

Abstract
The systemic autoinflammatory diseases are disorders of the innate immune system distinguished by severe inflammation resulting from dysregulation of the innate immune system. Hereditary fever syndromes, such as FMF, TNF receptor-associated periodic syndrome, cryopyrin-associated periodic syndromes and mevalonate kinase deficiency, were the first group of systemic autoinflammatory diseases for which a genetic basis was established, between 1999 and 2001. Currently according to the latest report of the international union of immunological societies, 37 separate monogenic disorders were classified as autoinflammatory. In addition to the abovementioned monogenic conditions, we describe Schnitzler’s syndrome, a well-defined, acquired autoinflammatory condition without a clear genetic basis. For the purposes of this review, we discuss several conditions defined by the latest consensus process as systemic autoinflammatory diseases. We focus on those disorders where recent studies have contributed to further phenotypic characterization or had an impact on clinical management.
Keywords: pyrin-associated autoinflammatory diseases, NLRP3-related autoinflammatory diseases, relopathies, A20 haploinsufficiency, RELA haploinsufficiency, Schnitzler’s syndrome
Rheumatology key messages
The number of monogenic SAIDs has grown significantly since their initial discovery in the late 1990s.
Improved understanding of key inflammatory pathways has led to development of effective targeted therapies.
Schnitzler’s syndrome is a rare acquired, IL1-blockade responsive, SAID whose immunopathogenesis remains elusive.
Introduction
The systemic autoinflammatory diseases (SAIDs) are disorders of the innate immune system distinguished by severe inflammation and fever and a propensity towards serositis, arthritis and rashes but often with poorly defined phenotypes beyond severe systemic upset. Hereditary fever syndromes, such as FMF, TNF receptor-associated periodic syndrome, cryopyrin-associated periodic syndromes (CAPS) and mevalonate kinase deficiency, were the first group of SAIDs for which a genetic basis was established, between 1999 and 2001. These disorders differ from typical autoimmune disorders in that dysregulation of the innate immune system, rather than the adaptive immune system, underpins their pathogenesis. The term autoinflammation was coined by McDermott et al. in 1999 [1] to emphasize this difference.
Increasing use of whole genome or exome sequencing in the investigation of patients with suspected SAID has led rapidly to the identification of many more monogenic disorders. According to the latest report of the International Union of Immunological Societies, 37 separate monogenic disorders were classified as autoinflammatory [2]. However, as the number of conditions identified as SAID increases, as well as the number of molecular pathways implicated in autoinflammatory dysregulation, the definition and classification of autoinflammatory diseases becomes more difficult. For example, it has been recognized for some time that the pathogenesis of some common inflammatory diseases, such as rheumatoid arthritis, involves dysregulation of both innate and adaptive immune systems. Consequently, a more complex classification of inflammatory conditions along an immunological disease continuum has been suggested [3]. Classical monogenic SAID lies at one end of this spectrum, autoimmune disorders at the other, and all other conditions somewhere in between depending on their predominant pathological process. Although this allowed for most of the immunologically mediated disorders to be classified in some way, it has been argued that in some cases this has also led to blurring of the boundaries between autoimmunity and autoinflammation. More recently, a number of disorders that have autoinflammatory features were also found to have an overlapping immunodeficiency phenotype, complicating matters further [4].
Consequently, the most recent definition of SAID aims to reaffirm the distinction between autoinflammatory and autoimmune diseases. According to the international expert group: ‘Autoinflammatory diseases are clinical disorders caused by defect(s) or dysregulation of the innate immune system, characterized by recurrent or continuous inflammation (elevated acute phase reactants) and the lack of a primary pathogenic role for the adaptive immune system (autoreactive T cells or autoantibody production)’ [5].
For the purposes of this review we will discuss several conditions defined by the latest consensus process as SAIDs. We will focus on those conditions where recent studies have contributed to further phenotypic characterization or had an impact on clinical management. It is beyond the scope of this review to discuss all conditions in detail and where appropriate we will direct the reader to other relevant literature. We will also only touch briefly on the immunopathogenesis of these conditions, mainly for the purposes of understanding clinical features of the diseases and rationale for their treatment. The innate immune pathways involved in the pathogenesis of SAID are reviewed in detail elsewhere in this edition. Lastly, we will provide an overview of Schnitzler’s syndrome (SchS), an acquired SAID, which has retained its original name since its pathogenesis remains obscure.
The inflammasomopathies
Inflammasomes are multimolecular intracellular complexes which, when activated in response to pathogens or danger-associated molecular patterns, catalyse conversion of the potent proinflammatory cytokines IL-1 and IL-18 into their mature forms. Gain-of-function mutations resulting in monogenic SAIDs have been reported in four different inflammasomes (Table 1). Here we provide an update on diseases associated with pyrin, NLR family pyrin domain containing 3 protein (NLRP3) and NLR family CARD domain-containing protein 4 (NLRC4) inflammasomes.
Table 1.
Monogenic autoinflammatory syndromes
| General pathway | Disease | Gene | Affected protein | Mode of inheritance | Age of onset | Key clinical features | Treatment |
|---|---|---|---|---|---|---|---|
| Inflammasomopathies | FMF | MEFV | Pyrin | AR/AD | 0–20 years | Peritonitis, joint attacks and joint pain | Colchicine/anti-IL-1 |
| PAAND | MEFV | Pyrin | AD | 0–10 years | Neutrophilic dermatosis (pyoderma gangrenosum) arthralgia, myalgia | Anti-IL-1/anti-TNF | |
| PAPA | PSTPIP1 | CD2 binding protein-1 | AD | 1–16 years | Juvenile-onset arthritis, painful ulcers and acne | Prednisone/anti-IL-1, anti-TNF | |
| MKD | MVK | Mevalonate kinase | AR | Variablea | Lymphadenopathy, abdominal pain, joint pain, diarrhoea, skin rashes and headache | NSAIDs/prednisone/anti-IL-1, anti-TNF | |
| NLRP3-AID (spectrum) CINCA/NOMID, MWS, FCAS | NLRP3 | NLRP3 | ADb | Variablea | Conjunctivitis, general malaise, headaches, rash, joint pain | Anti-IL-1 | |
| NLRP12-AIDc | NLRP12 | Monarch-1 protein | AD | Infancy | Skin rash, lymphadenopathy, aphthous ulcers, abdominal complaint | Anti-IL-1/corticosteroids | |
| NLRC4-AID | NLRC4 | NLRC4 | AD | Infancy | Infantile enterocolitis Macrophage activation syndrome | Anti-IL-1/anti-IL-18 | |
| NLRP1-AIDc | NLRP1 | NLRP1 | AD | 6 months–10 years | Dyskeratosis, arthritis | Acitretin, anti-IL-1 | |
| Interferonopathies | Aicardi–Goutières syndrome | TREX1 RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 | Exonuclease subunits of the RNase H2 endonuclease complex, control of dNTP pool | AD/AR | Infancy–childhood | Encephalopathy, hepatosplenomegaly, skin lesions | Symptomatic treatment JAK inhibition Reverse transcriptase inhibitors |
| Proteasome-associated autoinflammatory syndrome (PRAAS), CANDLE | PSMB3, PSMB4, PSMB8, PSMB9, POMP | Proteasome | AR | Infancy | Skin eruptions, progressive lipodystrophy, hepatosplenomegaly, myositis, | Glucocorticoids JAK inhibition | |
| SAVI | TMEM173 | Stimulator of interferon genes (STING) | AD | Infancy-childhood | Vasculopathy, skin lesions (leading to ulcers and necrosis), Raynaud phenomenon | JAK inhibition | |
| Relopathies | A20 haploinsufficiency | TNFAIP3 | NF-κB regulatory protein, A20 | ADa (some cases unclear) | Variable | Oral, gastrointestinal and genital ulcers; arthralgia | Colchicine, systemic corticosteroids, anti-IL-1, anti-IL-6, anti-TNF |
| Biallelic RIPK1 mutations | RIPK1 | Receptor-interacting serine/threonine kinase 1 | N/A | Infancy | Early-onset inflammatory bowel disease, and progressive polyarthritis | HSCT successful in one patient | |
| HOIL-1/HOIP deficiency | HOIL-1, HOIP | HOIP, HOIL-1 and SHARPIN (components of LUBAC) | AR | Infancy | Amylopectinosis, increased susceptibility to viral and bacterial infections | HSCT | |
| ORAS | OTULIN | Otulin (deubiquitinator protease) | AR | Infancy | Panniculitis, diarrhoea, swollen joints | Anti-TNF | |
| RELA (p65) haploinsufficiency | P65 | REL-associated protein | AD | Variable | Abdominal pain, mucocutaneous ulceration vomiting, leukocytosis | Anti-TNF | |
| Others | DADA2 | CECR1 | Adenosine deaminase 2 | AR | Variable | Mottled rash (livedo reticularis) anaemia, joint pain, fatigue | Anti-TNF, bone marrow transplantation |
| DIRAd | IL1RN | IL-1 Receptor antagonist | AR | Infancy | Painful joint swelling, pustular rash, hepatosplenomegaly | IL-1 blockade | |
| DITRA | IL36RN | IL-36 Receptor antagonist | AR | Infancy | Pustular psoriasis, asthenia | IL-1 blockade, anti-TNF | |
| TRAPSc | TNFRSF1A | TNF | AD | Variable | Skin rash, abdominal pain, myalgia | Corticosteroids, anti-IL-1/ anti-TNF | |
| NOD2- associated granulomatous disease, Blau syndromec | NOD2 | NOD2 inflammasome | ADe | Infancy-childhood | Granulomatous dermatitis, arthritis, uveitis | Anti-TNF |
Aside from apparent high fever, other key clinical features are stated.
Age of onset depends on disease severity.
Reports of sporadic cases as well.
These diseases are included for informational purposes, but not discussed in the review. Readers are encouraged to research these pathways in literature.
Fever is not a typical feature in this condition. AD: autosomal dominant; AID: associated inflammatory disease; AR: autosomal recessive; CAMPS: CARD14-mediated pustular psoriasis; CANDLE: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome; CINCA: chronic infantile neurological, cutaneous and articular syndrome; DADA2: deficiency of adenosine deaminase 2; DIRA: deficiency of interleukin-1 receptor antagonist; DITRA: deficiency of the IL-36 receptor antagonist; FCAS2: familial cold autoinflammatory syndrome 2; HOIL-1L: haem-oxidized IRP2 ubiquitin ligase 1L; HOIP: HOIL-1 interacting protein; HSCT: haematopoietic stem cell transplantation; LUBAC: linear ubiquitin chain assembly complex; MKD: mevalonate kinase deficiency; MWS: Muckle–Wells syndrome; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NOD2: Nucleotide-binding oligomerization domain-containing protein 2; NOMID: neonatal onset multisystem inflammatory disease; ORAS: ovarian tumor (OTU) deubiquitinase with linear linkage specificity (OTULIN)-related autoinflammatory syndrome; PAAND: pyrin-associated autoinflammation with neutrophilic dermatosis; PAPA: pyogenic arthritis, pyoderma gangrenosum and acne syndrome; RELA (p65): Transcription factor p65; SAVI: sting-associated vasculopathy with onset in infancy; SHARPIN: SHANK-associated RH-domain-interacting protein; TRAPS: TNF-associated periodic fever syndrome.
Pyrin (pathway)-associated autoinflammatory diseases
FMF became the first SAID with an identified genetic basis in 1997. However, the biological function of pyrin, the protein encoded by the MEFV gene, was only fully revealed recently [6]. This was helped by the identification of a novel SAID that had autosomal dominant inheritance, with clinical features atypical of FMF but was caused by gain of function mutations in MEFV. The condition was named pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND), and it is now classified together with FMF under an umbrella term, pyrin-associated autoinflammatory diseases [7]. There were prior clues to suggest that MEFV mutations associated with FMF can also result in gain of function, despite the apparently autosomal recessive inheritance of this condition. Firstly, around 30% of patients with FMF only have a heterozygous mutation in MEFV, and there were also previous reports of an apparent autosomal dominant mode of inheritance in some families [8, 9]. Murine studies have shown that the FMF phenotype can only be reproduced in experimental animals by ‘knocking in’ the causative MEFV mutations, whereas the pyrin-deficient mice do not have the typical phenotype [10]. Lastly, the vast majority of 125 variants in MEFV associated with FMF are missense mutations, while null mutations are extremely rare [11].
The molecular pathway governing the activation and regulation of the pyrin inflammasome has recently been mapped out (for a detailed review, see other articles in this edition). The pyrin inflammasome is maintained in its inactive state by interaction between inhibitory 14–3–3 protein with phosphorylated serine residues S242 and S208 found on pyrin. The pyrin inflammasome can be activated by bacterial toxins, such as Clostridium difficile toxin B (TcdB) via dephosphorylation of S242 and S208, which releases pyrin from 14–3–3 inhibition and eventually leads to release of IL-1β and IL-18.
The original series of patients with PAAND were all found to have heterozygous C-to-G substitution at c.726 in exon 2 of MEFV [12]. This results in a serine-to-arginine substitution at position 242 (S242R) in the pyrin protein. They presented with a clinical picture that was both atypical and more severe than that seen in FMF, including childhood-onset pyoderma gangrenosum, neutrophilic dermatosis, myalgia, prolonged fevers, arthralgia and persistently raised acute phase reactants. The S242R mutation resulted in constitutive activation of the pyrin inflammasome, since the loss of serine at position 242 prevents phosphorylation of pyrin and interaction with the inhibitory protein, 14–3–3. In contrast, mutations associated with FMF are thought to reduce the threshold for inflammasome activation, rather than causing constitutive activation [12].
Since the original description of PAAND, two additional reports have been published describing a new MEFV mutation. The index case in one of these reports was a 43-year-old male with 30-year history of chronic and severe pustular acne, recurrent pyoderma gangrenosum, severe hidradenitis suppurativa, recurrent long-lasting febrile episodes, neutrophilic panniculitis, as well as polyarthralgia and oligoarthritis of small and large joints [7]. A clinical diagnosis of pyogenic arthritis, pyoderma gangrenosum and acne syndrome was considered, but genetic testing later revealed a heterozygous mutation in MEVF c.730G>A leading to p.E244K substitution. Although this mutation does not affect the serine at position 242, the mutation is +2 from the phosphorylation site and affects the binding motif for 14–3–3 [RXX(pS)XP] resulting in constitutive activation of the pyrin inflammasome.
More recently three patients of Pakistani consanguineous background were found to have a homozygous mutation at the other serine phosphorylation site on pyrin (S208) important for interaction with 14–3–3. Two patients had homozygous c.623 G > C (p.S208T) and one was homozygous for c.622 A > T (p.S208C) MEFV mutation. The clinical phenotype here was somewhat different from the original description of PAAND. All patients had a remitting–relapsing course of disease, characterized by high fevers and acute phase reactants. Other features included eosinophilia, oral ulceration, intestinal inflammation and lymphadenopathy [13].
In all PAAND cases, functional ex vivo and in vitro studies clearly show spontaneous activation of the pyrin inflammasome with enhanced production of IL-1β and IL-18; IL-1β inhibition, while effective in colchicine resistant FMF [14], is not always effective for PAAND. The patient with a p.E244K substitution had the best response to anti-TNF therapy (good disease control for 8 years on infliximab) while anakinra was largely ineffective. There are several possible explanations for this, but one offered by the authors is intriguing. They suggest that the increased cell death observed in PAAND might result in enhanced release of danger-associated molecular patterns that trigger local production of cytokines including TNF in tissues such as skin [7].
Another monogenic condition under the umbrella of pyrin-associated autoinflammatory diseases is mevalonate kinase deficiency, where the lack of enzyme activity leads to impaired function of RhoGTPase. In the case of autoinflammatory periodic fever, immunodeficiency and thrombocytopenia, mutation of WD40 repeat protein 1 (WDR1), which is an actin-depolymerization cofactor, results in cytoskeletal abnormalities and pyrin-mediated autoinflammatory diseases, although the precise mechanism for this remains unknown [15, 16]. For more details, see [17]).
NLRP3-related autoinflammatory diseases
Previously also known as cryopyrin-associated periodic fever (CAPS), NLRP3-AID is an umbrella term for the continuum of three syndromes of varying severity, namely the mild familial cold autoinflammatory syndrome, the moderate Muckle–Wells syndrome and the severe early-onset chronic infantile neurological, cutaneous and articular syndrome/neonatal onset multisystem inflammatory disease (CINCA/NOMID). All these disorders are due to gain of function mutations in NLRP3, also known as cryopyrin.
It has long been known that low levels of mutated NLRP3, due to somatic mosaicism, were responsible for what appeared to be genetically negative cases of CINCA/NOMID [18]. However more recently, acquired forms of CAPS were found in adult patients, resulting from somatic mutations in NLRP3 that have arisen in later life within the bone marrow [19, 20]. The average age of disease onset in these patients is 50 years. In terms of symptomatology and response to treatment they were similar to patients with a germline form of CAPS. This disease mechanism is not limited to NLRP3, and there are reports of other genes associated with SAID acting in a similar fashion [21, 22].
A change of terminology was recently proposed to better reflect the biological and clinical characteristics of these conditions. The term ‘periodic’ was deemed inaccurate since in some forms of CAPS (particularly CINCA/NOMID) the conditions tend to have a chronic inflammatory course [5].
A novel consensus method was recently used to establish the diagnostic criteria for NLRP3-AID. These include elevated inflammatory markers (CRP/serum amyloid) and at least two of the following six clinical manifestations: urticaria-like rash, cold/stress-triggered episodes, sensorineural hearing loss, chronic aseptic meningitis, musculoskeletal symptoms (arthritis, arthralgia, myalgia) and skeletal abnormalities (frontal bossing, epiphyseal overgrowth). The proposed diagnostic criteria have a sensitivity of 81% and a specificity of 94% across the entire severity spectrum of NLRP3-AID [23].
There are frequent neurological manifestations of NLRP3-AID and several studies have investigated this aspect of the condition. Two studies examined the neurological and cognitive sequelae of NLRP3-AID, with learning difficulties affecting a large proportion of both the adult (61%, n = 11) and paediatric (50%, n = 3) population. Leptomeningeal or dural enhancement on MRI was associated with a lower IQ [24]. The second study assessed the neurological manifestations of 17 patients with variable-penetrance NLRP3 mutations of unknown significance (Q703K and V198M) in a cohort of patients presenting with limited autoinflammatory symptoms. Among these 65% suffered from severe headaches and 53% had a concomitant diagnosis of multiple sclerosis. Three additional relatives carrying the pathogenic gene mutation were also found to have multiple sclerosis [25].
Interestingly, a study by Rodriguez-Smith et al. established that among nine cytokines examined, only IL-6 and IP-10/CXCL10 (interferon-gamma-inducible 10kDa protein) levels were increased in the Cerebrospinal fluid (CSF) relative to their serum levels, indicating a future role for these cytokines as markers of CNS inflammation in CAPS. Anakinra was found to be more effective than canakinumab at decreasing CSF levels of IL-6, IP-10/CXCL10 and IL-18, as well as CSF granulocyte counts; however, complete normalization of CSF did not occur even in cases of complete clinical remission [26].
Lately, an ever increasing number of ‘common’ disorders are being found to have an important autoinflammatory component. A study describing NLRP3 mutations as the driving mechanism behind sensorineural hearing loss (through their effect on cochlear function), both within and without the context of a systemic autoinflammatory phenotype. What is more, IL-1 blockade in these cases has led to a reversal or reduction of hearing loss, indicating a possible role for anti-IL-1 therapy in a variety of ‘idiopathic’ hearing loss disorders [27].
Almost all clinical manifestations associated with NLRP3-AID are due to inappropriate and/or excessive release of IL-1β resulting from gain of function mutations in NLRP3. Targeted IL-1β blockade is therefore the mainstay of treatment, and its remarkable efficacy is well established; biological therapies such as anakinra (recombinant IL-Ra) and canakinumab (monoclonal anti-IL-1β antibody) are both licenced for NLRP3-AID. The first retrospective analyses of the real-life effectiveness of canakinumab among 68 CAPS patients revealed that a treat-to-target approach with regard to drug dosing is needed, as only 53% of the affected patients achieved complete remission on the standard 150 mg 8-weekly dose, with a significant proportion of patients requiring a 2–4-fold dose increase [28].
There are several recent studies that have looked at other aspects of treatment including safety. A recent retrospective study examined the effect of IL-1 blockade on breastfeeding and pregnancy, also taking into account paternal exposure to IL-1-blocking medication at conception, in pregnancy and breastfeeding. The study found that both anakinra and canakinumab were safe in pregnancy, with anakinra being the therapeutic of choice due to its shorter half-life and homology to naturally occurring IL-1Ra. Paternal exposure and breastfeeding were both found to be safe with either canakinumab or anakinra therapy. There were no cases of rilonacept exposure in pregnancy and its use remains contraindicated due to evidence of teratogenicity in animals [29].
A retrospective study assessed the safety of tetanus/diphtheria, influenza and pneumococcal vaccines in patients with CAPS; 70% were able to mount a response to pneumococcal vaccine, with an odds ratio to mount a response to influenza and tetanus/diphtheria vaccine of 31.0 (95% CI: 8, 119) and 10.8 (95% CI: 2, 74), respectively. The vaccination responses tended to be of a longer duration and also included instances of severe autoinflammatory reactivation. It was suggested that an in-depth, individual risk–benefit assessment be made prior to vaccinating to pneumococcus and that the pneumococcal conjugate vaccine might be safer than the pneumococcal polysaccharide vaccine [30].
NLRC4-related autoinflammatory diseases
The early reports of NLRC4-related autoinflammatory diseases (NLRC4-AID) described clinical phenotypes ranging from cold-induced urticaria to early-onset enterocolitis and macrophage activation syndrome [31–33]. The gain of function mutations in these cases were all confined to the nucleotide binding and oligomerization domain of the protein, a domain similar to that found in NLRP3-AID. Mutated NLRP3 and NLRC4 have an increased tendency towards oligomerization and can activate spontaneously or at least require only partial upstream signalling for full activation. When activated, NLRC4, like NLRP3, associates with ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domains) resulting in the assembly of a caspase-1-activating inflammasome. However, the predominant cytokine produced by NLRC4 inflammasome is IL-18. Although IL-1β and IL-18 were both elevated in patients with NLRC4-AID compared with healthy controls, the levels of IL-18 were several-fold higher and this was also the case when compared with patients with NOMID/CINCA [25].
A report describing novel mutations affecting the leucine-rich-repeat domains of the NLRC4 molecule reveals how point mutations in the leucine-rich-repeat domain can affect interactions between adjacent leucine-rich-repeat domains, leading to enhanced oligomerization and activation of the NLRC4 inflammasome. This in turn leads to caspase-1-driven cleavage of pro-IL-1β and pro-IL-18 into their activated states, as well as increased ASC speck formation, resulting in potentially lethal macrophage activation syndrome [34].
There have been multiple reports on the use of immunomodulatory drugs and biologics in NLRC4-AID-associated macrophage activation syndrome, with anakinra, infliximab, high-dose corticosteroids and vedolizumab all proving to be largely ineffective. It wasn’t until the implementation of an experimental IL-18 binding protein that clinical and/or biochemical improvement was seen, confirming the importance of this cytokine in the pathogenies of NLRC4-AID [34, 35].
Relopathies
‘Relopathy’ is a newly coined term for autoinflammatory diseases caused by inappropriate activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB; Fig. 1) [17]. NK-κB plays a key role in the regulation of various immunological responses, and is found in almost all cell types. Among other functions, NF-κB regulates B cell survival (non-canonical pathway), but it is also critical for generating inflammatory responses, and can be activated by TNF, IL-1 and pattern recognition receptors (classical or canonical pathway). NF-κB activation is largely governed by the process of ubiquitination whereby certain proteins are targeted for proteasome- or lysosome-mediated degradation. Ubiquitination results in in release of NF-κB inhibition and transmigration of the transcription factor to the nucleus [16]. Loss of NF-κB activity has historically been linked to the pathogenesis of certain human primary immunodeficiencies (PIDs) [36]. More recently several monogenic diseases resulting from dysregulation of NF-κB have been linked with purely autoinflammatory phenotypes, or have been shown to have mixed PID and autoinflammatory clinical features.
Fig. 1.

Disease-associated enzymes involving TNF and IL-1 pathways leading to NF-κB activation
Red proteins are involved in TNF and blue in IL-1 signalling. Purple indicates a shared pathway. Known relopathies marked with lightning symbol. Ubiquitin ligase activity indicated by dotted lines, deubiquitinase activity by scissor symbols. A20: TNF-induced protein 3; cIAPs: cellular inhibitor of apoptosis proteins; HOIL-1L: haem-oxidized IRP2 ubiquitin ligase 1L; HOIP: HOIL-1 interacting protein; IκBa: NF-κB inhibitor α; IKKα/b: inhibitor of NF-κB kinase a/b; IRAK 4/1: IL-1 receptor-associated kinase 4/1; MyD88: myeloid differentiation primary gene 88; NEMO: NF-κB essential modulator; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; OTULIN: ovarian tumor (OTU) deubiquitinase with linear linkage specificity; RIPK1: receptor-interacting serine/threonine protein kinase 1; SHARPIN: SHANK-associated RH-domain-interacting protein; TAB1/2/3: TAK binding protein 1/2/3; TAK1: TGF-β activated kinase 1; TNFR: TNF receptor; TRADD: TNF receptor-associated death domain; TRAF2: TNF receptor-associated factor 2; TRAF6: TNFR-associated factor 6.
A20 haploinsufficiency
Multiple mutations of the Tumor necrosis factor, alpha-induced protein 3 (TNFAIP3) gene, also know as A20, have been reported in a novel disease entity—haplosinsufficiency of A20 (HA20). Most of the disease-causing mutations in A20 (a negative regulator of NFκB) result in a truncated protein, which is usually not expressed, leading to reduced (50%) expression of the wild-type functional A20. Consequently there is increased activation of NFκB in stimulated cells, resulting in enhanced release of various cytokines including IL-1β, TNF, IL-6, IL-18 and IL-17 [37]. Murine models have also hinted at the existence of a direct effect of A20 function on NLRP3 functioning, with HA20 resulting in NFκB-independent increase in ASC speck formation and IL-1β release [38]. Interestingly, cell-specific ablation of A20 in mice results in different clinical phenotypes. The mice with homozygous A20 deficiency have severe, early-onset systemic autoinflammatory diseases [39]. However, selective knock-out in myeloid cells results in an RA-like phenotype [40], while enterocyte-specific deficiency results in selective intestinal inflammation [41]. Lastly, deficiency of A20 in B or dendritic cells results in production of autoantibodies with tissue-dependent responses [42].
It is not therefore surprising that HA20 can be associated with a wide array of clinical presentations from a Behçet’s-like syndrome characterized by systemic inflammation, oral and genital ulceration, arthralgia/arthritis and ocular symptoms, to autoinflammatory conditions such as sJIA and adult-onset Still’s disease, to classic autoimmune conditions including SLE [38, 43–45]. Patients with autoimmune lymphoproliferative syndrome-like condition and a label of undifferentiated complex autoimmune disease have also been described, illustrating the complexity of phenotypes associated with this condition.
With such disease heterogeneity, treatment options must be tailored to the patient; these include colchicine, anti-IL-1 and anti-IL-6 therapy [37]. One patient underwent bone marrow transplant with most of the clinical features resolving following the procedure.
OTULIN-related autoinflammatory syndrome/otulipenia
Like A20, ovarian tumor (OTU) deubiquitinase with linear linkage specificity (OTULIN) functions as a deubiquitinase and loss of function mutations result in accumulation of ubiquitinated target proteins such as receptor-interacting serine/threonine protein kinase 1 (RIPK1), NF-κB essential modulator (NEMO) and TNF receptor (TNFR) with associated elevation of pro-inflammatory cytokines, IL-6 and TNF. The condition is characterized by early-onset systemic inflammation and fevers, sterile neutrophilia, failure to thrive, neutrophilic dermatosis, lipodystrophy and panniculitis [46]. To date patients with OTULIN-related autoinflammatory syndrome have not shown clinical features associated with PID. Biological therapies targeting IL-1β and TNF have been used successfully in selected cases.
RELA (p65) haploinsufficiency
A monogenic disorder phenotypically resembling HA20, but with a different underlying abnormality of the NF-κB pathway is RELA haploinsufficiency. NF-κB is typically a hetero- or homodimer made up of several subunits—RelA, RelB, c-Rel, NF-κB1, NF-κB2—in different combinations. A heterodimer consisting of RelA and NF-κB1 is the most frequent form of NF-κB.
Badran et al. [47] described a single family in which the index case presented at 3 years old with a history of recurrent mouth ulceration, fevers, vomiting, acute ileitis and elevated inflammatory markers. The patient eventually responded to a combination of methotrexate and infliximab (anti-TNF). Their mother and sibling also had mucocutaneous lesions responsive to infliximab. Genetic testing of the family eventually revealed a heterozygous mutation in RELA c.559 + 1G>A, resulting in a premature stop codon and reduction in wild-type RELA mRNA expression by 50%.
In the context of TNF signalling, which can promote both apoptosis and cell survival, NF-κB has a role in promoting the latter and therefore limiting the toxic effects of TNF. Testing of the fibroblasts from patients with RELA haploinsufficiency showed increased apoptosis and impaired NF-κB activation in response to TNF. RELA haploinsufficiency therefore leads to impaired stromal and epithelial cell recovery following a mucosal injury; this has been demonstrated in murine models. The authors suggest that the systemic autoinflammatory features of this condition might result from effects of the local microbiome on the ulcerated oral, gastrointestinal and vaginal mucosa acting as inflammatory stimuli for further TNF release.
The clinical phenotypes associated with RELA haploinsufficiency have recently been expanded. There is a report of a 5-year-old boy who was misdiagnosed with autoimmune lymphoproliferative syndrome [48]. The initial presentation was with splenomegaly, refractory immune thrombocytopenic purpura, anaemia and neutropenia. Other features included headaches and aseptic meningitis, but no oro-genital ulcers or recurrent fevers. The immune thrombocytopenic purpura responded to rituximab, but there was no effect on headaches. Genetic testing revealed a de novo heterozygous nonsense mutation (NM_021975.3: c.736C>T, p.Arg246*) in RELA resulting in reduced p65 mRNA and protein expression.
It is fascinating that both potentiation and reduction of NF-κB activation can lead to monogenic SAIDs with overlapping clinical phenotypes. It is likely that in conditions such as HA20 and RELA haploinsufficiency, other genetic variants and environmental factors are important in determining the overall phenotypes.
HOIL-1L/HOIP deficiency
Loss of function mutations in haem-oxidized IRP2 ubiquitin ligase 1L (HOIL-1L) and HOIL-1 interacting protein (HOIP) have been described, manifesting as systemic autoinflammation with increased propensity for viral and bacterial infections, as well as amylopectinosis [4].
HOIL-1L, SHANK-associated RH-domain-interacting protein (SHARPIN) and HOIP form an oligomer known as the linear ubiquitin chain assembly complex. This is responsible for the attachment of Lys63-linked ubiquitin chains to various proteins; one of these target proteins inhibits the NF-κB kinase subunit γ/NF-κB essential modulator (IKK γ/NEMO) responsible for inhibiting IKKα/IKKβKB. Ubiquitination of IKKγ/NEMO leads to increased IKK activity and results in the translocation of NF-κB components p50 and p65 into the nucleus, leading to increased transcription of various pro-inflammatory cytokines.
Loss of function mutations lead to absent NF-κB activity in fibroblasts and B cells (partly explaining the immune deficiency), whereas NF-κB activity was increased in monocytes and was likely responsible for the autoimmune phenotype [4].
Biallelic RIPK1 mutations
RIPK1 is a critical regulator of many signalling pathways including toll-like receptor 3 and 4 and TNF-mediated NF-κB activation. Typically, upon TNF binding to TNFR, RIPK1, TNF receptor-associated death domain protein and E3 ubiquitin ligases (e.g. TNF receptor-associated factor 2 and cellular inhibitor of apoptosis proteins) are recruited to the tumor necrosis factor receptor 1 and associate to form the TNF receptor complex I. With the help of RIPK1, linear ubiquitin chain assembly complex is also recruited resulting in eventual phosphorylation and activation of IKK kinase and NF-κB activation.
A recent report describes four patients from three unrelated families who presented with a combination of PID and systemic autoinflammatory features, which included early-onset inflammatory bowel disease and progressive polyarthritis [49]. All four patients were found to have biallelic RIPK1 mutations (deletions or frameshift) that resulted in RIPK1 deficiency. Functional studies using patients’ fibroblasts demonstrated marked reduction in mitogen-activated protein kinase p38 and activator protein 1 phosphorylation, and partial reduction in NF-κB activation. This was associated with reduced production of proinflammatory cytokines (TNF, IL-6 and IL-12) by patients’ monocytes following stimulations with lipopolysaccharide (LPS). However, production of IL-1β was preserved or significantly increased using various cell sources (whole blood and monocytes) and stimuli such as phytohaemagglutinin and LPS. Further studies demonstrated that LPS stimulation of RIPK1-deficient monocytes resulted in increased necroptosis (inflammatory cell death) and IL-1β release; this has also been observed in animal models.
As with RELA haploinsufficiency, the proinflammatory clinical features of RIPK1 deficiency result from reduced cell viability; however, there is additional release of a potent inflammatory mediator. Overall, RIPK1 deficiency therefore results in a more pronounced negative effect on inflammatory pathways, leading to clinically evident PID. One case was treated successfully with haematopoietic stem cell transplantation and this might be considered for similar cases in the future.
Schnitzler’s syndrome
SchS is a rare, acquired SAID with two major defining features; IgM-κ (rarely IgG) paraprotein and recurrent urticarial rash (Fig. 2). Clinical features constituting minor diagnostic criteria include recurrent fever, leucocytosis and/or elevated CRP, neutrophilic dermal infiltrate on skin biopsy, and bone remodelling with or without bone pain [50]. Symptoms of headache, myalgia, arthralgia, fatigue, peripheral neuropathy, weight loss and lymphadenopathy are frequently reported but are not included in the Strasbourg diagnostic criteria for SchS (Fig. 3). Patients typically present in the fifth decade, without positive family history [51]. Around 15–20% develop overt lymphoproliferative disease, specifically Waldenstrom’s macroglobulinaemia [52–54].
Fig. 2.
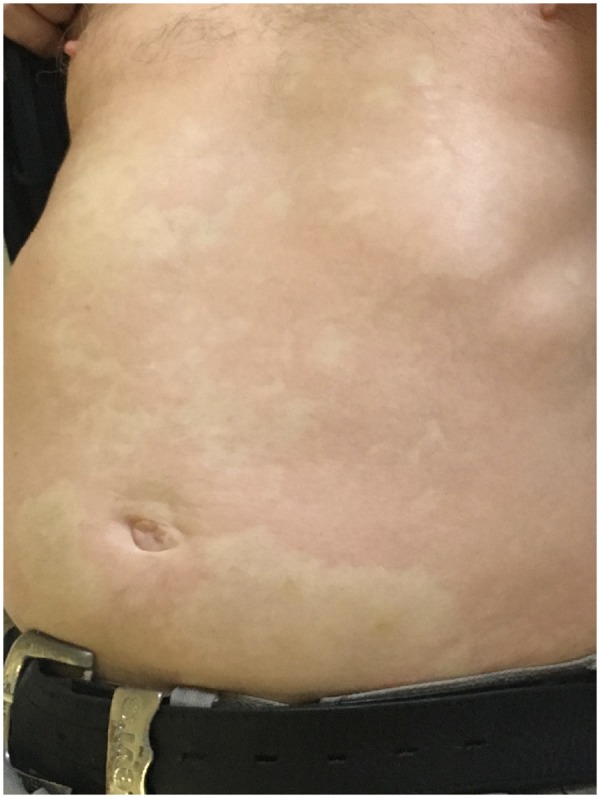
Urticarial-like rash typical of Schnitzler’s syndrome
Fig. 3.

Strasbourg diagnostic criteria of Schnitzler’s syndrome adapted from Simon et al. [50]
aMust be >38°C, and otherwise unexplained. Occurs usually—but not obligatorily—together with the skin rash. bAs assessed by bone scintigraphy, MRI or elevation of bone alkaline phosphatase. cCorresponds usually to the entity described as ‘neutrophilic urticarial dermatosis’ (Medicine 2009; 88: 23–31); absence of fibrinoid necrosis; and significant dermal oedema. dNeutrophils >10 000/mm3 and/or CRP >30 mg/l.
The aetiology of SchS remains largely unknown, but there are clues regarding molecular mechanisms that might be relevant for the disease pathogenesis. Phenotypically, SchS resembles NLRP3-AIDs; there are overlapping clinical features and both show an excellent response to IL-1 inhibition. The fact that acquired forms of NLRP3-AID have recently been identified suggests that similar mechanisms could be responsible for some cases of SchS. A large study of 21 patients with SchS failed to identify any patients with germline or mosaic pathogenic variants in NLRP3 nor in 32 other genes typically associated with SAID [51]. A follow-on study also failed to find any germline or somatic NLRP3 mutations in another 11 patients [55]. It is therefore highly unlikely that the gain of function mutations in NLRP3 play an important role in the pathogenesis of SchS. In fact, it is more likely that the previous reports of SchS patients with somatic NLRP3 mutations and transient IgG paraprotein [56], or SchS patients without paraprotein [57, 58], are all cases of acquired forms of NLRP3-AID.
Other studies have found increased secretion of IL-1β and IL-6 by peripheral blood mononuclear cells following stimulation with LPS [59, 60]. In addition, IL-6 but not IL-1β is increased in serum and might correlate with disease activity. A recent study found higher levels of ASC aggregates (typically released following inflammasome activation), IL-6 and IL-18 in the sera of 21 patients, comparing with controls [61]. Interestingly, levels were similar to those measured in CAPS patients, again clearly suggesting activation of inflammasomes although the underlying mechanism remains obscure. The latest study to examine specific biomarkers of SchS found that CCL2, a monocytic attractant, was significantly higher in serum of SchS patients comparing with healthy controls [62]. However, levels of this chemokine were decreased upon IL-1 inhibition, suggesting that this finding is a consequence of elevated IL-1β levels rather than a manifestation of SchS itself.
The link between SchS and Waldenstrom’s macroglobulinaemia suggest another mechanism. Around 90% of patients with Waldenstrom’s macroglobulinaemia are reported to have a somatic MYD88 L265P mutation [63]. When present in B cells this gain of function mutation drives development of lymphoid malignancy via toll-like receptor signalling pathways. However, MYD88 is also an adaptor protein involved in IL-1R activation. If this same variant was present in cells of myeloid origin this could result in excessive IL-1 release. This notion is supported by the recent description of an inflammatory disorder caused by a germline gain-of-function mutation in MYD88 characterized by a skin rash resembling the rash seen in SchS [55]. However, a recent study showed that the presence of MYD88 L265P mutation was found only in 9/30 patients, implying that if the L265P variant indeed had a role in the pathogenesis of SchS, this did not appear to be a universal mechanism [55].
Lastly the role of IgM paraprotein itself remains unclear. For example, there appears to be no relationship between the quantity of IgM paraprotein and severity of SchS or response to IL-1 inhibition [61]. There is a report of a patient where paraprotein only became visible 4 years after the first clinical symptoms were reported, suggesting that the paraprotein was a consequence rather than cause of the disease. However, decreases in the IgM paraprotein even after prolonged successful anti-IL-1 treatment have not been reported and a recent article reports a patient successfully treated with ibrutinib, a Burton tyrosine kinase inhibitor, which targets B cell receptor signalling and is known to reduce the burden of IgM paraprotein [65].
At present IL-1 inhibition remains the mainstay therapy for SchS. The support for this treatment comes from case reports, case series and randomized trials [28, 66–70]. In the UK anakinra has been approved by NHS England following adoption of the clinical commissioning policy for periodic fever syndromes and SchS (NHS England Reference: 170062P). Tocilizumab (RoActemra™) targeting IL-6 has also been used successfully in selected SchS patients with an apparently incomplete response to IL-1 blockers [71]. There is currently one active study testing the effects of Dapansutrile (Clinical Trial ID: NCT03595371), an oral NLRP3 inflammasome inhibitor that theoretically halts the processing and release of IL-1β and IL-18.
Conclusions
The continued discovery of monogenic SAIDs provides fascinating insights into functioning of the immune system. For disorders such as NLRP3-AID, future developments will focus on optimization of already existing and effective therapies. New therapeutics such as oral NLRP3 inhibitors are in development and trials in patients should start soon. Relopathies demonstrate how a fine balance between proinflammatory and compensatory mechanisms is maintained using the same signalling pathways. Some of the pathological mechanisms, clinical manifestations and outcomes seen in these conditions were predictable based on animal models and the known functions of the proteins; however, the diverse clinical phenotypes associated with relopathies suggest an important role for other genetic and environmental modifiers that we are yet to fully understood. Finally, the pathogenesis of SchS largely remains unresolved and the role of anti-IL-1 therapy in reducing complications such as progression to lymphoid malignancy needs to be systematically studied. As always for rare diseases, such questions are best addressed by collaborative international studies.
Funding: This paper was published as part of a supplement funded by an educational grant from Sobi. The authors were entirely independent, and received no remuneration for their work.
References
- 1. McDermott MF, Aksentijevich I, Galon J. et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999;97:133–44. [DOI] [PubMed] [Google Scholar]
- 2. Bousfiha A, Jeddane L, Picard C. et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol 2018;38:129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McGonagle D, McDermott MF.. A proposed classification of the immunological diseases. PLoS Med 2006;3:e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boisson B, Laplantine E, Dobbs K. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med 2015;212:939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ben-Chetrit E, Gattorno M, Gul A. et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDS): a Delphi study. Ann Rheum Dis 2018;77:1558–65. [DOI] [PubMed] [Google Scholar]
- 6. Heilig R, Broz P.. Function and mechanism of the pyrin inflammasome. Eur J Immunol 2018;48:230–8. [DOI] [PubMed] [Google Scholar]
- 7. Moghaddas F, Llamas R, De Nardo D. et al. A novel pyrin-associated autoinflammation with neutrophilic dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to familial Mediterranean fever. Ann Rheum Dis 2017;76:2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sánchez Ferrer F, Martinez Villar M, Fernández Bernal A, Martín De Lara I, Paya Elorza I.. Cardiac tamponade as first manifestation in Mediterranean fever with autosomal dominant form. An Pediatr 2015; 82:e82–5. [DOI] [PubMed] [Google Scholar]
- 9. Suzuki T, Nakamura A, Yazaki M, Ikeda S.. A Japanese case of familial Mediterranean fever with homozygosity for the pyrin E148Q mutation. Intern Med 2005;44:765–6. [DOI] [PubMed] [Google Scholar]
- 10. Chae JJ, Cho Y-H, Lee G-S. et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity 2011;34:755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Infevers. The registry of Hereditary Auto-inflammatory Disorders Mutations. https://infevers.umai-montpellier.fr/web/ (11 February 2019, date last accessed).
- 12. Masters SL, Lagou V, Jéru I. et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med 2016;8:332ra45. [DOI] [PubMed] [Google Scholar]
- 13. Hong Y, Standing ASI, Nanthapisal S. et al. Autoinflammation due to homozygous S208 MEFV mutation. Ann Rheum Dis 2019;78:571–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Özen S, Bilginer Y, Ayaz NA, Calguneri M.. Anti-interleukin 1 treatment for patients with familial Mediterranean fever resistant to colchicine. J Rheumatol 2011;38:516–8. [DOI] [PubMed] [Google Scholar]
- 15. Kuhns DB, Fink DL, Choi U. et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood 2016;128:2135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Standing ASI, Malinova D, Hong Y. et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med 2017;214:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steiner A, Harapas CR, Masters SL, Davidson S.. An update on autoinflammatory diseases: relopathies. Curr Rheumatol Rep 2018;20:39. [DOI] [PubMed] [Google Scholar]
- 18. Saito M, Nishikomori R, Kambe N. et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood 2008;111:2132–41. [DOI] [PubMed] [Google Scholar]
- 19. Zhou Q, Aksentijevich I, Wood GM. et al. Cryopyrin-associated periodic syndrome caused by a myeloid-restricted somatic NLRP3 mutation. Arthritis Rheumatol 2015;67:2482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rowczenio DM, Gomes SM, Aróstegui JI. et al. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience. Front Immunol 2017;8:1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saito M, Fujisawa A, Nishikomori R. et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum 2005;52:3579–85. [DOI] [PubMed] [Google Scholar]
- 22. Labrousse M, Kevorkian-Verguet C, Boursier G. et al. Mosaicism in autoinflammatory diseases: cryopyrin-associated periodic syndromes (CAPS) and beyond. A systematic review. Crit Rev Clin Lab Sci 2018;55:432–42. [DOI] [PubMed] [Google Scholar]
- 23. Kuemmerle-Deschner JB, Ozen S, Tyrrell PN. et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis 2017;76:942–7. [DOI] [PubMed] [Google Scholar]
- 24. Mamoudjy N, Maurey H, Marie I, Koné-Paut I, Deiva K.. Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS). Orphanet J Rare Dis 2017;12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuh E, Lohse P, Ertl-Wagner B. et al. Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm 2015;2:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodriguez-Smith J, Lin YC, Tsai WL. et al. Cerebrospinal fluid cytokines correlate with aseptic meningitis and blood–brain barrier function in neonatal-onset multisystem inflammatory disease: central nervous system biomarkers in neonatal-onset multisystem inflammatory disease correlate with central nervous system inflammation. Arthritis Rheumatol 2017;69:1325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakanishi H, Kawashima Y, Kurima K. et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci USA 2017;114:E7766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuemmerle-Deschner JB, Hofer F, Endres T. et al. Real-life effectiveness of canakinumab in cryopyrin-associated periodic syndrome. Rheumatology 2016;55:689–96. [DOI] [PubMed] [Google Scholar]
- 29. Youngstein T, Hoffmann P, Gül A. et al. International multi-centre study of pregnancy outcomes with interleukin-1 inhibitors. Rheumatology 2017;56:2102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaeger VK, Hoffman HM, van der Poll T. et al. Safety of vaccinations in patients with cryopyrin-associated periodic syndromes: a prospective registry based study. Rheumatology 2017;56:1484–91. [DOI] [PubMed] [Google Scholar]
- 31. Canna SW, de Jesus AA, Gouni S. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014;46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K.. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med 2014;211:2385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romberg N, Al Moussawi K, Nelson-Williams C. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 2014;46:1135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moghaddas F, Zeng P, Zhang Y. et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)-LRR oligomerization interface. J Allergy Clin Immunol 2018;142:1956–67.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Canna SW, Girard C, Malle L. et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol 2017;139:1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pannicke U, Baumann B, Fuchs S. et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N Engl J Med 2013;369:2504–14. [DOI] [PubMed] [Google Scholar]
- 37. Aksentijevich I, Zhou Q.. NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol 2017;8:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lawless D, Pathak S, Scambler TE. et al. A case of adult-onset Still’s disease caused by a novel splicing mutation in TNFAIP3 successfully treated with tocilizumab. Front Immunol 2018;9:1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee EG, Boone DL, Chai S. et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 2000;289:2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matmati M, Jacques P, Maelfait J. et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 2011;43:908–12. [DOI] [PubMed] [Google Scholar]
- 41. Vereecke L, Sze M, Guire CM. et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med 2010;207:1513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chu Y, Vahl JC, Kumar D. et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 2011;117:2227–36. [DOI] [PubMed] [Google Scholar]
- 43. Sato S, Fujita Y, Shigemura T. et al. Juvenile onset autoinflammatory disease due to a novel mutation in TNFAIP3 (A20). Arthritis Res Ther 2018;20:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franco-Jarava C, Wang H, Martin-Nalda A. et al. TNFAIP3 haploinsufficiency is the cause of autoinflammatory manifestations in a patient with a deletion of 13Mb on chromosome 6. Clin Immunol 2018;191:44–51. [DOI] [PubMed] [Google Scholar]
- 45. Aeschlimann FA, Batu ED, Canna SW. et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis 2018;77:728–35. [DOI] [PubMed] [Google Scholar]
- 46. Zhou Q, Yu X, Demirkaya E. et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA 2016;113:10127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Badran YR, Dedeoglu F, Leyva Castillo JM. et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J Exp Med 2017;214:1937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Comrie WA, Faruqi AJ, Price S. et al. RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. J Allergy Clin Immunol 2018;141:1507–10.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cuchet-Lourenço D, Eletto D, Wu C. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 2018;361:810–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simon A, Asli B, Braun-Falco M. et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy 2013;68:562–8. [DOI] [PubMed] [Google Scholar]
- 51. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy 2014;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mulla E, Neame R.. Delayed development of the IgM paraprotein in Schnitzler’s syndrome. Scand J Rheumatol 2015;44:521–2. [DOI] [PubMed] [Google Scholar]
- 53. Willekens I, Walgraeve N, Goethals L, De Geeter F.. Correlative bone imaging in a case of Schnitzler’s syndrome and brief review of the literature. Hell J Nucl Med 2015;18:71–3. [PubMed] [Google Scholar]
- 54. Dingli D, Camilleri M.. Schnitzler syndrome: clinical features and histopathology. Pathol Lab Med Int 2015;7:39. [Google Scholar]
- 55. Pathak S, Rowczenio DM, Owen RG. et al. Exploratory study of MYD 88 L265P, rare NLRP3 variants and clonal hematopoiesis prevalence in patients with Schnitzler’s syndrome. Arthritis Rheumatol 2019, doi: 10.1002/art.41030. [DOI] [PubMed] [Google Scholar]
- 56. De Koning HD, Van Gijn ME, Stoffels M. et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol 2015;135:561–4. [DOI] [PubMed] [Google Scholar]
- 57. Husak R, Nestoris S, Goerdt S, Orfanos CE.. Severe course of chronic urticaria, arthralgia, fever and elevation of erythrocyte sedimentation rate: Schnitzler’s syndrome without monoclonal gammopathy? Br J Dermatol 2000;142:581–2. [DOI] [PubMed] [Google Scholar]
- 58. Cristina T, Varella N, Nishimura MY. et al. Schnitzler’s syndrome without monoclonal gammopathy. Acta Derm Venereol 2005;85:272–3. [DOI] [PubMed] [Google Scholar]
- 59. Ryan JG, de Koning HD, Beck LA. et al. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol 2008;121:260–2. [DOI] [PubMed] [Google Scholar]
- 60. de Koning HD, Schalkwijk J, Stoffels M. et al. The role of interleukin-1 beta in the pathophysiology of Schnitzler’s syndrome. Arthritis Res Ther 2015;17:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rowczenio DM, Pathak S, Arostegui JI. et al. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood 2018;131:974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krause K, Sabat R, Witte‐Händel E. et al. Association of CCL2 with systemic inflammation in Schnitzler syndrome. Br J Dermatol 2019;180:859–68. [DOI] [PubMed] [Google Scholar]
- 63. Varettoni M, Arcaini L, Zibellini S. et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood 2013;121:2522–8. [DOI] [PubMed] [Google Scholar]
- 64. Sikora KA, Bennett JR, Vyncke L. et al. Germline gain-of-function myeloid differentiation primary response gene-88 (MYD88) mutation in a child with severe arthritis. J Allergy Clin Immunol 2018;141:1943–7.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Treon SP, Gustine J, Meid K. et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J Clin Oncol 2018;36:2755–61. [DOI] [PubMed] [Google Scholar]
- 66. Néel A, Henry B, Barbarot S, Masseau A, Perrin F, Bernier C. et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: A French multicenter study. Autoimmun Rev 2014;13:1035–41. [DOI] [PubMed] [Google Scholar]
- 67. Vanderschueren S, Knockaert D.. Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum 2013;42:413–6. [DOI] [PubMed] [Google Scholar]
- 68. Krause K, Tsianakas A, Wagner N. et al. Efficacy and safety of canakinumab in Schnitzler syndrome: a multicenter randomized placebo-controlled study. J Allergy Clin Immunol 2017;139:1311–20. [DOI] [PubMed] [Google Scholar]
- 69. Krause K, Weller K, Stefaniak R. et al. Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy 2012;67:943–50. [DOI] [PubMed] [Google Scholar]
- 70. Pesek R, Fox R.. Successful treatment of Schnitzler syndrome with canakinumab. Cutis 2014; 94:E11–2. [PubMed] [Google Scholar]
- 71. Krause K, Feist E, Fiene M, Kallinich T, Maurer M.. Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol 2012;129:848–50. [DOI] [PubMed] [Google Scholar]
- 72. Rigante D, Vitale A, Lucherini OM, Cantarini L. The hereditary autoinflammatory disorders uncovered. Autoimmun Rev 2014;13:892–900. [DOI] [PubMed] [Google Scholar]
- 73. Berteau F, Rouviere B, Delluc A. et al. Autosomic dominant familial Behcet disease and haploinsufficiency A20: a review of the literature. Autoimmun Rev 2018;17:809–15. [DOI] [PubMed] [Google Scholar]
- 74. Li C, Zhang J, Li S. et al. Gene mutations and clinical phenotypes in Chinese children with Blau syndrome. Sci China Life Sci 2017;60:758–62. [DOI] [PubMed] [Google Scholar]
- 75. Cowen EW, Goldbach-Mansky R. DIRA, DITRA, and new insights into pathways of skin inflammation: what’s in a name? Arch Dermatol 2012;148:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu Y, Jesus A, Marrero B. et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014;371:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rossi-Semerano L, Piram M, Chiaverini C. et al. First clinical description of an infant with interleukin-36-receptor antagonist deficiency successfully treated with anakinra. Pediatrics 2013;132:e1043–7. [DOI] [PubMed] [Google Scholar]