

SUMMARY
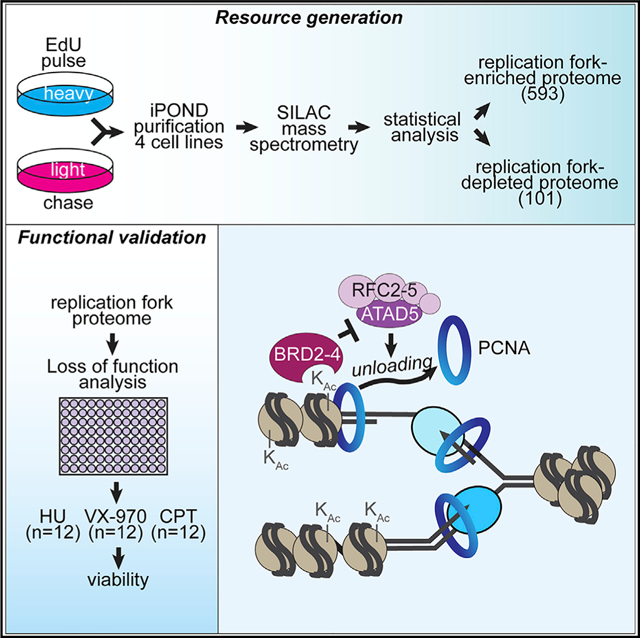
Identifying proteins that function at replication forks is essential to understanding DNA replication, chromatin assembly, and replication-coupled DNA repair mechanisms. Combining quantitative mass spectrometry in multiple cell types with stringent statistical cutoffs, we generated a high-confidence catalog of 593 proteins that are enriched at replication forks and nascent chromatin. Loss-of-function genetic analyses indicate that 85% yield phenotypes that are consistent with activities in DNA and chromatin replication or already have described functions in these processes. We illustrate the value of this resource by identifying activities of the BET family proteins BRD2, BRD3, and BRD4 in controlling DNA replication. These proteins use their extra-terminal domains to bind and inhibit the ATAD5 complex and thereby control the amount of PCNA on chromatin.
Graphical Abstract
In Brief
Wessel et al. use iPOND purifications and loss-of-function analyses to identify and characterize a replication fork and nascent chromatin proteome of 593 proteins. They demonstrate the resource’s utility by showing that the BET proteins BRD2, BRD3, and BRD4 inhibit ATAD5-dependent PCNA unloading from chromatin.
INTRODUCTION
Maintaining genome integrity requires complete and faithful replication of the genome every cell division cycle. In addition to accurate duplication, the DNA must be properly packaged into chromatin to facilitate chromosome segregation, gene expression, and many other processes. Replication stress, in the form of DNA lesions, difficult-to-replicate sequences, or conflicts with transcription, is an unavoidable threat to dividing cells and can impede fork progression. Cancer cells are particularly susceptible to drugs that interfere with DNA replication, DNA damage signaling, or DNA repair mechanisms (Forment and O’Connor, 2018). Understanding how genome stability is maintained, the diseases that arise from defects in these processes, and how to develop therapeutic intervention requires a complete description of DNA and chromatin replication proteins.
Although a eukaryotic replisome capable of performing DNA synthesis can be reconstituted with purified proteins (Yeeles et al., 2015, 2017), new replication and replication stress regulators continue to be identified, suggesting there is much we still do not know.
Isolation of proteins on nascent DNA (iPOND) identifies proteins associated with replication forks and nascent chromatin (Sirbu et al., 2011). An advantage of this approach is that it can be combined with quantitative mass spectrometry (MS) and serve as an unbiased protein discovery tool (Cortez, 2017). Multiple studies have used iPOND and related methods to characterize the replication fork and nascent chromatin proteome (Alabert et al., 2014; Aranda et al., 2014; Dungrawala et al., 2015; Lecona et al., 2016; Lopez-Contreras et al., 2013; Lossaint et al., 2013; Sirbu et al., 2013). Several reported a list of replication-fork-associated proteins in a single cell line with the two largest datasets using stable isotope labeling by amino acids in cell culture (SILAC)-MS methodologies (Alabert et al., 2014; Dungrawala et al., 2015). However, of the hundreds of proteins identified in these two studies, only 67 are shared, raising significant concerns about the accuracy and reliability of these datasets.
We aimed to generate a high-confidence catalog of the replication fork and nascent chromatin proteome that can serve as a robust resource for genome maintenance studies. To this end, we used iPOND combined with SILAC-MS to identify the nascent-DNA-associated proteomes of multiple cell lines and performed loss-of-function genetic analyses to validate the resource. We report the identification of 593 proteins that are enriched at replication forks or nascent chromatin, of which 85% have known activities or yield loss-of-function phenotypes consistent with a function in DNA and chromatin replication or replication stress responses. These proteins include the core replisome proteins that are present at every fork and accessory factors that may be utilized in only a subset of forks experiencing specific replication challenges. Furthermore, we utilize this resource to identify the bromodomain and extra-terminal domain (BET) family proteins BRD2, BRD3, and BRD4 as important regulators of DNA replication by acting as inhibitors of the ATAD5-replication-factor C-like PCNA unloading complex.
RESULTS
Identification of a Nascent-DNA-Associated Proteome
To identify proteins localized to replication forks and nascent chromatin, we performed iPOND coupled with SILAC-MS. Cells treated with 10 min of 5-ethynyl-2′-deoxyuridine (EdU) were compared to cells treated with 10 min of EdU followed by a 1-h chase in media without EdU (Figure 1A). The result is a ratio of the relative abundance of each protein purified with nascent DNA compared to mature chromatin. Ten experiments in four separate cell lines (HCT-116, HeLa, HEK293T, and RPE-hTERT) were combined with three previously published datasets in HEK293T cells by using the same methodology (Dungrawala et al., 2015). The median Pearson correlation coefficient for pairwise comparisons of all experiments across cell lines is 0.78. An average of approximately 1,600 proteins per experiment were quantified, for a total of 3,826 individual proteins (Table S1). Of these, 3,051 were observed in at least two purifications (Figure 1B). By using a 5% false discovery rate (FDR) adjusted two-tailed t test, we found 713 of these proteins are significantly enriched on nascent DNA (Table S2A). A total of 132 proteins are significantly enriched in the chase sample, which indicates depletion from sites of DNA replication compared to the mature chromatin (Table S2B). Further filtering of this list by using an absolute median log2 ratio ≥ 0.5 cutoff results in a list of 593 proteins enriched on nascent DNA and 101 proteins enriched in matured chromatin (Tables S3A and S3B).
Figure 1. 593 Proteins Are Significantly Enriched at Replication Forks.

(A) Schematic for identifying proteins enriched at nascent DNA. Cells treated with 10 min of EdU were compared to cells treated with 10 min of EdU followed by a 1-h chase in thymidine by iPOND-SI-LAC-MS.
(B) A total of 3,826 proteins were quantified in at least one experiment and 3,051 in at least two. A total of 713 of the 3,051 proteins were significantly enriched by a 5% FDR-adjusted t test, and applying an abundance ratio cutoff of log2 ≥ 0.5 yielded 593 proteins.
(C) Venn diagram depicting how many of the 593 proteins were identified with a median log2 ratio ≥ 0.5 in each cell type.
(D) Proteins were assigned to categories based on median log2 abundance ratios.
See also Figure S1.
A Gene Ontology (GO) analysis indicates that nuclear-envelope-organizing proteins, including components of the nuclear pore and nuclear lamina, are more abundant on mature chromatin compared to nascent DNA (Table S4A). These complexes make chromosome attachments to help organize the nucleus and regulate gene expression (Ibarra and Hetzer, 2015; van Steensel and Belmont, 2017). Furthermore, nuclear envelope proteins control genome stability by generating a favorable environment for DNA repair (Géli and Lisby, 2015). Their identification as enriched in the “chase” sample compared to nascent DNA indicates the chromosome-nuclear envelope attachments are released during replication and then re-form significantly after DNA synthesis is completed (>10 min), which is consistent with the observation that active genes lose peripheral nuclear localization for approximately 30 min after the onset of DNA replication in budding yeast (Brickner and Brickner, 2011).
Filtering the 593 nascent-DNA-associated proteins for those that have an enrichment ratio of median log2 ≥ 0.5 in each individual cell type revealed that 578 (97.5%) were identified in at least two cell types and 462 (77.9%) were identified in at least three (Figure 1C). The RPE-hTERT datasets had significantly fewer proteins enriched at forks than the other three cell lines (254 versus 508 in HCT116, 522 in HeLa, and 548 in HEK293T cells). The difference between the RPE-hTERT and the transformed cell lines may be due to the reduced number of replicating RPE-hTERT cells that were analyzed and reduced overall numbers of replication forks captured. Consistent with this interpretation, fewer total proteins were quantitated, and the average size of the fork proteins identified in the RPE-hTERT cells was 93 kDa versus 76 kDa for the other cell types. Larger proteins are easier to quantitate by MS because they produce more tryptic peptides. From this analysis, we conclude that most nascent-DNA-associated proteins are shared among divergent human cell types.
A notable exception to this conclusion is p53, which is enriched at replication forks specifically in HEK293T cells. p53 is stabilized in HEK293T cells due to E1A and T-antigen expression, whereas it is degraded in HeLa cells by the papillomavirus E6 protein and expressed at low levels in undamaged HCT116 and RPE-hTERT cells. Consistent with our data, p53 has previously been identified at replication forks likely due to its interaction with Replication Protein A (RPA) (Dutta et al., 1993; Li and Botchan, 1993).
We next compared the list of 593 proteins to the previously published datasets of fork/nascent-chromatin-associated proteins. A total of 79% (172 proteins) of the previous iPOND-SI- LAC-MS dataset (Dungrawala et al., 2015), 26% (112 proteins) of the NCC-SILAC-MS dataset (Alabert et al., 2014), 49% (41 proteins) of the Sirbu et al. (2013) iPOND-spectral counting dataset, and 92% (44 proteins) of the Lopez-Contreras et al. (2013) iPOND dataset are shared with our nascent-DNA-associated proteome (Figure S1A). However, our dataset contains 355 proteins that are not in any of the previous studies. They include several proteins with well-described functions in DNA replication, repair, or damage responses including ATM, TRAIP, FANCJ, and TOP2B.
GO Analysis and Identification of Protein Complexes
GO analysis of the 593 nascent DNA proteins finds enrichment for many expected biological pathways, including DNA replication and repair and response to DNA damage (Figure S1B; Table S4B). We next stratified our data into three enrichment categories based on median log2 abundance ratios to provide quantitative information about the amount of enrichment and a confidence factor that each protein is localized to forks or nascent chromatin (Figure 1D).
Category 1 includes 236 (40%) proteins that had a median SILAC-quantified enrichment on nascent DNA of at least 2-fold. This category is highly enriched for DNA replication, repair, and damage response proteins (Figure S1C; Table S4C). Category 2 proteins, with median log2 ratios between 0.75 and 1, are enriched for chromatin and chromosome organization (Figure S1D; Table S4D), whereas those in category 3 are enriched for RNA metabolic pathways (Figure S1E; Table S4E). This stratification based on fold enrichment likely reflects functions of category 2 and 3 proteins in gene expression, placing them both in nascent and mature chromatin.
Proteins that are part of a single complex should all exhibit similar changes in nascent DNA abundance in response to any experimental perturbation. Thus, in our previous iPOND analyses, we found that unsupervised hierarchical clustering of multiple iPOND datasets is sufficient to identify new protein complexes (Dungrawala et al., 2015). The more experimental samples, the stronger this clustering should be in defining physical and functional relationships. Therefore, we performed an unsupervised hierarchical clustering analysis on the 593 proteins enriched on nascent DNA by using data from a total of 71 iPOND-SILAC-MS experiments from this study and three prior studies (Dungrawala et al., 2015, 2017; Mohni et al., 2019). This analysis identifies many well-characterized protein complexes, including the majority of subcomplexes of the replisome, RPA-linked replication stress response complexes, and replication-coupled DNA repair complexes (Figure 2). For example, the ATR/ATRIP/TOPBP1 signaling, MRE11/RAD50/NBN nuclease, mismatch repair, RNaseH2 ribonucleotide removal, Cohesin, and Condensin II complexes all are readily identified (Figure 2).
Figure 2. Hierarchical Clustering Identifies Protein Complexes Enriched on Nascent DNA.

Unsupervised hierarchical clustering of the 593 proteins enriched on nascent DNA identifies protein complexes. Selected portions of the heatmap that depict the abundance of proteins (row) in each experiment (column) with black being unchanged are shown. Red and green indicate an increase or decrease in abundance and gray indicates the protein was not observed. Selected complexes are diagramed. Subunits of the complexes in gray were not significantly enriched at forks or were not observed; those outlined with a solid line are category 1, dotted outline are category 2, and no outline are category 3.
See also Figure S2.
The clustering analysis will not be useful for proteins that are either part of more than one complex at forks or are transient interactors that can function independently of other proteins. For example, RBBP4 is a subunit in multiple chromatin regulatory complexes that behave differently across these datasets, so it does not cluster with any of them. Because this clustering analysis can be a powerful tool for generating hypotheses about uncharacterized proteins within the dataset, we included a searchable table of the Euclidean distances between each protein in the 71 published datasets in Table S5.
Surprisingly, the ORC2–6 replication initiation complex but not ORC1 is enriched on nascent DNA compared to mature chromatin (Figure 2; Table S3). ORC1 is degraded in S-phase cells as part of a mechanism to prevent re-licensing of origins, explaining its absence (Li and DePamphilis, 2002; Méndez et al., 2002). The enrichment of ORC2–6 on nascent DNA compared to bulk chromatin suggests that it rapidly rebinds to DNA after replication, but then some proportion of the complexes must be removed over time, perhaps as a consequence of chromatin deposition and maturation.
Many complexes involved in chromatin replication through histone deposition, modification, and nucleosome positioning are enriched on nascent DNA. Notably, the entire NuRD histone de-acetylation complex; the G9a/GLP histone methyltransferase complex; DNMT1 DNA methyltransferase complex; nucleosome remodeling factor (NURF), remodeling and spacing factor (RSF), and WSTF-ISWI (WICH) chromatin remodeling complexes; and the ASF1, FACT, CAF1, and MMS22-TONSL histone chaperone complexes are present (Figures 2 and S2; Table S3). Although some SWI/SNF and INO80 complex subunits were observed, many had high variability or did not have median abundance ratios above 0.5 and, therefore, did not meet our stringent cutoffs.
Seven subunits of the 14-subunit integrator complex (INT) were also enriched on nascent DNA (Figure S2A). Furthermore, four additional subunits had median log2 enrichment ratios greater than 0.75 (INTS2 = 1.14, INTS5 = 1.13, INTS6 = 0.78, INTS8 = 1.71) but were excluded because variability in the measurements increased the false discovery p value above our statistical threshold. INTS3 also is part of the SOSS (sensor of single-stranded DNA) complex (Huang et al., 2009), but other SOSS subunits were not identified as enriched. INT is a U-rich small nuclear RNA processing factor that may have additional functions in transcription initiation and termination. Specifically, INT is needed for RNAPII to escape pausing at mRNA-coding genes and progress into productive elongation (Baillat and Wagner, 2015). This function is tied to an interaction with the NELF (negative elongation factor) complex. Intriguingly, two of the four NELF complex proteins are also enriched on nascent DNA (Figure S2A), and the other two subunits had median log2 enrichment scores greater than 0.7 (NELFB = 0.86, NELF = 0.72) but were excluded because the measurements did not meet the statistical threshold. The INT and NELF complexes may have a function in genome maintenance because their presence at transcriptional pause sites has been linked to an increase in genome instability at these open regions of chromatin. These regions have a propensity to form R-loop structures that can generate replication stress (Baillat and Wagner, 2015). The DRB-sensitivity-inducing factor (DSIF) complex of SPT4 and SPT5 also regulates transcriptional pausing and is enriched at nascent DNA (Yamaguchi et al., 2013). Furthermore, some RNA polymerase III transcription events can stall replication forks (Deshpande and Newlon, 1996; Ivessa et al., 2003), and five of the six subunits of the transcription factor IIIC (TFIIIC) RNA polymerase III pre-initiation complex are enriched on nascent DNA. This TFIIIC complex is also observable by hierarchical clustering (Figure 2).
Several complexes involved in ubiquitination, sumoylation, and neddylation are enriched on nascent DNA. All of these post-translational modifications regulate replication and replication stress responses. For example, the CUL4A/B ubiquitin ligase complexes are involved in cell cycle control, DNA damage response, and translesion synthesis (O’Connell and Harper, 2007). Likewise, UBC9/SUMO2 complexes, which are involved in responding to DNA damage or stress, are enriched (Moschos and Mo, 2006). Other proteins we identified include SKP1 and CUL1, core components of SCF (SKP1-CUL1-F-box protein) complexes, which mediate cell cycle progression and other processes depending on the specific F-box protein. For example, LRR1 mediates ubiquitylation and unloading of the CMG helicase during termination and is enriched with nascent DNA (Dewar et al., 2017; Sonneville et al., 2017). Another important regulator of these cullin complexes, CAND1, is also present on nascent DNA. Furthermore, neddylation is a required modification for Cullin activity, and 5 of the 9 subunits of the COP9 signalosome, which mediates the deneddylation (Chung and Dellaire, 2015), were identified (Figure S2A). Of the four subunits not identified, three were enriched (COPS7A = 0.95, COPS3 = 0.77, COPS8 = 0.38) but were not significant due to variability across experiments. The final subunit, COPS9, is 6.2 kDa and was not observed. The missing subunits of the COPS9, INT, and NELF complexes that were excluded largely based on statistical cutoffs illustrate that the 593-protein list is not yet a complete inventory of the proteins that act at nascent DNA.
Assessment of False Negatives
Our 593-protein dataset is missing five proteins thought to be core components of the replisome (POLD4, DNA2, POLE3, POLE4, and TIPIN). Of these, four had median log2 ratios ≥ 0.5 but did not meet the statistical threshold (Table S1). POLD4, which is approximately 12 kDa, was not observed in any of the experiments. Thus, these proteins and several others that are part of category 1 nascent DNA protein complexes shown in gray in Figures 2 and S2A, such as RAD1, HUS1, RMI1, RMI2, and CTF8, are all likely false negatives. In many of these cases, the reason these proteins did not meet the significant enrichment cutoff is apparent when examining the values in Table S1. For example, the median log2 enrichment for TIPIN is 1.6, making it a very strong “hit” that would have gone into category 1. However, of the seven values in the dataset, there is one outlier that introduced variation increasing the FDR-adjusted p value. This issue is particularly noticeable when a protein is small and only observed by the mass spectrometer a few times. For example, POLE4, with a mass of only 12 kDa, was only observed in two datasets, with enrichment values of 2.07 and 2.16, placing it in the top 80 enriched proteins. But even this small amount of variability was sufficient to increase the FDR-adjusted p value above 0.05 because it was only detected twice. To assist in identifying these types of cases, we cataloged 850 proteins with a median log2 ratio ≥ 0.5 that could be possible false negatives (Table S6). Proteins in this list that have previously been implicated in DNA replication or replication stress responses include RTEL1, BOD1L, ETAA1, RAD17, FANCC, SOSB1, DONSON, REV3L, RNF4, FANCL, POLE4, FAAP100, TRESLIN, TIPIN, PMS2, FAN1, RAD1, FANCB, RMI1, DNA2, RADX, POLE3, POLK, FANCE, WRN, HUS1, LIG4, TDP2, CHTF8, FANCM, XRCC4, CHK1, MBD2, BRCA1, and RIF1.
Notably, most of the proteins listed above are recruited to damaged or stalled forks and many were identified in previous iPOND experiments that utilized hydroxyurea to stall replication forks (Dungrawala et al., 2015; Higgs et al., 2015; Reynolds et al., 2017). Unsupervised hierarchical clustering of these 850 possible false negatives and the 593 proteins significantly enriched on nascent DNA with additional published iPOND datasets is useful in evaluating these proteins and generating hypotheses. This analysis clearly identifies proteins like RADX (cxorf57), ETAA1, WRN, and RMI1 as functionally related to RPA and stress responses, as previously shown (Figure S2B). It also identifies RAD51C correlating with XRCC3, WRNIP1, and FANCA, as well as a cluster showing SIRT6 correlates with ATM (Figure S2B). ATM and SIRT6 were previously shown to function together at double-strand breaks (Atsumi et al., 2015). In addition, there are multiple additional candidate false negatives that behave similarly to known replication fork and replication stress response proteins. These include OTX1 that correlates with UNG, CCDC94 and C1orff112 that correlate with SLX4, TSSC4 that correlates with GINS4 and POLE2, VPS36 that correlates with PNKP and CHD1L, and SUB1 that correlates with DNA-PKcs (Figure S2B).
Functional Analyses of the Replication Fork Proteome
To increase the value of the 593-protein fork proteome dataset, we assessed loss-of-function phenotypes in U2OS cells, a cell line that is often utilized in the replication and DNA damage response fields. We utilized four individual small interfering RNAs (siRNAs) per gene to knock down expression and then measured cell growth, sensitivity to the fork stalling agent hydroxyurea (HU), sensitivity to replication-associated DNA damage with camptothecin (CPT), and dependency on the replication checkpoint kinase ATR for growth and viability (Figure 3A). These conditions were selected to identify proteins important for cell division and viability, needed to overcome acute replication stress and DNA damage, or that prevent accumulation of replication stress that creates a dependency on replication stress responses. Twelve biological replicates were performed for untreated, ATR-inhibited, and CPT-treated samples. Six replicates of low-dose (0.2 mM) and six replicates of high-dose (2 mM) HU treatments were also performed. Of the 593 tested, 34 (5.7%) gene products were essential for viability, defined as having at least 2 of the 4 siRNAs yield less than 5% viability. Category 3 is enriched for these compared to the other two categories and contains all 9 with 4/4 siRNAs yielding less than 5% viability (Figures 3 and S3).
Figure 3. Loss-of-Function Screens Identify Proteins Important for the Response to Replication Stress.

(A) Schematic for performing loss-of-function screens. Genes were considered hits if 2 or more of the 4 siRNAs yielded the same effect.
(B) Number of gene knockdowns resulting in viability less than 5% of control.
(C) Numbers of gene knockdowns resulting in resistance or sensitivity to ATRi, CPT, and HU.
(D) Heatmaps of the average viability of cells transfected with each siRNA as compared to a control siRNA in the absence of added replication stress. Each row is a single gene and each column is one of four siRNAs.
(E) Average viability of cells transfected with each siRNA and treated with the indicated drugs as compared to a control siRNA.
See also Figure S3.
Knockdown of 316 of the 593 gene products caused significant changes in cell viability in response to at least one drug when compared to untreated cells, with a requirement that at least two of the four individual siRNAs yield an FDR-adjusted significant difference (Figures 3 and 4A; Table S7). Of the 316, 264 knockdowns resulted in sensitivity to at least one drug and 90 caused resistance (Table S8). There were 38 instances where knockdown caused resistance in one or more conditions and sensitivity in another. The 316 gene knockdowns with changes in drug sensitivity account for 53% of the dataset. Knockdown of 28 genes resulted in hyper-sensitivity to all three drugs (Figures 4B and 4C). These include multiple subunits of the replication checkpoint pathway, including ATR, ATRIP, RAD9A, and TIMELESS; and several replisome subunits, including POLD1, RFC5, and POLA2.
Figure 4. 85% of the Replication Fork Proteome Validates with Loss-of-Function Phenotypes or Known Roles in DNA Replication and Repair.

(A) Pie chart depicting phenotypes observed from the drug sensitivity analyses and existing literature. Loss-of-function (LOF) phenotype includes sensitivity, resistance, and toxicity. Literature indicates published evidence for a function in DNA replication, repair, or related process.
(B) Venn diagram comparing proteins that when depleted result in sensitivity to ATRi, CPT, and HU. Low- and high-dose HU lists are combined for this analysis.
(C) List of the 28 proteins with loss of function resulting in sensitivity to ATRi, CPT, and HU.
See also Figure S3.
Interestingly, despite apparent functional differences between enrichment categories based on the GO analysis, the percentage of genes with a loss-of-function phenotype is relatively consistent across all three because 65% of category 1, 50% of category 2, and 61% of category 3 genes generated phenotypes in these screens (Figure S3). GESS (genome-wide enrichment of seed sequences) (Sigoillot et al., 2012) analysis did not detect off-targeted transcripts from the screen data. In total, knockdown of 350 of the 593 nascent-DNA proteins (59%) generated a phenotype (resistance, sensitivity, or toxicity) in at least one of the screened conditions (Figures 3 and 4; Table S9A).
A GO term analysis of the remaining 243 proteins identified an additional 77 with already-documented functions in DNA repair, replication, chromatin maturation, or related processes (Table S9B). A total of 45 of the remaining 166 proteins were identified in other nascent DNA proteomes (excluding Dungrawala et al., 2015 as those data were used in the generation of this dataset) or in PCNA interaction datasets (Table S9C) (Alabert et al., 2014; Lopez-Contreras et al., 2013; Sirbu et al., 2013; Srivastava et al., 2018). An additional 32 do not have relevant GO terms associated with them but have reported activities or binding partners linked with nascent DNA-related processes (Table S9D). This leaves 89 proteins of the 593-protein dataset that could be considered possible false positives (Table S9E). However, this group includes subunits of the INT, TFIIIC2 RNA polymerase III pre-initiation, and NELF complexes. Because other subunits of these complexes exhibited phenotypes consistent with activities in DNA replication or repair, their lack of loss-of-function phenotypes could be due to insufficient siRNA-mediated inactivation. Thus, the actual number of true positives is likely higher than 504. In summary, our functional validation tests combined with the published literature indicate that at least 504 of the 593 proteins (85%) we identified as enriched on nascent DNA likely do function to promote faithful DNA and/or chromatin replication. Thus, this is a robust inventory of proteins that act at replication forks or on the newly forming chromatin.
BRD2, BRD3, and BRD4 Are Enriched on Nascent Chromatin and Regulate PCNA Unloading
To provide a proof of principle that this proteomic resource can be useful to identify regulators of DNA replication, we examined the 28 genes whose silencing caused hypersensitivity to ATRi, HU, and CPT. One of these is the BET family protein BRD3. BRD3 is one of several proteins including BRD2 and BRD4 that contain highly similar BET domains and function as transcriptional regulators. For example, BRD4 is needed to mediate c-Myc-dependent transcription (Delmore et al., 2011). Therefore, BET domain inhibitors such as JQ1 are being developed as cancer therapies. These inhibitors do not distinguish between BRD2, BRD3, and BRD4, which have highly similar bromo and extra-terminal (ET) domains.
BRD2, BRD3, and BRD4 are all significantly enriched on nascent DNA (Table S3). There are no reported functions for BRD3 in DNA replication, but both BRD2 and BRD4 were reported to regulate DNA replication or DNA damage responses previously (Bowry et al., 2018; Gursoy-Yuzugullu et al., 2017; Li et al., 2018; Maruyama et al., 2002; Sansam et al., 2018; Zhang et al., 2018). Loss of BRD4, but not BRD2 or BRD3, causes replication stress through increased RNA synthesis (Bowry et al., 2018). BRD4 also interacts with the pre-replication complex protein CDC6 and is reported to affect replication checkpoint signaling (Zhang et al., 2018). BRD2 and BRD4 are reported to interact with the replication factor TICRR and control replication initiation (Sansam et al., 2018). Additionally, BRD2 is recruited to double-strand breaks to regulate repair (Gursoy-Yuzugullu et al., 2017). JQ1 has been reported to slow DNA synthesis and cause hypersensitivity to ATR inhibitors (Bowry et al., 2018; Da Costa et al., 2013; Muralidharan et al., 2016, 2017; Pericole et al., 2019), which we confirmed (Figure S4A).
Although depletion of BRD3 resulted in hyper-sensitivity to ATR inhibition, HU, and CPT, silencing BRD2 with siRNA did not yield any hypersensitive phenotypes (Table S7). Silencing BRD4 caused sensitivity to CPT and also significantly reduced cellular viability (Tables S7 and S8). BRD3 knockdown caused ATR inhibitor hyper-sensitivity over a large range of doses with no overall change in cell cycle distribution (Figures 5A and S4B). Furthermore, BRD3 knockdown caused a significant decrease in replication fork elongation rates as compared to controls in both U2OS and RPE-hTERT cells (Figures 5C and 5D). We did not observe an elongation defect with BRD2 depletion in RPE-hTERT cells but did in U2OS cells (Figures 5D and 5E). BRD4 depletion also has been reported to cause slower replication elongation, which can be rescued by treatment with the transcription inhibitor DRB (Bowry et al., 2018). In contrast, DRB is unable to rescue the elongation defect associated with BRD3 loss (Figure 5F).
Figure 5. BRD2, 3, and 4 Are Replication Fork Proteins Required for Fork Progression.

(A) U2OS cells were transfected with non-targeting (siNT) or BRD3 siRNAs, treated with increasing concentrations of ATR inhibitor (VX-970) and measured for viability after 72 h. Mean ± SD, n = 3, two-way ANOVA with Dunnett post-test.
(B) Immunoblot analysis to verify siRNA knockdown.
(C–F) Fork speeds were measured in (C, E, and F) U2OS or (D) RPE-hTERT cells transfected with siRNAs and labeled with IdU and CldU as indicated. Mean, ANOVA with Dunnett post-test (C, D, and F) or two-tailed t test (E). In (F) cells were pre-incubated with DRB for 1 h prior to labeling and maintained during labeling. siRNA pools were used in (D) and (E).
See also Figure S4.
To better understand how BRD3 may be functioning at replication forks, we analyzed BRD3-interacting proteins by MS. Consistent with previous BRD3 protein interaction studies (Lambert et al., 2019; Wai et al., 2018), we observed an interaction with BRD9, transcription initiation factors (TAF1–10), and chromatin remodeling complexes (INO80 and Swi/Snf complexes) (Figure S4C). However, these interactions are un-likely to explain its function at replication forks because these proteins are not enriched on nascent DNA. The BRD3 immunoprecipitates also contained all the subunits of the ATAD5 replication factor C-like complex, including ATAD5, RFC2, RFC3, RFC4, and RFC5. This ATAD5 complex unloads the PCNA sliding clamp from DNA (Bellaoui et al., 2003; Kang et al., 2019; Lee et al., 2013, 2010). We did not observe an interaction with RFC1 that forms the RFC1–5 PCNA loading complex (Figures 6A and 6B). ATAD5 immunoprecipitates contain BRD2, BRD3, and BRD4, indicating that ATAD5 can interact with all three proteins (Figure 6C).
Figure 6. BET Proteins Interact with the ATAD5/RFC2–5 Complex.

(A) FLAG-HA-BRD3 was immunopurified from 293T cells and interacting proteins identified by mass spectrometry. The table indicates the number of peptides identified for each protein. The control is 293T cells that do not express any tagged protein.
(B and C) FLAG-HA-BRD3 (B) or FLAG-ATAD5 (C) was immunopurified from 293T cells and immunoblotted as indicated.
(D) Schematic of BET family proteins and fragments utilized. The ability of each GFP-HA-NLS-tagged protein to interact with ATAD5 as measured in (C) and (E) is indicated.
(E and F) GFP-HA-NLS-tagged fragments (E) or constructs (F) were immunopurified from 293T cells and immunoblotted as indicated.
See also Figure S4.
To map the interaction region on BRD3 for ATAD5, we tested the ability of several BRD3 fragments to co-immunoprecipitate ATAD5 (Figures 6D and 6E). All fragments containing the ET domain formed a complex with ATAD5, whereas a fragment lacking the ET domain did not (Figure 6E). The ET domains of BRD2, BRD3, and BRD4 are highly similar, and the ET domains of BRD2 and BRD4 also interact with ATAD5 (Figure 6E). To further test this interaction, we mutated conserved residues within the BRD3 ET domain that are homologous to BRD4 residues important for binding to interacting partners (Crowe et al., 2016). Both the BRD3 V596S/I614S and BRD3 E615R/E617R ET proteins failed to co-immunoprecipitate ATAD5 (Figure 6F).
To test the idea that the interaction between BRD3 and ATAD5 regulates the ability of ATAD5 to unload PCNA, we measured PCNA levels on chromatin in S-phase cells. In chromatin fractionation experiments, we were unable to detect a change of PCNA on chromatin simply by silencing BRD3 (Figures 7A–7C). However, BRD3 inactivation in cells that have partially compromised ATAD5 function revealed significant differences. Depletion of ATAD5 dramatically increases the amount of PCNA on chromatin even at low concentrations of ATAD5 siRNA in cells containing functional BRD3. In cells where BRD3 is inactivated, PCNA levels on chromatin were significantly lower at intermediate levels of ATAD5 siRNA (Figures 7A–7C). As less PCNA is indicative of more ATAD5 activity, this suggests that BRD3 is acting as an inhibitor of PCNA unloading. This phenotype is not dependent on fork movement, as similar results are obtained in the presence of 2 mM HU (Figures 7D–7F).
Figure 7. BET Proteins Regulate the Levels of PCNA on Chromatin.

(A–F) The soluble and chromatin fractions of S-phase synchronized U2OS cells transfected with the indicated siRNAs were immunoblotted for PCNA and H2B.
(A) Representative immunoblot from untreated cells transfected with the indicated siRNAs.
(B) Quantification of the percent of PCNA in the chromatin fraction in (A).
(C) Quantification of three independent experiments. Mean ± SD, two-way ANOVA corrected for multiple comparisons using the Bonferroni method.
(D–F) Repeat of experiments as in (A)–(C) but with 2-mM HU treatment for 1 h prior to harvesting cells.
(G) Intensity of detergent-resistant PCNA immunofluorescence in S-phase cells transfected with the indicated siRNA pools. Number of cells analyzed in each column is indicated (representative of n = 6 replicates, mean indicated by bar, ANOVA with Dunnett post-test).
(H) Immunoblot analysis to verify siRNA knockdown efficiencies.
(I and J) Intensity of detergent-resistant PCNA in S-phase cells transfected with plasmids encoding the indicated (I) fragments or (J) mutants. Only GFP-positive cells were included in the analysis. Number of cells analyzed in each column is indicated (n = 3 technical replicates, representative of n = 2 biological replicates, mean indicated by bar, ANOVA with Dunnett post-test).
See also Figures S4 and S5.
To further test this hypothesis and investigate whether BRD2 and BRD4 have similar activities, we used a more quantitative assay measuring detergent-resistant PCNA intensity by immunofluorescence. In this assay, we were able to observe that silencing BRD2, BRD3, or BRD4 resulted in a small but significant decrease in PCNA levels on chromatin in S-phase cells compared to controls (Figures 7G and S4F–S4H). Conversely, overexpression of BRD3 proteins that contain the ET domain or the wild-type BRD2, BRD3, or BRD4 ET domain alone causes retention of PCNA on S-phase chromatin (Figures 7I–7J and S5). In contrast, fragments and mutants of BRD3 that cannot interact with ATAD5 do not alter PCNA levels, as would be predicted by a model in which these BET proteins inhibit ATAD5-dependent PCNA unloading.
Together, these data provide validation to our replication fork proteome resource and indicate that the BRD2, BRD3, and BRD4 proteins have an important function at replication forks to regulate PCNA. All three proteins likely have additional functions that promote efficient DNA replication because BRD3 inactivation yields strong drug hypersensitivity phenotypes distinct from BRD2 and BRD4, and yet all display similar PCNA regulation phenotypes. Nonetheless, these results demonstrate that the replication fork proteome dataset will be a useful resource for investigators interested in DNA and chromatin replication.
DISCUSSION
By using iPOND-SILAC-MS, we identified 593 proteins that are significantly enriched at replication forks or nascent DNA in human cells. We coupled this dataset with loss-of-function analyses and identified 350 proteins that yielded phenotypes consistent with activities at replication forks or nascent chromatin. When our dataset is combined with the existing literature, we find evidence that 85% of the identified proteins likely function in DNA replication, repair, or other related processes. These data provide a functionally validated and statistically robust resource of the nascent DNA proteome. Most of these proteins are likely only present at a subset of replication forks under specific conditions. Thus, rather than view this fork proteome dataset as defining a static replisome, it should be interpreted as identifying components of a dynamic machine with subunits that come and go as needed to ensure faithful DNA and chromatin replication.
In addition to the majority of the core replisome subunits, we identified many DNA repair proteins and proteins involved in DNA damage signaling (Figures 2 and S2). DNA repair complexes are enriched on nascent DNA even though no added replication stress agents were added to the cells. It is possible that EdU itself may be recognized as a damaged DNA base. However, that would not explain why the repair proteins would not be equally recruited to mature chromatin containing EdU. More likely, the presence of these proteins reflects the substantial amounts of replication stress and DNA damage always present in cells, especially when cultured in vitro. Thus, repair and replication stress responses serve housekeeping functions during replication (Cox et al., 2000).
We find that chromatin modifying and positioning complexes are highly enriched on nascent DNA, which is consistent with a need to remove nucleosomes ahead of the fork and properly re-chromatinize the newly replicated DNA. There are also complexes on nascent DNA that are involved in regulating RNA polymerase II (Pol II) pausing, including the INT and NELF complexes. These complexes may be present at nascent DNA to limit R-loop formation and the resulting replication stress. It is also possible that these complexes are traveling with the fork to help any RNA Pol II paused ahead of the fork escape and prevent collisions with the replisome.
A comparison of our resource to previous studies finds 355 proteins unique to our dataset. The differing methods and thresholds utilized in each study can impact the resulting data. Despite the large percentage of proteins not previously identified, 266 (75%) of the 355 proteins unique to this resource were found to have loss-of-function phenotypes or reports in the literature consistent with activities in DNA replication or repair.
Interestingly, despite differences in functional enrichment across categories, we observed a similar percentage of proteins causing loss-of-function phenotypes in each (Figures 3 and S3). The differences in enrichment are likely due to the category 2 and 3 proteins having important functions in mature chromatin in addition to activities on nascent DNA. This fits with our observation that the lower abundance categories are more enriched for chromatin- and histone-related processes and RNA metabolic pathways.
A total of 90 gene depletions resulted in increased resistance to one or more of the drugs tested. This accounts for a much lower percentage of the dataset than those causing sensitivity. Some of these proteins may have specific functions in making cells more sensitive to DNA replication stress or damage (therefore, their loss would make them resistant). For example, CUL4A sets a DNA repair threshold that can be increased by inactivating this ubiquitin ligase (Liu et al., 2009). Inactivating other proteins may decrease the number of actively replicating cells. The effect of the drugs is largely dependent on DNA replication, so reductions in S-phase populations would make them appear resistant in a short-term viability or proliferation assay. A GO analysis of this set of genes indicates enrichment for DNA metabolic processes and cell cycle regulation, consistent with these explanations.
One of the 28 genes whose silencing caused hypersensitivity to ATRi, HU, and CPT encodes the BET family protein BRD3. The BET family proteins BRD2, BRD3, and BRD4 are all significantly enriched on nascent DNA (Table S3). All bind H4K5Ac and H4K12Ac (LeRoy et al., 2008; Wu et al., 2006), which are marks associated with newly synthesized H4, and this may serve as a mechanism to recruit these BRD proteins to nascent chromatin. In addition to its function as a transcriptional regulator, BRD4 has recently been implicated in the DNA damage response, replication, and the repair of DNA double-strand breaks (Bowry et al., 2018; Li et al., 2018; Zhang et al., 2018). Our results suggest that these proteins act on nascent chromatin to inhibit PCNA unloading by binding the ATAD5/RFC2–5 complex. PCNA is a regulator of DNA polymerases, Okazaki fragment maturation, DNA repair, and chromatin deposition (Moldovan et al., 2007). Thus, BRD protein-dependent regulation could be a means of coupling H4 de-acetylation with PCNA unloading. As the histone acetylation levels on nascent chromatin decrease, BRD proteins would be released and the ATAD5/RFC complex would unload any remaining PCNA to recycle it for continued replication elongation (Figure S5). These proteins also have multiple additional functions that could contribute to proper DNA and chromatin replication. For example, BRD3 regulates gene expression and ribosome biogenesis that could indirectly impact DNA replication (Lambert et al., 2019).
In agreement with our data, protein interaction screens previously suggested interactions between BRD2 and BRD4 with replication factor C subunits (Lambert et al., 2019; Maruyama et al., 2002; Rahman et al., 2011). Cell type differences in expression, differences in silencing efficiency, and additional functions beyond PCNA regulation could contribute to the differences we observed in the loss-of-function assays for these proteins. This complexity means that the mechanism of action of non-selective BET inhibitors is likely multi-factorial and complicates their development as anti-cancer therapeutics.
In summary, our study provides a robust and functionally validated resource of the nascent DNA proteome that will be useful to identify and possibly predict functions for proteins needed for DNA replication, DNA repair, and chromatin maturation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu). Plasmids generated in this study are available without restriction upon request. No other unique reagents were generated.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
U2OS and RPE-hTERT cells were cultured in DMEM with 7.5% FBS. Kasumi-1 cells were cultured in RPMI with 10% FBS. For iPOND experiments, HEK293T, HCT-116, HeLa, and RPE-hTERT cells were cultured in DMEM for SILAC supplemented with 10% dialyzed FBS, 400mg/L L-Proline, 0.79mM L-Lysine, and 0.39mM L-Arginine. All media for RPE-hTERT cells was also supplemented with 0.25% sodium bicarbonate. The heavy isotopes used for heavy SILAC growth were L-Lysine 13C6 15N2 and L-Arginine 13C6 15N4. All cell lines were purchased from ATCC, tested for mycoplasma, and authentication verified using short tandem repeat profiling. U2OS, HEK293T, HeLa, and RPE-hTERT are female. HCT-116 and Kasumi-1 are male.
METHOD DETAILS
iPOND-SILAC Mass Spectroscopy and Data Analysis
iPOND-SILAC-MS and data analysis was performed as previously described (Dungrawala et al., 2015). For HEK293T, HCT-116, and HeLa experiments, an average of 3×108 asynchronous cells for each pulse and chase sample was used. For RPE-hTERT experiments, one replicate was performed with asynchronous cells, and two replicates were performed following a 16hr thymidine block and a 5-hour release into fresh media to maximize the number of cells in S-phase. An average of 2.6×108 cells for each RPE-hTERT pulse and chase sample was used. Cells were labeled with EdU for 10 minutes. For pulse-chase experiments with thymidine (Sigma), EdU-labeled cells were washed once with temperature- and pH-equilibrated medium containing 10 μM thymidine to remove the EdU, then chased into 10 μM thymidine for 1 hour. After labeling, cells were cross-linked in 1% formaldehyde/PBS for 10 min at room temperature, quenched using 1.25 M glycine, and washed three times in PBS. Collected cell pellets were frozen at −80°C, then resuspended in 0.25% Triton-X/PBS to permeabilize. Pellets were washed once with 0.5% BSA/PBS and once with PBS prior to the click reaction.
Light and heavy labeled cells were mixed 1:1. The click reaction was completed in 1 hr. and the cells were lysed by sonication. Capture of DNA-protein complexes utilized streptavidin-coupled C1 magnabeads for 1 hr. Beads were washed with lysis buffer (1% SDS in 50 mM Tris [pH 8.0]), low salt buffer (1% Triton X-100, 20 mM Tris [pH 8.0], 2 mM EDTA, 150 mM NaCl), high salt buffer (1% Triton X-100, 20 mM Tris [pH 8.0], 2 mM EDTA, 500 mM NaCl), lithium chloride wash buffer (100 mM Tris [pH 8.0], 500 mM LiCl, 1% Igepal), and twice in lysis buffer. Captured proteins were eluted and cross-links were reversed in SDS sample buffer by incubating for 30 min at 95°C.
iPOND samples were separated by SDS-PAGE. Gel regions above and below the streptavidin band were excised and treated with 45 mM DTT for 30 min, and available cysteine residues were carbamidomethylated with 100 mM iodoacetamide for 45 min. After destaining the gel pieces with 50% acetonitrile (MeCN) in 25 mM ammonium bicarbonate, proteins were digested with trypsin (Promega) in 25 mM ammonium bicarbonate at 37°C. Peptides were extracted by gel dehydration (60% MeCN, 0.1% trifluoroacetic acid [TFA]), vacuum dried, and reconstituted in 0.1% formic acid.
MudPIT mass spectrometry analysis was performed with an eight-step salt gradient. Peptides were introduced via nano-electrospray into a Q Exactive mass spectrometer (Thermo Scientific) operating in the data-dependent mode acquiring higher energy collisional dissociation tandem MS (HCD MS/MS) scans (R = 17,500) after each MS1 scan (R = 70,000) on the 20 most abundant ions using an MS1 ion target of 106 ions and an MS2 target of 105 ions. The maximum ion time for MS/MS scans was set to 100 ms, the HCD-normalized collision energy was set to 28, dynamic exclusion was set to 30 s, and peptide match and isotope exclusion were enabled.
MS/MS spectra were searched against a human subset database created from the UniprotKB protein database (https://www.uniprot.org). Precursor mass tolerance was set to 20 ppm for the first search, and for the main search, a 10-ppm precursor mass tolerance was used. The maximum precursor charge state was set to 7. Variable modifications included carbamidomethylation of cysteines (+57.0214) and oxidation of methionines (+15.9949). Enzyme specificity was set to Trypsin/P, and a maximum of two missed cleavages were allowed. The target-decoy FDR for peptide and protein identification was set to 1% for peptides and 2% for proteins. A multiplicity of 2 was used, and Arg10 and Lys8 heavy labels were selected.
For SILAC protein ratios, a minimum of two unique peptides and one or more ratio counts were required for protein group inclusion in the analysis. SILAC protein ratios for all datasets were analyzed within the Perseus software (Tyanova et al., 2016). Only proteins identified in at least 2 datasets were included in the statistical analysis in Perseus using a two-tailed t test with an FDR-adjusted significance level of 5 percent by the Benjamini-Hochberg method. Hierarchical clustering was performed within Perseus software using Euclidean distance measurements and complete linkage on proteins observed in at least 2 datasets (Tyanova et al., 2016). Euclidean distances for Table S5 were calculated in R (R Core Team, 2018). The clustering and Euclidean distance analyses were inclusive of SILAC ratio counts from one or more unique peptides.
Drug Sensitivity Screens and Assays
Sensitivity screens were performed in U2OS cells as previously described (Mohni et al., 2014, 2015). The custom siRNA library targets 593 genes with four unique siRNAs per gene in individual wells arrayed in 384 well plates. Cells were transfected with the siRNA and split into four 384 well plates 72 hours post-transfection and either mock treated or treated with 100nM ATRi (VX-970) or 0.2mM HU for 72 hours. Alternatively, cells were treated with 5nM camptothecin (CPT) or 2mM HU for 24 hours and released into fresh media for an additional 48 hours. Cell viability was measured with alamar blue (Invitrogen). Twelve biological replicates were performed for the mock, ATRi, and CPT treatments, and 6 biological replicates were performed for each of the HU treatments.
After subtracting background absorbance, the alamar blue absorbance of the mock treated sample siRNA was divided by the mock treated non-targeting siRNA to determine the viability of each sample siRNA. The drug sensitivity for each sample siRNA was determined by dividing the alamar blue absorbance of the drug treated siRNA by the mock treated siRNA. These values were then normalized for plate-to-plate and replicate-to-replicate variation by dividing by the average viability (drug/mock) of the non-targeting controls for each plate. Individual siRNAs with a viability of less than 5% were defined as toxic and removed from further analysis. The normalized drug sensitivities were then log2 transformed and each individual siRNA compared to the non-targeting controls in Prism by a two-tailed t test using an FDR-adjusted significance level of one percent by the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli.
Validation sensitivity assays were performed with either short-term alamar blue assays or long-term colony forming assays. For the short-term viability assays, cells were plated in 96-well plates 72 hours after siRNA transfection, the ATR inhibitor was added to the media for an additional 72 hours and cell viability was measured as previously described (Mohni et al., 2014). For long-term viability assays cells were treated with 100nM ATRi (VX-970) for 24 hours. Cells were plated for single colonies and scored after two weeks of growth. All viability measurements are presented as a percentage of the untreated control. For ATRi and JQ1 combination treatments, cells were plated in 2mM thymidine overnight, released for two hours in fresh media, and then treated with JQ1 for 4 hours. ATRi was added for the last two hours and then cells were placed in fresh media and allowed to form colonies. In all cell viability assays the values represent the mean (n = 3) and error bars represent the standard deviation of one experiment. All experiments were performed at least twice.
Plasmids
BRD3 cDNA was reverse transcribed from total RNA isolated from U2OS cells and then cloned into pENTR221 and a destination vector containing an N-terminal FLAG-HA tag using Gateway technology (Invitrogen). BRD2 was amplified from plasmid #65376 (Addgene) (Gong et al., 2015). BRD4 was amplified from plasmid #14441 (Addgene) (You et al., 2004). BRD3, BRD2, and BRD4 constructs were cloned into a pLEGFP-C1 backbone containing an N-terminal HA and NLS. ATAD5 plasmids were a gift from Kyungjae Myung. All plasmids were sequenced prior to use.
Flow Cytometry
Cells were labeled with 10 μM BrdU for 30 minutes, fixed with 70% ethanol, denatured with 2N HCl for 30 minutes, stained with anti-BrdU antibody, and then treated with propidium iodide and RNase A. Cells were analyzed on a BD Biosciences FACS Calibur.
siRNA and Antibodies
All siRNA transfections were performed as previously described (Mohni et al., 2014) with 10nM siRNA and Dharmafect 1 (Invitrogen). siRNAs for the library screen were purchased from Dharmacon and catalog numbers are listed in Table S7. The siRNA pool for BRD3 was a combination of BRD3–2 and BRD3–17. Antibodies used were as follows: BRD3, Abcam ab50818; GAPDH, Millipore MAB374; ATAD5 (Lee et al., 2010), gift from Kyungjae Myung; RFC1, Abcam ab3853; RFC4, Abcam ab192021; PCNA, Santa Cruz SC-056; H2B, Abcam ab1790; BRD2, Abcam ab139690; BRD4, Bethyl A301–985A50; GFP, Santa Cruz sc-8334.
Co-immunoprecipitation and Mass Spectrometry
HEK293T cells were transfected with FLAG-HA-BRD3, FLAG-ATAD5, or GFP-HA-NLS tagged constructs and nuclear extracts prepared as previously described (Dignam et al., 1983). FLAG-BRD3 was immunoprecipitated using EZview Red FLAG M2 affinity gel. FLAG-ATAD5 was immunoprecipitated using Anti-FLAG M2 magnetic beads (Sigma). GFP-tagged constructs were immunoprecipitated using GFP-Trap magnetic agarose beads (Chromotek). BRD3 and interacting proteins were eluted by addition of FLAG peptide, TCA precipitated, and analyzed by two-dimensional liquid chromatography tandem mass spectrometry. For immunoblot analysis of co-precipitating proteins immunoprecipitation was performed as above. Eluted proteins were analyzed by SDS-PAGE and immunoblotting.
Single Molecule Analysis of Replication
DNA fiber spreading experiments were performed as described previously (Couch et al., 2013). Cells were labeled with 20 μM IdU followed by 100 μM CldU for twenty minutes each. Following stretching and fixation on glass slides, DNA was denatured in 2.5M HCl for 80 min, washed 3 times with PBS, and blocked in 10% goat serum/PBS with 0.1% Triton X-100 for 1 hour. For DNA combing experiments, cells were labeled with 20 μM CldU followed by 100 μM IdU for 20 minutes each. Approximately 3×105 cells were embedded in agarose plugs and processed according to the manufacturer’s suggested protocol (Genomic Vision). For both spreading and combing assays, the DNA was stained with antibodies recognizing IdU and CldU for 1 hour, washed 3 times with PBS, and probed with secondary antibodies for 30 minutes. Images were obtained using a 40X oil objective (Nikon Eclipse Ti) and fiber lengths analyzed using NI-elements software. Normalization of fiber lengths was performed by dividing each value by the mean of the control in each experiment.
Chromatin Fractionation
Triton X-100 soluble and insoluble (Chromatin enriched) fractions were generated as previously described (Lee et al., 2010). Cells were incubated with 2mM Thymidine for 16 hours and then released for 2 hours into fresh media. After harvesting by trypsinization, cells were resuspended in Buffer A (100mM NaCl, 300mM sucrose, 3mM MgCl2, 10mM PIPES pH 6.8, 1mM EGTA, 0.2% Triton X-100, 100uM NaVO4, 50mM NaF, and protease inhibitors) and incubated on ice for 5 minutes. Insoluble material was pelleted, and supernatant was saved as the soluble fraction. The pellet (chromatin fraction) was washed once with Buffer A and then resuspended in Buffer B (50mM Tris-HCl (pH 7.5), 150mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 100uM NaVO4, 50mM NaF, and protease inhibitors) for 10 minutes on ice. The isolated chromatin fraction was then boiled for 10 minutes and sonicated. Equivalent volumes of buffer A and B were used for isolation of the soluble and chromatin fractions and proteins in each sample were visualized by immunoblotting. For analysis of PCNA levels on chromatin, the intensity of PCNA in the chromatin fraction was divided by total intensity of PCNA (soluble plus chromatin fractions).
Immunofluorescence
Cells were plated in 96 well clear-bottom plates. Experiments with siRNA were performed 72 hours post-transfection while experiments with overexpression plasmids were performed 48 hours post-transfection. Cells were incubated with media containing 10uM EdU for 30 minutes, pre-extracted for 5 minutes on ice in 20mM HEPES, pH 7.0, 50mM NaCl, 3mM MgCl2, 300mM sucrose, and 0.5% Triton X-100 followed by fixation in 3% paraformaldehyde. Cells were blocked for 1 hour in PBS containing 1mg/mL BSA, 5% goat serum, 0.1% Triton X-100, and 1mM EDTA. EdU was labeled by addition of 2mg/mL sodium ascorbate, 2mM copper sulfate, and 5uM Alexa Fluor conjugated azide in PBS for 30 minutes. Primary antibody incubation was performed for 1 hour in 1% BSA in PBS followed by a 45-minute incubation in secondary antibody. Nuclei were stained with a 5-minute incubation with DAPI in PBS. Plates were imaged on a Molecular Devices ImageXpress system. The integrated nuclear intensity of PCNA of EdU positive cells was analyzed. In overexpression experiments, cells were co-transfected with 0.1ug of a vector expressing GFP-H2B to visualize transfected cells and the integrated nuclear intensity of PCNA in EdU and GFP positive cells was analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of iPOND data was performed using Perseus. All other statistical analyses were completed using Prism. An ANOVA test was used when comparing more than two groups followed by a Dunnett multiple comparison post-test. A two-tailed t test was used to compare two samples with normally distributed data. For the proteomic and siRNA screens, p values were adjusted for 5% and 1% false discovery rates using the Benjamini-Hochberg method or two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli respectively. No statistical methods or criteria were used to estimate sample size or to include/exclude samples. Multiple siRNAs were analyzed to confirm results were not caused by off-target effects. Statistical details of individual experiments can be found in the figure legends and Results section. Unless otherwise stated all experiments were performed at least twice and representative experiments are shown.
DATA AND CODE AVAILABILITY
The publication includes all datasets generated and analyzed during this study. All original data are available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti PCNA | Santa Cruz Biotechnology | Cat#sc-56; RRID:AB_628110 |
| Mouse monoclonal anti FLAG M2 | Sigma-Aldrich | Cat#F3165; RRID:AB_259529 |
| Mouse monoclonal anti BRD3 | Abeam | Cat#ab50818; RRID:AB_868478 |
| Mouse monoclonal anti GAPDH | Millipore | Cat#MAB374; RRID:AB_2107445 |
| Rabbit anti ATAD5 | Gift from Kyungjae Myung | Lee et al., 2010 |
| Rabbit polyclonal anti RFC1 | Abeam | Cat#ab3853; RRID:AB_2238314 |
| Rabbit monoclonal anti RFC4 | Abeam | Cat#ab192021 |
| Rabbit polyclonal anti H2B | Abeam | Cat#ab1790; RRID:AB_302612 |
| Rabbit monoclonal anti BRD2 | Abeam | Cat#ab139690; RRID:AB_2737409 |
| Rabbit polyclonal anti BRD4 | Bethyl | Cat#A301–985A50; RRID:AB_2631449 |
| Rabbit polyclonal anti GFP | Santa Cruz | Cat#se-8334; RRID:AB_641123 |
| Rat monoclonal anti BrdU | Abeam | Cat#ab6326; RRID:AB_305426 |
| Mouse monoclonal anti BrdU | BD Bioscienees | Cat#347580; RRID:AB_400326 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EdU | VICB Synthesis Core | N/A |
| VX-970 | Selleck | Cat#S7102 |
| Camptothecin | Sigma Aldrich | Cat#C9911 |
| 13C615N2 L-Arginine | Cambridge Isotope Laboratories | CNLM-539-H |
| 13C615N4 L-Lysine | Cambridge Isotope Laboratories | CNLM-291-H |
| JQ1 | Selleck | Cat#S7110 |
| GFP-Trap Magnetic Agarose | Chromotek | Cat#gtma-10 |
| IdU | Sigma | Cat#l7125 |
| CldU | Sigma | Cat#C6891 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| HEK293T | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| hTERT-RPE | ATCC | Cat#CRL-4000; RRID:CVCL_4388 |
| HCT-116 | ATCC | Cat#CCL-247; RRID:CVCL_0291 |
| HeLa | ATCC | Cat#CCL-2; RRID:CVCL_0030 |
| Kasumi-1 | ATCC | Cat#CRL-2724; RRID:CVCL_0589 |
| Oligonucleotides | ||
| Screen siRNAs | Dharmacon | Refer to Table S7 |
| BRD3 siRNA 2 | Dharmacon | D-004936–02 |
| BRD3 siRNA 17 | Dharmacon | D-004936–17 |
| BRD2 siRNA pool | Dharmacon | M-004935–02 |
| BRD4 siRNA pool | Dharmacon | M-004937–02 |
| ATAD5 siRNA pool | Dharmacon | M-004738 |
| BRD2 siRNA 8 | Dharmacon | J-004935–08 |
| BRD2 siRNA 9 | Dharmacon | J-004935–09 |
| BRD4 siRNA 8 | Dharmacon | J-004937–08 |
| BRD4 siRNA 9 | Dharmacon | J-004937–09 |
| Recombinant DNA | ||
| BRD3 plasmid | This paper | N/A |
| BRD2 plasmid | Addgene | Addgene Cat#65376; Gong et al., 2015 |
| BRD4 plasmid | Addgene | Addgene Cat#14441; You et al., 2004. |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad Software |
https://www.graphpad.com/scientific-software/prism/
RRID: SCR_000306 |
| Perseus | Perseus | Tyanova et al., 2016; RRID:SCR_015753 |
Highlights.
iPOND proteomics identifies 593 proteins enriched on nascent DNA
101 proteins are found to be depleted from nascent DNA compared to bulk chromatin
Loss-of-function and bioinformatics analyses provide functional validation
The BET proteins BRD2, BRD3, and BRD4 bind ATAD5 and regulate PCNA unloading
ACKNOWLEDGMENTS
This research was supported by grants to D.C. from the NIH (R01GM116616 and R01CA239161) and the Breast Cancer Research Foundation. K.N.M. was supported by the Susan G. Komen fellowships PDF14302198 and 5T32CA009582. S.R.W. was supported by NIH F32 GM126646 and 5T32CA009582. Additional support came from the Vanderbilt-Ingram Cancer Center. We thank Kyungjae Myung for providing ATAD5 reagents.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.051.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Rutishauser D, and Ernfors P (2014). Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Res. 42, 6972–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi Y, Minakawa Y, Ono M, Dobashi S, Shinohe K, Shinohara A, Takeda S, Takagi M, Takamatsu N, Nakagama H, et al. (2015). ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient γH2AX foci formation. Cell Rep. 13, 2728–2740. [DOI] [PubMed] [Google Scholar]
- Baillat D, and Wagner EJ (2015). Integrator: surprisingly diverse functions in gene expression. Trends Biochem. Sci 40, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaoui M, Chang M, Ou J, Xu H, Boone C, and Brown GW (2003). Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 22, 4304–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowry A, Piberger AL, Rojas P, Saponaro M, and Petermann E (2018). BET inhibition induces HEXIM1-and RAD51-dependent conflicts between transcription and replication. Cell Rep 25, 2061–2069.e2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner DG, and Brickner JH (2011). Gene positioning is regulated by phosphorylation of the nuclear pore complex by Cdk1. Cell Cycle 10, 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D, and Dellaire G (2015). The role of the COP9 signalosome and neddylation in DNA damage signaling and repair. Biomolecules 5, 2388–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2017). Proteomic Analyses of the Eukaryotic Replication Machinery. Methods Enzymol. 591, 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, and Cortez D (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, and Marians KJ (2000). The importance of repairing stalled replication forks. Nature 404, 37–41. [DOI] [PubMed] [Google Scholar]
- Crowe BL, Larue RC, Yuan C, Hess S, Kvaratskhelia M, and Foster MP (2016). Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. USA 113, 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Costa D, Agathanggelou A, Perry T, Weston V, Petermann E, Zlatanou A, Oldreive C, Wei W, Stewart G, Longman J, et al. (2013). BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 3, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AM, and Newlon CS (1996). DNA replication fork pause sites dependent on transcription. Science 272, 1030–1033. [DOI] [PubMed] [Google Scholar]
- Dewar JM, Low E, Mann M, Räschle M, and Walter JC (2017). CRL2Lrr1 promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 31, 275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, and Cortez D (2017). RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 67, 374–386.e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Ruppert JM, Aster JC, and Winchester E (1993). Inhibition of DNA replication factor RPA by p53. Nature 365, 79–82. [DOI] [PubMed] [Google Scholar]
- Forment JV, and O’Connor MJ (2018). Targeting the replication stress response in cancer. Pharmacol. Ther 188, 155–167. [DOI] [PubMed] [Google Scholar]
- Géli V, and Lisby M (2015). Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. BioEssays 37, 1287–1292. [DOI] [PubMed] [Google Scholar]
- Gong F, Chiu L-Y, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. (2015). Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev 29, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Yuzugullu O, Carman C, and Price BD (2017). Spatially restricted loading of BRD2 at DNA double-strand breaks protects H4 acetylation domains and promotes DNA repair. Sci. Rep 7, 12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ, et al. (2015). BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol. Cell 59, 462–477. [DOI] [PubMed] [Google Scholar]
- Huang J, Gong Z, Ghosal G, and Chen J (2009). SOSS complexes participate in the maintenance of genomic stability. Mol. Cell 35, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, and Hetzer MW (2015). Nuclear pore proteins and the control of genome functions. Genes Dev 29, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, and Zakian VA (2003). The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 12, 1525–1536. [DOI] [PubMed] [Google Scholar]
- Kang M-S, Ryu E, Lee S-W, Park J, Ha NY, Ra JS, Kim YJ, Kim J, Abdel-Rahman M, Park SH, et al. (2019). Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun 10, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-P, Picaud S, Fujisawa T, Hou H, Savitsky P, Uusküla-Reim-and L, Gupta GD, Abdouni H, Lin Z-Y, Tucholska M, et al. (2019). Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 73, 621–638.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecona E, Rodriguez-Acebes S, Specks J, Lopez-Contreras AJ, Ruppen I, Murga M, Muñoz J, Mendez J, and Fernandez-Capetillo O (2016). USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol 23, 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Yang K, Cohn MA, Sikdar N, D’Andrea AD, and Myung K (2010). Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J. Biol. Chem 285, 10362–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Fu H, Aladjem MI, and Myung K (2013). ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol 200, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Rickards B, and Flint SJ (2008). The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 30, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, and Botchan MR (1993). The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell 73, 1207–1221. [DOI] [PubMed] [Google Scholar]
- Li C-J, and DePamphilis ML (2002). Mammalian Orc1 protein is selectively released from chromatin and ubiquitinated during the S-to-M transition in the cell division cycle. Mol. Cell. Biol 22, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Baek G, Ramanand SG, Sharp A, Gao Y, Yuan W, Welti J, Rodrigues DN, Dolling D, Figueiredo I, et al. (2018). BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 22, 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, Yin Y, Koff A, Ma L, and Zhou P (2009). CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 34, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Muñoz J, and Fernandez-Capetillo O (2013). A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 3, 1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, and Constantinou A (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol. Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, Sciortino S, Shuman J, Hurwitz J, and Ozato K (2002). A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell. Biol 22, 6509–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J, Zou-Yang XH, Kim S-Y, Hidaka M, Tansey WP, and Stillman B (2002). Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 9, 481–491. [DOI] [PubMed] [Google Scholar]
- Mohni KN, Kavanaugh GM, and Cortez D (2014). ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 74, 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Thompson PS, Luzwick JW, Glick GG, Pendleton CS, Lehmann BD, Pietenpol JA, and Cortez D (2015). A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS One 10, e0125482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019). HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan G-L, Pfander B, and Jentsch S (2007). PCNA, the maestro of the replication fork. Cell 129, 665–679. [DOI] [PubMed] [Google Scholar]
- Moschos SJ, and Mo Y-Y (2006). Role of SUMO/Ubc9 in DNA damage repair and tumorigenesis. J. Mol. Histol 37, 309–319. [DOI] [PubMed] [Google Scholar]
- Muralidharan SV, Bhadury J, Nilsson LM, Green LC, McLure KG, and Nilsson JA (2016). BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 35, 4689–4697. [DOI] [PubMed] [Google Scholar]
- Muralidharan SV, Einarsdottir BO, Bhadury J, Lindberg MF, Wu J, Campeau E, Bagge RO, Stierner U, Ny L, Nilsson LM, and Nilsson JA (2017). BET bromodomain inhibitors synergize with ATR inhibitors in melanoma. Cell Death Dis. 8, e2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BC, and Harper JW (2007). Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr. Opin. Cell Biol 19, 206–214. [DOI] [PubMed] [Google Scholar]
- Pericole FV, Lazarini M, de Paiva LB, Duarte ADSS, Vieira Ferro KP, Niemann FS, Roversi FM, and Olalla Saad ST (2019). BRD4 inhibition enhances azacitidine efficacy in acute myeloid leukemia and myelodysplastic syndromes. Front. Oncol 9, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2018). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, and Howley PM (2011). The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JJ, Bicknell LS, Carroll P, Higgs MR, Shaheen R, Murray JE, Papadopoulos DK, Leitch A, Murina O, Tarnauskaitė Ž, et al. (2017). Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat. Genet 49, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CG, Pietrzak K, Majchrzycka B, Kerlin MA, Chen J, Rankin S, and Sansam CL (2018). A mechanism for epigenetic control of DNA replication. Genes Dev. 32, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, and King RW (2012). A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 9, 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, and Cortez D (2013). Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem 288, 31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneville R, Moreno SP, Knebel A, Johnson C, Hastie CJ, Gartner A, Gambus A, and Labib K (2017). CUL-2LRR−1 and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol 19, 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava M, Chen Z, Zhang H, Tang M, Wang C, Jung SY, and Chen J (2018). Replisome Dynamics and Their Functional Relevance upon DNA Damage through the PCNA Interactome. Cell Rep. 25, 3869–3883.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai DCC, Szyszka TN, Campbell AE, Kwong C, Wilkinson-White LE, Silva APG, Low JKK, Kwan AH, Gamsjaeger R, Chalmers JD, et al. (2018). The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators. J. Biol. Chem 293, 7160–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, Lee A-Y, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, and Chiang C-M (2006). Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Shibata H, and Handa H (2013). Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim. Biophys. Acta 1829, 98–104. [DOI] [PubMed] [Google Scholar]
- Yeeles JT, Deegan TD, Janska A, Early A, and Diffley JF (2015). Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, and Diffley JFX (2017). How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 65, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Croyle JL, Nishimura A, Ozato K, and Howley PM (2004). Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117, 349–360. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, et al. (2018). BRD4 facilitates replication stress-induced DNA damage response. Oncogene 37, 3763–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The publication includes all datasets generated and analyzed during this study. All original data are available upon request.