

Abstract
Objective
A 2-stage genome-wide association was conducted to explore the genetic etiology of amyotrophic lateral sclerosis (ALS) in the Chinese Han population.
Methods
Totally, 700 cases and 4,027 controls were genotyped in the discovery stage using Illumina Human660W-Quad BeadChips. Top associated single nucleotide polymorphisms from the discovery stage were then genotyped in an independent cohort with 884 cases and 5,329 controls. Combined analysis was conducted by combining all samples from the 2 stages.
Results
Two novel loci, 1p31 and 12p11, showed strong associations with ALS. These novel loci explained 2.2% of overall variance in disease risk. Expression quantitative trait loci searches identified TYW/CRYZ and FGD4 as risk genes at 1p13 and 12p11, respectively.
Conclusions
This study identifies novel susceptibility genes for ALS. Identification of TYW3/CRYZ in the current study supports the notion that insulin resistance may be involved in ALS pathogenesis, whereas FGD4 suggests an association with Charcot-Marie-Tooth disease.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease1 characterized clinically by paralysis and amyotrophy that ultimately progresses to fatal respiratory muscle failure. Numerous genes have been linked to ALS susceptibility. The gene encoding superoxide dismutase 1 (SOD1) is a well-known gene involved in familial ALS, and SOD1 mutations are also reported in a small portion of sporadic ALS cases.2,3 Tar DNA binding protein 43 (TDP-43) is an important component of protein deposits observed in the postmortem ALS brain,4 and TDP-43 gene mutations have been identified in both familial and sporadic ALS.5,6 The RNA/DNA-binding protein fused in sarcoma was also identified in ALS protein deposits.7 Recently, many studies have reported that hexanucleotide repeat expansion in the C9orf72 gene is associated with ALS with frontotemporal dementia.8,9 Furthermore, haploinsufficiency of TBK1 can also cause ALS and FTD.10
Numerous genome-wide association studies (GWASs) have been conducted to elucidate the genetic etiology of ALS. Several such studies have identified 9p21, the location of C9orf72, as a susceptibility locus for ALS.11,12 In our previous study, the first GWAS for ALS in the Chinese Han population, 2 single nucleotide polymorphisms (SNPs) located at CAMK1G and SUSUD2 were indentified.13
The genes cited above explain only a smaller fraction of ALS risk, so the pathomechanisms leading to ALS remain elusive. To further explore the genetic etiology of ALS, we analyzed genotyping data from a 2-stage GWAS of 1,584 cases and 9,356 controls (figure e-1, links.lww.com/NXG/A193).
Methods
Participants
Two cohorts of participants were included in the discovery stage of the current study. The first cohort with 533 cases and 1,892 controls was genotyped in our previous GWAS,13 whereas the second cohort with 167 cases and 2,135 controls was recruited by the First Affiliated Hospital of Anhui Medical University, West China Hospital, and other hospitals in China. The validation stage was conducted in an independent cohort of 884 cases and 5,329 controls also recruited by of the First Affiliated Hospital of Anhui Medical University, West China Hospital, and other hospitals in China. All participants in both the discovery and validation stages were of Han Chinese ancestry. All cases fulfilled the criteria for definite or probable ALS according to the revised El Escorial criteria.14 The diagnosis was conducted by at least 2 neurologists. Controls were matched with cases for age, sex, and geographic distribution (birthplace) and had no medical or family history of neurologic diseases.
Standard protocol approvals, registrations, and patient consents
Written informed consent was obtained from all participants. The study was approved by the Ethics Committee of Anhui Medical University.
Genotyping in the discovery stage
Genomic DNA was extracted for genotyping. Genotyping was performed according to the Infinium Human DNA protocol from Illumina.15 All samples were whole-genome amplified, fragmented, precipitated, and resuspended in appropriate hybridization buffer. Denatured samples were hybridized on prepared Illumina Human660W-Quad BeadChips. The BeadChip oligonucleotides were extended by a single labeled base, which was detected by fluorescence imaging using an Illumina Bead Array Reader. SNP genotypes of each samples were converted from fluorescence intensities using Illumina GenomeStudio software.
Quality controls in the discovery stage
In the discovery analysis, SNPs were excluded if they had a call rate <98% in cases or controls, minor allele frequency (MAF) < 5% in the population, or deviation from Hardy-Weinberg equilibrium in the controls (phwe < 1 × 10−4). Samples were excluded if the genotyping call rate was <98%. Case and control samples were then examined for potential genetic relatedness by identity by state analysis using PLINK 1.07 software. Cases and controls in the second cohort were assessed independently by principal component analysis (PCA) using EIGENSTRAT.16 Outliers of PCA were removed.
SNP selection for replication
The top associated common SNPs (p < 5.0 × 10−4 and MAF > 0.05) were chosen for replication. We excluded SNPs validated in a previous study.13 Two SNPs (rs8141797 and rs6703183) reported in our previous study were also excluded from the current validated stage. In total, 60 SNPs were selected for replication.
Genotyping in the replication study and Quality Control in the validation study
Genomic DNA was extracted from blood samples and amplified using a PCR touch-down method. Purified PCR products were used as templates for micro-sequencing PCR (SNaPshot PCR). The purified SNaPshot products were used for electrophoresis. Allele detection was performed using an ABI 3730XL automatic sequencer. Genotype was analyzed using Genemapper 4.0. SNPs with a call rate <98% in cases or controls or deviation from Hardy-Weinberg equilibrium in the controls (p < 1 × 10−4) were excluded. Samples were exclude with a call rate <98%.
Statistical analysis
PCA was performed twice using EIGENSTRAT.16 Retained cases and controls after quality control (QC) in the discovery stage were assessed by first PCA. A quantile-quantile plot was constructed, and the genomic control value was calculated to evaluate the potential impact of population stratification. To minimize the adverse impact of population stratification, association analysis was performed using logistic regression including significant PCAs as covariates. The final joint analysis of the combined discovery and validation samples was performed using meta-analysis.17 Heterogeneity between the discovery and validation samples was tested using the Cochran Q-test.18 SNPs showing heterogeneity (phet < 0.05) were excluded from the genome-wide results. The variance in liability to ALS that could be explained by the identified SNPs was estimated by the restricted maximum likelihood method using the genome-wide complex trait analysis (GCTA) tool.19
We conducted functional annotation of the 2 confirmed SNPs by investigating the potential regulatory functions from HaploReg, SNiPA, and RegulomeDB, as well as expression quantitative trait loci (eQTL) effects in BRAINEAC (braineac.org/).20
The network analysis was conducted using the online database resource Search Tool for the Retrieval of Interacting Genes (STRING) (string-db.org).21 The genes that encode the protein products are from amyotrophic lateral sclerosis online genetics database (ALSOD) (alsod.org).22 The first STRING analysis presented a complex network centered on ubiquitin C (UBC) and ubiquitin B (UBB). Genes widely verified as ALS risk factors (SOD1, TARDBP, FUS, C9orf72, and TBK1)23 or identified by GWASs in the Chinese Han population (TYW3, CRYZ, FGD4, H3F3C, SUSD2, and CAMK1G) were selected to construct a subnetwork.
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
Discovery of novel risk loci by a 2-stage GWAS
In total, 700 cases and 4,027 controls were genotyped using Illumina Human660W-Quad BeadChips, including 533 cases and 1,892 controls genotyped in a previous GWAS13 and 167 cases and 2,135 controls newly genotyped (table e-1, links.lww.com/NXG/A193). After QC (see Methods), 666 cases and 3,988 controls with 473,080 SNPs were retained for further association analysis. The first PCA results confirmed that all samples were of Han Chinese ancestry (figure e-2), and the second PCA showed a good genetic match between all cases and controls (figure e-3). The quantile-quantile plot and genomic control value (λGC = 1.04) of the genome-wide association results suggested minimal population stratification (figure 1). Genome-wide association analysis was conducted by logistic regression. After Bonferroni correction, several SNPs showed association with ALS (p < 0.05) (figure 2). Two SNPs (rs8141797 and rs6703183) reported in our previous study showed obvious association with ALS in the current discovery stage (p = 2.05 × 10−5 for rs6703183 and p = 6.82 × 10−8 for rs8141797). The combined p value of the 2 SNPs was conducted by combining the samples from the current discovery stage and previous validation stage. Both of the 2 SNPs showed association with ALS with p < 5.0 × 10−8 (table e-2).
Figure 1. Quantile-quantile (QQ) plot of the genome-wide association results from the discovery analysis.
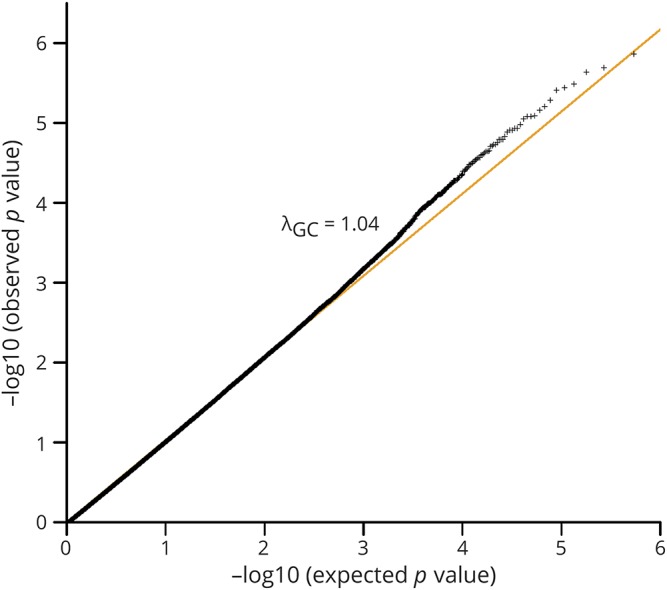
The quantile-quantile (Q–Q) plots of the p values (adjust PC1-4) from the expanded discovery analysis of 666 ALS cases and 3,988 controls. The x-axis is the expected p value (–log10 p value), and the y-axis is the observed p value (–log10 p value). Displayed are observed vs expected –log10 p values for all variants. The yellow line indicates the expected distribution of –log10 p values under the null hypothesis. The plot in black is for the p values from all SNPs. λGC shown in black by all the SNPs.
Figure 2. Manhattan plot of the genome-wide association results from the discovery analysis.

The genome-wide p values from logistic regression analysis of 456,414 SNPs in 666 cases and 3,988 controls are presented. The x-axis represents the chromosomal position, and the y-axis represents the −log 10 of the p value for each SNP.
After the discovery stage, 60 associated common SNPs (p < 5.0 × 10−4, MAF > 0.05) (table e-3, links.lww.com/NXG/A193) were selected for the following validation stage (SNP selection standards are described in the Methods). These SNPs were genotyped in an independent cohort of 884 cases and 5,329 controls (table e-1) using SNaPshot. Fifty-seven of the 60 selected SNPs were successfully genotyped, of which 31 showed association (p < 0.05) with ALS in the independent validation samples (table e-3).
The final joint analysis of the discovery and validation samples was performed using a meta-analysis.17 We tested heterogeneity between the discovery and validation samples using the Cochran Q-test.18 After combining the samples from both stages, 2 SNPs surpassed genome-wide significance, rs12145183 at 1p31 (pcombined = 2.10 × 10−14, OR = 1.60) and rs1419311 at 12p11 (pcombined = 5.19 × 10−9, OR = 1.27) (table 1). Both SNPs showed consistent association in the discovery and validation samples without genetic heterogeneity (phet > 0.05). The regional association plots of the detected SNPs were generated by LocusZoom24 (figure 3). We calculated the risk variance explained by the 2 SNPs using the restricted maximum likelihood method of the GCTA tool.19 These SNPs explained 2.2% of the overall variance in disease risk.
Table 1.
Association results of significant SNPs in the discovery, validation, and combined samples

Figure 3. Regional plots for 2 SNPs with genome-wide significant association.
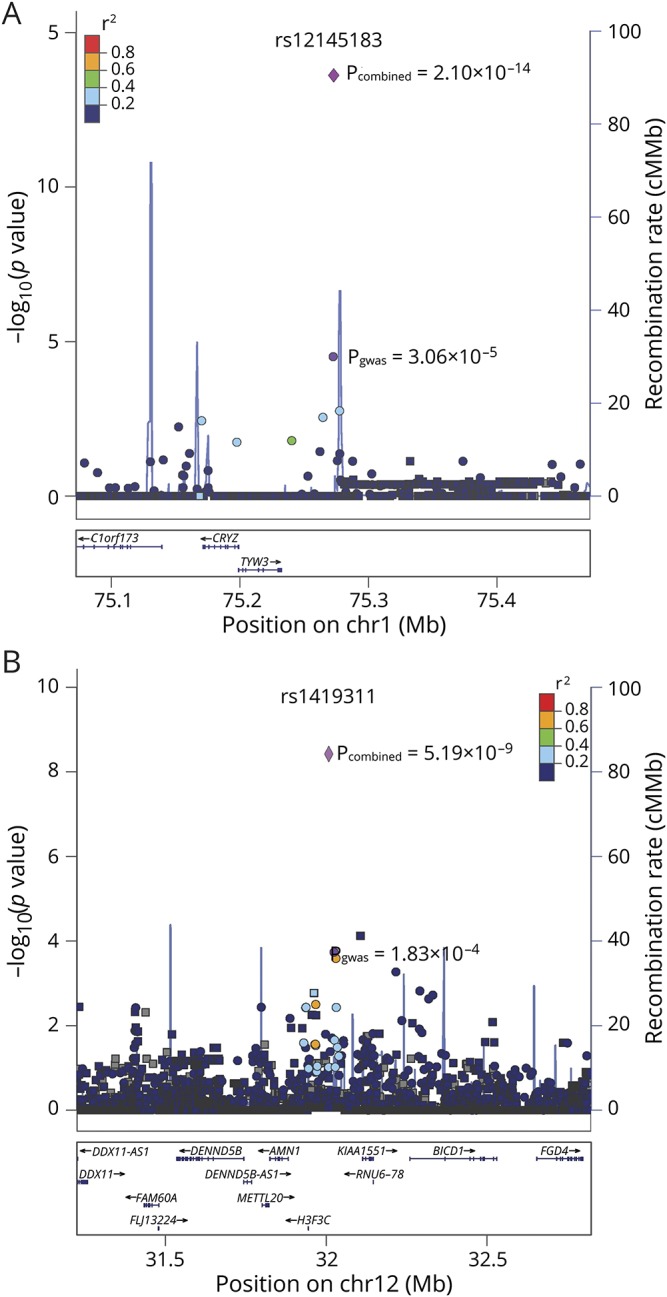
Regional association plots are p values (−log10 scale) of the association tests for genotyped SNPs (circles) and imputed SNPs (squares) in the discovery samples. The GWAS p value and combined p value of the 2 significant genotyped SNPs are shown by the purple circle and diamond. The top significant SNPs were given rsIDs. The color of each SNP spot reflects its r2 value with the rsID (from blue to red). Genetic recombination rates (based on 1000 Genomes Project reference data) are represented by light-blue lines, and genes within the regions are depicted by blue arrows. (A) Regional plot of 1p31. (B) Regional plot of 12p11.
Locus annotation
Both SNPs are intergenetic (table e-4, links.lww.com/NXG/A193). We fine mapped these SNPs to susceptibility genes by searching eQTL in the brain using BRAINEAC, which contains expression data of 1,231 brain tissue samples from 134 neurologically health controls across 10 brain regions.20
One of the identified SNPs, rs12145183, is located at 1p31 and is conserved during evolution (PhyloP: 0.023, genomic evolutionary rate profiling programs+: 1.84) (table e-4). The linkage disequilibrium (LD) block encompassing rs12145183 contains 2 genes, TYW3 and CRYZ. The regional association plot showed that a few SNPs in LD with rs12145183 (r2 > 0.2) are located at TYW3 and CRYZ (figure 3A). Furthermore, the cis-eQTL genes identified by searching BRAINEAC for rs12145183 were TYW3 (p = 3.10 × 10−5) and CRYZ (p = 4.30 × 10−5) (table e-5). Therefore, TYW3 and CRYZ were identified as candidate ALS susceptibility genes.
The other one, rs1419311, is located at 12p11 in an LD block with many genes, of which the nearest is H3F3C (table e-4, links.lww.com/NXG/A193). A regional association plot showed that many SNPs having high LD with this SNP (r2 > 0.6) are located at H3F3C (figure 3B). However, only 1 cis-eQTL gene at rs1419311 in the brain is FGD4 (p = 2.30 × 10−4) (table e-5). FGD4 is highly expressed in the spinal cord, frontal cortex, peripheral nerve, and some other tissues, whereas H3F3C is specifically expressed in the testis according to the Genotype-Tissue Expression pilot (figure e-4).25 Thus, FGD4 is the most likely susceptibility gene for ALS at 12p11.
Protein-protein interaction network results
Most cases of ALS likely arise from a complex interaction among many different susceptibility genes. To obtain a global view of the organization of these genes in ALS, we used protein-protein interaction (PPI) analysis of genes from ALSOD26 to map the relations among proteins expressed by these genes. Then, Retrieval of Interacting Genes (STRING) was used to cluster an expanded global network based on known PPIs using the genes from ALSOD.21 STRING analysis revealed a complex network centered on UBC and UBB (figure e-5, links.lww.com/NXG/A193). To clarify the relationship between UBC/UBB and ALS, widely verified ALS risk genes (SOD1, TARDBP, FUS, C9orf72, and TBK1)23 and those identified by GWASs of the Chinese Han population (TYW3, CRYZ, FGD4, H3F3C, SUSD2, and CAMK1G) were selected to construct a subnetwork. The subnetwork indicated that most of the selected genes were strongly linked to the UBC node (table e-6).
Discussion
The current study identifies CRYZ or TYW3 and FGD4 as strong candidate susceptibility genes for ALS in the Chinese Han population. Further network analysis of PPIs placed these and other susceptibility genes in a network with UBC as a hub, suggesting that ubiquitination is a common pathomechanism for ALS with distinct genetic etiologies.
CRYZ encodes zeta-crystallin, an nicotinamide-adenine dinucleotide phosphate-dependent quinone reductase reported to act as a trans-acting factor in the regulation of certain mRNAs such as Bcl-2.26 TYW3 encodes tRNA-wybutosine synthesis protein 3 homolog. Wybutosine is a hyper-modified phenylalanine tRNA.27 A recent study reported that TYW3/CRYZ is associated with insulin resistance.28 The role of insulin in aging has received increasing attention, and insulin resistance has also been implicated in neurodegenerative disease.29 Indeed, multiple studies have reported a high risk of Alzheimer disease in patients with type 2 diabetes.30–32 It has also been reported that the prevalence of diabetes is higher in patients with Parkinson disease than the general population.33 Furthermore, the mechanisms and consequences of insulin resistance may be involved in neurodegeneration, including reduced cerebral glucose metabolism, increased inflammation, oxidative stress, accumulation of advanced glycation end products, vascular dysfunction, reduced neurogenesis, and disrupted neuronal repair.29 Moreover, ALS has been directly associated with insulin resistance,34 in accord with our findings that TYW3/CRYZ is associated with ALS.
FGD4 encodes the Rho guanosine diphosphate/guanosine triphosphate exchange factor F-actin binding protein, mutations of which can cause Charcot-Marie-Tooth (CMT) type 4H.35 CMT, characterized by distal muscle weakness and atrophy, is the most common degenerative disorder of the peripheral nerves. Accumulating evidence suggests that CMT and ALS share certain genetic mechanisms. Indeed, mutation of SPG11, which causes autosomal recessive juvenile ALS,36 has also been reported in autosomal recessive juvenile axonal CMT.37 VCP and CHCHD10 have also been identified in both ALS and CMT.38,39 Our finding that FGD4 is associated with ALS is consistent with shared genetic mechanisms between CMT and ALS.
The PPI network analysis revealed a central role for UBC among the network of identified ALS genes (SOD1, TARDBP, FUS, C9orf72, TBK1, TYW3, CRYZ, FGD4, H3F3C, SUSD2, and CAMK1G). Ubiquitination is a critical endogenous pathway for protein degeneration, but pathologic ubiquitination can induce the formation of protein aggregates that ultimately form neurotoxic inclusions in the nucleus and cytoplasm. Extracellular and intracellular inclusions are key features of neurodegenerative diseases,40 and ubiquitin-positive inclusions are common in ALS.41 For instance, pathologically ubiquitinated TDP-43 aggregates are found in the nucleus and cytoplasm across diverse brain regions in FTD and motor neurons in ALS.4 Two pathology studies have also confirmed the presence of TDP-43-negative ubiquitin inclusions in ALS with C9orf72 mutation.42,43 Mutations in the gene encoding ubiquitin 2 (UBQLN2) have been detected in dominantly inherited chromosome X-linked ALS and ALS/dementia.44 Functional analysis suggests that abnormal ubiquilin 2 leads to impaired protein degradation and abnormal protein aggregation. Thus, ubiquitin-positive inclusions may be attributed to dysfunctional ubiquitination. The ubiqutin-proteasome system is regulated by ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligase E3. Increasing evidence indicates that E3 is involved in the development of insulin resistance,45 suggesting that TYW3/CRYZ may contribute to ALS pathogenesis through the ubiquitin pathway. Thus, multiple convergent lines of evidence suggest that abnormal ubiquitination may be a common pathogenic mechanism for ALS cases with otherwise distinct genetic etiologies.
Two SNPs showed associated with ALS at the whole-genome level in the Chinese Han population. These 2 SNPs explain 2.2% of overall variance in disease risk. The identification of TYW3/CRYZ and FGD4 at these loci suggests novel pathogenic mechanisms for ALS. Furthermore, PPI analysis revealed UBC as a central hub in the network of ALS-associated genes, suggesting that aberrant ubiquitination is a shared pathogenic mechanism for ALS.
Acknowledgment
The authors thank all participants in this study.
Glossary
- ALS
amyotrophic lateral sclerosis
- CMT
Charcot-Marie-Tooth
- eQTL
expression quantitative trait loci
- GCTA
genome-wide complex trait analysis
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- MAF
minor allele frequency
- PCA
principal component analysis
- PD
Parkinson disease
- PPI
protein-protein interaction
- QC
quality control
- SNP
single nucleotide polymorphism
- SOD1
superoxide dismutase 1
- STRING
Search Tool for the Retrieval of Interacting Genes
- UBB
ubiquitin B
- UBC
ubiquitin C
- UPS
ubiquitin-proteasome system
Appendix. Authors


Study funding
The study was funded by the National Key Research and Development Program of China (2016YFC1306400 and 2016YFC1300600), the National Natural Science Foundation of China (81401049), and Anhui Collaborative Innovation Center of Neuropsychiatric Disorder and Mental Health.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:639–649. [DOI] [PubMed] [Google Scholar]
- 2.Deng HX, Hentati A, Tainer JA, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993;261:1047–1051. [DOI] [PubMed] [Google Scholar]
- 3.Bowling AC, Schulz JB, Brown RH Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem 1993;61:2322–2325. [DOI] [PubMed] [Google Scholar]
- 4.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 5.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574. [DOI] [PubMed] [Google Scholar]
- 6.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 8.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 2015;18:631–636. [DOI] [PubMed] [Google Scholar]
- 11.Fogh I, Ratti A, Gellera C, et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum Mol Genet 2014;23:2220–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Es MA, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 2009;41:1083–1087. [DOI] [PubMed] [Google Scholar]
- 13.Deng M, Wei L, Zuo X, et al. Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat Genet 2013;45:697–700. [DOI] [PubMed] [Google Scholar]
- 14.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 15.Zhang XJ, Huang W, Yang S, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet 2009;41:205–210. [DOI] [PubMed] [Google Scholar]
- 16.Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 2009;41:1234–1237. [DOI] [PubMed] [Google Scholar]
- 17.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–1558. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramasamy A, Trabzuni D, Guelfi S, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014;17:1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011;39:D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abel O, Powell JF, Andersen PM, Al-Chalabi A. Credibility analysis of putative disease-causing genes using bioinformatics. PLoS One 2013;8:e64899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017;13:96–104. [DOI] [PubMed] [Google Scholar]
- 24.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapucci A, Lulli M, Amedei A, et al. zeta-Crystallin is a bcl-2 mRNA binding protein involved in bcl-2 overexpression in T-cell acute lymphocytic leukemia. FASEB J 2010;24:1852–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez MR, Porte S, Crosas E, et al. Human and yeast zeta-crystallins bind AU-rich elements in RNA. Cell Mol Life Sci 2007;64:1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi Q, Menzaghi C, Smith S, et al. Genome-wide association analysis identifies TYW3/CRYZ and NDST4 loci associated with circulating resistin levels. Hum Mol Genet 2012;21:4774–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004;3:169–178. [DOI] [PubMed] [Google Scholar]
- 30.Curb JD, Rodriguez BL, Abbott RD, et al. Longitudinal association of vascular and Alzheimer's dementias, diabetes, and glucose tolerance. Neurology 1999;52:971–975. [DOI] [PubMed] [Google Scholar]
- 31.Peila R, Rodriguez BL, Launer LJ; Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes 2002;51:1256–1262. [DOI] [PubMed] [Google Scholar]
- 32.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 2004;61:661–666. [DOI] [PubMed] [Google Scholar]
- 33.Pressley JC, Louis ED, Tang MX, et al. The impact of comorbid disease and injuries on resource use and expenditures in parkinsonism. Neurology 2003;60:87–93. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed RM, Irish M, Piguet O, et al. Amyotrophic lateral sclerosis and frontotemporal dementia: distinct and overlapping changes in eating behaviour and metabolism. Lancet Neurol 2016;15:332–342. [DOI] [PubMed] [Google Scholar]
- 35.Delague V, Jacquier A, Hamadouche T, et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet 2007;81:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010;133:591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montecchiani C, Pedace L, Lo Giudice T, et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 2016;139:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez MA, Feely SM, Speziani F, et al. A novel mutation in VCP causes Charcot-Marie-Tooth type 2 disease. Brain 2014;137:2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auranen M, Ylikallio E, Shcherbii M, et al. CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol Genet 2015;1:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 2004;10:1055–1063. [DOI] [PubMed] [Google Scholar]
- 41.Leigh PN, Whitwell H, Garofalo O, et al. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991;114:775–788. [DOI] [PubMed] [Google Scholar]
- 42.Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 2011;122:673–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012;135:693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011;477:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi JS, Park JS, Ham YM, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun 2013;4:2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.