

Abstract
Major contributors to therapeutic resistance in pancreatic cancer (PDAC) include Kras mutations, a dense desmoplastic stroma that prevents drug delivery to the tumor, and activation of redundant signaling pathways. We have previously identified a mechanistic rationale for targeting STAT3 signaling to overcome therapeutic resistance in PDAC. In this study, we investigate the molecular mechanisms underlying the heterogeneous response to STAT3 and RAS pathway inhibition in PDAC. Effects of JAK/STAT3 inhibition (STAT3i) or MEK inhibition (MEKi) were established in Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice and patient-derived xenografts (PDX). Amphiregulin (AREG) levels were determined in serum from human PDAC patients, LSL-KrasG12D/+;Trp53R172H/+;Pdx1Cre/+ (KPC), and PKT mice. MEKi/STAT3i-treated tumors were analyzed for integrity of the pancreas and the presence of cancer stem cells (CSC). We observed an inverse correlation between ERK and STAT3 phosphorylation. MEKi resulted in immediate activation of STAT3, while STAT3i resulted in TACE-induced, AREG-dependent activation of EGFR and ERK. Combined MEKi/STAT3i sustained blockade of ERK, EGFR, and STAT3 signaling, overcoming resistance to individual MEKi or STAT3i. This combined inhibition attenuated tumor growth in PDX and increased survival of PKT mice while reducing serum AREG levels. Furthermore, MEKi/STAT3i altered the PDAC tumor microenvironment by depleting tumor fibrosis, maintaining pancreatic integrity, and downregulating CD44+ and CD133+ CSC. These results demonstrate that resistance to MEKi is mediated through activation of STAT3, while TACE-AREG-EGFR-dependent activation of RAS pathway signaling confers resistance to STAT3 inhibition. Combined MEKi/STAT3i overcomes these resistances and provides a novel therapeutic strategy to target the RAS and STAT3 pathway in PDAC.
Significance:
This report describes an inverse correlation between MEK and STAT3 signaling as key mechanisms of resistance in PDAC and shows that combined inhibition of MEK and STAT3 overcomes this resistance and provides an improved therapeutic strategy to target the RAS pathway in PDAC.
Keywords: STAT3 signaling, RAS pathway, EGFR, amphiregulin, pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is currently the third leading cause of cancer mortality in the US and estimated to become the second leading cause of cancer death by 2020(1). Despite advances in understanding the biology of PDAC, the clinical course is still hampered by therapeutic resistance and lack of progress in improving survival outcomes. Activating Kras mutations, present in over 90% of PDAC tumors (2), are difficult to target due to the inter-dependence of redundant signaling pathways and feedback loops. Another hallmark of PDAC is the presence of a dense desmoplastic reaction composed of stromal cells, including stellate cells, immune cells and cancer “stem” cells (CSCs) that support the tumor microenvironment (TME), acts as barrier to drug delivery and also contributes to therapeutic resistance of PDAC (3,4). There remains an incomplete understanding of interactions among the different TME components that support cancer growth and promote resistance to therapy.
In PDAC, Kras activating mutations promote proliferation and inhibit apoptosis through the RAF/MEK/ERK and PIK3/AKT pathways (5–7). Since targeting RAS has remained elusive, efforts have focused on targeting downstream effectors of RAS through MEK inhibition (5,8). The clinical success of MEK inhibitors in Kras mutant melanoma and lung cancer implicate their therapeutic potential in PDAC (9,10). Unfortunately, clinical trials of MEK inhibition (MEKi), have been unsuccessful in PDAC (7,9,11), likely due to the activation of resistance pathways.
Shedding of EGFR ligands from PDAC cells results in autocrine feedback and subsequently facilitates Kras activation to promote the onset of pancreatic neoplasia (12,13). EGFR ligands amphiregulin (AREG) and transforming growth factor-α (TGF-α) reside as transmembrane glycoproteins in their precursor form and are enzymatically shed via metalloproteinase TNFα-converting enzyme (TACE, ADAM17) to regulate EGFR signaling (12,14–16). We have shown that TACE and AREG, but not TGF-α, are overexpressed in colorectal cancer and PDAC (14). AREG appears to be the primary ligand for promoting TACE-mediated activation of EGFR, MAPK and STAT3 signaling.
The poor response of PDAC patients to targeted and systemic therapies is also partially due to the dense desmoplastic reaction, which impedes drug delivery into tumors (17–20). We have shown that targeted inhibition of STAT3 (STAT3i) in combination with gemcitabine enhances drug delivery into tumors by remodeling collagen fibers and increasing microvessel density (19,20). Therapeutic resistance in PDAC is also conferred by increased percent of CSCs in the TME following chemotherapy (21).
In this study, we have provided proof that STAT3 activation is a novel molecular mechanism underlying the therapeutic resistance of PDAC to RAS pathway inhibition. We show a direct inverse correlation between ERK and STAT3 signaling in PDAC and provide evidence that a major resistance mechanism impairing MEKi occurs through TACE-induced, AREG-mediated EGFR pathway activation. We further show combined MEKi/STAT3i results in sustained inhibition of both ERK and STAT3 signaling, overcoming therapeutic resistance associated with the AREG-EGFR mediated parallel feedback. In addition, we show MEKi/STAT3i inhibits tumor fibrosis and downregulates CSCs to enhance therapeutic response in PDAC. Furthermore, elevated serum AREG levels are decreased with MEKi/STAT3i in PKT mice, suggesting a role for serum AREG as a prognostic biomarker of therapeutic response as well as a biomarker of therapeutic resistance to EGFR, MEK and STAT3 inhibition.
Materials and Methods
Cell Lines and Chemicals
Mouse PanIN, PDA and LMP cell lines were derived from the LSL-KrasG12D/+; Pdx1Cre/+ (KC) and LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) genetically engineered mouse models (GEM) of PDAC (22,23) (kindly provided by Dr. Andrew Lowy) and maintained as previously described (20). Authenticated human PDAC cell lines MiaPaCa2, PANC1, CFPAC, Capan2, Capan1, AsPC1, SW1990, Panc04, Panc02, Panc10 and BxPC3 were obtained and maintained according to American Type Culture Collection (ATCC, Manassas, VA) guidelines. The PC-13 and PC-17 cell lines were derived from patient derived xenografts (PDXs) grafts (kindly provided by Dr. Anthony Capobianco) with known mutational status (PC-13: KrasQ61H/+/PIK3CAM/+/p53R72P/+; PC-17: APCM/M/KrasG12D/+/p53R72P/R72P).
Cell authentication was performed by using STR DNA profiling (latest date: June 16, 2016) and cell lines tested negative for Mycoplasma via Genetica cell line testing (Burlington, NC) using eMYCO plus kit (iNtRON Biotechnology). Cells with relative low passage numbers (< 20) were used in the study.
Primary antibodies used for Western blot analysis (Supplementary Table S1), immunohistochemistry (Supplementary Table S2) and flow cytometry analysis (Supplementary Table S3) were summarized in the Supplementary Tables S1-S3. The chemical agents used are presented at Supplementary Table S4.
Tissue Microarray of Human Pancreatic Tissues
Previous constructed tissue microarray (TMA)(20) slides were concurrently evaluated by 2 of the authors (C.S. and N.B.M.). Nuclear and cytoplasmic staining was scored as follows: staining index was considered as the sum of the intensity score (0, no staining; 1+, weak; 2+, moderate; 3+, strong) and the distribution score (0 – no staining; 1+ – staining of <33% of cells; 2+ – between 33% and 66% of cells; and 3+ – staining of >66% of cells). Staining indices were classified as follows: 3+ or higher – strong staining; 1+ to 2+ – weak staining; and 0 – negative staining. pERK and pSTAT3 were scored as positive if any detectable nuclear or cytoplasmic staining was present.
Mice
Female athymic nude mice – Foxn1 nu/nu (4–5 weeks old) – were purchased from Harlan Sprague Dawley, Inc. Ptf1acre/+;Tgfbr2flox/flox and LSL-KrasG12D/+;Tgfbr2flox/flox mice were provided by Dr. Hal Moses (Vanderbilt University Medical Center, Nashville, TN). These 2 lines were intercrossed to generate Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox mice (PKT) on a C57Bl/6 background. Genotyping of alleles was performed using oligonucleotide primers as described previously (20,24). LSL-KrasG12D/+, Pdx1Cre/+and p53R273H/+mice were intercrossed to generate indicated LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) animals (22,23).
Xenograft Models
Subcutaneous tumors were established by injecting 2×106 PANC1, MiaPaCa2 or BxPC3 cells into the flank of 6-week-old Fox1-nu/nu mouse [n=4 (PANC1) or n=3 (MiaPaCa2, BxPC3) in each group]. Treatment was initiated when the subcutaneous tumors reached 75 – 100 or 200–250 (BxPC3) mm3 size. Drug treatment was initiated at the same time point. AZD1480 (30 mg/kg/day), AZD6244 (25 mg/kg/day), both drugs together or vehicle (Hydroxypropyl methyl cellulose/Tween 80) was administered by oral gavage for 41(MiaPaCa2), 49 (PANC1) and 27 (BxPC3) days. The data was obtained as described in the Supplementary Materials and Methods.
Treatment of Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) Mice
PKT mice were treated with vehicle, AZD1480 (JAK/STAT3 inhibitor), AZD6244 (MEK inhibitor) or a combination of AZD6244 and AZD1480. Mice in the AZD1480 (30 mg/kg/day) and AZD6244 (25 mg/kg/day) arm received by oral gavage 5 days/week, starting at 4 weeks of age. Mice were euthanized and dissected after 2 weeks unless they were part of the survival arm. Due to the irregularity of the tumor dimensions, size was determined by weighing the entire tumor. Tumor tissue was processed for further immunohistochemical examination. Overall survival was determined by log-rank analysis using statistical software package R (version 3.3.2).
Statistical Analysis
Descriptive statistics were calculated using Microsoft Excel and Prism software (Graphpad Software Inc., La Jolla, CA). Results are shown as values of mean ± s.d. unless otherwise indicated. Statistical analyses of immunohistochemistry data were performed using the ANOVA followed by Tukey’s multiple comparisons test to determine P-values. The 2-sided Student’s t test was used for statistical analysis, with P < 0.05 taken as significant, except when indicated otherwise. Kaplan-Meier survival analysis was performed, and survival differences between groups were assessed with the log-rank test. Pearson correlation coefficient or Pearson’s r analysis was conducted using Prism software.
Study Approval
All animal experiments were carried out using protocols approved by the Institutional Animal Care and Use Committees at the Vanderbilt University Medical Center and the University of Miami (#15–099).
For additional experimental procedures please refer to Supplementary Materials and Methods.
Results
Inverse Correlation of ERK and STAT3 signaling in PDAC
A direct inverse correlation in expression levels of phosphorylated ERK (pERK) and STAT3 (pSTAT3) was observed both in vitro and in vivo (Fig. 1). Inverse baseline expression of pERK and pSTAT3 was confirmed in eleven human PDAC cell lines (Fig. 1A, left panel). Cell lines with low or minimal pERK expression showed high levels of pSTAT3 expression while cell lines with high pERK expression showed little or no pSTAT3 expression. This pattern was also seen in a pancreatic intraepithelial neoplasia (PanIN)-derived cell line from the LSL-KrasG12D/+; Pdx1Cre/+ (KC) GEM, and from primary PDAC (PDA) and liver metastasis (LMP) cell lines derived from the LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) GEM (Fig. 1A, right panel) (22). Results from a human tumor tissue microarray (TMA) confirmed that the majority of patient tumors with strong pERK expression had low pSTAT3 expression while tumors with high pSTAT3 expression had low pERK expression (Fig. 1B and Supplementary Fig. S1A). Knockdown of STAT3 in PANC1 cells (sh-STAT3) (20) showed increased expression of pERK when compared to sh-scrambled cell lysate (Fig. 1C). In addition, pancreatic tumors from Ptf1acre/+;LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice (Fig. 1D and Supplementary Fig. S1B) treated with the JAK/STAT3 inhibitor (STAT3i) AZD1480 showed increased pERK levels while mice treated with the MEK inhibitor (MEKi) AZD6244 showed increased pSTAT3 expression. Both STAT3i and MEKi showed increased pEGFR expression (Fig. 1D).
Figure 1.
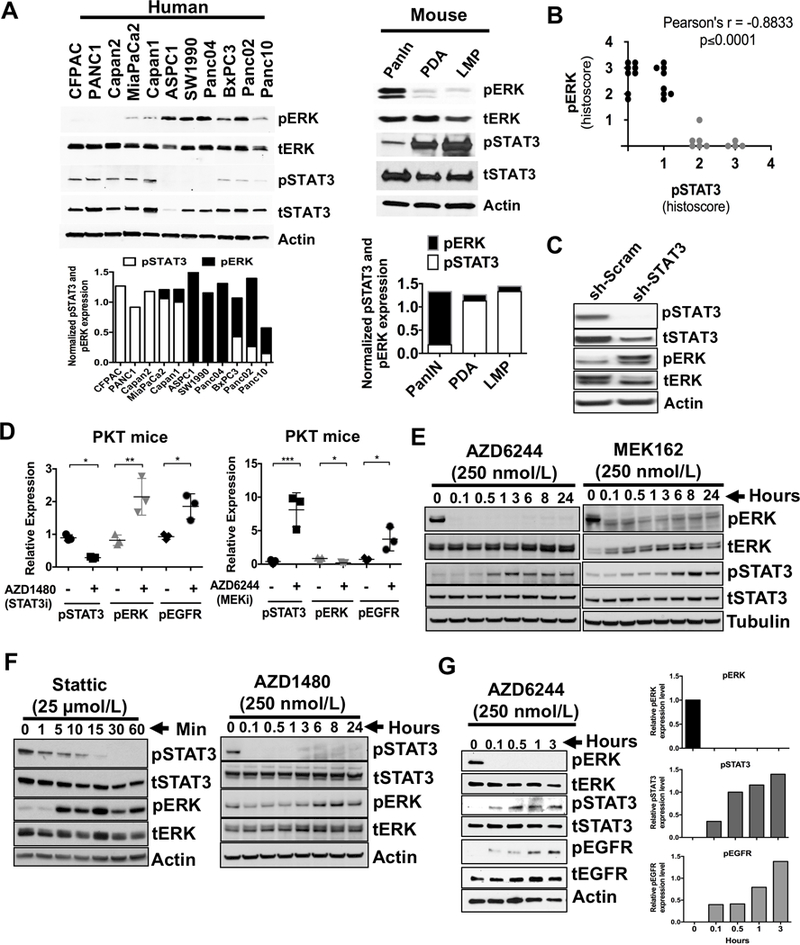
Inverse correlation of ERK and STAT3 in PDAC. A, expression of activated and total levels of ERK and STAT3 in cell lines from human PDAC (left panel) or cells generated from pancreatic intraepithelial neoplasia (PanIN), primary PDAC (PDA) and liver metastatic (LMP) lesions from LSL-KrasG12D/+;Pdx1Cre/+ (PanIN) and LSL-KrasG12D/+;Trp53R172H/+;Pdx1Cre/+ (PDA and LMP) mice (right panel) are demonstrated. Densitometry analyses of pSTAT3 and pERK normalized to tSTAT3 and tERK respectively were shown in the bottom panels of A. B, selected human PDAC tumor samples were stained for pERK and pSTAT3 expression. Pearson correlation showed negative correlation for pERK and pSTAT3 expression. r = −0.8833, p ≤ 0.0001. C, sh-STAT3 and sh-scrambled (sh-Scram) control cell lysates were analyzed for pSTAT3 and pERK expression by Western blot. D, tumors tissues from PKT mice treated with STAT3 inhibitor AZD1480 (left panel) or MEK inhibitor AZD6244 (right panel) were analyzed for pERK, pEGFR or pSTAT3 expression respectively by immunohistochemistry and analyzed using Image J. E, Western blot of lysates from MiaPaCa2 cells were treated with AZD6244 or MEK162 (MEK inhibitor) in time-dependent manner. F, Western blot of lysates from MiaPaCa2 cells were treated with STAT3 inhibitors Stattic (left panel) or AZD1480 (right panel) in time dependent manner. G, Western blot of lysates from SW1990 cells treated with MEKi (AZD6244) in time (left panel) -dependent manner and analyzed for the levels of pERK, pSTAT3 and pEGFR (right panels). Densitometry analyses of pERK, pSTAT3 and pEGFR normalized to tERK, tSTAT3 and tEGFR respectively. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Treatment with the MEK inhibitors AZD6244 or MEK162 effectively suppressed pERK while inducing pSTAT3 activation in PDAC cells in a time-dependent manner (Fig. 1E). Conversely, STAT3 inhibitors Stattic and AZD1480 resulted in pERK activation in a time- (Fig. 1F and Supplementary Fig. S2) and dose-dependent manner (Supplementary Fig. S3A-C). Even SW1990 cells, which have very low basal levels of pSTAT3 (Fig. 1A, left panel), increased pSTAT3 and pEGFR levels with MEKi in a time- (Fig. 1G) and dose-dependent manner (Supplementary Fig. S3D).
To determine the effects of cytotoxic chemotherapy on STAT3 or ERK signaling, we treated MiaPaCa2 cells with gemcitabine with or without AZD1480 (Supplementary Fig. S4A and B) or Erlotinib (Supplementary Fig. S4C). Gemcitabine treatment alone resulted in a dose-dependent activation of both pSTAT3 and pERK. Cells treated with both gemcitabine and AZD1480 exhibited similar effects on STAT3 and ERK signaling compared to AZD1480 (STAT3i) alone, demonstrating that gemcitabine did not alter the reciprocal activation of STAT3 or ERK signaling. EGFR inhibition with erlotinib attenuated EGFR phosphorylation.
Combined MEKi and STAT3i Inhibits Pancreatic Tumorigenicity
To confirm this reciprocal activation of MEK and STAT3 as a mechanism of resistance, we targeted both STAT3 and MEK in PDAC cell lines. MiaPaCa2 (Fig. 2A) cells treated in combination with U0126, a highly selective inhibitor of MEK1/2 and Stattic, a small molecule that specifically inhibits the SH2 domain of STAT3 or PANC1 and MiaPaCa2 (Fig. 2B) with combined AZD6244/AZD1480 showed sustained inhibition of both ERK and STAT3 activation. Combined MEKi/STAT3i in MiaPaCa2 cells resulted in enhanced apoptosis (Fig. 2C), reduced colony formation (Fig. 2D, left panel), decreased cell invasion (Fig. 2D, right panel) and reduced spheroid growth (Fig. 2E). Furthermore, spheroid growth of patient derived xenograft (PDX) cells PC-13 and PC-17 (Fig. 2F) were significantly reduced with combined STAT3i/MEKi when compared to control cells. These results show that combined MEKi/STAT3i suppresses pancreatic tumorigenicity.
Figure 2.
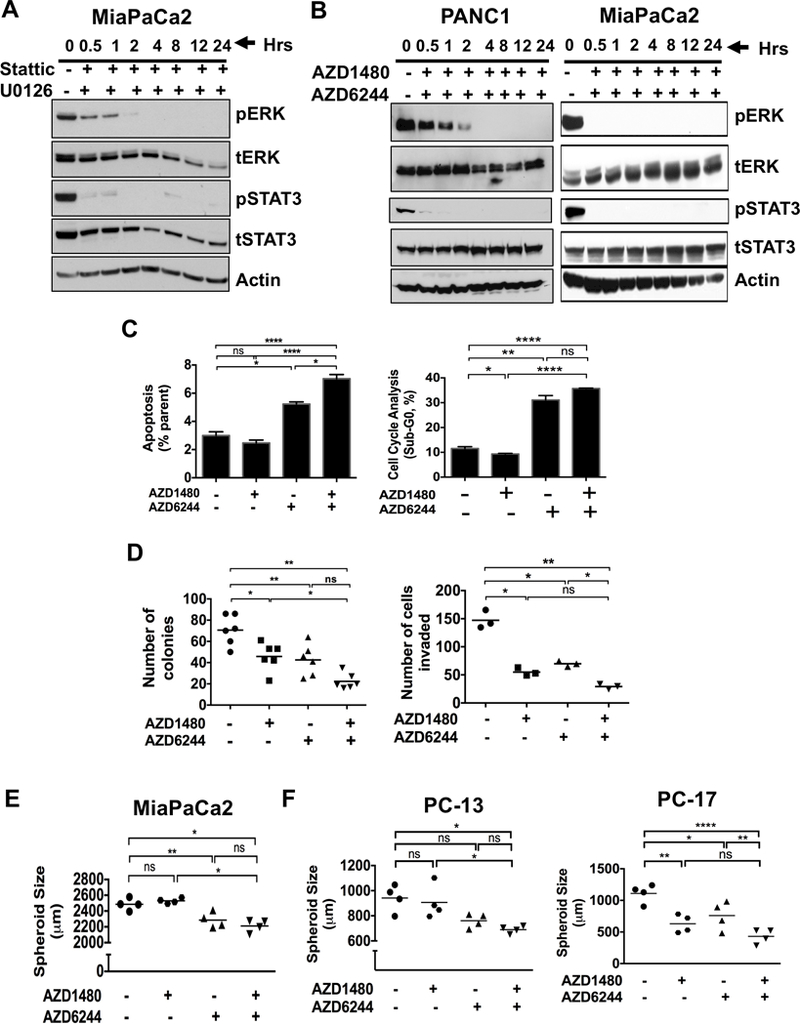
Combined MEKi and STAT3i abrogates tumorigenicity, spheroid growth, and cell invasion while enhancing apoptosis in PDAC cells. Expression of pERK and pSTAT3 in human MiaPaCa2 cells treated with MEK inhibitor (U0126) and STAT3 inhibitor (Stattic) (A) or PANC1 and MiaPaCa2 (B) cells treated with AZD6244 (MEK inhibitor) and AZD1480 (JAK/STAT3 inhibitor) in time-dependent manner for up to 24 hours. C, MiaPaCa2 cells were treated with AZD1480 with or without AZD6244 for 24 hours, stained with annexin V-FITC and propidium iodide (PI) (left panel) or PI (right panel) and analyzed by flow cytometry. Cells with AZD1480 and AZD6244 treatment resulted in an increase in apoptosis (annexin positive and PI negative) and Sub-G0 level (PI positive). D, MiaPaCa2 cells treated with the combination of AZD6244 and AZD1480 showed decreased number of colonies (left panel) and cell invasion (right panel). E, Combination of AZD1480 and AZD6244 drug treatment decreased spheroid size in MiaPaCa2 cells when compared to either control treatment or AZD1480. F, combination of AZD1480 and AZD6244 decreased spheroid size in PDXs PC-13 (left panel) and PC-17 (right panel) cells. *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, P > 0.05.
STAT3i Results in ERK Activation Through TACE-Dependent Shedding of AREG and EGFR Activation
RAS signaling pathway is a known mediator of tumor progression and survival which is activated downstream of the epidermal growth factor receptor (EGFR) (25). Having shown, both in vitro and in vivo, that reciprocal reactivation of pERK or pSTAT3 upon STAT3i or MEKi, respectively (Fig. 1C-G), we hypothesized that STAT3i would also result in EGFR activation, upstream of RAS signaling. As seen in Fig. 3A, STAT3i with AZD1480 resulted in pSTAT3 inhibition, with subsequent activation of pERK and pEGFR in both time- and dose-dependent manner (Fig. 3A and Supplementary Fig. S3C). Interestingly, over time, pSTAT3 becomes reactivated following activation of EGFR even with STAT3i. Again, as pSTAT3 is reactivated, pERK levels decrease, further confirming the inverse correlation of pSTAT3 and pERK signaling. These data suggest STAT3i-mediated ERK activation may be dependent on EGFR signaling. To further explore the mechanism by which STAT3i promotes EGFR tyrosine phosphorylation, we examined the upstream activators of EGFR signaling.
Figure 3.
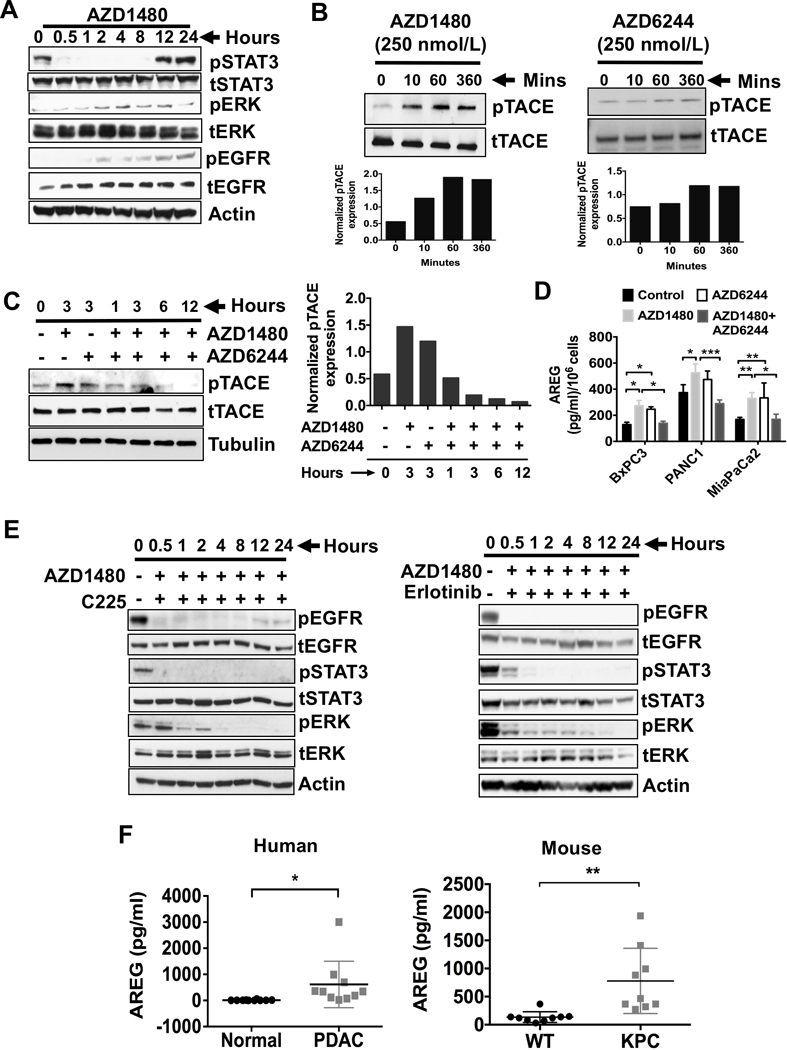
STAT3i results in ERK phosphorylation, which occurs through TACE-AREG-EGFR signaling. A, Western blots of human MiaPaCa2 cells treated with JAK/STAT3 inhibitor (AZD1480, 100 nmol/L) and analyzed for the expression of pERK, pSTAT3 and pEGFR in time dependent manner. B, MiaPaCa2 Cells were treated with AZD1480 (250 nmol/L, left panel) or AZD6244 (250 nmol/L, right panel) for up to 360 minutes, immunoblotted for TACE and pTACE (T735). Densitometry analyses of pTACE normalized to tTACE was shown in the bottom panels of B. C, MiaPaCa2 cells were treated with AZD1480 (250 nmol/L) and/or AZD6244 (250 nmol/L) for up to 12 hours, immunoblotted for pTACE (left panel). Densitometry analyses for the immunoblots from pTACE after normalization to total TACE protein are shown (right panel). Densitometry analyses of pTACE normalized to tTACE was shown in the bottom panels of C. D, PDAC cells were treated with AZD1480 (100 nmol/L) and/or AZD6244 (250 nmol/L) for 48 hours and analyzed for AREG release in the culture media (concentrations per 1 × 106 cells). Each value represents the mean and SD (n = 3). E, MiaPaCa2 cells were treated with AZD1480 (100 nmol/L) with or without Cetuximab (C225), monoclonal antibody to EGFR (4 microgram/ml) (left panel) or EGFR kinase inhibitor erlotinib (1 microgram/ml) (right panel) for up to 24 hours, lysed and immunoblotted for pSTAT3 and pERK. F, Serum AREG (pg/ml) levels from normal and PDAC patients were analyzed (left panel). Serum AREG (pg/ml) levels from wildtype (WT) and KPC mice were analyzed (right panel). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Previously, we and others have shown that TACE is regulated by G-protein-coupled receptors (GPCRs) which act as mediators of EGFR transactivation (14,26,27). TACE, when activated, induces cleavage and shedding of soluble forms of the EGFR ligands, amphiregulin (AREG) and TGF-α. Importantly, we have shown TACE and AREG, but not TGF-α, are overexpressed in pancreatic tumors when compared to normal human pancreatic tissues and are involved in EGFR/ERK/STAT3 signaling activation (14). First, we confirmed EGFR inhibition with erlotinib activates STAT3 phosphorylation (Supplementary Fig. S4C). To determine the mechanism by which STAT3i leads to EGFR activation, we analyzed the effects of STAT3i or MEKi on TACE phosphorylation (Fig. 3B). Interestingly, STAT3i increased TACE phosphorylation in a time-dependent manner. Similarly, MEKi also showed increase in TACE phosphorylation. Combined MEKi/STAT3i attenuated TACE activation (Fig. 3C), suggesting that TACE phosphorylation mediates the reciprocal activation of STAT3 and ERK through EGFR. We then assessed MEKi/STAT3i effects on AREG shedding in conditioned media (CM) from BxPC3, PANC1 and MiaPaCa2 cells (Fig. 3D). STAT3i with AZD1480 and MEKi with AZD6244 resulted in significantly increased AREG levels in the CM from all three cell lines. This observation further confirms AREG involvement in MEKi and STAT3i-mediated activation of EGFR signaling.
We then sought to determine if AREG is the ligand that activates EGFR signaling in this process. First, we confirmed AREG activates ERK phosphorylation (Supplementary Fig. S5). To further delineate the role of STAT3 in AREG-mediated EGFR-ERK activation, MiaPaCa2 cells were treated with AZD1480 and cetuximab (C225), a mAb that inhibits EGFR ligand binding (Fig. 3E, left panel) or erlotinib, a tyrosine kinase inhibitor of EGFR (Fig. 3E, right panel). Combined treatment with AZD1480 and C225 or erlotinib completely inhibited ERK and STAT3 activation along with EGFR signaling. Taken together, these data suggest EGFR tyrosine kinase activity mediates STAT3-dependent ERK activation.
Since TACE activation correlates with AREG-dependent ERK activation (14), we sought to address the role of AREG release during in vivo PDAC progression. Serum was collected from PDAC patients and normal individuals (Fig. 3F, left panel). Serum from PDAC patients had significantly higher AREG levels when compared to normal patients, suggesting increased TACE activity in PDAC patients as we have previously shown.(14) Furthermore, we compared AREG serum levels between KPC and wildtype (WT) mice (Fig. 3F, right panel). Serum AREG levels were significantly higher in KPC mice compared to WT mice, further corroborating the roles of AREG and activated TACE in PDAC progression. Collectively these data show TACE-AREG-mediated EGFR pathway activation is a major resistance mechanism that impairs the efficacy of individual MEKi or STAT3i. These results suggest combined EGFR and STAT3 or MEK and STAT3 inhibition may be therapeutically effective in PDAC. Furthermore, AREG may serve as a potential circulating biomarker of resistance to MEK or STAT3 inhibition.
Combined MEKi and STAT3i Reduces Tumor Burden, Retains Pancreatic Integrity and Improves Survival in PKT Mice
To determine the effects of MEKi/STAT3i on tumor growth in vivo, PANC1 (Fig. 4A) and MiaPaCa2 (Fig. 4B) xenografts were subjected to vehicle, AZD1480, AZD6244 or combined treatment by daily oral gavage. Combined MEKi/STAT3i significantly suppressed tumor growth compared to vehicle or individual agent treatment. We did not observe any significant changes in animal body weight, suggesting there is no added toxicity from the combined treatment (Supplementary Fig. S6A and B). Immunohistochemical (IHC) staining of xenograft tumors showed decreased Ki67 (Fig. 4C) and increased cleaved caspase 3 (Fig. 4D) staining in the combined treated tumors compared to treatment with either agent alone.
Figure 4.
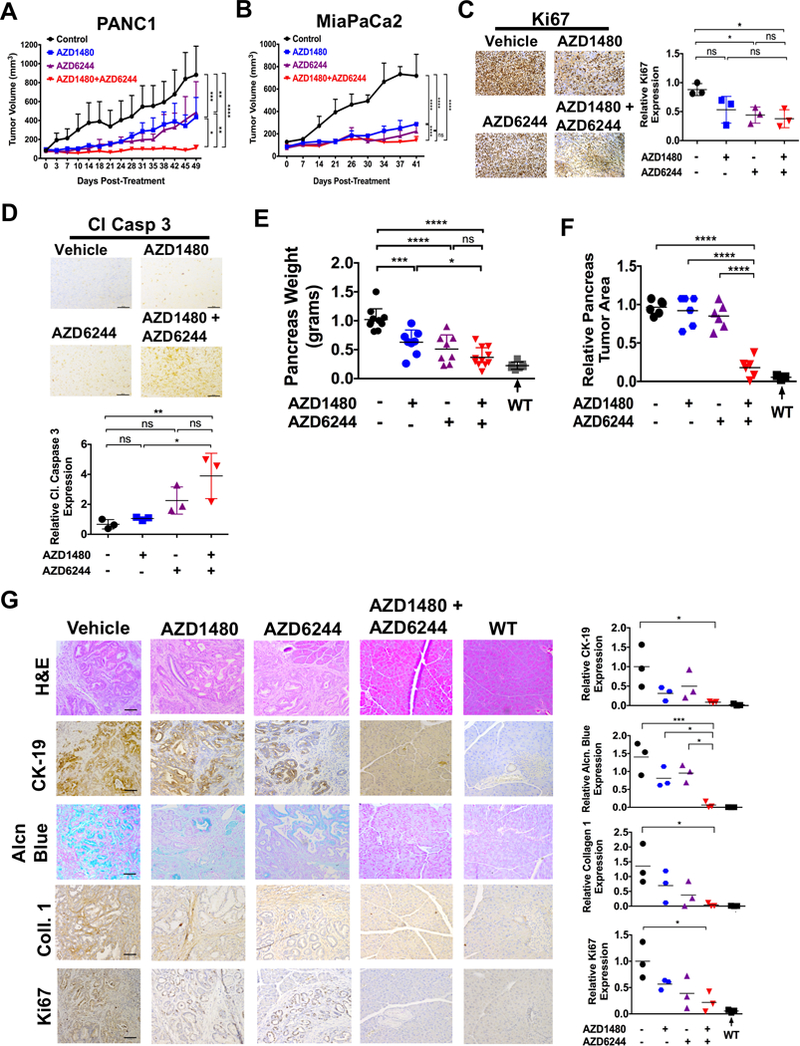
Combined AZD1480/AZD6244 treatment decreases tumor growth, tumor burden and proliferation while retaining the pancreatic integrity. Tumor growth rate of PDAC cells PANC1 (A) and MiaPaCa2 (B) flank xenografts in Fox 1-nu/nu mice treated with vehicle, AZD1480 (30 mg/kg/day), AZD6244 (25 mg/kg/day) or AZD1480/AZD6244. Error bars indicate SD of mean; n = 4 (PANC1) or n = 3 (MiaPaCa2) per group. (C and D) Immunohistochemistry of pancreatic tumor xenograft tissues show decreased Ki-67 (C) and increased cleaved caspase 3 (Cl Casp 3) expression with MEKi/STAT3i treatment and analyzed using Image J (lower panels). (E, F and G) PKT mice received AZD1480, AZD6244 or AZD1480/AZD6244 by oral gavage 5 days/week, starting at 4 weeks of age for 2 weeks. E, total pancreatic tumor weight was measured at the end of treatment at 6 weeks of age. Treatment of MEKi and MEKi/STAT3i showed a significantly reduced weight compared to vehicle treated mice. (F and G) The relative tumor area (F) was measured and the histological sections of pancreatic tissue were stained for Cytokeratin 19 (CK-19), alcian blue, collagen 1 and Ki67 (G, left panels), and analyzed for the PDAC tumor integrity. The expression of the CK-19, alcian blue, Collagen 1 and Ki67 stains were calculated and analyzed using Image J (right panels). WT: wild type; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, P > 0.05.
To determine therapeutic efficacy in a GEM of PDAC, we treated PKT mice with the same treatment regimen (vehicle, AZD1480, AZD6244 or combination) beginning at 4 weeks of age when pancreatic tumors are firmly established in this GEM (Fig. 4E-G). There was a significant reduction in pancreatic tumor weight with combined AZD1480/AZD6244 treatment compared to vehicle or treatment with either agent alone (Fig. 4E). Furthermore, while pancreata of vehicle or monotherapy treated mice were completely replaced with tumor burden, mice treated with combined MEKi/STAT3i had little or no tumor burden and maintained their pancreatic integrity (Fig. 4F and G). Again, there were no significant changes in animal body weight, suggesting no added toxicity from the combined treatment (Supplementary Fig. S7A). To further analyze pancreatic integrity, PKT tumors were assessed for cytokeratin 19 (CK-19), alcian blue, collagen 1 and Ki67 staining by IHC (Fig. 4G). Vehicle-treated PKT mice had abundant staining with the proliferation marker Ki67, extracellular matrix protein collagen type 1 and alcian blue as well as CK-19 staining for adenocarcinoma. Pancreata of PKT mice treated with AZD1480 or AZD6244 alone had a slight reduction in staining of these markers compared to vehicle-treated mice. However, PKT mice treated with combined AZD1480/AZD6244 had significantly more normal pancreatic architecture and histology, demonstrating a staining profile similar to their wild type littermates (Fig. 4G). These results show that combined MEKi/STAT3i significantly reduced tumor burden, reduced tumor cell proliferation and retained pancreatic integrity in this aggressive PDAC GEM.
In a separate study, PKT mice were analyzed for survival under the same treatment arms. Again, tumor growth was significantly reduced in PKT mice treated with combined MEKi/STAT3i (Fig. 5A). Furthermore, survival of PKT mice treated with MEKi/STAT3i was significantly improved when compared to vehicle-treated and STAT3i treated mice (53 vs 85 or 63 vs 85 days respectively; p=0.0002, log-rank test). While there was an improvement in survival of mice treated with combined MEKi/STAT3i compared to MEKi treated mice (85 vs 76 days, Fig. 5B), these results were not statistically significant. Western blot analysis of whole tumor lysates showed high pERK and pSTAT3 but low pEGFR expression in vehicle-treated mice (Fig. 5C). While STAT3i completely inhibited pSTAT3 expression, it increased expression of both pERK and pEGFR. MEKi inhibited pERK expression, but had limited effect on pSTAT3 expression. In contrast, combined MEKi/STAT3i significantly reduced expression of both pSTAT3 and pERK without activating pEGFR. IHC staining showed significantly decreased in Ki67 staining (Fig. 5D) and enhanced cleaved caspase 3 (Fig. 5E) expression in tumors treated with combined MEKi/STAT3i compared to tumors treated with vehicle or single agents alone. These results confirm the enhanced in vivo effects of combined MEKi/STAT3i in overcoming therapeutic resistance in an aggressive PDAC GEM.
Figure 5.
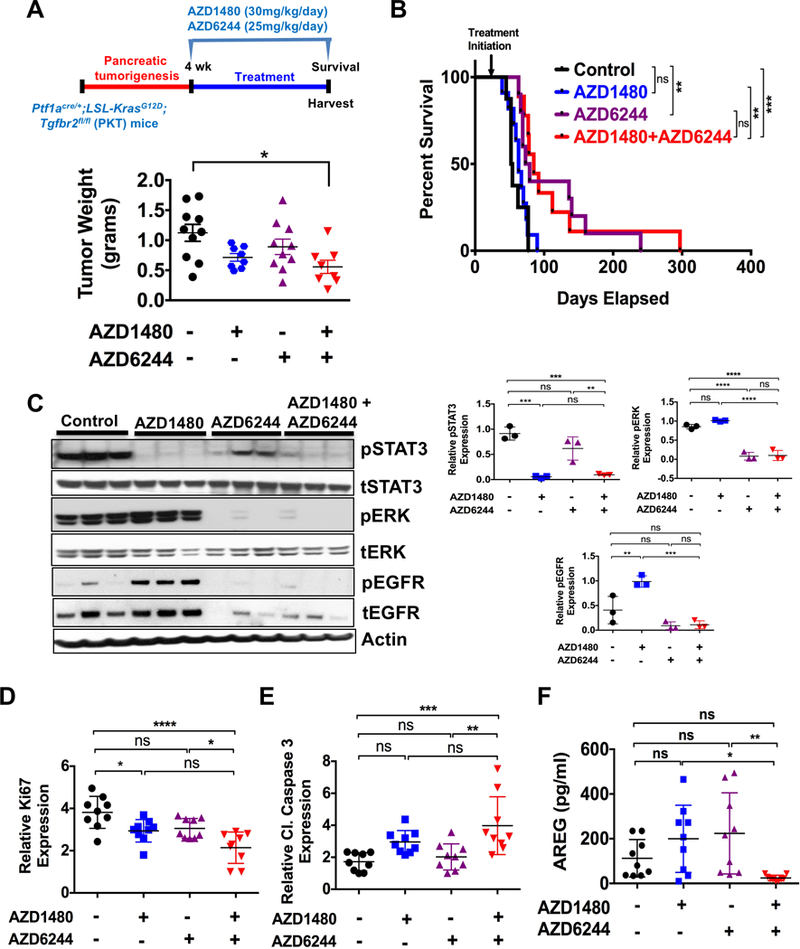
Combined MEKi/STAT3i treatment improves survival of PKT mice. For survival study, PKT mice were treated with vehicle, AZD1480 (30 mg/kg/day), AZD6244 (25 mg/kg/day) or combined AZD1480/AZD6244 by oral gavage 5 days/week, starting at 4 weeks of age. A, tumor weight in the AZD1480/AZD6244 treated mice was significantly decreased compared with vehicle-treated controls. B, Kaplan-Meier survival analysis shows significantly improved overall survival with AZD1480/AZD6244 (median 85 days) compared with vehicle control (median 53 days, p=0.0002, log-rank test). C, Western blot analysis of whole tumor lysates demonstrated decreased expression of pSTAT3 and pERK in mice treated with AZD1480/AZD6244 compared to vehicle or single drug treated mice (left panels) and analyzed using Image J (right panels). D, Proliferation (Ki67 staining) was significantly decreased and apoptosis (cl caspase 3) (E) was significantly increased with AZD1480/AZD6244 treatment when compared to either vehicle or AZD6244 treated mice. F, AREG amount was significantly decreased in the serum of AZD1480/AZD6244 treated mice when compared to either AZD1480 or AZD6244 treated mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, P > 0.05.
Combined MEKi/STAT3i attenuated TACE activation (Fig. 3C and Supplementary Fig. S7B), suggesting that TACE phosphorylation mediates the reciprocal activation of STAT3 and ERK through EGFR. Furthermore, to assess the effect of MEKi/STAT3i on systemic AREG levels, we measured AREG in the serum of treated PKT mice. Serum AREG levels were negligible and significantly lower in PKT mice treated with combined MEKi/STAT3i compared to treatment with single agents alone (Fig. 5F). These results further corroborate the effect of combined MEKi/STAT3i in overcoming AREG-EGFR-mediated resistance to RAS and STAT3 signaling inhibition in PDAC.
Combined MEKi/STAT3i Decreases CSCs in PDAC
CSCs are self-renewing cells in the TME that are resistant to cytotoxic and targeted therapies and are often the cause of tumor recurrence despite initial therapeutic response. STAT3 signaling is active in tumor-associated CSCs (3,28,29). To determine if MEKi/STAT3i affects CSCs in PDAC, PKT mouse tumors treated with MEKi and/or STAT3i were evaluated for CSC cell markers by flow cytometry. Analysis showed significantly decreased EpCAM+CD44+ (Fig. 6A) and EpCAM+CD133+ (Fig. 6B) CSCs in tumors treated with combined MEKi/STAT3i when compared to vehicle treatment. These results indicate that the enhanced in vivo effects of combined MEKi/STAT3i further overcome therapeutic resistance by targeting CSCs in PDAC. To further assess the impact on CSCs, BxPC3 xenografts were treated with vehicle or combined MEKi/STAT3i for 27 days and then treatment was stopped and tumor growth was assessed. There was regrowth of tumors from day 6 to day 17 after stopping MEKi/STAT3i treatment (Supplementary Fig. S7C), suggesting that MEKi/STAT3i suppress CSCs, but does not completely eliminate the CSC population.
Figure 6.
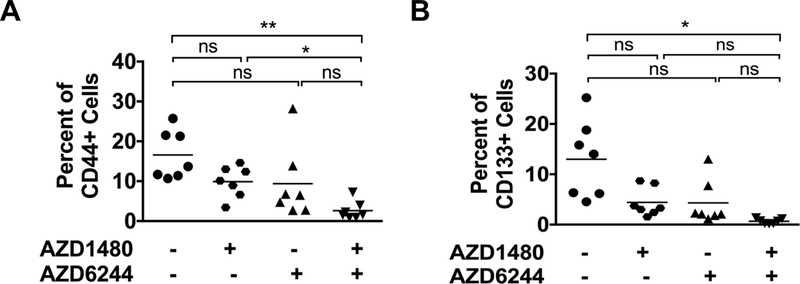
Effects of MEKi and STAT3i on cancer stem cells (CSCs) in PKT mice. Single cell suspension isolated from pancreatic tumors of PKT mice treated with AZD1480, AZD6244 or AZD1480/AZD6244 were analyzed for CSC population by flow cytometry. A, CD44+ tumor cell populations (CD45NegEpCam+CD44+) were significantly decreased in the combined MEKi/STAT3i treatment compared to STAT3i alone or the vehicle groups. B, CD133+ tumor cell populations (CD45NegEpCam+CD133+) were significantly decreased in the combination MEKi/STAT3i compared to the vehicle treated group. *, P < 0.05; **, P < 0.01; ns, P > 0.05.
Discussion
A major therapeutic challenge in PDAC is its oncogene-driven innate and acquired chemoresistance. KRAS is the key oncogenic driver of tumorigenesis and malignant progression. Direct inhibition of KRAS has yet to be achieved, however, due to the molecular characteristics of the RAS protein which currently render it “undruggable”. We and others have developed strategies to inhibit RAS indirectly by targeting its downstream effectors in the RAF/MEK/ERK signaling cascade. MEK is a key node in this axis and serves as the primary mediator for KRAS signal transduction. However, MEK inhibition (MEKi) has not achieved clinical efficacy in PDAC due to redundant signaling pathways and molecular crosstalk that induce compensatory survival signaling. Furthermore, we have also previously identified activated STAT3 signaling as a key biomarker of resistance in PDAC (20,30).
Our current study helps define the heterogeneous response and therapeutic resistance of PDAC to RAS and STAT3 pathway inhibition. We have identified a novel molecular mechanism whereby STAT3i leads to reciprocal activation of MEK-ERK signaling while MEKi results in activation of STAT3 signaling. Furthermore, we show that STAT3i results in TACE-AREG-EGFR dependent activation of ERK. These results provide a mechanistic rationale of acquired resistance to downstream RAS and STAT3 pathway inhibition. This mechanism of ERK reactivation is relevant to other studies that have shown ERK signaling reactivation plays a critical role in primary and acquired resistance to targeted therapies (8,9,31). Preclinical studies have identified distinct mechanisms by which cells acquire resistance to MEKi, including amplification of mutant BRAF (31,32), PI3K upregulation (8,33) or EGFR activation (8,34). Dual inhibition of these pathways has provided benefit in some patients (10,25,35).
STAT3 activation has been observed in a variety of human tumors (20,28) through engagement of cytokine or growth factor receptors. We have previously shown that STAT3i with gemcitabine remodels the tumor stroma and enhances in vivo drug delivery to the tumor thereby improving survival in PKT mice (20). However, STAT3i results in reciprocal activation of ERK signaling thereby preventing a sustained therapeutic response. The results of our current study support targeting two components of the EGFR-RAS-MEK-JAK/STAT3 pathway to overcome primary and acquired resistance of targeted monotherapies.
Combined MEKi/STAT3i results in sustained inhibition of both ERK and STAT3 signaling. Importantly, the combination of MEKi/STAT3i induces robust apoptosis in vitro and tumor regression in vivo in PDAC tumors. In PDAC cells, EGFR is activated upon STAT3i, perhaps as part of an initial effort by the cell to escape apoptosis or re-engage the cell cycle. Furthermore, EGFR activation is suppressed only when STAT3 is inhibited in combination with ERK. With combined MEKi/STAT3i, the cellular escape mechanisms are shut down and growth of PDAC cells is effectively suppressed.
TACE and AREG play a primary role in activating EGFR signaling and are overexpressed during PDAC progression (14). The pro-EGFR ligand, AREG, has been implicated in resistance to other therapies (36,37) and is required to induce EGFR-mediated ERK signaling (14,27,36). We have previously shown knock down of AREG prevents STAT3 and ERK activation in response to deoxycholic acid in PDAC cells (14). Previous studies have also suggested inhibiting AREG activity may be necessary to overcome resistance to EGFR-targeted therapies in NSCLC and hepatocellular carcinoma (38,39), which is consistent with recent reports showing that inhibition of AREG is associated with a better response to cetuximab therapy in colorectal cancer (40,41).
Our results show AREG levels are increased following STAT3i treatment of PDAC cells, leading to EGFR phosphorylation and subsequent re-activation of ERK and STAT3 signaling. We found STAT3i results in increased EGFR activity through TACE activation (Fig. 3A, B and C). Sera from KPC mice and human PDAC patients have significantly elevated levels of AREG (Fig. 3F), suggesting that serum AREG levels may serve as a noninvasive prognostic biomarker. Furthermore treatment with MEKi/STAT3i result in a significant decrease in serum AREG levels, suggesting its role as a biomarker of therapeutic response to EGFR, MEK or STAT3 targeted therapies. We now demonstrate that STAT3i in combination with specific blockade of the AREG-mediated EGFR receptor using cetuximab or erlotinib, completely inhibits ERK reactivation. Therefore, our study supports the potential of targeting TACE-AREG-EGFR axis as a potent approach to overcome resistance to MEKi or STAT3i.
Combined MEKi/STAT3i results in enhanced therapeutic efficacy not only through sustained inhibition of the RAS pathway-associated redundant feedback loop reactivation, but also by suppressing the TME through reducing CD44+ and CD133+ CSCs. The interplay between tumor cells and surrounding supportive cells such as fibroblasts, immune cells and CSCs, clearly indicates that targeting a single component will not result in sustained inhibition of tumor growth. Targeting multiple signaling pathways and different components of the TME is the means of overcoming either primary or acquired resistance to targeted monotherapy. This approach increases the likelihood of a sustained response by concurrently affecting multiple diverse mechanisms of action associated with cancer development and therapeutic resistance.
We have identified a novel mechanism associated with resistance to RAS-MEK-ERK pathway inhibition through activation of STAT3 signaling and resistance to STAT3 pathway inhibition resulting in TACE-AREG mediated EGFR and ERK reactivation. We provide strong evidence supporting the role of targeting two components of the AREG-EGFR-MEK-STAT3 pathway to overcome therapeutic resistance associated with RAS pathway reactivation in PDAC. In addition combined MEKi and STAT3i inhibits CSCs and maintains pancreatic integrity, providing a secondary mechanisms by which this combination therapy may enhance anti-tumor response and prevent recurrence of disease. In addition, our results suggest that serum AREG levels may serve as a key circulating prognostic biomarker of PDAC and a potential biomarker of therapeutic resistance and response to EGFR, MEK and STAT3 inhibition.
Supplementary Material
Acknowledgements
The authors thank Mr Frank Revetta, Ms Jennifer Barretta, Dr. Alexander Gaidarski III and Ms Yanhua Xiong for their technical assistance and Dr. Nilesh Kashikar (pathologist) for evaluating stained tissues.
Financial Support: This work was supported by the NIH R01 CA161976, Pancreatic Cancer Action Network (PanCan)-AACR Translational Research Grant (15–65-25-MERC), NIH T32 CA211034, James & Ether King Biomedical Research Program, Florida Department of Health 8JK08 and Sylvester Comprehensive Cancer Center to N. B. Merchant, NIH NCI R21 CA209536, American Cancer Society IRG 98–277-13 and Stanley Glaser Foundation Research Award (UM SJG 2017–24) to N.S. Nagathihalli, and American Cancer Society IRG 98–277-13 to M. VanSaun. Flow Cytometry Core Service was performed through the Sylvester Comprehensive Cancer Center (SCCC) support grant (N.B. Merchant and N. Nagathihalli).
Abbreviations:
- (STAT3)
Signal transducer and activator of transcription 3
- (MEK/MAPK)
mitogen activated protein kinase kinase
- (AREG)
amphiregulin
- (EGFR)
epidermal growth factor receptor
- (PanIN)
pancreatic intraepithelial neoplasia
- (PDAC)
pancreatic ductal adenocarcinoma
- (TME)
tumor microenvironment
Footnotes
Conflict of Interest: The authors declare they have no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29 [DOI] [PubMed] [Google Scholar]
- 2.Shibata D, Almoguera C, Forrester K, Dunitz J, Martin SE, Cosgrove MM, et al. Detection of c-K-ras mutations in fine needle aspirates from human pancreatic adenocarcinomas. Cancer Res 1990;50:1279–83 [PubMed] [Google Scholar]
- 3.Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol 2013;29:537–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013;144:1210–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007;26:3291–310 [DOI] [PubMed] [Google Scholar]
- 6.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013;23:406–20 [DOI] [PubMed] [Google Scholar]
- 7.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wicki A, Mandala M, Massi D, Taverna D, Tang H, Hemmings BA, et al. Acquired Resistance to Clinical Cancer Therapy: A Twist in Physiological Signaling. Physiol Rev 2016;96:805–29 [DOI] [PubMed] [Google Scholar]
- 9.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 2015;15:577–92 [DOI] [PubMed] [Google Scholar]
- 10.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88 [DOI] [PubMed] [Google Scholar]
- 11.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 2014;50:2072–81 [DOI] [PubMed] [Google Scholar]
- 12.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012;22:304–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013;144:1220–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, et al. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res 2014;74:2062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest 2007;117:337–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merchant NB, Voskresensky I, Rogers CM, Lafleur B, Dempsey PJ, Graves-Deal R, et al. TACE/ADAM-17: a component of the epidermal growth factor receptor axis and a promising therapeutic target in colorectal cancer. Clin Cancer Res 2008;14:1182–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009;324:1457–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015;149:1932–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dauer P, Nomura A, Saluja A, Banerjee S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017;17:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83 [DOI] [PubMed] [Google Scholar]
- 23.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50 [DOI] [PubMed] [Google Scholar]
- 24.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20:3147–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov 2015;5:960–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J 2003;22:2411–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol 2006;291:C1–10 [DOI] [PubMed] [Google Scholar]
- 28.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014;14:736–46 [DOI] [PubMed] [Google Scholar]
- 29.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013;23:839–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj NS, Washington MK, Merchant NB. Combined Blockade of Src Kinase and Epidermal Growth Factor Receptor with Gemcitabine Overcomes STAT3-Mediated Resistance of Inhibition of Pancreatic Tumor Growth. Clin Cancer Res 2011;17:483–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, Hampson M, et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal 2011;4:ra17. [DOI] [PubMed] [Google Scholar]
- 33.Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A 2009;106:18351–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov 2013;3:158–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res 2015;21:396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Q, Chiao P, Sun Y. Amphiregulin in Cancer: New Insights for Translational Medicine. Trends in Cancer 2016;2:111–3 [DOI] [PubMed] [Google Scholar]
- 37.Ishikawa N, Daigo Y, Takano A, Taniwaki M, Kato T, Hayama S, et al. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res 2005;65:9176–84 [DOI] [PubMed] [Google Scholar]
- 38.Hopfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherubl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol 2004;41:1008–16 [DOI] [PubMed] [Google Scholar]
- 39.Busser B, Sancey L, Josserand V, Niang C, Favrot MC, Coll JL, et al. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Molecular therapy : the journal of the American Society of Gene Therapy 2010;18:528–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 2009;27:5068–74 [DOI] [PubMed] [Google Scholar]
- 41.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230–7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.