lu K, Liu XS, and Jin C (2017) Regulation of Chromatin Assembly and Cell Transformation by Formaldehyde Exposure in Human Cells. Environ. Health Perspect 125, 097019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Rager JE, Moeller BC, Doyle-Eisele M, Kracko D, Swenberg JA, and Fry RC (2013) Formaldehyde and epigenetic alterations: microRNA changes in the nasal epithelium of nonhuman primates. Environ. Health Perspect 121, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, and Impey S (2005) A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. U. S. A 102, 16426–16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, and Greenberg ME (2006) A brain-specific microRNA regulates dendritic spine development. Nature 439, 283–289. [DOI] [PubMed] [Google Scholar]
- (126).Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, Hubel K, Dekker F, Hedberg C, Rengarajan B, Drepper C, Waldmann H, Kauppinen S, Greenberg ME, Draguhn A, Rehmsmeier M, Martinez J, and Schratt GM (2009) A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol 11, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Packer AN, Xing Y, Harper SQ, Jones L, and Davidson BL (2008) The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci 28, 14341–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Haramati S, Chapnik E, Sztainberg Y, Eilam R, Zwang R, Gershoni N, McGlinn E, Heiser PW, Wills AM, Wirguin I, Rubin LL, Misawa H, Tabin CJ, Brown R Jr., Chen A, and Hornstein E (2010) miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. U. S. A 107, 13111–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, Sano T, O’Tuathaigh C, Waddington JL, Prenter S, Delanty N, Farrell MA, O’Brien DF, Conroy RM, Stallings RL, DeFelipe J, and Henshall DC (2012) Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat. Med 18, 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Hebert SS, Horre K, Nicolai L, Bergmans B, Papadopoulou AS, Delacourte A, and De Strooper B (2009) MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis 33, 422–428. [DOI] [PubMed] [Google Scholar]
- (131).Emery DC, Shoemark DK, Batstone TE, Waterfall CM, Coghill JA, Cerajewska TL, Davies M, West NX, and Allen SJ (2017) 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer’s Post-Mortem Brain. Front. Aging Neurosci 9, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, and Peter K (2018) Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol 9, 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Seow WJ, Zhang L, Vermeulen R, Tang X, Hu W, Bassig BA, Ji Z, Shiels MS, Kemp TJ, Shen M, Qiu C, Reiss B, Beane Freeman LE, Blair A, Kim C, Guo W, Wen C, Li L, Pinto LA, Huang H, Smith MT, Hildesheim A, Rothman N, and Lan Q (2015) Circulating immune/inflammation markers in Chinese workers occupationally exposed to formaldehyde. Carcinogenesis 36, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, and Swaab DF (1999) Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J. Clin. Endocrinol. Metab 84, 323–327. [DOI] [PubMed] [Google Scholar]
- (135).Mei Y, Duan C, Li X, Zhao Y, Cao F, Shang S, Ding S, Yue X, Gao G, Yang H, Shen L, Feng X, Jia J, Tong Z, and Yang X (2016) Reduction of Endogenous Melatonin Accelerates Cognitive Decline in Mice in a Simulated Occupational Form-aldehyde Exposure Environment. Int. J. Environ. Res. Public Health 13, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]