

Abstract
Small open reading frames (smORFs) encode previously unannotated polypeptides or short proteins that regulate translation in cis (eukaryotes) and/or are independently functional (prokaryotes and eukaryotes). Ongoing efforts for complete annotation and functional characterization of smORF-encoded proteins have yielded novel regulators and therapeutic targets. However, because they are excluded from protein databases, initiate at non-AUG start codons, and produce few unique tryptic peptides, unannotated small proteins cannot be detected with standard proteomic methods. Here we outline a procedure for mass spectrometry-based detection of translated smORFs in cultured human cells, from protein extraction, digestion, and LC-MS/MS, to database preparation and data analysis. Following proteomic detection, translation from a unique smORF may be validated via siRNA-based silencing or overexpression and epitope tagging. This is necessary to unambiguously assign a peptide to a smORF within a specific transcript isoform or genomic locus. Provided that sufficient starting material is available, this workflow can be applied to any cell type/organism and adjusted to study specific (patho)physiological contexts including, but not limited to, development, stress, and disease.
Keywords: Small open reading frame, short open reading frame, upstream open reading frame, microprotein, small protein, peptidomics, proteomics, proteogenomics, mass spectrometry, genomics, transcriptomics
INTRODUCTION:
Proteogenomics, or use of proteomic data to re-annotate genomes (Jaffe, Berg, & Church, 2004), has enabled detection of classes of gene products that were previously systematically missed by genome annotation technologies (Jaffe et al., 2004; Kim et al., 2014). This includes smORF-encoded “microproteins” (Andrews & Rothnagel, 2014; Storz, Wolf, & Ramamurthi, 2014) of fewer than 100 amino acids (Harrison, Kumar, Lang, Snyder, & Gerstein, 2002). Thousands of small and non-annotated proteins have been reported to date using ribosome footprinting and deep sequencing (RIBO-seq) (Ingolia, Ghaemmaghami, Newman, & Weissman, 2009; Ingolia, Lareau, & Weissman, 2011) as well as two-dimensional tandem liquid chromatography-mass spectrometry (LC-MS/MS) proteomics coupled with genomic or transcriptomic libraries (Menschaert et al., 2013; Slavoff et al., 2013). Ever-increasing numbers of these novel gene products from bacteria, humans, and other organisms have been shown to be functional (Couso & Patraquim, 2017; Ruiz-Orera & Alba, 2019; Saghatelian & Couso, 2015; Storz et al., 2014), motivating the need to generate a comprehensive catalog of translated smORFs. However, many novel smORFs initiate at non-AUG start codons (Kozak, 1997; Oyama et al., 2004) and map to regions of RNA previously designated as non-coding (i.e. long non-coding RNAs, non-coding transcript isoforms, and 5’ and 3’ untranslated regions) (Slavoff et al., 2013). As a result, many smORFs are impossible to detect using standard proteomic methods, which generally utilize databases of known proteins and/or in silico translation of nucleic acid sequences using canonical, AUG start codons (Gundry et al., 2009).
RIBO-seq and mass spectrometry offer complementary approaches to smORF annotation, though sources of false positive and false negative identifications via each method must be considered. In some cases, RIBO-seq signal can be generated by RNA-protein interactions in addition to ribosomal footprints (Ji, Song, Regev, & Struhl, 2015), potentially complicating detection of bona fide smORF translation. Furthermore, some translated smORFs may represent proto-genes that generate non-functional polypeptides (Carvunis et al., 2012), which may be unstable and therefore fail to accumulate to sufficient concentration in cells to be detectable via proteomics. In contrast, smORFs are likely under-detected by mass spectrometry because they generate few tryptic peptides, and sequencing low-abundance species during data-dependent acquisition remains limited by sample complexity. LC-MS/MS proteomic smORF discovery also requires careful validation because the large databases required for proteogenomics may result in elevated numbers of false positive peptide-spectral matches (Jeong, Kim, & Bandeira, 2012). Overall, while RIBO-seq reveals many more putatively translated smORFs than does mass spectrometry (Calviello et al., 2016), proteomics offers direct evidence of smORF-encoded protein products.
This protocol delineates a proteogenomic microprotein discovery pipeline applied to human cells in culture (Ma et al., 2014b; Slavoff et al., 2013). First, small proteins and peptides are enriched from the whole proteome, then profiled with LC-MS/MS. Resulting peptide spectra are matched against a three-frame translation database generated from cell-specific RNA-seq transcriptomic data, and known proteins computationally excluded. Peptide identifications produced by searching mass spectrometry data against RNA-seq databases are filtered to exclude peptides that match exactly or with a <2 amino acid difference to known human proteins. The remaining list of peptides is stringently analyzed for high quality MS/MS identification including requirements for more than four consecutive ion identifications and a significant, unique sequence match. Several follow-up experiments are recommended to rigorously confirm smORF translation. First, isotopically labeled peptide standards can be used to verify the initial spectrum identifications based on retention time and fragmentation patterns. Second, expression from a given transcript isoform or locus can be confirmed by gene silencing in tandem with targeted proteomic detection. Alternatively, smORF expression can be confirmed by transiently overexpressing a putative transcript with an inserted epitope tag. This approach has proved powerful for identifying hundreds of translated smORFs across a variety of cell lines and experimental conditions (Ma et al., 2014b; Slavoff et al., 2013).
BASIC PROTOCOL 1. PROTEIN EXTRACTION, SIZE SELECTION, AND TRYPSIN DIGESTION
This protocol describes a gel-based method for size selection of small proteins from whole cell lysates using a tricine buffer SDS-PAGE system followed by standard in-gel digestion protocols (Ma et al., 2014a; Rinehart et al., 2011; Shevchenko, Tomas, Havlis, Olsen, & Mann, 2006; Slavoff et al., 2013). Tricine gels are designed for high resolution separation of low molecular weight proteins greater than 2 kilodaltons (kDa). Tricine sample buffer includes Coomassie blue tracking dye, since bromophenol blue migrates more slowly than some low molecular weight proteins. The in-gel size selection protocol requires at least 107-108 cells as starting material; if less material is available, the in-solution protocol described in Alternate Protocol 1 is recommended (Figure 1). Methanol/chloroform precipitation described in Support Protocol 1 can be used to concentrate lysate prior to gel loading (Wessel & Flugge, 1984). Using molecular weight cutoff filters for the purposes of concentration or size selection is not advised due to the potential for variable recovery of small proteins.
Figure 1:
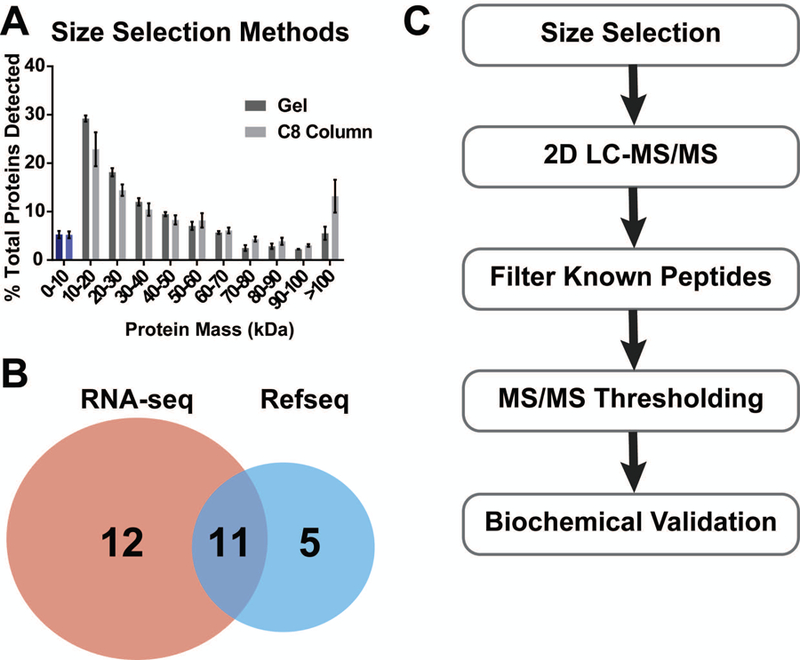
A. Comparison of size selection performed in mammalian cells using either the gel-based or in-solution methods. B. Number of non-annotated peptides detected in the same set of fractionated samples from mammalian cells using either a RefSeq transcriptomics database from NCBI or a cell-specific RNA-seq database for searching. C. General workflow for micropeptide discovery with proteomics described in this protocol.
Materials:
107-108 cells of interest (see Support Protocol 1)
Lysis buffer (see Recipe)
Dulbecco’s phosphate buffered saline (dPBS) (Sigma, D8537–100ML)
16% Tricine gels (Thermo Fisher, EC6695BOX)
2X Tricine SDS sample buffer (Invitrogen, LC1676)
10X Tricine SDS running buffer (Invitrogen, LC1675)
1M Tris (pH 8.0)
Precision Plus Dual Xtra Prestained Protein Standard (Bio-Rad, #1610377)
Coomassie stain (see Recipe)
Coomassie destain (see Recipe)
Acetonitrile (ACN), HPLC-grade
50 mM ammonium bicarbonate (AMBIC) solution
50 mM AMBIC/ACN wash solution (see Recipe)
10 mM AMBIC/ACN wash solution (see Recipe)
Digestion buffer (see Recipe)
0.5 mg/mL trypsin, MS-Grade (Promega, V511X)
Extraction buffer (see Recipe)
Water (ultrapure)
0.1% Trifluoroacetic acid (TFA), 99.9% ultrapure water
1.5 mL plastic tubes
50 mL conical tubes
Heat block (set to 90–100 °C)
Refrigerated microcentrifuge (set to 4 °C)
Gel tank and power supply adapters (Thermo Fisher, A25977)
Power supply (Thermo Fisher, EC300XL2)
Clean razor blades
Clean 150 mM petri dishes
Rocking platform
Tube rotator
Vortex mixer
SpeedVac vacuum concentrator (Thermo Scientific, Savant SPD10)
37 °C Incubator
-
Harvest 107-108 cells, or thaw flash frozen cell pellets stored at –80 °C. Thoroughly wash cells with ice-cold dPBS and pellet by centrifuging at 2,000g for 5 min at 4 °C prior to freezing and/or lysis.
Specific harvesting, washing and lysis procedures may vary depending on source of biological material; the following steps are appropriate for mammalian cells grown in culture.
-
Add 300 µL lysis buffer (see Recipe) directly to cell pellets and boil at 100 °C for 20 min.
Rapid lysis and boiling eliminate proteolysis, which creates unwanted protein fragments in the small molecular weight range.
Place lysates on ice for 1 min and centrifuge at 14, 000g for 20 min at 4 °C.
Remove supernatant to a new tube and either proceed with further extraction (Support protocol 1) or continue directly to Step 5.
-
Combine lysate 1:1 (v/v) with Tricine SDS sample buffer.
If buffer turns color from blue to green or yellow, add 1 µL 1 M Tris (pH 8.0).
Set 16% Tricine gel within running chamber. Fill chamber with 1X Tricine SDS running buffer.
-
Load 5 µL Precision Plus Dual Xtra size marker and maximal volume of protein solution into individual wells until entire sample is loaded on the gel.
If sample volume is greater than the volume of a single well, the sample may be divided into multiple wells, or concentrated as described in Support Protocol 1 (chloroform/methanol precipitation) and loaded in a single well.
If loading different samples on a single gel, skip wells between each sample to prevent cross contamination.
Run gel at 200V for 30 min, or until the dye front reaches the bottom edge of the gel.
-
For steps 9–15, perform all procedures in a clean laminar flow hood with new, clean, dust-free materials and supplies while wearing clean gloves. Carefully disassemble the gel plates and remove the gel to a clean plastic 150 mM petri dish.
Introduction of dust will contaminate proteomics samples with keratin and should be carefully avoided.
-
Mix 30 mL Coomassie destain and 400 µL Coomassie stain in a 50 mL tube, add mixture to the gel, and rock for a maximum of 20 min.
In order to avoid contamination by dust and other samples, use fresh Coomassie stain and destain.
-
Carefully remove and discard staining solution. Cover gel with 30 mL Coomassie destain and rock for 10–30 min at room temperature.
Do not destain by boiling or microwaving.
-
Remove and discard Coomassie destain. Add enough fresh Coomassie destain to just cover the gel, without allowing it to float freely in the container.
All solutions that contact gel pieces after destaining must be kept in glass bottles/vials, never plastic, to avoid plasticizer contamination during LC-MS/MS. These glass bottles and vials must be washed with only acid or mass spectrometry-compatible detergent, never soap, in order to avoid contamination with polyethylene glycol. Maintaining a separate set of glassware intended for mass spectrometry prevents contamination.
Use clean razor blades to excise gel bands between 2–5, 5–10, and 10–15 kDa size marks. Use a fresh razor blade for each cut to avoid cross-contamination.
Further cut each of the three bands into 5–6 smaller pieces and place pieces into three 1.5 mL plastic tubes marked with the size range.
Add 1 mL Coomassie destain to each tube and incubate on a rotator for an additional 30 min.
Replace Coomassie destain with 200 µL (or enough to completely immerse the gel pieces) 50 mM AMBIC/ACN wash solution and rotate for 2 min.
Discard first wash, and replace with 200 µL fresh 50 mM AMBIC/ACN and incubate 30 min on the rotator.
Discard second wash, replace with 200 µL 10 mM AMBIC/ACN and wash for 30 min.
Discard third wash, and replace with 500 µL ACN, vortex 10s and allow the gel pieces to shrink for 2 min.
Spin down and carefully remove all of the ACN wash, without disturbing the gel pieces. Allow the gel pieces to air dry for an additional 10 min.
Add enough digestion buffer to cover the gel pieces (normally about 100–200 µL) and incubate on ice for 1 h.
After 1 h supplement digestion buffer as needed to fully cover the gel pieces and incubate tubes in a 37 °C incubator for 12–14 h.
Spin down and carefully remove the digestion buffer to a clean 1.5 mL tube.
To the gel pieces, add 200 µL extraction buffer and rotate for 15 min to maximally extract peptides from the gel. Spin down briefly.
-
Combine extraction buffer with the digestion buffer solution and dry using a vacuum concentrator using the following parameters: Auto Run, Run Time: 1h, Heat time: 0 h, Vacuum Ramp Setting: 4 (approximately 50 torr/min).
Peptides and other precipitates may or may not be visible at this stage. Proceed even if no pellet is visible.
Resuspend dried peptides in 100 µL 0.1% TFA. Proceed to de-salting (Support Protocol 3).
ALTERNATE PROTOCOL 1. IN-SOLUTION C8 COLUMN SIZE SELECTION
Reverse phase C8 cartridge-based isolation of small proteins and peptides was developed several decades ago to enrich peptide hormones from plasma (Vale et al., 1986). Recently, this method was successfully applied for discovery of non-annotated small proteins in humans and bacteria (D’Lima et al., 2017; Ma et al., 2014a). This protocol may be used in lieu of gel-based size selection to maximize protein recovery if sample is limiting. However, size selection using the C8 column may result in increased detection of undesired high molecular weight proteins (Figure 1A).
Materials:
107-108 cells of interest (see Support Protocol 1)
Lysis buffer (see Recipe)
Dulbecco’s phosphate buffered saline (Sigma, D8537–100ML)
Bradford reagent (Sigma, B6916)
Methanol, HPLC grade
0.25 M triethylammonium formate (TEAF) (pH 3.0)
C8 elution buffer (see Recipe)
1.5 mL plastic tubes
15 mL conical tubes
32 mm syringe filter with 5 μm membrane (Pall Life Sciences, 4650)
5 mL syringe
UV-Vis spectrophotometer
Bond Elut C8 column (Agilent Technologies, 12105028)
SpeedVac vacuum concentrator (Thermo Scientific, Savant SPD10)
Prepare cell lysates as described in Basic Protocol 1 Steps 1–4.
-
Filter the supernatant through a 5 μm filter using a 5 mL syringe.
This step eliminates cell debris which may block the column.
Quantify protein concentration using Bradford assay (Bradford, 1976) or other appropriate protein quantification method according to instructions from the manufacturer.
Precondition a Bond Elut C8 column with 1 column volume (350–400 µL) of methanol followed by 2 column volumes of 0.25 M TEAF (pH 3.0).
Load column with approximately 5 mg total protein per 50 mg of bed resin and wash with 2 column volumes of 0.25 M TEAF (pH 3.0).
Elute size-selected (low molecular weight) proteins with two column volumes of C8 elution buffer.
Dry protein mixture on a vacuum concentrator using the following parameters: Auto Run, Run Time: 5 min, Heat time: 0 h, Vacuum Ramp Setting: 4 (approximately 50 torr/min).
Proceed with Support Protocol 1 (chloroform/methanol precipitation).
SUPPORT PROTOCOL 1. CHLOROFORM/METHANOL PRECIPITATION
This protocol may be done prior to loading on a tricine gel, or following in-solution size selection with a C8 column. Protein precipitation may be used to concentrate a dilute protein sample before SDS-PAGE, minimizing loss of material during in-gel digestion due to inefficient extraction. Following the C8 column protocol, chloroform/methanol precipitation is essential to remove detergents (up to 5% SDS), β-mercaptoethanol, and lipids from the sample before mass spectrometry.
Materials:
Water (ultrapure)
Methanol, HPLC grade
Chloroform, HPLC grade
1.5 mL plastic tubes
Refrigerated microcentrifuge (set to 4 °C)
Vortex mixer
SpeedVac vacuum concentrator (Thermo Scientific, Savant SPD10)
-
Resuspend dried protein pellet from Alternate Protocol 1 step 7 in 100 µL water.
This protocol may be scaled up as necessary. If using larger volumes separate sample such that each 1.5 mL tube contains no more than 100 µL sample.
Add 400 µL of methanol, vortex for 10 s at maximal speed.
Add 100 µL chloroform, vortex for 10 s at maximal speed.
-
Add 300 µL water, vortex for 10 s at maximal speed.
The sample will appear milky.
Spin in a microcentrifuge for at 14,000 g for 1 min at 4 °C.
Observe the protein at the interface between the top (aqueous) and bottom (organic) layer. The protein layer looks like a white plaque. Immediately remove and discard the top layer, taking care to avoid disturbing the protein interface.
Add 400 µL methanol to precipitate the protein, vortex 10 s at maximal speed.
Spin in a microcentrifuge at 14,000g for 1min at 4 °C.
Remove as much methanol as possible without disturbing the protein pellet.
Air dry for 10 min.
-
Proceed with reduction/alkylation (Support Protocol 2), or freeze at –80 °C.
Samples can be stored for several weeks at –80 °C at this point.
SUPPORT PROTOCOL 2. REDUCTION, ALKYLATION, AND IN-SOLUTION PROTEASE DIGESTION
This is a standard protocol to reduce disulfide bonds prior to addition of trypsin for greater protease accessibility and efficient protein digestion. Iodoacetamide alkylation is used to prevent disulfide bonds from re-forming.
Materials:
Resuspension solution (see Recipe)
Bradford reagent (Sigma, B6916)
Water (ultrapure)
45 mM dithiothreitol (DTT) in ultrapure water (Roche, 10197777001), freshly prepared
100 mM iodoacetamide (IAA) in ultrapure water <99%, (Sigma, 16125–5G), freshly prepared
1 M Tris (pH 8.0)
0.5 mg/mL trypsin, MS-Grade (Promega, V511X)
20% TFA solution in ultrapure water
20% TFA, 80% ultrapure water
Vortex mixer
UV-Vis Spectrophotometer
Heat block (set to 60 °C)
Mini Centrifuge
pH strips
37 °C Incubator
Resuspend the protein pellet (from Support protocol 1, step 11) in 22 µL of resuspension solution, vortexing as necessary. A larger volume may be added, if required to completely dissolve the pellet.
-
Use 2 µL of sample to calculate protein concentration using Bradford (Bradford, 1976) or equivalent protein quantification assay according to instructions of the manufacturer.
Nanodrop protein quantification is not recommended.
Dilute to a concentration of 1 mg/mL with ultrapure water.
To reduce disulfides, add 2 µL of freshly prepared 45 mM DTT per 20µL of resuspension solution. Amounts can be scaled depending on sample volume.
Incubate the solution for 10 min in a 60 °C heat block.
Place the reaction on ice for 30 s, then return to room temperature.
Add 2 µL of freshly prepared 100 mM IAA solution, and incubate in the dark at room temperature for 30 min.
Add another 1 µL of the 45 mM DTT solution.
Add 115 µL ultrapure water to dilute the urea in the resuspension buffer. Concentrations of urea above 1 M are incompatible with trypsin digestion.
Add 20 µL 1M Tris (pH 8.0).
Add freshly thawed trypsin solution. Trypsin should be added at a ratio of 1:50, w/w, relative to total protein. For example, to 1 mg of protein, add 20 µg trypsin (40 µL of a 0.5 mg/mL trypsin solution).
Incubate 12–14 h, or overnight at 37 °C.
Acidify the sample by adding 1:20 (v/v) 20% TFA.
Vortex for 10 s at maximal speed, then spin down in a mini centrifuge for 10 s.
Make certain the pH of the sample is below 3 by pipetting 1 µL of solution onto a pH strip.
Continue to de-salting (Support Protocol 3).
SUPPORT PROTOCOL 3. PEPTIDE DE-SALTING
Prior to analysis by mass spectrometry, peptide samples must be free of detergents, salts, and any other reagents that interfere with chromatography or peptide ionization. Components of the digestion buffer such as 8M urea and Tris salts used for protein solubilization must be removed. There are many methods to achieve peptide desalting; the following commercial de-salting columns work well for <15 µg peptides.
Materials:
2–15 µg digested peptides (Basic Protocol 1)
80% ACN, 0.1% TFA, 19.9% ultrapure water
0.1% TFA, 99.9% ultrapure water
1.5 mL plastic tubes
2 mL plastic tubes (optional)
Ultramicrospin columns (washers and tubes are provided, Nest group, SUM SS18V)
Refrigerated microcentrifuge (set to 4 °C)
SpeedVac vacuum concentrator (Thermo Scientific, Savant SPD10)
Place the Ultramicrospin column within a washer and fit the washer inside a 1.5- or 2-mL clean plastic tube.
Condition each column with 400 µL 80% ACN/0.1% TFA by centrifuging at 200g for 1 min at 4 °C. Perform all subsequent centrifugations using these settings. Discard the flow-through.
Wash the column with 200 µL 0.1% TFA and spin as in Step 2. Discard the flow-through.
Add 100 µL of the acidified digest to the column and centrifuge. Discard the flow-through.
Wash the column with 400 µL 0.1% TFA and centrifuge. Discard the flow-through.
Repeat Step 5.
Replace the collection tube with a clean 1.5 mL tube, and add 200 µL 80% ACN/0.1% TFA and elute by centrifuging.
Repeat the elution with 100 µL 80% ACN/0.1% TFA.
Discard the column and rinse the re-usable washer with methanol prior to storing for further use.
Separate elution into two tubes: one tube containing 30 µL of the elution, marked 10%, and the other tube containing 270 µL which will be used for fractionation and LC-MS/MS proteomics.
-
Use the vacuum concentrator to dry down the eluted sample(s).
Dried peptides can be stored at –80 °C for no more than 1 month.
BASIC PROTOCOL 2. TWO-DIMENSIONAL LC-MS/MS WITH ERLIC FRACTIONATION
Complementary (“2-dimensional”) chromatographic techniques are used to achieve maximal coverage of proteins in complex peptide mixtures. Although more than 10,000 peptides may be detected per two-hour gradient on the Q Exactive, non-abundant peptides are less well sampled during data-dependent acquisition (Michalski, Cox, & Mann, 2011). Therefore, orthogonal fractionation of the peptide mixture is beneficial for increased sensitivity. “Offline” electrostatic repulsion hydrophilic interaction chromatography (ERLIC) fractionates peptides based on charge (Alpert, 2008; Hao et al., 2010) prior to reversed-phase chromatography of each ERLIC fraction during LC-MS/MS. It is expected that fractions at the end of the ERLIC gradient will contain relatively fewer peptides than initial fractions, so fraction volume is recommended to be scaled accordingly (see Step 5 below).
To assess sample quality, a 10% fraction of the de-salted peptide digest should be analyzed by LC-MS/MS without prior ERLIC fractionation. For in-gel digests, 10% of the desalted peptides from each gel band should be analyzed. At least two hundred proteins should be identified in every 10% sample when whole cell lysates are used as input. Fewer protein identifications may indicate issues with sample preparation such as degradation or insufficient starting material.
Materials:
De-salted peptides (see Support Protocol 3)
MS sample buffer (see Recipe)
ERLIC mobile phase A (see Recipe)
ERLIC mobile phase B (see Recipe)
ACQUITY mobile phase A (see Recipe)
ACQUITY mobile phase B (see Recipe)
polyWAX LP column (150 × 1.0-mm; 5um 300 Å; PolyLC)
Agilent 1100 HPLC or equivalent
High recovery vials (Agilent, 5183–2030)
Screw top caps (Agilent, 5185–5863)
75 μm × 50 cm PicoFrit column
1.9 μm ReproSil-Pur 120Å C18-AQ resin (Dr. Maisch)
Q Exactive Plus (Thermo) mass spectrometer coupled with an ACQUITY UPLC M-Class (Waters)
-
Dissolve 10% of the total de-salted peptide sample (0.4–4 µg) in 10 μl MS sample buffer for unfractionated LC-MS/MS analysis. Transfer to a high recovery glass vial. These solutions will be analyzed by LC-MS/MS without fractionation for sample quality assessment (see introduction to Basic Protocol 2, above).
This volume is appropriate for a 5 µL injection onto the column. No more than 2 µg peptide should be injected each LC-MS/MS run to avoid overloading the column.
Dissolve remaining dried peptides in 50 μl of ERLIC Solvent A prior to fractionation.
Perform fractionation. Inject up to 50 μg peptides onto the Agilent 1100 HPLC connected to a polyWAX LP column using the HPLC autosampler.
Maintain the flow rate at 50 µL/min. Run a 80 min gradient of ERLIC mobile phase A and B, beginning with 5 min at 100% A, 17 min to 8% B, 25 min linear gradient 8% to 45% B, 10 min of 45% to 100% B, followed by 100% B for 5 min, 10 min back to 100% A and 8 min at 100% A.
Collect fractions throughout the length of the gradient. Smaller 3–5 min fractions should be taken during the first 25 min of the gradient followed by larger 10–20 min fractions. A total of 10–15 fractions is recommended.
Dry each fraction using a vacuum concentrator (see Basic Protocol 1 step 25).
Resuspend each fraction in 10 µL MS sample buffer and transfer to a glass vial.
Inject 5 μl sample (each 10% sample and each ERLIC fraction) onto an analytical PicoFrit column packed with 1.9 μm ReproSil-Pur 120Å C18-AQ resin using ACQUITY UPLC M-Class connected to a Q Exactive Plus.
Set Q Exactive with a nanospray source at an electrospray potential of 1.5 kV. MS: 70,000 resolution, 3 × 106 AGC target, 300–1,700 m/z scan range; dd-MS2: top10 method, 17,500 resolution, 1 × 106 AGC target, 10 loop count, 1.6 m/z isolation window, 27 normalized collision energy.
Run 130 min gradients using ACQUITY solvents A and B at a flow rate of 0.25 µL/min. Begin gradient with 99% A for 40 min, 2 min linear gradient to 94% A, 58 min to 76% A, 5 min to 52% A, 5 min to 26% A, 5 min to 20% A, 5 min back to 99% A, and final 10 min hold at 99% A.
Save resulting .raw files for data analysis.
BASIC PROTOCOL 3. TRANSCRIPTOMIC DATABASE CONSTRUCTION
Peptide spectrum matches (PSMs) are generated by proteomics search algorithms based on statistical comparison of theoretical peptide fragmentation spectra from a database to empirically collected MS/MS spectra (Gundry et al., 2009; Henzel et al., 1993). To identify non-annotated proteins, their amino acid sequence must be included in a searchable database; because standard proteomics searches rely on annotated protein databases, non-annotated protein databases must be custom generated. To curb search times and avoid false positives, the smallest possible database that encompasses all translated sequences must be used. If possible, cell- and condition-specific RNA-seq databases should therefore be curated for each experiment. Alternatively, publicly available transcriptomic data for the organism of interest can be downloaded from National Center for Biotechnology Information (NCBI) repositories (Figure 1B). Since eukaryotic translation proceeds in the 5’−3’ direction only, a three-frame (not six-frame) translation of all expressed transcripts is compiled into a searchable database (Figure 2).
Figure 2:

A. Workflow for creating a three-frame translated transcriptomics database compatible with Mascot. Transcript sequences are translated using CLC SequenceViewer. Then, Remove_stops_from_fasta.py designates each ORF with a unique identifier and removes all ORFs of fewer than seven amino acids.
Materials:
CLC SequenceViewer Version 7.7 (Qiagen)
Transcriptomics database in fasta format (see Alternate Protocol 2).
A machine that runs python
Remove_stops_from_fasta.py
-
Download an organism-specific transcriptome assembly from RefSeq in fasta format.
Alternatively, use an in-house generated database (See Alternate Protocol 2).
Import into CLC Sequence viewer using the Import button in the top toolbar.
Select the desired fasta file and select “Automatic Import” and click “Next”.
Select the desired folder for import within the CLC console and click “Finish”.
The import may take several minutes. The transcriptome file should load in one of the tabs in the center of the CLC Console. Otherwise, open the file by clicking on it in the left side bar.
To create a three-frame translation, navigate the “Toolbox” tab at the bottom left corner.
Click on “Nucleotide” and select “Translate to Protein”.
Choose your transcriptome file and click “Next”.
Select Reading frames +1, +2, and +3 and Genetic Code “1 Standard”. Click Next.
Either “Open” and directly “Save” the result and click “Finish”.
-
Save in fasta format. If using CLC SequenceViewer, this file will contain three amino acid sequences for each transcript with asterisks (*) denoting stop codons.
Depending on the software that is used to search mass spectrometry data this three-frame translated database may need to be cleaned up by removing all asterisks and designating each ORF (amino acid sequence between adjacent stop codons) with a separate identifier. If uploading the database to Mascot, the following step is mandatory.
-
Clean up the three-frame translation fasta by running Remove_stops_from_fasta.py (python must be installed). This removes asterisks from the file and separates each ORF using a unique identifier. Additionally, ORFs smaller than 7 amino acids are eliminated. Run the script using the following command:
python Remove_stops_from_fasta.py Name_of_input.fasta Name_of_output.fasta
ALTERNATE PROTOCOL 2. TRANSCRIPTOMICS DATABASE GENERATION WITH GFFREAD
This protocol describes how to generate a cell-specific fasta format transcriptomic database: a text file that contains transcript sequence data separated by single line identifiers specific to each entry. To reduce search space, only transcripts which are expressed in the cell type of interest should be included. Using assembled transcript expression data generated by Cufflinks (Trapnell et al., 2012), a full list of genomic sequences, and a python script, FPKM_filter.py, it is possible to construct a transcript database using expression level cutoffs. Expression level in RNA-seq data is calculated as the number of sequenced fragments per kilobase transcript per million reads (FPKM). A cutoff of FPKM>1 is recommended. This setting results in a database of about 50 million amino acids for experiments with human cell lines; this number can vary depending on organism.
Materials:
A machine that runs python
Cufflinks gffread utility
FPKM_filter.py
Reference fasta file with genomic sequences (example: hg19.fa)
Expression file generated by Cufflinks, isoforms.fpkm_tracking
Assembled transcripts gtf file generated by Cufflinks, transcripts.gtf*
*Alternatively, download a previously assembled transcriptome (see Step 1 below)
(optional) Previously assembled transcriptomics data is available for download from Refseq FTP (See Internet Resources) and other public repositories (e.g. GEO Datasets).
Process RNA-seq data using the Tuxedo suite (Trapnell et al., 2013) according to standard protocols(Trapnell et al., 2012). Cufflinks generates a transcript assembly file and expression file transcripts.gtf and isoforms.fpkm_tracking, respectively.
-
Place Cufflinks-generated files and a file containing the genomic sequence in fasta format in the same directory or define their paths in FPKM_filter.py.
Note: Cufflinks gffread utility must be installed prior to use. Gffread is included with the Cufflinks package.
-
Define the FPKM cutoff by editing the following two lines:
cutoff=1useCutoff = True
An FPKM cutoff of >1 is recommended. All transcripts with expression levels below the cutoff will not be included in the generated fasta file.
-
Run FPKM_filter.py using the command:
python FPKM_filter.py
If using a cutoff of 1, a fasta file transcripts_filtered1.fa should be generated. If no cutoff is applied (useCutoff=False) then transcripts_nofilter.fa should be generated. These fasta files can be imported into CLCSequenceViewer to generate a three-frame translation (see Basic Protocol 3).
BASIC PROTOCOL 4. NON-ANNOTATED PEPTIDE IDENTIFICATION FROM LC-MS/MS DATA
This protocol explains how to find non-annotated peptides in mass spectrometry data by first searching a database using ProteoWizard MSConvert (Chambers et al., 2012) and Mascot Daemon (Perkins, Pappin, Creasy, & Cottrell, 1999) software, then filtering out peptides that align to known proteins using Peptide_filter.pl. Remaining MS/MS spectra are inspected manually to remove false positives. Typically, between 10–100 putative non-annotated peptides are identified from each experiment using this approach following 2D LC-MS/MS.
Materials:
Mass spectrometry data files (e.g. Thermo Fisher .raw files)
Mascot Daemon version 2.5.1 (Matrix Science, Inc., London, UK), configured with ProteoWizard MS Convert version 3 and custom database
Microsoft Excel (2019)
Notepad or other word processing software
A machine that runs perl
Peptide_filter.pl
Uniprot database of all known protein sequences for organism of interest
Contaminants database in fasta format
Open Mascot Daemon. Navigate to the Parameter Editor tab.
-
Select both the contaminants database and your custom database from the drop-down list. Check the box next to Decoy database and use the Monoisotopic mass option.
Instructions for custom database upload are available in Mascot documentation.
-
Select modifications. If using the in-solution digest method select Carbamidomethyl (C) and move to Fixed modifications.
Fixed modifications will be applied to each residue (e.g., carbamidomethylation of all cysteines).
-
Select Oxidation (M) and Acetyl (N-term) and add to “Variable modifications”.
These settings may differ based on the source of biological material; choosing only the most abundant naturally occurring protein modifications is strongly recommended to reduce false positive identifications (Nielsen, Savitski, & Zubarev, 2006).
Set maximum missed cleavages to 2 and the enzyme to semi-Trypsin.
-
Choose peptide charges +2, +3 and +4 and set peptide tolerance ±0.6 Da with # 13C set to 1.
This allows for the identification of a 13C peak while maintaining a tight mass tolerance window.
-
Save the parameter file and navigate to the Task Editor tab. Load the desired LC-MS/MS raw files and choose Proteowizard as the data import filter. ProteoWizard is a peak picking software which may be installed and configured with Mascot Daemon.
See Mascot documentation for Proteowizard configuration instructions. Alternatively, use the interactive Proteowizard feature MSConvertGUI to convert files to .mzmL format and upload to Mascot Daemon directly.
-
Change the parameter set to the desired parameter file and click Run.
Due to large database size, searches may take overnight to several days to finish.
Once the run is finished open the Result File URL.
Set the FDR (false discovery rate) to 1% under the tab “Sensitivity and FDR (reversed protein sequences)”.
Under the tab Protein Family Summary set the Max. number of families to 1. This dumps all of the individual peptide sequences into the “Unassigned” category.
Open the Unassigned tab and copy all peptide entries with a score above 50.
Paste the list as text into Microsoft Excel and sort by the “Expect” Column. The expectation value designates the probability of detecting a peptide match of the same or higher score by chance. Filter out all values with an expectation value over 0.01.
Now order the list by score. The score is a measure of the likelihood that a peptide match is a random event. Higher scores represent higher confidence peptide matches. Filter out any peptides with a score lower than 50.
Select the sequences of all remaining peptides and copy into Notepad as a tab-delimited list.
Save the file in the same folder as the Peptide_filter.pl.
Download a fasta file containing all known organism-specific protein sequences from Uniprot.
Download a fasta file containing all known contaminant protein sequences. One can be found when downloading the free Maxquant software under MaxQuant/bin/conf/contaminants.fasta
Append the contaminant sequences to your Uniprot database using copy and paste.
Save the Uniprot fasta file in the same directory as the Peptide_filter.pl script.
-
Open command line (perl must already be installed) and run the script as follows:
perl Peptide_filter.pl -pep yourpeptidelist.txt -fasta Uniprotdatabase.fasta -out outfile.txt
Open the output file and copy the contents into Excel.
The list should consist of two columns, the first column contains the peptide sequences and the second column the names of the protein match. Sort the list alphabetically according to the second column.
Find any peptide sequences that were “Not_Found” and save as a separate list. These are the putative small ORF-encoded peptide matches.
Use NCBI Protein Blast to search peptide sequences against the proteome of your organism of interest.
Remove any peptides that contain only one amino acid difference compared with a known protein sequence. These can arise through single nucleotide polymorphisms and may be more likely to be false positives.
- The MS/MS spectra of the remaining unmatched peptides should be inspected one by one in Mascot by clicking on each peptide sequence to open a new “Peptide View” window with the MS/MS spectrum. Peptide-spectral matches with the following characteristics should be eliminated:
- Peptides which are not significant matches (not represented in bold red).
- Uncertain peptide sequence assignments where multiple sequences can be attributed to the same MS/MS with equivalent scores (click on the peptide sequence to navigate to the Peptide View page, a table at the bottom of the page shows all query matches).
- Peptides with fewer than five identified consecutive b- or y- ions.
- Peptides with either only b- or y- ions identified in MS/MS.
-
Peptides with at least two of the following: a missed cleavage, >1 variable modification, a charge state that does not match calculated charge state at pH=3.Retain peptides that pass this filter as tryptic fragments of candidate novel smORF-encoded polypeptides.
Extract transcript sequences corresponding to the remaining unaligned peptides to match them to specific genomic locations and isoforms. This can be done by matching the identifier in the three-frame translated database (which can be found at the top of the Peptide View page) to the identifier in the original transcriptome database. Identify small open reading frames encoding the unaligned tryptic peptides that include a start codon and stop codon, or a non-canonical start codon within a Kozak consensus sequence (Kozak, 1989)
BASIC PROTOCOL 5. VALIDATION USING ISOTOPICALLY LABELED SYNTHETIC PEPTIDE STANDARDS AND siRNA
Following LC-MS/MS data analysis, newly identified proteins must be validated using an additional method. To show that peptide identification during proteomic profiling was not a false positive, an isotopically labeled peptide standard which is chemically indistinguishable can be purchased. Due to isotope labeling, the peptides should elute at the same retention time, but the different masses of the endogenous versus isotopically labeled peptides should be resolved in the MS1 spectrum (Figure 3). This experiment is best performed in targeted mode with parallel reaction monitoring (PRM) (Peterson, Russell, Bailey, Westphall, & Coon, 2012). Both the endogenous and isotopically labeled peptide masses should be entered into an inclusion list to trigger fragmentation and allow greater sensitivity.
Figure 3:
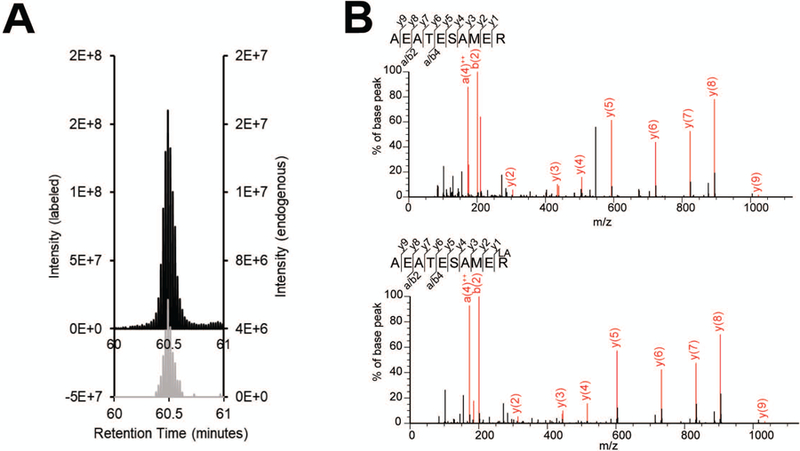
A. Example of targeted validation using isotopically labeled standards. MS1 peaks corresponding to target peptide (grey) and spiked in isotopically labeled standard (black) co-eluting at the same retention time (x-axis). B. MS/MS fragmentation spectra of a target peptide from cell lysate (top) and the corresponding labeled peptide (bottom).
If using cultured mammalian cells, assignment of the peptide to a transcript is possible using siRNA-based silencing in conjunction with the targeted MS and peptide standards. Several (at least two) specific siRNAs should be designed against the transcript of interest to which the novel peptide putatively maps. Knockdown efficiency over 48–96 hours should be assessed via RT-qPCR and quantitative targeted proteomics using the isotopically labeled standard peptide in lieu of Western blotting for novel proteins, against which antibodies have not yet been raised (Figure 4).
Figure 4:
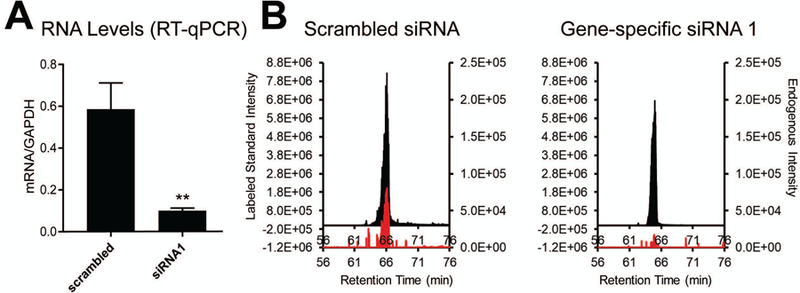
A. Levels of target transcript after transfection either with a scrambled control or gene-specific siRNA. Relative RNA levels are quantified with RT-qPCR and normalized to non-changing control GAPDH. B. MS1 peaks of targeted proteotypic peptide after transfection with scrambled or gene-specific siRNA. Equal concentrations of peptide standard were spiked into each sample. 2 µg cell lysate were used per injection.
Materials:
Complete medium (cell-specific)
Complete medium, antibiotic-free
Dulbecco’s phospate buffered saline (Sigma, D8537–100ML)
DharmaFECT reagent, (Dharmacon, T-2001–01)
At least two targeting siRNAs, custom prepared (Sigma Aldrich)
Control scrambled siRNA, 1 nmol (IDT, 51–01-14–03)
Isotopically labeled peptide standard (SpikeTides, JPT Peptide Technologies)
MS Sample buffer (see Recipe)
Trizol Reagent (Thermo Fisher, 15596026)
Agarose (Alfa Aesar, J66501–18)
50x Tris-acetate- EDTA running buffer (Thermo Fisher Scientific, J66501–18)
Ethidium bromide (Sigma Aldrich, E7637)
Isopropanol (≥99.5%)
70% ethanol (molecular biology grade)
Water (nuclease-free)
DNase I, RNase-free (NEB, M0303S)
10X DNase I reaction buffer (supplied with DNase I, RNase-free, NEB, M0303S)
Transcript-specific primers (Sigma Aldrich)
iScript cDNA synthesis kit (15596026)
iScript Reverse Transcription Supermix, (BioRad,1708840)
Steri Cycle 37 °C, CO2 Incubator (Thermo Fisher, TH-370N)
Sterile Vacuum Filter Units (MilliporeSigma, SCGVU02RE)
12-well plates (treated for cell culture)
T100 Thermal Cycler, (Bio-Rad, #1861096)
Heat block/water bath set to 37 °C/65 °C
Nanodrop Spectrophotometer
High recovery vials, (Agilent, 5183–2030)
Screw top caps, (Agilent, 5185–5863)
75 μm × 50 cm PicoFrit column
1.9 μm ReproSil-Pur 120Å C18-AQ resin (Dr. Maisch)
Q Exactive Plus (Thermo) mass spectrometer coupled with an ACQUITY UPLC M-Class (Waters)
XCalibur 3.1 (Thermo Fisher, OPTON-30382)
Order labeled proteotypic (unique to your protein of interest) peptides from JPT Peptide Technologies corresponding to non-annotated tryptic fragments of interest (Basic protocol 4, step 27). Peptides should be labeled with arginine (Arg10) or lysine (Lys8) isotopes at the C-terminal residue. Dissolve crude peptide in water and store in 1 mg/mL aliquots at –80 °C until further use.
Dilute aliquot to 100 ng/mL in MS Sample buffer.
-
Add the mass of the isotopically labeled peptide and the mass of the endogenous peptide to an inclusion list in your MS method. Instructions for this are specific to the instrument and software system. Refer to manufacturer’s guidelines.
For best results, no more than five peptides may be targeted within a single run. Once the retention time is determined, set a 5–10 min retention time window in the inclusion list. This is especially useful if targeting more than one pair of peptides in a single run.
-
Perform a test LC-MS/MS run with mixture of crude isotopically labeled peptides obtained from JPT.
Several concentrations may need to be tested: if the standard concentration is too low, MS/MS fragmentation may not be triggered while a high concentration may increase unwanted, false-positive detection of any unlabeled peptide that is present in the crude mixture. Make serial dilutions to optimize, if necessary. At the ideal concentration, the targeted peptide is reliably targeted for fragmentation and generates high quality MS/MS spectra (score>50), while no unlabeled peptide is detected.
-
Perform Mascot search as described above in Basic Protocol 4 steps 1–10, making certain that no unlabeled (“light”, identical to endogenous) peptide signal is detected from the crude peptide mixture.
If unlabeled peptide is identified at a different retention time than the isotopically labeled peptide this is likely due to misidentification of one of the impurities in the crude mixture. Further purification of the labeled standard will solve this issue.
Low levels (≤ 1%) of unlabeled peptide may be present in the unpurified mixture (this will perfectly co-elute with the labeled standard). Scaling down amount of peptide mixture per injection should eliminate detection of the undesired peptide.
-
Prepare peptide digests from your cells of interest, as above (Basic Protocols 1 and 2).
Because the specific mass of the peptide triggers fragmentation in targeted mass spectrometry experiments, less enrichment may be required than in data-dependent acquisition. Fractionation and size selection may be therefore omitted in some cases.
Dissolve up to 4 µg trypsin digest (Support Protocol 3, Step 11) in 10 µL MS sample buffer, and add labeled peptide to a final concentration of 100 ng mL−1 (or adjust concentration based on previous runs).
-
Observe retention time and fragmentation of the isotopically labeled and endogenous peptides. Perfect co-elution and similar fragmentation patterns should be observed to confirm correct identification (Figure 3).
Optimization may be required to detect endogenous peptides. If endogenous peptide is not detected, add fractionation and size-selection steps to the workflow to achieve reproducible detection.
Compare the intensity of endogenous and isotopically labeled peptides. Intensities should be similar (within about an order of magnitude); if not, repeat run with adjusted concentration of labeled standard.
Proceed to siRNA experiments. If no enrichment is necessary for reproducible fragmentation of endogenous peptide, cells may be plated in a 12-well plate. This will produce enough RNA and protein for RT-qPCR and targeted proteomics.
-
Optimize siRNA silencing conditions (i.e. siRNA concentration, length of incubation) according to instructions of the DharmaFECT transfection reagent manufacturer. Perform transfection in complete medium without antibiotic. Replace with complete medium after 24 h.
Reproducible siRNA knockdown with greater than 2-fold efficiency should be obtained before proceeding with LC-MS/MS. However, because mRNA and protein levels are not strictly correlated, results may differ by target (Liu, Beyer, & Aebersold, 2016).
Use 12-well plate to generate three replicates of each the following samples using previously optimized conditions: 1) cells untreated with siRNA; 2) cells transfected with scrambled siRNA (negative control); 3) cells transfected with first targeting siRNA; 4) cells transfected with second targeting siRNA.
Wash cells with cold PBS and harvest by centrifuging 5 min at 2,000 g/4 °C in 1.5 mL tubes.
Add 1 mL Trizol reagent directly to cells and extract RNA and protein according to instructions from the manufacturer.
-
Resuspend RNA pellets in 22 µL nuclease-free water. Use 1 µL sample to quantify RNA concentration by Nanodrop.
If purifying RNA for the first time, RNA quality and integrity should be assessed on a 1% agarose gel stained with ethidium bromide, or via Bioanalyzer according to the manufacturer’s instructions.
Intact RNA from eukaryotic cells will have two distinct ribosomal bands; the upper 28S band should be about twice as bright as the lower 18S band. Any visible smearing may indicate RNA degradation.
Add 1.5 µL DNase I and 2.5 µL DNase I buffer to RNA samples. Incubate 30 min at 37 °C in a heat block or thermal cycler.
-
Inactivate the DNase by incubating 10 min at 65 °C in a heat block or thermal cycler.
RNA quality and integrity can again be assessed by agarose gel electrophoresis or Bioanalyzer after this step, according to the same criteria as in Step 15.
-
Reverse transcribe 1 µg total RNA using the iScript Reverse Transcription mix and perform RT-qPCR with iScript cDNA synthesis kit and custom RT-qPCR primers according to instructions from the manufacturer.
Samples may be kept at –20 °C for up to several weeks after reverse transcription. Due to instability, it is not recommended to freeze RNA samples.
Upon confirmation of successful mRNA-level knockdown with RT-qPCR, proceed with protein digestion and targeted proteomics, as in Basic Protocols. Perform each step in parallel for all samples, adding equal amounts of isotopically labeled peptide standard to each.
Compare MS1 peak intensity ratios for endogenous: standard peptide in each sample. Retention time and m/z are required to find the peak in the total ion chromatogram (TIC). Each peptide identified by Mascot search will have this information listed at the top of the page in Peptide View.
Note down the retention time and m/z of the isotopically labeled standard from the Mascot search results in Peptide View. Use these metrics to find the MS1 peak of the isotopically labeled standard using mass spectrometer-specific software, for example Xcalibur (Thermo Fisher).
Export chromatogram, and paste to Excel.
Using the retention time of the isotopically labeled standard as reference, find the MS1 peak corresponding to the m/z of the unlabeled peptide in the MS Software console.
-
Export chromatogram and paste in Excel. Plot chromatograms corresponding to each peptide m/z using the same retention time range (± 2 minutes to the MS1 peak of interest).
Endogenous:labeled ratio should be consistent between control replicates. If siRNA-mediated silencing is highly efficient, endogenous peptide signal may not be detected in siRNA-treated samples. This must be confirmed by detection of isotopically labeled peptide signal in these samples.
BASIC PROTOCOL 6. TRANSCRIPT VALIDATION USING TRANSIENT OVEREXPRESSION
Basic Protocol 5 enables mapping of a tryptic fragment of a putative smORF-encoded protein to a transcript. However, bottom-up proteomics rarely affords full sequence coverage of a given protein; therefore, additional experiments are required to define the full coding sequence of a putative smORF and to demonstrate smORF translation. Many smORFs are co-translated with other proteins on a single mRNA or map to non-coding genes (lncRNAs). Therefore, biochemical confirmation that these transcripts can support translation - or can be translated in multiple open reading frames - is required. While there are many protocols that can be implemented, transient overexpression of epitope-tagged full-length transcript is one of the fastest and most facile options. This protocol covers how to insert a FLAG epitope tag in a transcript of interest using pcDNA3.1 vector. In 24 h following transfection, cells are harvested, lysed, and analyzed by Western blot. This approach can be especially useful for identifying the translated transcript isoform, or identifying the start codon in non-AUG initiated proteins via mutagenesis (Slavoff et al., 2013).
Materials:
HEK293T cells (ATCC® CRL-3216™)
HEK293T complete growth medium (See Recipe)
Full length cDNA (obtained by cloning or synthesized by GenScript)
pcDNA3.1 expression vector (Addgene, V790–20)
Restriction enzymes (New England Biolabs)
Lipofectamine 2000 (Invitrogen, 11668030)
Opti-MEM Reduced Serum Media, (Thermo Fisher, 31985062)
Precision Plus Dual Xtra Prestained Protein Standard (Bio-Rad, #1610377)
16% Tricine gels (Thermo Fisher, EC6695BOX)
2X Tricine SDS sample buffer (Invitrogen, LC1676)
10X Tricine SDS running buffer (Invitrogen, LC1675)
Anti-FLAG M2 antibody (Sigma Aldrich, F3165)
Rabbit anti-mouse HRP-conjugate (Abcam, ab97046)
10X SDS Running Buffer (Rockland, MB-017)
10X Transfer Buffer (VWR, 10128–706)
Western ECL Substrate (Bio-Rad, 1705060S)
Steri Cycle 37 °C, CO2 Incubator (Thermo Fisher, TH-370N)
Sterile Vacuum Filter Units (MilliporeSigma, SCGVU02RE)
Sterile 1.5 mL plastic tubes
Six-well plates (treated for cell culture)
Gel tank and power supply adapters (Thermo Fisher, A25977)
Power supply (Thermo Fisher, EC300XL2)
Rocking platform
Nitrocellulose and filter paper (Invitrogen, LC2000)
Mini Blot Module for Western blot transfers (Invitrogen, B1000)
-
Order cDNA clone synthesized by Genscript that represents the sequence of the RNA transcript to which your tryptic peptide of interest has been mapped. Encode a C-terminal FLAG (DYKDDDDK) epitope tag in the smORF nucleotide sequence. If other translated sequences are co-encoded in this cDNA clone, a second epitope, such as a MYC epitope tag (EQKLISEEDL), may be included at the C-terminus of the annotated sequence to confirm co-expression.
If the smORF overlaps another coding ORF, make certain that FLAG insertion at the C-terminus will not introduce any stop codons into the other coding frame.
Clone epitope-tagged cDNA into pcDNA3.1 expression vector using appropriate restriction enzymes according to instructions from supplier.
Purify at least 1 µg each of 1) pcDNA3.1-cDNA expression construct and 2) empty pcDNA3.1 vector as a negative control using standard protocols and resuspend in nuclease-free, sterile water (Birnboim & Doly, 1979).
Prepare HEK293T cells for transfection by growing in six-well plates until they reach 50–75% confluency.
Prepare vector. Combine 100 µL Opti-MEM with 1 µg vector in a sterile 1.5 mL tube. Prepare at least one control with empty vector.
Prepare transfection reagent. Obtain two additional 1.5 mL tubes with 100 µL Opti-MEM. To each tube, add 4 µL Lipofectamine 2000. Mix and incubate for an additional 5 min.
While incubating, replace media from cells in two of the wells in the six-well plates with 800 µL Opti-MEM. Return to incubator.
Mix each vector with transfection reagent (approximately 200 µL total volume). Wait an additional 20 min.
Add each vector/transfection reagent mixture to cells and incubate 6 h.
After 6 h, aspirate Opti-MEM and replace with complete media to avoid cytotoxicity.
Harvest cells after 24 h and check for protein expression using standard Western blotting procedures (Mahmood & Yang, 2012).
REAGENTS AND SOLUTIONS:
ACQUITY mobile phase A (500 mL)
500 µL 98–100% (v/v) formic acid (FA)
5 mL ACN
494.5 mL ultrapure water
Store several months at room temperature.
ACQUITY mobile phase B (500 mL)
500 µL 98–100% (v/v) FA
400 mL ACN
99.5 mL ultrapure water
Store several months at room temperature.
50 mM AMBIC (10 mL)
40 mg ammonium bicarbonate (AMBIC)
10 mL ultrapure water
Make fresh.
50 mM AMBIC/ACN wash (10 mL)
5 mL 50 mM AMBIC
5 mL ACN
Store up to one month at 4 °C.
10 mM AMBIC/ACN wash (10 mL)
1 mL 50 mM AMBIC
4 mL ultrapure water
5 mL ACN
Store up to one month at 4 °C.
C8 Elution buffer (12 mL)
9 mL 250 mM TEAF (pH 3.0)
3 mL ACN
Store at room temperature.
Coomassie stain (50 mL)
100 mg Coomassie brilliant blue
25 mL methanol
20 mL deionized water
5 mL glacial acetic acid
Store up to one year at room temperature.
Coomassie destain (1L)
500 mL methanol
400 mL deionized water
100 mL glacial acetic acid
Store up to one year at room temperature.
Digestion buffer (10 mL)
36 mg AMBIC
9 mL ultrapure water
1 mL ACN.
Store up to one month at 4 °C. Add 1 µL 0.5 mg/mL trypsin per 29 µL buffer immediately prior to digestion.
ERLIC mobile phase A (1 L)
1 mL 98–100% (v/v) FA
199 mL ultrapure water
800 mL ACN
Store up to one year at room temperature.
ERLIC mobile phase B (1 L)
1 mL 98–100% (v/v) FA
300 mL ACN
699 mL ultrapure water
Store up to one year at room temperature
Extraction buffer (10 mL)
6 mL ACN
3 mL ultrapure water
1 mL FA
Store for up to several months at room temperature.
HEK293T complete growth medium (560 mL)
500 mL DMEM
50 mL (10% v/v) fetal bovine serum
5 mL L-glutamine (200 mM)
5 mL penicillin streptomycin
Sterile filter using a 0.22 µM membrane. Store several months at 4 °C. Pre-warm medium in a 37 °C water bath 15 min before use.
Lysis buffer (10 mL)
5 µL 37% (12 M) HCl
10 µL 98% (v/v) β-mercaptoethanol (BME)
5 µL Triton X-100
9.98 mL ultrapure water
Make fresh.
MS Sample buffer (10 mL)
1.9 mL FA
10 µL TFA
8.09 mL ultrapure water
Store up to several months at room temperature.
Resuspension solution (1 mL)
400 µL 1M Tris (pH 8.0)
480 mg urea (8 M)
2.2 mg CaCl2∙2H2O (20 mM)
Add ultrapure water to 1 mL
Make fresh.
COMMENTARY
BACKGROUND INFORMATION:
Proteomic screens for smORF-encoded protein discovery in human cells date back to 2004, when an initial analysis of the K562 cell line using LC-MS/MS revealed four novel translated smORFs embedded in 5’ leader sequences upstream of canonical ORFs (Oyama et al., 2004). Improvements in small protein enrichment, LC-MS/MS technology and optimized database searching subsequently enabled mass spectrometric identification of many more hundreds of micropeptides across human cell lines and tissue samples (Ma et al., 2014b; Slavoff et al., 2013; Vanderperre et al., 2013). Further advances have improved coverage of small proteins and to allow for semiquantitative comparison of various cellular conditions (D’Lima et al., 2017; Ma et al., 2016; Yuan, D’Lima, & Slavoff, 2018).
A number of newly annotated smORFs have been implicated in various physiological and pathological processes (Couso & Patraquim, 2017; Khitun, Ness, & Slavoff, 2019; Saghatelian & Couso, 2015). Meanwhile, many more newly reported smORF-encoded proteins and “micropeptides” or “microproteins” remain uncharacterized. Detection of micropeptides within specific cellular environments can help reveal their possible biological significance. Methods that enable robust detection of micropeptides across many sample types and conditions can be implemented for this purpose. In addition to MS-based detection, ribosome profiling may be used to profile non-annotated translation events transcriptome-wide. By comparison with proteomics, ribosome profiling is more high-throughput and less biased with respect to peptide sequence composition, one of many biochemical factors that influence ionization and detection by mass spectrometry (Brar et al., 2012; Calviello et al., 2016). However, ribosome profiling may miss some micropeptides deriving from overlapping open reading frames. Furthermore, ribosome profiling is more likely to capture translational “noise” from proto-genes which do not generate stable proteins (Carvunis et al., 2012).
smORFs may also be recognized by conservation analyses. Randomly occurring ORFs are unlikely to be preserved through evolution, whereas highly conserved regions are likely to be functional. While bioinformatic analyses have successfully identified functional proteins, many recently published studies report on thousands or even millions of conserved non-annotated regions (Mat-Sharani & Firdaus-Raih, 2019). However, translated smORFs exhibit a lower degree of evolutionary conservation than known genes (Storz et al., 2014).
CRITICAL PARAMETERS:
For maximal identification of proteins less than 10 kDa in size (Basic Protocol 1), cell pellets must be quickly boiled in lysis buffer after centrifugation. Boiling eliminates proteolysis, a process that generates contaminating polypeptides in the 2–10 kDa molecular weight range (Slavoff et al., 2013). Excessive amounts of proteolysis products from abundant, larger proteins may suppress signal from less abundant peptides and result in fewer novel peptide identifications. Other chemical contaminants such as salts, plasticizers, and detergent may impede detection, and must therefore be rigorously eliminated from all mass spectrometry samples, solutions and glassware (Yeung & Stanley, 2010). Solutions which are stored for more than two days should be kept in clean glassware which has never contacted detergent and not in plastic tubes.
Mass spectrometric identification of proteins typically requires multiple tryptic peptide-spectral matches (PSMs) that map to the same open reading frame, due to the statistical nature of individual PSMs. Because small proteins often produce only one proteotypic tryptic peptide, the additional confidence derived from multiple peptide identifications is not possible for this class of proteins. Therefore, special care must be taken to examine the MS/MS spectra for smORF-derived peptides individually (Basic Protocol 4). Highly scoring PSMs sometimes match to several similar peptide sequences with nearly equal confidence. Specifically, leucine and isoleucine are indistinguishable by mass spectrometry and can be mis-assigned as a result. Therefore, peptide matches with multiple leucine or isoleucine residues may contain any unique combination thereof, and therefore must be treated as uncertain.
During biochemical validation with isotopically labeled standards (Basic Protocol 5), it is critical to inject neat isotopically labeled peptides for LC-MS/MS analysis prior to spiking in to cell lysates. Sometimes, impurities generated during peptide synthesis may be misidentified as the unlabeled peptide during searching, or small amounts of unlabeled peptide may be present. It is important to be aware of the amounts of these species within your standard to avoid false positive identifications.
UNDERSTANDING RESULTS:
Numbers of newly identified smORFs will vary vastly between organisms, cell lines, tested conditions, as well as the state of micropeptide annotation in the organism or cell of interest at the time of the experiment. Factors that will contribute to MS data quality are the amount of starting material and the LC-MS/MS platform used. The 10% sample is expected to contain nearly one thousand proteins while each fractionated sample is expected to contain at least several hundred proteins. Close to a third of the proteins should be less than 20 kDa in size, if size selection is performed successfully in mammalian cells (Figure 1).
TIME CONSIDERATIONS:
Protein isolation, size selection and trypsin digestion described in Basic Protocol 1 or Alternate Protocol 1 will take at least 2 days to complete. Desalting peptides according to the procedure in Support Protocol 3 will take approximately 3 h, where 2 h is allotted for peptide drying in the vacuum centrifuge. Database setup can be done in parallel with the previously described protocols. Basic Protocol 3 can be completed within 10 minutes; similarly Alternate Protocol 3 can be completed within several minutes given availability of transcriptomic data and prior installation of all necessary software. Timing for data analysis described in Basic Protocol 4 will vary depending on quantity of putative peptide queries. This step can take up to several days. Validation with isotopically labeled standards (Basic Protocol 5) can take from 1 week to several weeks if optimization of transfection conditions is necessary. Validation by transient overexpression (Basic Protocol 6) will take five days: three days for cloning and two days for transfection and Western blotting.
SIGNIFICANCE STATEMENT:
Unannotated small open reading frames (smORFs) that encode functional proteins exist in every genome from bacteria to humans; this protocol outlines a specialized, widely applicable proteomic approach to detect and validate smORF-encoded proteins.
ACKNOWLEDGEMENTS:
We would like to acknowledge Michael Rutenberg-Schoenberg and Gedas Bertasius for assistance with writing the python scripts. We would like to thank Amir D. Karger for writing the perl string searching script. This work was supported in part by the Searle Scholars Program, the Leukemia Research Foundation, a Smith Family Foundation Odyssey Award, the NIH (R01GM122984), and Yale University West Campus start-up funds (to S.A.S.). A. K. was supported in part by an NIH training grant (5T32GM06754).
Footnotes
INTERNET RESOURCES:
Scripts referenced in this manuscript, FPKM_filter, Peptide_filter, and Remove_stops_from_fasta can be downloaded from:
Transcript and genomic databases are publicly available from RefSeq FTP:
https://www.ncbi.nlm.nih.gov/refseq/
National Center for Biotechnology Information (NCBI) Protein Blast tool can be accessed at:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins
ProteoWizard Version 3 Peak Picking Software can be downloaded from:
http://proteowizard.sourceforge.net/download.html
Mascot Daemon Client Software Tool can be purchased from Matrix Science:
LITERATURE CITED:
- Alpert AJ (2008). Electrostatic repulsion hydrophilic interaction chromatography for isocratic separation of charged solutes and selective isolation of phosphopeptides. Anal Chem, 80(1), 62–76. doi: 10.1021/ac070997p [DOI] [PubMed] [Google Scholar]
- Andrews SJ, & Rothnagel JA (2014). Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet, 15(3), 193–204. doi: 10.1038/nrg3520 [DOI] [PubMed] [Google Scholar]
- Birnboim HC, & Doly J (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res, 7(6), 1513–1523. doi: 10.1093/nar/7.6.1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72, 248–254. doi: 10.1006/abio.1976.9999 [DOI] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, & Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science, 335(6068), 552–557. doi: 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviello L, Mukherjee N, Wyler E, Zauber H, Hirsekorn A, Selbach M, … Ohler U (2016). Detecting actively translated open reading frames in ribosome profiling data. Nat Methods, 13(2), 165–170. doi: 10.1038/nmeth.3688 [DOI] [PubMed] [Google Scholar]
- Carvunis AR, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, … Vidal M (2012). Proto-genes and de novo gene birth. Nature, 487(7407), 370–374. doi: 10.1038/nature11184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, … Mallick P (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol, 30(10), 918–920. doi: 10.1038/nbt.2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couso JP, & Patraquim P (2017). Classification and function of small open reading frames. Nat Rev Mol Cell Biol, 18(9), 575–589. doi: 10.1038/nrm.2017.58 [DOI] [PubMed] [Google Scholar]
- D’Lima NG, Khitun A, Rosenbloom AD, Yuan P, Gassaway BM, Barber KW, … Slavoff SA (2017). Comparative Proteomics Enables Identification of Nonannotated Cold Shock Proteins in E. coli. J Proteome Res, 16(10), 3722–3731. doi: 10.1021/acs.jproteome.7b00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, & Van Eyk JE (2009). Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol, Chapter 10, Unit10 25. doi: 10.1002/0471142727.mb1025s88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao P, Guo T, Li X, Adav SS, Yang J, Wei M, & Sze SK (2010). Novel application of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) in shotgun proteomics: comprehensive profiling of rat kidney proteome. J Proteome Res, 9(7), 3520–3526. doi: 10.1021/pr100037h [DOI] [PubMed] [Google Scholar]
- Harrison PM, Kumar A, Lang N, Snyder M, & Gerstein M (2002). A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res, 30(5), 1083–1090. doi: 10.1093/nar/30.5.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, & Watanabe C (1993). Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci U S A, 90(11), 5011–5015. doi: 10.1073/pnas.90.11.5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, & Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science, 324(5924), 218–223. doi: 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, & Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell, 147(4), 789–802. doi: 10.1016/j.cell.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Berg HC, & Church GM (2004). Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics, 4(1), 59–77. doi: 10.1002/pmic.200300511 [DOI] [PubMed] [Google Scholar]
- Jeong K, Kim S, & Bandeira N (2012). False discovery rates in spectral identification. BMC Bioinformatics, 13 Suppl 16, S2. doi: 10.1186/1471-2105-13-S16-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Song R, Regev A, & Struhl K (2015). Many lncRNAs, 5’UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife, 4, e08890. doi: 10.7554/eLife.08890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khitun A, Ness TJ, & Slavoff SA (2019). Small open reading frames and cellular stress responses. Mol Omics, 15(2), 108–116. doi: 10.1039/c8mo00283e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, … Pandey A (2014). A draft map of the human proteome. Nature, 509(7502), 575–581. doi: 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1989). Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol Cell Biol, 9(11), 5073–5080. doi: 10.1128/mcb.9.11.5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1997). Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J, 16(9), 2482–2492. doi: 10.1093/emboj/16.9.2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, & Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell, 165(3), 535–550. doi: 10.1016/j.cell.2016.03.014 [DOI] [PubMed] [Google Scholar]
- Ma J, Diedrich JK, Jungreis I, Donaldson C, Vaughan J, Kellis M, … Saghatelian A (2016). Improved Identification and Analysis of Small Open Reading Frame Encoded Polypeptides. Anal Chem, 88(7), 3967–3975. doi: 10.1021/acs.analchem.6b00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ward CC, Jungreis I, Slavoff SA, Schwaid AG, Neveu J, … Saghatelian A (2014a). The Discovery of Human sORF-Encoded Polypeptides (SEPs) in Cell Lines and Tissue. J Proteome Res doi: 10.1021/pr401280w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ward CC, Jungreis I, Slavoff SA, Schwaid AG, Neveu J, … Saghatelian A (2014b). Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res, 13(3), 1757–1765. doi: 10.1021/pr401280w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood T, & Yang PC (2012). Western blot: technique, theory, and trouble shooting. N Am J Med Sci, 4(9), 429–434. doi: 10.4103/1947-2714.100998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mat-Sharani S, & Firdaus-Raih M (2019). Computational discovery and annotation of conserved small open reading frames in fungal genomes. BMC Bioinformatics, 19(Suppl 13), 551. doi: 10.1186/s12859-018-2550-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menschaert G, Van Criekinge W, Notelaers T, Koch A, Crappe J, Gevaert K, & Van Damme P (2013). Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteomics, 12(7), 1780–1790. doi: 10.1074/mcp.M113.027540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A, Cox J, & Mann M (2011). More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res, 10(4), 1785–1793. doi: 10.1021/pr101060v [DOI] [PubMed] [Google Scholar]
- Nielsen ML, Savitski MM, & Zubarev RA (2006). Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol Cell Proteomics, 5(12), 2384–2391. doi: 10.1074/mcp.M600248-MCP200 [DOI] [PubMed] [Google Scholar]
- Oyama M, Itagaki C, Hata H, Suzuki Y, Izumi T, Natsume T, … Sugano S (2004). Analysis of small human proteins reveals the translation of upstream open reading frames of mRNAs. Genome Res, 14(10B), 2048–2052. doi: 10.1101/gr.2384604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, & Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis, 20(18), 3551–3567. doi: [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, & Coon JJ (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics, 11(11), 1475–1488. doi: 10.1074/mcp.O112.020131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Vazquez N, Kahle KT, Hodson CA, Ring AM, Gulcicek EE, … Lifton RP (2011). WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J Biol Chem, 286(34), 30171–30180. doi: 10.1074/jbc.M111.222893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Orera J, & Alba MM (2019). Translation of Small Open Reading Frames: Roles in Regulation and Evolutionary Innovation. Trends Genet, 35(3), 186–198. doi: 10.1016/j.tig.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Saghatelian A, & Couso JP (2015). Discovery and characterization of smORF-encoded bioactive polypeptides. Nat Chem Biol, 11(12), 909–916. doi: 10.1038/nchembio.1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, & Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc, 1(6), 2856–2860. doi: 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, … Saghatelian A (2013). Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol, 9(1), 59–64. doi: 10.1038/nchembio.1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Wolf YI, & Ramamurthi KS (2014). Small proteins can no longer be ignored. Annu Rev Biochem, 83, 753–777. doi: 10.1146/annurev-biochem-070611-102400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, & Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol, 31(1), 46–53. doi: 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, … Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc, 7(3), 562–578. doi: 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale W, Vaughan J, Jolley D, Yamamoto G, Bruhn T, Seifert H, … Rivier J (1986). Assay of growth hormone-releasing factor. Methods Enzymol, 124, 389–401. [DOI] [PubMed] [Google Scholar]
- Vanderperre B, Lucier JF, Bissonnette C, Motard J, Tremblay G, Vanderperre S, … Roucou X (2013). Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS One, 8(8), e70698. doi: 10.1371/journal.pone.0070698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, & Flugge UI (1984). A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem, 138(1), 141–143. [DOI] [PubMed] [Google Scholar]
- Yeung YG, & Stanley ER (2010). Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr Protoc Protein Sci, Chapter 16, Unit 16 12. doi: 10.1002/0471140864.ps1612s59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, D’Lima NG, & Slavoff SA (2018). Comparative Membrane Proteomics Reveals a Nonannotated E. coli Heat Shock Protein. Biochemistry, 57(1), 56–60. doi: 10.1021/acs.biochem.7b00864 [DOI] [PMC free article] [PubMed] [Google Scholar]