

Globally, more people die annually from tuberculosis than from any other single infectious agent. Unfortunately, there is no commercially available vaccine that is sufficiently effective at preventing the acquisition of pulmonary tuberculosis in adults. In this study, preexposure prophylactic pulmonary delivery of active aerosolized antituberculosis bacteriophage D29 was evaluated as an option for protection against Mycobacterium tuberculosis infection.
Keywords: experimental therapeutics, global health, in vivo murine model, nose-only inhalation device, phage prophylaxis, vibrating mesh nebulizer
ABSTRACT
Globally, more people die annually from tuberculosis than from any other single infectious agent. Unfortunately, there is no commercially available vaccine that is sufficiently effective at preventing the acquisition of pulmonary tuberculosis in adults. In this study, preexposure prophylactic pulmonary delivery of active aerosolized antituberculosis bacteriophage D29 was evaluated as an option for protection against Mycobacterium tuberculosis infection. An average bacteriophage concentration of approximately 1 PFU/alveolus was achieved in the lungs of mice using a nose-only inhalation device optimized with a dose simulation technique and adapted for use with a vibrating mesh nebulizer. Within 30 min of bacteriophage delivery, the mice received either a low dose (∼50 to 100 CFU) or an ultralow dose (∼5 to 10 CFU) of M. tuberculosis H37Rv aerosol to the lungs. A prophylactic effect was observed, with bacteriophage aerosol pretreatment significantly decreasing M. tuberculosis burden in mouse lungs at 24 h and 3 weeks postchallenge (P < 0.05). These novel results indicate that a sufficient dose of nebulized mycobacteriophage aerosol to the lungs may be a valuable intervention to provide extra protection to health care professionals and other individuals at risk of exposure to M. tuberculosis.
INTRODUCTION
The World Health Organization (WHO) has classified the bacterium Mycobacterium tuberculosis as the leading infectious killer globally for a fourth consecutive year (1). Aside from being a significant comorbidity in persons living with human immunodeficiency virus (HIV), in both 2016 and 2017, tuberculosis (TB) was the cause of death for 1.3 million persons without HIV infection (1). Not only do low-income countries continue to suffer from endemic TB, but so do some populations in developed countries such as Indigenous peoples in Nunavut (NU), Canada (2).
The global community is increasingly concerned about the increase of drug-resistant M. tuberculosis strains. The WHO estimated that in 2017, roughly 0.5 million M. tuberculosis-infected individuals developed rifampin resistance, and over 80% of those cases were considered multidrug resistant (1). As global treatment success for drug-resistant M. tuberculosis cases remains unacceptably low, at roughly 55%, alternative interventions that block transmission and subsequent new infections are urgently needed (1). Vaccines against M. tuberculosis are an active area of research (1, 3–6), and indeed, much of the global community receives Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccination against TB as youth. BCG and other vaccine candidates can induce limited prophylactic protection through adolescence (7); however, BCG provides limited-to-no prophylactic effect if given to adults, does not prevent reactivation of latent TB, and does not prevent M. tuberculosis transmission (8, 9). Indeed, there is no vaccine that effectively prevents the acquisition of M. tuberculosis and progression to TB disease in adults (1). The lengthy, complex, multidrug treatment regimens available to target active TB often result in poor side effects for patients. This limits adherence and perpetuates the development of drug resistance.
Epidemiologic vulnerability to TB disease correlates with proximity to an active case or intensity of exposure over time. This is evidenced in highly exposed health care workers who have a 2- to 4-fold higher infection risk than do medical students with low exposure (10). Indeed, many studies document the increased risk to health care workers (11, 12), which justifies research toward the development of interventions against M. tuberculosis infection for this high-risk population. Bacteriophage (phage) delivery may be leveraged as an adjunct to current preventative strategies, which include personal protective equipment and administrative and environmental controls (13, 14) for health care professionals who are regularly exposed to infectious active cases of TB.
Phages are diverse viruses that have coevolved with bacteria and represent the most common biologic on earth (15). Phage strains are host and receptor restricted and therefore only capable of infecting a narrow spectrum of bacteria, which notably results in minimal harm to host microbiomes such as gut flora (16). Furthermore, antibiotic resistance does not influence bacterial susceptibility to phage lysis (16), making phages an attractive tool against drug-resistant organisms. Phages predicted to use obligatory lytic life cycles can be distinguished from temperate phages through genetic analysis (17). The lytic cycle encompasses the injection of phage DNA into the cell, phage replication, and lysis of the bacterial cell wall to release the progeny. Lytic phage therapy is considered safe and regularly utilized clinically in Eastern Europe, where some phage cocktails are available without prescription (18). Indeed, studies have repeatedly demonstrated that phages are not inherently harmful to humans (19).
Phage therapy is a viable option for compassionate use in the United States, with a recent study demonstrating successful treatment of a patient with disseminated multidrug-resistant Acinetobacter baumannii infection (20). In the United Kingdom, a cystic fibrosis patient with disseminated Mycobacterium abscessus recently showed objective clinical improvement with an intravenous three-phage cocktail treatment (21). The treatment of pulmonary infections in humans using phages has been reviewed elsewhere (22). Advanced research of aerosol phage therapy has become prevalent, including in vitro studies evaluating phage delivery with nebulizers, dry powder inhalers, and pressurized metered-dose inhalers (23–31). As more than 80% of TB cases originate from M. tuberculosis infections in the lungs, aerosol delivery of phage may be an ideal mechanism for enabling activity at the primary site of M. tuberculosis infection (32). Note that phage aerosol delivery without the use of additional vectors is unlikely to have efficacy against M. tuberculosis already harbored within a granuloma; even phage infection of M. tuberculosis within a macrophage, the primary target cell of M. tuberculosis, has low efficiency (33). However, prophylactic delivery of phages to the alveoli may allow the phage to infect the mycobacteria before macrophage uptake (17, 27, 34). Since the lungs contain millions of alveoli, a high dose of active phage is likely required for prophylaxis. Of particular interest for prophylactic protection against TB is Siphoviridae mycobacteriophage D29 (Fig. 1), which can effectively infect and lyse a range of mycobacteria, including M. tuberculosis (35).
FIG 1.
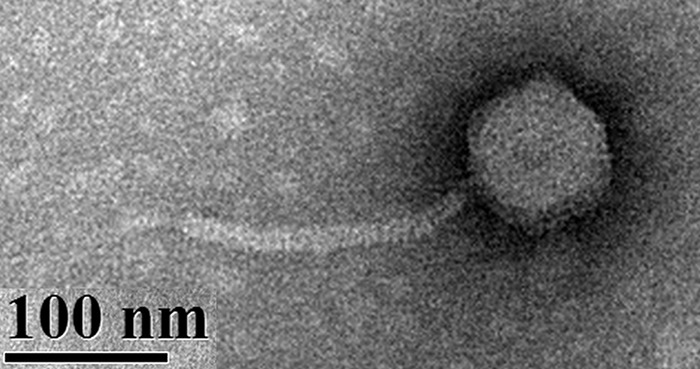
Transmission electron micrograph of phage D29. The icosahedral capsid contains double-stranded DNA. The tail is flexible and does not contract during infection. The method for imaging is described elsewhere (27).
Before testing the efficacy of phage D29 aerosol in humans, animal studies are of interest. Laboratory mice, commonly used for studying drug efficacy, have the advantages of low cost, short growth time, and small size, allowing for many mice to be tested simultaneously (36). Different methods for delivering aerosol to mice include a nose-only inhalation device (NOID), whole-body exposure system, nose drip, and intranasal or intratracheal instillation. The use of a NOID is the most common exposure method for rodents, as it allows for a uniform distribution of aerosol in the lungs via the nasal inhalation route, which is applicable to rodents, as they are obligate nasal breathers (37–39). Whole-body exposure systems require larger doses, as they are inefficient and cause aerosol deposition on the body of the mouse (36, 38, 40). Nose drip methods do not simulate natural aerosol inhalation and have variable inhaled droplet size. Instillation leads to nonuniform, patchy deposition, primarily near the site of instillation, and little or no alveolar deposition (37). Furthermore, inhalation methods are preferable to injection, as the aerosol is delivered directly to the site of infection, the lungs, and hence is available there at higher concentrations (41). Semler et al. (42) demonstrated that phage aerosol delivery by inhalation with a NOID was superior to intraperitoneal delivery, as evidenced by a greater reduction in bacterial burden and phage replication in the lungs.
Effective phage delivery to the lungs requires a prudent choice of aerosol delivery device to avoid phage inactivation (27). Factors that may inactivate phage are described elsewhere and include shear stress, osmotic shock, and thermal stress, among others (43, 44). In a recent study, it was demonstrated that the use of a vibrating mesh nebulizer resulted in less phage D29 inactivation and a higher active phage D29 aerosol delivery rate than with the use of a jet nebulizer (27). However, traditional NOID designs use jet nebulizers and have very low delivery efficiency. An adequate delivery method that allows retention of lytic capacity and the ability to deliver high titers of phage are current hurdles to testing prophylactic delivery of phages to the lungs of mice. In this study, a NOID was modified for use with a vibrating mesh nebulizer to deliver high doses of active phage D29 to the lungs of mice.
In order to advance phage D29 as a candidate therapy for TB, we leveraged our well-established mouse model of low-dose aerosol challenge of M. tuberculosis. We hypothesized that sufficient prophylactic pulmonary delivery of phage D29 would reduce M. tuberculosis bacterial burden at 24 h postchallenge. In order to evaluate our hypothesis, delivery parameters maximizing the inhaled dose in a repeatable fashion were experimentally simulated and subsequently used throughout the challenge studies. Measurements were then performed to quantify the number of phage reaching the lungs of mice and their clearance kinetics. Finally, phage D29 aerosol was delivered to mice prior to M. tuberculosis aerosol challenge to evaluate the prophylactic protection afforded by this treatment.
RESULTS
Lung homogenization does not reduce phage activity.
Naive mouse lungs were collected and spiked with an established titer of phage D29 and subsequently homogenized to determine if this process, used routinely to evaluate M. tuberculosis CFU ex vivo, would result in any loss of phage activity. The phage D29 titer after lung homogenization was 12.08 ± 0.03 log10(PFU/ml) compared to the control titer before homogenization of 12.12 ± 0.04 log10(PFU/ml), with no significant difference (P > 0.5; n = 3 each). These data indicate that the lung homogenization did not cause phage D29 inactivation, nor did any innate tissue factor influence phage activity or the properties of the plaque assay used to quantify phage in a sample. This demonstrated the viability of this approach for quantifying pulmonary phage delivery.
Nose-only inhalation device: dose simulation matches in vivo experiment.
Tryptophan tracer deposition at different locations within two different versions of the NOID quantified by assay of rinsate is presented in Table 1, as is the predicted dose to the lungs of a mouse, Tm/A, calculated using Equation 2 (see Materials and Methods). As shown, the use of the smaller width of the first plenum achieved the target dose, with a Tm/A of 1.0 ± 0.1 PFU/alveolus, meaning that the total lung dose of active phage in the lungs of a mouse was predicted to be equal to the total number of alveoli in a mouse. The predicted dose of phage D29 to the lungs was 7.6 ± 0.1 log10(PFU/mouse). Hence, this NOID configuration was chosen for in vivo experiments.
TABLE 1.
Tryptophan tracer deposition within two different versions of the NOID as a simulation of phage D29 delivery
| First plenum width (mm) | % tryptophan delivery (mean ± SD) ina: |
Predicted dose to a mouse Tm/A (PFU/alveolus) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Nebulizer reservoir | First plenum | Mixing tube | Back plenum | Nosepiece and adapter | Mouse filter | Exit filter | Unaccounted | ||
| 95 | 1.0 ± 0.8 | 61.2 ± 7.5 | 1.9 | 5.1 ± 1.2 | 0.062 ± 0.008 | 0.013 ± 0.005 | 1.07 ± 0.05 | 30 ± 6 | 0.4 ± 0.2 |
| 32 | 1.2 ± 0.6 | 60.3 ± 0.4 | 3.2 | 6.3 ± 0.6 | 0.073 ± 0.004 | 0.033 ± 0.004 | 1.50 ± 0.023 | 27 ± 3 | 1.0 ± 0.1 |
Results are from 3 replicate experiments, except for the mixing tube, which was measured once. The components are labeled in Fig. 5 (see Materials and Methods).
It is important to note that the ratio of flow rate entering the exit filter (478 ml/min) to the flow rate entering the mouse filter and nosepiece with adapter (22 ml/min) was 22, whereas the ratio of dose on the exit filter (1.50%) to dose on the surrogate mouse filter and nosepiece with adapter (0.11%) was 14. If a uniform aerosol concentration were present, the ratios would be equal. The latter ratio was lower (14 < 22), as expected, due to aerosol deposition in the front plenum.
A fraction of 0.044 = (22 ml/min)/(500 ml/min) of the total dose reaching all 12 nose ports deposited on the single tested mouse filter and nosepiece with adapter, on which a dose of 0.11% was measured. Relative to the use of the unmodified NOID presented by Nadithe et al. (36), the amount of tryptophan reaching the mouse filter was improved by a factor of 1.8. Considering that approximately 6,000 times more active phage D29 was delivered per unit time with the vibrating mesh nebulizer than with the jet nebulizer (27), an improvement by a factor of approximately 11,000 was achieved over the unmodified NOID that used a jet nebulizer in terms of the predicted number of active phage D29 reaching the lungs of the mice per unit of time. The small standard deviation indicated that the dosing was repeatable and consistent and provided confidence in this modified NOID setup.
We next optimized phage D29 aerosol delivery to mice in the modified NOID setup with the parameters described above. As phage D29 was amplified in Mycobacterium smegmatis, the resulting lysate input to the nebulizer clogged the mesh and hence had to be diluted 1:1 in isotonic saline prior to delivery, i.e., 3 ml of lysate at 11.8 ± 0.1 log10(PFU/ml) was added to 3 ml of isotonic saline, and a combined total of 6 ml was delivered. This resulted in lower delivery than the 6 ml of 12.2 ± 0.1 log10(PFU/ml) which was assumed during dose simulation. For the lower-delivery conditions, the predicted dose in the lungs of mice was 6.9 ± 0.1 log10(PFU/mouse), which is within the range of doses measured in vivo in the lungs of mice after phage D29 aerosol delivery, shown in Table 2. This indicates that the method of dose simulation and the model given by Equation 2 were accurate for predicting in vivo phage dose to the lungs of mice. This also demonstrates the accuracy and reliability of the dose simulation and the NOID setup in general.
TABLE 2.
Phage D29 dose in the lungs of mice post-NOID delivery
| Time between exposure and euthanasia (min) | Phage D29 dose in mouse lungs (mean ± SD) (log10[PFU/mouse])a |
|---|---|
| 0 | 6.6 ± 0.3 |
| 30 | 7.3 ± 0.1 |
| 90 | 7.0 ± 0.4 |
n = 3 mice per time point.
Along with total immediate delivery, potential pulmonary clearance of phage D29 was evaluated at 30 and 90 min postdelivery (see Table 2), and no statistically significant difference was observed (P > 0.05). Hence, phage D29 was not quickly cleared from the lungs of mice, providing confidence to proceed with M. tuberculosis exposure.
M. tuberculosis H37Rv is susceptible to phage D29.
Before commencing bacterial aerosol challenge studies, we confirmed prior data (45) indicating that M. tuberculosis strain H37Rv, a common laboratory strain, was susceptible to phage D29 lysate in vitro. A control plate (no phage added) contained 38 CFU, whereas 2 replicates with D29 addition resulted in 1 CFU and 2 CFU. Therefore, H37Rv lysis via phage D29 was 92 to 95% effective. The lysis may have not been 100% effective due to phage not coming into contact with every bacterium during plating, or potentially due to phage resistance. The high lysis effectiveness provided confidence to proceed with M. tuberculosis exposure.
Inhaled phage D29 provides in vivo prophylactic protection against TB.
In order to evaluate the potential application of phage D29 aerosol as a prophylactic tool against M. tuberculosis infection, we next quantified the bacterial burden (CFU) of M. tuberculosis H37Rv in mouse lungs at 24 h postinfection with or without phage D29 pretreatment less than 30 min prior to M. tuberculosis exposure. An average of 7.7 ± 0.3 log10(PFU/mouse) of phage D29 was delivered to the lungs in replicate 2, which corresponded to ∼1 PFU/alveolus on average, indicating that the target dose of phage D29 was achieved. A significant reduction (P < 0.05) in bacterial burden in the lungs at 24 h postchallenge was observed (Fig. 2). These data suggest that with a target dose of 1 PFU/alveolus, a significant level of prophylactic protection against inhaled M. tuberculosis aerosol is indeed possible.
FIG 2.
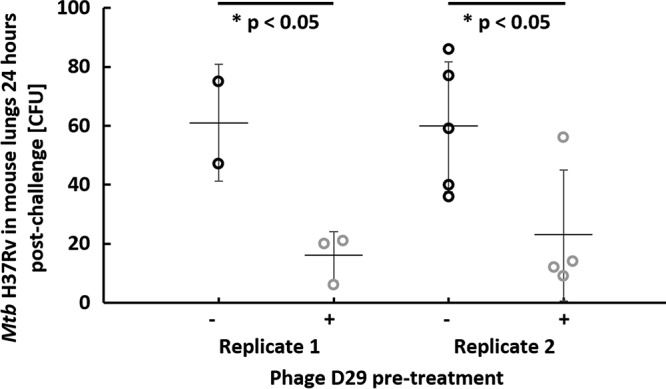
Pretreatment with phage D29 aerosol delivered by nose-only inhalation significantly reduces pulmonary bacterial burden 24 h postchallenge with low-dose M. tuberculosis (Mtb) H37Rv. On the x axis, “−” indicates no phage D29 pretreatment, and “+” indicates phage D29 pretreatment. Each circle represents a single mouse, and error bars span the standard deviation around the mean indicated by the horizontal line.
A separate cohort of mice from replicate 1 was followed out to 3 weeks postchallenge to determine if the effects of prophylactic phage application would persist over time. Bacterial burden was measured in the lungs and the spleen 3 weeks postchallenge (Fig. 3). Interestingly, at 3 weeks, phage D29-pretreated mice sustained a significantly lower bacterial burden than did mice that did not receive phage pretreatment in the lungs (P < 0.05), although bacterial burden was not significantly different in the spleen (P > 0.1). The bacterial burden at 3 weeks was of a high magnitude.
FIG 3.

Log10 of bacterial burden 3 weeks postchallenge in the lungs (left) and spleen (right), without and with phage D29 pretreatment. On the x axis, “−” indicates no phage D29 pretreatment, and “+” indicates phage D29 pretreatment. Each circle represents a single mouse, and error bars span the standard deviation around the mean indicated by the horizontal line.
In order to more closely simulate M. tuberculosis infections in humans, we next optimized and utilized an ultralow-dose aerosol challenge of H37Rv calibrated to deliver 5 to 10 CFU of bacteria. An average of 7.4 ± 0.1 log10(PFU/mouse) of phage D29 was delivered to the lungs. At 24 h postchallenge, a significant reduction (P < 0.05) in M. tuberculosis in the lungs was observed in the group that received phage D29 aerosol pretreatment, relative to the group that did not (Fig. 4), providing important further evidence of prophylactic efficacy.
FIG 4.
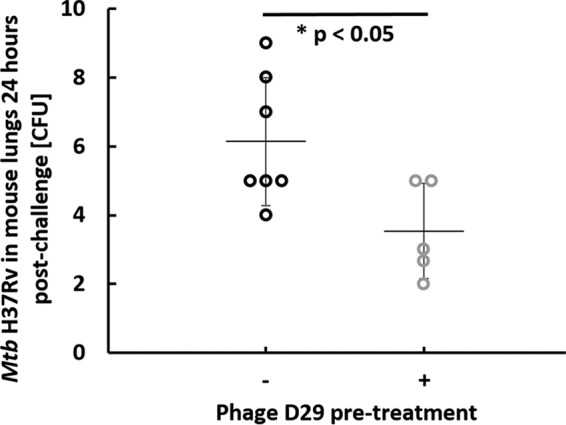
Pretreatment with phage D29 aerosol delivered by nose-only inhalation significantly reduces pulmonary bacterial burden 24 h postchallenge with ultralow-dose M. tuberculosis H37Rv. On the x axis, “−” indicates no phage D29 pretreatment and “+” indicates phage D29 pretreatment. Each circle represents a single mouse, and error bars span the standard deviation around the mean indicated by the horizontal line.
DISCUSSION
The results of this study demonstrate that inhalation of phage D29 aerosol prior to challenge with M. tuberculosis aerosol can significantly decrease the pulmonary bacterial burden in mice 24 h postinfection. These data suggest that inhaled mycobacteriophage aerosol resulted in M. tuberculosis lysis in the lungs prior to macrophage uptake and granuloma formation. This proof-of-principle study may have implications for the development of prophylactic aerosol treatments for health care professionals exposed to patients with active TB and reduction in M. tuberculosis transmission rates in this setting. This is important because many health care professionals are relatively unwilling to work in areas of hospitals with a high risk of tuberculosis transmission (46). Additionally, more protection could potentially be offered to individuals in areas in which TB is endemic, or to other individuals at high risk, such as household contacts and visiting family members at hospitals. This treatment is intended to complement normal precautions, which includes the use of administrative controls, environmental controls, and personal respiratory protection (13, 14).
We are only aware of one other study regarding prophylactic inhalation of phages prior to the inhalation of bacteria (47), and that study demonstrated 4-day prophylaxis against multidrug-resistant (MDR) Pseudomonas aeruginosa in mice, but it used intranasal instillation rather than an aerosol delivery device. Instillation results in localized deposition and is not as representative of natural inhalation to the different regions of the lungs as nose-only inhalation (37, 48). Therefore, we believe this work, the first study demonstrating prophylactic protection with phage using nose-only inhalation of aerosol, represents a significant advancement in this area of research. Additionally, to our knowledge, a much higher titer of phage was delivered to the lungs of mice by nose-only inhalation in this study than in any previous study. Relative to the use of a jet nebulizer in a previous NOID design (36), approximately 11,000 times more active phage D29 could be delivered to the lungs of mice, and with repeatable results, indicating that a major improvement of dosing to mice was achieved with this novel approach. Furthermore, Liu et al. (41) only delivered 102 to 103 PFU of phage D29 to the lungs of mice using a Collison jet nebulizer with a NOID, which is orders of magnitude lower than in this study, in which >107 PFU was delivered to the lungs of mice. This improvement allowed, for the first time, an average dose of ∼1 PFU/alveolus to be achieved. It is important to consider that some alveoli are poorly perfused, and hence, >1 PFU per accessible alveolus may have been achieved on average. Another factor to consider is that phage progeny released after M. tuberculosis lysis would offer further protection in their vicinity but are unlikely to be transferred to nearby alveoli, as they are nonmotile. As M. tuberculosis is also nonmotile, it is unlikely to move between alveoli and come into contact with phage in that manner prior to uptake by immune cells.
The prophylactic delivery of a calculated average of 1 PFU/alveolus allowed for a significant reduction in M. tuberculosis levels in the lungs, demonstrating a prophylactic effect. Whether such a decrease in M. tuberculosis levels is sufficient to decrease mortality rates is not known. While it is difficult to deliver more than 1 PFU/alveolus on average to a mouse using a NOID, such levels may be necessary to achieve complete bacterial eradication. Poisson statistics can be used to estimate the probability of a specific random occurrence during an interval of interest knowing the average number of occurrences in that interval (49). Using this method, the probability, y, that an alveolus will contain a certain number of phage, x, can be predicted knowing the average number of PFU/alveolus, λ, according to Equation 1:
| (1) |
Using the values λ = 1 and x = 0 gives a y value of 0.37. This indicates that if on average 1 PFU/alveolus is delivered to the lungs, the probability that an alveolus does not contain a phage is still 37%. Hence, the chance that a bacterium encounters at least one phage in an alveolus is about 63%. In prophylactic phage administration experiments (see Fig. 2), the average reduction in bacterial burden was from approximately 60 CFU to approximately 20 CFU, or about 67%. This is in close agreement with the theoretically calculated prediction of 63% reduction in bacterial burden that assumes that all bacteria that deposit in an alveolus in the presence of at least one phage are inactivated.
To achieve at least one phage in 99% of the alveoli of a mouse, the average number of phage per alveolus (λ) would need to be 4.6. For 99.9% coverage, λ is 6.9. A potential indicator of the dose required for complete prophylaxis is when the probability that an alveolus contains no phage becomes less than the inverse of the number of alveoli in the lungs, which for a mouse occurs with an λ of >17.5.
Achieving a high dose of active phage in human lungs is not as difficult as in mice because deposition losses in the NOID would not be present, and humans do not require equally small droplets for efficient lung deposition. Human lungs contain ∼4.8 × 108 alveoli (50), ∼10 times the number that mouse lungs have. An average number of phage per alveolus of approximately 20 may be required for complete prophylaxis in a human according to the above-mentioned indicator. A recent study suggested that in humans, a respirable dose of active phage D29 of ∼1.3 × 109 PFU could be achieved with delivery of 6 ml of diluted lysate using a vibrating mesh nebulizer (27). This corresponds to a dose to human lungs of ∼2.7 PFU/alveolus on average, or 93% alveolar coverage. In that study, the phage D29 lysate was diluted 1:100 in isotonic saline prior to aerosolization, as without dilution, the lysate purification level was not sufficient to prevent mesh clogging. Potentially, without dilution, i.e., with better purification techniques, orders of magnitude higher titers may reach the lungs, on the order of 102 PFU/alveolus on average. This could be sufficient for complete prophylaxis, according to the above-described Poisson statistics argument. The chance of noncontact between phage and bacteria would be substantially decreased at this dose level. Additionally, with daily prophylactic doses, the likelihood that a specific alveolus receives a sufficient number of phages to eradicate intruding bacteria increases. As it may take days for granulomas to form (51), it is possible that phages delivered soon after M. tuberculosis exposure may still offer protection, but this depends on macrophage uptake dynamics (33) and awaits further exploration.
The development of regulatory-approved, commercial phage formulations for inhalation will likely require the use of cocktails containing various phages that target different receptors to ensure that M. tuberculosis does not become phage resistant (17). The development of anti-TB cocktails is an active area of research (17). Phage D29 and others that may be included in a therapeutic cocktail would be applicable for the prevention of drug-sensitive or drug-resistant strains of M. tuberculosis. Further development of phage cocktail therapy will require efficacy testing against clinical and drug-resistant M. tuberculosis strains and, importantly, measurement of potential phage-induced host immune responses. It should be determined whether daily prophylactic doses of phage cocktails lead to an immune response that inactivates the phages or to the development of bacteria which are resistant to all of the phages in the cocktail, although it is important to note that phages are capable of mutating to overcome bacterial resistance, and new phages targeting different receptors can be isolated relatively rapidly. Some clinical applications of phage therapy have reported humoral immune responses to phage (anti-phage IgG and IgM antibodies); however, the magnitude of the anti-phage antibody response did not correlate with a decrease in clinical efficacy (52). This suggests that even if an adaptive immune response to phage is induced, it may not be detrimental to the host nor inhibit phage efficacy. Although recent evidence suggests that mucosal delivery (aerosol) does not induce a robust or detrimental anti-phage immune response (53), future work should specifically examine the effects of repeated phage exposure to determine the feasibility of protection in high-risk populations likely to regularly encounter M. tuberculosis.
In therapeutic TB treatment, for example, with antibiotics, drug-resistant M. tuberculosis persisters may arise if not all bacteria are eradicated (54). Persisters may arise to some extent in prophylaxis using phages if there are bacteria that are less likely to be lysed by phages and hence have a higher probability to establish an infection. However, therapeutic treatment typically requires eradicating a large population in which a small proportion may be resistant, whereas prophylactic treatment typically requires eradicating a much smaller population, perhaps even as low as a single bacterium. Therefore, the probability of establishing resistant M. tuberculosis is expected to be smaller for prophylaxis than for therapy.
For future development of phage cocktails, it will be important to ensure that each type of phage is capable of surviving the nebulization process and can effectively reach the lungs. In this study, experimental dose simulation of phage delivery to mice using a tryptophan tracer, simulated breathing, and filter aerosol capture with a NOID resulted in accurate prediction of phage dose reaching the lungs of mice in vivo. Hence, assuming the same droplet size distribution, it is possible that one could simply measure the phage activity retention from the nebulizer by aerosolization to a filter, as described elsewhere (27), and predict the in vivo dose reaching the lungs of mice in the present NOID setup using the modeling approach (see Equation 2) and aerosol delivery efficiency results (Table 1) presented in this study. This would allow the lung dose of different phages in a cocktail to be predicted prior to in vivo experiments, saving time and resources, reducing the risk of failed animal work, and expediting the development process.
In summary, inhalation of anti-TB mycobacteriophage D29 aerosol is a promising and novel approach to provide prophylactic protection against primary infection of inhaled M. tuberculosis aerosol. A significant reduction in bacterial burden was achieved with prophylactic delivery of an average dose of active phage D29 of ∼1 PFU/alveolus to the lungs. Complete prophylaxis may be achievable with larger or repeated doses of active phages. The number of active phages reaching the alveoli can be predicted prior to animal studies with a NOID by aerosolizing a tracer, simulating breathing through a filter, assaying the recovered aerosol on the filter, and applying the developed mathematical modeling approach. The development of a high-titer phage cocktail against TB is recommended over monophage therapy. The cocktail may provide extra protection to health care professionals regularly exposed to patients with active TB and to individuals in areas with high rates of TB transmission while limiting the likelihood of resistance to phages. Additionally, countries with a high burden of TB and MDR-TB are often involved in military engagement, with resulting additional risk of exposure. Given that the risk of M. tuberculosis transmission is higher in congregate settings, a single individual with TB disease exerts an immediate and disruptive impact upon patients’ lives, military operations, and daily functioning at military and civil treatment facilities. Delivery of high doses of active phages to human lungs for prophylactic purposes appears to be achievable, proceeding to human clinical trials is of interest, and compliance to delivery of high doses of phage aerosol on a daily basis should be studied.
MATERIALS AND METHODS
Mice.
The mice used in this study were female C57BL/6 mice 4 to 6 weeks of age, weighing 14 to 16 g, purchased from Charles River Laboratories (Wilmington, MA, USA). Mice were housed at the Infectious Disease Research Institute (IDRI) biosafety level 3 animal facility under pathogen-free conditions and were handled in accordance with the specific guidelines of IDRI’s Institutional Animal Care and Use Committee. The reported minute ventilation rate for CD-1 mice, similar to the C57BL/6 strain used here, was 1.46 ml/gram of body weight (55). For an average mass of 15 g, this corresponds to an average minute ventilation of ∼22 ml/min per mouse, and this value was used for calculations for nose-only aerosol delivery.
Phage D29 amplification, shipping, and plaque assay.
Phage D29 was prepared to a titer of 1.6 × 1012 PFU/ml via replication with M. smegmatis strain mc2155 using solid medium, sterile filtration, centrifugation, and pellet resuspension in buffer, as described elsewhere (https://phagesdb.org/workflow/) (27). The amplified phage lysate was shipped to the IDRI (Seattle, WA, USA) from the University of Alberta (Edmonton, Alberta, Canada) using cold packs and a Styrofoam container. This shipment did not result in a reduction of the titer of the lysate. The titer of phage D29 was measured using full-plate plaque assay, as described elsewhere (https://phagesdb.org/workflow/) (27).
Lung homogenization.
It was necessary to homogenize the lung tissue of the mice to generate a representative liquid sample to assay and to determine the number of active phage and bacterial burden in the lungs. To verify that the phage remained active after the high-shear homogenization process, a 20-μl sample of phage lysate was spiked in lung tissue within 2 ml of buffer in a 15-ml Eppendorf tube, homogenized (Omni Prep multisample homogenizer; Omni International, Kennesaw, GA, USA) for 1 min, and centrifuged for 2 min at 1,800 rpm. The titer after homogenization was compared to the titer before homogenization to determine if the homogenization process inactivated the phages.
Nose-only inhalation device: device design and dose simulation versus in vivo experiment.
A schematic of the developed NOID, a modified version of the device described by Nadithe et al. (36), set up for dose simulation experiments, is shown in Fig. 5. Nadithe et al. (36) reported that their NOID design had substantial losses of aerosol. Only 0.108% ± 0.027% of the dose input to the jet nebulizer reached the mice, and only 8.19% ± 3.56% of that amount reached the lungs of the mice. This corresponded to 0.0087% ± 0.0021% (870 ppm) of the input dose reaching the lungs of all 12 mice combined. Much of this loss was attributed to the compressor of the jet nebulizer delivering 4.5 liters/min of airflow into the system, resulting in a substantial amount of aerosol convecting by the noses of the mice and exiting the back of the device unused. This is because the combined total minute volume for 12 mice is only 0.264 liters/min; hence, about 4.2 liters/min of airflow, or 93% of aerosol available at the nosepieces, bypassed the mice. Therefore, in our modified design, the minimum airflow rate into the system that could safely be used without developing a hypoxic environment (38), i.e., 0.5 liters/min, was used. The negative pressure induced by a vacuum pump (model UN726FTP; KNF Neuberger, Inc., Trenton, NJ, USA) past the exit of the device caused the airflow into the NOID. The pressure difference between the front plenum, back plenum, and atmosphere was measured with manometers to rule out leaks in the system. A rotameter (catalog no. 5079K63; McMaster-Carr, Elmhurst, IL, USA), calibrated with a thermal mass flow meter (TSI 4043; TSI Incorporated, Shoreview, MN, USA), was used to measure the airflow rate into the NOID, which was controlled with valves past the exit filter and in the rotameter. An inlet air filter (Respirgard II 303; Vital Signs Incorporated, Englewood, CO, USA) prevented foreign virus or bacterial contamination from the airflow into the device.
FIG 5.

Schematic of a modified NOID adapted for use with a vibrating mesh nebulizer.
The NOID was modified to incorporate a vibrating mesh nebulizer (Aerogen Solo with Pro-X controller; Aerogen Ltd., Dangan, Galway, Ireland) to produce the aerosol. The aerosol entered the first plenum, which was developed because preliminary experiments demonstrated that use of the commercial wye connector for the vibrating mesh nebulizer resulted in a large extent of aerosol recirculation, droplet coalescence, and deposition losses. This is because the small-volume wye connector was designed for the use of >3 liters/min airflow rate, whereas a lower airflow rate, 0.5 liters/min, was used in this study.
The operation of the vibrating mesh nebulizer has been described by Carrigy et al. (27). The mesh consists of ∼1,000 orifices, each with a diameter of ∼3 μm. The generated aerosol has been measured at the exit of a T-piece by laser diffraction to have a volume median diameter of ∼5.5 μm and geometric standard deviation of ∼1.8 (56). The vibrating mesh nebulizer has a liquid droplet production rate of about 0.36 ml/min (27), and hence, with the chosen airflow in of 500 ml/min, liquid entered the system at a concentration of 720 g/m3. Considering that room temperature air can hold ∼17.3 g/m3, the nebulizer produces approximately 42 times the amount of liquid required to fully saturate the airflow in, assuming it is initially dry. Hence, at a maximum, only about 2% of the mass of the droplets, corresponding to about 1% of the diameter assuming sphericity, is required to fully saturate the NOID. Therefore, the droplets essentially maintained their size after atomization as they transited through the device, neglecting any coalescence. However, mice require small droplets for deposition in the lungs (36, 57). In the present NOID system, the large droplets were filtered out by the first plenum and the smaller droplets produced by the nebulizer, recalling that the geometric standard deviation is ∼2, followed the airflow streamlines, due to a lower Stokes number, into the ∼12-mm diameter mixing tube. This mixing tube ensured that a uniform aerosol concentration entered near the top of the back plenum. Mice inhale through nosepieces attached to mouse restraint tubes, which used an airtight plunger to hold the mice in place with their noses at the nose ports. The mice inhaled the aerosol from the back plenum through the nose ports, and the excess airflow and exhaled air from the mice entered the front plenum and subsequently were filtered and exited the device.
Before proceeding to in vivo experiments, the dose was simulated to verify that a biologically relevant amount of aerosol would reach the lungs of mice. For dose simulation experiments, a mouse restraint tube was replaced with a filter and attached to the nosepiece by an adapter, rapid-prototyped out of an acrylic compound (Objet VeroGray RGD850; Stratasys Ltd., Eden Prairie, MN, USA) using a PolyJet three-dimensional (3D) printer (Objet Eden 350 V high-resolution 3D printer; Stratasys, Ltd.). This filter was termed a mouse filter due to its surrogate mouse function and location within the setup and was attached to a syringe pump (PHD Ultra syringe pump with push/pull mechanism, model no. 70-3008; Harvard Apparatus, Holliston, MA, USA). The syringe pump initiated a constant flow rate of 22 ml/min through the mouse filter, which is equivalent to the average minute volume for a mouse, as described previously. Only one port was evaluated because previous data with the unmodified NOID demonstrated a reasonably even distribution of aerosol between nose ports (36). Due to the >10-ml filter dead volume, the steady flow equivalent of tidal flow was used, and the effect of exhalation on lung deposition was neglected. l-Tryptophan (catalog no. 93659; Sigma-Aldrich, St. Louis, MO, USA) tracer in isotonic saline was atomized and captured on the mouse filter, as well as at various points within the NOID (nebulizer reservoir, first plenum, mixing tube, back plenum, nosepiece and adapter, and exit filter) to determine where deposition occurred. The tracer concentration was assayed using UV-Vis spectrophotometry (8452A diode array spectrophotometer; Hewlett-Packard, Mississauga, Ontario, Canada). The deposition was quantified for two different versions, where different widths of the developed first plenum, 32 mm and 95 mm, were tested. The length of the first plenum was kept fixed at 152 mm, and the depth was kept fixed at 211 mm, as this was approximately the distance at which bulk aerosol flow emitted from the nebulizer stopped after horizontal spray. Three replicate dose simulation experiments were performed for each of the two first plenum widths. As the interior volume of the NOID was ∼4 liters, the airflow through the system was allowed to continue for ∼8 min after nebulization was complete to allow time for the aerosol to transit through the system.
A model was developed to predict the average number of active phage per alveolus reaching the lungs of one mouse in the NOID, Tm/A, from the tryptophan experiments and literature data, and is given by
| (2) |
where T0 is the initial titer of the lysate input into the vibrating mesh nebulizer in PFU, fn is the fraction of the phage not inactivated by the nebulizer measured in a previous study to be 0.319 (27), fi is the fraction of the breathing cycle spent inhaling approximated as 0.5, fm is the fraction of the aerosol emitted from the nebulizer that is inhaled by a single mouse from tryptophan tracer dose simulation experiments, fl is the fraction of aerosol that is inhaled by a mouse that reaches its lungs taken as 0.08 (36), and Am is the number of alveoli per mouse taken as 4 × 107 (58).
For comparison to dose simulation, phage D29 was delivered to the mice, and the lungs of the mice were removed following euthanasia and tissues homogenized within 5 ml of buffer. The number of active phage in the homogenate was determined by plaque assay. Three mice were taken down at each of 0, 30, and 90 min after phage exposure to obtain a preliminary measure of lung clearance.
Host susceptibility.
Prior to performing in vivo experiments, it was confirmed that M. tuberculosis H37Rv is susceptible to phage D29 lysis. A volume of 100 μl of 1011 PFU/ml phage D29 lysate was added to a sample of M. tuberculosis H37Rv and subsequently plated on agar plates. Bacterial CFU were determined after incubating plates at 37°C and 5% CO2 for 21 days. Plates with or without D29 application were compared.
In vivo prophylactic protection.
The mice were acclimatized to remain calm in the restraint tubes of the NOID, as per previous methods (57), therefore retaining a normal breathing pattern to maximize peripheral lung deposition. Briefly, mice were “trained” in the NOID tubes without treatment 3 to 5 times for 5 min each in the week leading up to phage delivery. This training significantly reduced visible stress-induced changes in breathing. Within 30 min of receiving phage D29 aerosol with the NOID, mice were challenged with M. tuberculosis H37Rv aerosol using a previously described Wisconsin-Madison aerosol chamber (6, 59) calibrated to deliver ∼50 to 100 bacteria (low dose) or ∼5 to 10 bacteria (ultralow dose). The dose nomenclature was based on previous studies (6, 60–62). After M. tuberculosis aerosol inhalation and euthanasia, the lung tissue was isolated and homogenized in 5 ml of phosphate-buffered saline (PBS) plus Tween 80 (Sigma-Aldrich, St. Louis, MO, USA) buffer, and the entire lung homogenate sample was plated on Middlebrook 7H10 agar plates and subsequently incubated at 37°C and 5% CO2 for 3 weeks before colonies were counted. The bacterial burden of M. tuberculosis was evaluated at 24 h (n = 2 experiments) and 21 days (n = 1 experiment) postchallenge for the low-dose model and at 24 h (n = 1 experiment) postchallenge for the ultralow-dose model. The ultralow-dose bacterial challenge is expected to better reflect human infection conditions where relatively few bacteria are able to establish a pulmonary infection in the host. The NOID was disassembled, and components were disinfected with ethanol between all experiments. Following aerosol challenge with M. tuberculosis H37Rv, the Wisconsin-Madison aerosol chamber was thoroughly sprayed with Lysol aerosol and after 10 min cleaned with 70% ethanol and paper towels. Nebulizer components were sterilized between runs with 10% bleach.
Statistics.
Significance was evaluated using Student’s t tests, assuming equal variance at a significance level of 0.05. Two-sided t tests were used to determine if results were significantly different, and one-sided t tests were used to determine whether a result was significantly greater than or less than another result.
ACKNOWLEDGMENTS
N.B.C. thanks the Killam Trusts, the Natural Sciences and Engineering Research Council of Canada, Alberta Innovates, and the University of Alberta for scholarship funding. N.B.C. also thanks Bernie Faulkner for manufacturing the first plenum. The vibrating mesh nebulizer equipment was kindly provided by Jim Fink and Ronan MacLoughlin (Aerogen Ltd.).
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI125160, and S.E.L. is supported through the University of Washington Diseases of Public Health Importance T32 training grant number A1075090.
The funders and equipment providers had no role in the study design, data collection and interpretation, or the decision to submit the work for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.World Health Organization. 2018. Global tuberculosis report 2018. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Orr P. 2013. Tuberculosis in Nunavut: looking back, moving forward. CMAJ 185:287–288. doi: 10.1503/cmaj.121536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contreras GL, Awashthi S, Hanif SNM, Hickey AJ. 2012. Inhaled vaccines for the prevention of tuberculosis. J Mycobact Dis S1:002. doi: 10.4172/2161-1068.S1-002. [DOI] [Google Scholar]
- 4.Tyne AS, Chan JGY, Shanahan ER, Atmosukarto I, Chan H-K, Britton WJ, West NP. 2013. TLR2-targeted secreted proteins from Mycobacterium tuberculosis are protective as powdered pulmonary vaccines. Vaccine 31:4322–4329. doi: 10.1016/j.vaccine.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Aerosol Vaccines for Tuberculosis Workshop Summary Group. 2015. Developing aerosol vaccines for Mycobacterium tuberculosis: workshop proceedings National Institute of Allergy and Infectious Diseases, Bethesda, Maryland, USA, April 9, 2014. Vaccine 33:3038–3046. doi: 10.1016/j.vaccine.2015.03.060. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin SL, Reese VA, Huang PD, Beebe EA, Podell BK, Reed SG, Coler RN. 2016. Protection and long-lived immunity induced by the ID93/GLA-SE vaccine candidate against a clinical Mycobacterium tuberculosis isolate. Clin Vaccine Immunol 53:137–147. doi: 10.1128/CVI.00458-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemes H, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, Mabwe S, Makhethe L, Erasmus M, Toefy A, Mulenga H, Hanekom WA, Self SG, Bekker L-G, Ryall R, Gurunathan S, DiazGranados CA, Andersen P, Kromann I, Evans T, Ellis RD, Landry B, Hokey DA, Hopkins R, Ginsberg AM, Scriba TJ, Hatherill M. 2018. Prevention of M. tuberculosis infection with H4:IC31 vaccine or BCG revaccination. N Engl J Med 379:138–149. doi: 10.1056/NEJMoa1714021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues LC, Mangtani P, Abubakar I. 2011. How does the level of BCG vaccine protection against tuberculosis fall over time? BMJ 343:d5974. doi: 10.1136/bmj.d5974. [DOI] [PubMed] [Google Scholar]
- 9.Rowland R, McShane H. 2014. Current transmission prevention methods: vaccination, p 33–52. In Zellweger J-P. (ed), Clinical insights: tuberculosis prevention. Future Medicine Ltd., London, United Kingdom. [Google Scholar]
- 10.Toujani S, Cherif J, Mjid M, Hedhli A, Ouahchy Y, Beji M. 2017. Evaluation of tuberculin skin test positivity and early tuberculin conversion among medical intern trainees in Tunisia. Tanaffos 16:149–456. [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi R, Reingold AL, Menzies D, Pai M. 2006. Tuberculosis among health-care workers in low- and middle-income countries: a systematic review. PLoS Med 3:e494. doi: 10.1371/journal.pmed.0030494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uden L, Barber E, Ford N, Cooke GS. 2017. Risk of tuberculosis infection and disease for health care workers: an updated meta-analysis. Open Forum Infect Dis 4:ofx137. doi: 10.1093/ofid/ofx137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox GJ, Marks GB, Britton WJ. 2014. Current transmission prevention methods: reducing disease spread from infected individuals, p 53–76. In Zellweger J-P. (ed), Clinical insights: tuberculosis prevention. Future Medicine Ltd., London, United Kingdom. [Google Scholar]
- 14.Verkuijl S, Middelkoop K. 2016. Protecting our front-liners: occupational tuberculosis prevention through infection control strategies. Clin Infect Dis 62:S231–S237. doi: 10.1093/cid/civ1184. [DOI] [PubMed] [Google Scholar]
- 15.Hatfull GF. 2015. Dark matter of the biosphere: the amazing world of bacteriophage diversity. J Virol 89:8107–8110. doi: 10.1128/JVI.01340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loc-Carrillo C, Abedon ST. 2011. Pros and cons of phage therapy. Bacteriophage 1:111–114. doi: 10.4161/bact.1.2.14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatfull GF, Vehring R. 2016. Respirable bacteriophage aerosols for the prevention and treatment of tuberculosis, p 277–292. In Hickey AJ, Misra A, Fourie PB (ed), Drug delivery systems for tuberculosis prevention and treatment. John Wiley & Sons, Ltd., Chichester, United Kingdom. [Google Scholar]
- 18.Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. 2011. Phage treatment of human infections. Bacteriophage 1:66–85. doi: 10.4161/bact.1.2.15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordillo Altamirano FL, Barr JJ. 2019. Phage therapy in the postantibiotic era. Clin Microbiol Rev 32:e00066-18. doi: 10.1128/CMR.00066-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones JV, Salka S, Bishop-Lilly KA, Young R, Hamilton T. 2017. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob Agents Chemother 61:e00954-17. doi: 10.1128/AAC.00954-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, Hatfull GF, Spencer H. 2019. Engineered bacteriophages for treatment of a patient with a disseminated drug resistant Mycobacterium abscessus. Nat Med 25:730–733. doi: 10.1038/s41591-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abedon ST. 2015. Phage therapy of pulmonary infections. Bacteriophage 5:e1020260. doi: 10.1080/21597081.2015.1020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golshahi L, Seed KD, Dennis JJ, Finlay WH. 2008. Toward modern inhalational bacteriophage therapy: nebulization of bacteriophages of Burkholderia cepacia complex. J Aerosol Med Pulm Drug Deliv 21:351–360. doi: 10.1089/jamp.2008.0701. [DOI] [PubMed] [Google Scholar]
- 24.Golshahi L, Lynch KH, Dennis JJ, Finlay WH. 2011. In vitro lung delivery of bacteriophages KS4-M and ϕKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J Appl Microbiol 110:106–117. doi: 10.1111/j.1365-2672.2010.04863.x. [DOI] [PubMed] [Google Scholar]
- 25.Matinkhoo S, Lynch KH, Dennis JJ, Finlay WH, Vehring R. 2011. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J Pharm Sci 100:5197–5205. doi: 10.1002/jps.22715. [DOI] [PubMed] [Google Scholar]
- 26.Hoe S, Boraey MA, Ivey JW, Finlay WH, Vehring R. 2014. Manufacturing and device options for the delivery of biotherapeutics. J Aerosol Med Pulm Drug Deliv 27:315–328. doi: 10.1089/jamp.2013.1090. [DOI] [PubMed] [Google Scholar]
- 27.Carrigy NB, Chang RY, Leung SSY, Harrison M, Petrova Z, Pope WH, Hatfull GF, Britton WJ, Chan H-K, Sauvageau D, Finlay WH, Vehring R. 2017. Anti-tuberculosis bacteriophage D29 delivery with a vibrating mesh nebulizer, jet nebulizer, and soft mist inhaler. Pharm Res 34:2084–2096. doi: 10.1007/s11095-017-2213-4. [DOI] [PubMed] [Google Scholar]
- 28.Leung SY, Parumasivam T, Gao FG, Carrigy NB, Vehring R, Finlay WH, Morales S, Britton WJ, Kutter E, Chan H-K. 2016. Production of inhalation phage powders using spray freeze drying and spray drying techniques for treatment of respiratory infections. Pharm Res 33:1486–1496. doi: 10.1007/s11095-016-1892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leung SSY, Parumasivam T, Gao FG, Carter EA, Carrigy NB, Vehring R, Finlay WH, Morales S, Britton WJ, Kutter E, Chan H-K. 2017. Effect of storage conditions on the stability of spray dried, inhalable bacteriophage powders. Int J Pharm 521:141–149. doi: 10.1016/j.ijpharm.2017.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung SSY, Parumasivam T, Nguyen A, Gengenbach T, Carter EA, Carrigy NB, Wang H, Vehring R, Finlay WH, Morales S, Britton WJ, Kutter E, Chan H-K. 2018. Effect of storage temperature on the stability of spray dried bacteriophage powders. Eur J Pharm Biopharm 127:213–222. doi: 10.1016/j.ejpb.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung SSY, Carrigy NB, Vehring R, Finlay WH, Morales S, Carter EA, Britton WJ, Kutter E, Chan H-K. 2019. Jet nebulization of bacteriophage with different tail morphologies–structural effects. Int J Pharm 664:322–326. doi: 10.1016/j.ijpharm.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 32.Muttil P, Wang C, Hickey AJ. 2009. Inhaled drug delivery for tuberculosis therapy. Pharm Res 26:2401–2416. doi: 10.1007/s11095-009-9957-4. [DOI] [PubMed] [Google Scholar]
- 33.Xiong X, Zhang HM, Wu TT, Xu L, Gan YL, Jiang LS, Zhang L, Guo SL. 2014. Titer dynamic analysis of D29 within MTB-infected macrophages and effect on immune function of macrophages. Exp Lung Res 40:86–98. doi: 10.3109/01902148.2013.873841. [DOI] [PubMed] [Google Scholar]
- 34.Hatfull GF. 2014. Mycobacteriophages: windows into tuberculosis. PLoS Pathog 10:e1003953. doi: 10.1371/journal.ppat.1003953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Froman S, Will DW, Bogen E. 1954. Bacteriophage active against virulent mycobacterium tuberculosis I. Isolation and activity. Am J Public Health Nations Health 44:1326–1333. doi: 10.2105/ajph.44.10.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nadithe V, Rahamatalla M, Finlay WH, Mercer JR, Samuel J. 2003. Evaluation of nose-only aerosol inhalation chamber and comparison of experimental results with mathematical simulation of aerosol deposition in mouse lungs. J Pharm Sci 92:1066–1076. doi: 10.1002/jps.10379. [DOI] [PubMed] [Google Scholar]
- 37.Leong BKJ, Coombs JK, Sabaitis CP, Rop DA, Aaron CS. 1998. Quantitative morphometric analysis of pulmonary deposition of aerosol particles inhaled via intratracheal nebulization, intratracheal instillation or nose-only inhalation in rats. J Appl Toxicol 18:149–160. doi:. [DOI] [PubMed] [Google Scholar]
- 38.Wolff RK. 2015. Toxicology studies for inhaled and nasal delivery. Mol Pharm 12:2688–2696. doi: 10.1021/acs.molpharmaceut.5b00146. [DOI] [PubMed] [Google Scholar]
- 39.Phillips JE. 2017. Inhaled efficacious dose translation from rodent to human: a retrospective analysis of clinical standards for respiratory diseases. Pharmacol Ther 178:141–147. doi: 10.1016/j.pharmthera.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Phalen RF. 1976. Inhalation exposure of animals. Environ Health Perspect 16:17–24. doi: 10.1289/ehp.761617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu K-Y, Yang W-H, Dong X-K, Cong L-M, Li N, Li Y, Wen Z-B, Yin Z, Lan Z-J, Li W-P, Li J-S. 2016. Inhalation study of mycobacteriophage D29 aerosol for mice by endotracheal route and nose-only exposure. J Aerosol Med Pulm Drug Deliv 29:393–405. doi: 10.1089/jamp.2015.1233. [DOI] [PubMed] [Google Scholar]
- 42.Semler DD, Goudie AD, Finlay WH, Dennis JJ. 2014. Aerosol phage therapy efficacy in Burkholderia cepacia complex respiratory infections. Antimicrob Agents Chemother 58:4005–4013. doi: 10.1128/AAC.02388-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kutter E, Sulakvelidze A. 2005. Bacteriophages: biology and applications. CRC Press, Boca Raton, FL. [Google Scholar]
- 44.Hoe S, Semler DD, Goudie AD, Lynch KH, Matinkhoo S, Finlay WH, Dennis JJ, Vehring R. 2013. Respirable bacteriophages for the treatment of bacterial lung infections. J Aerosol Med Pulm Drug Deliv 26:317–335. doi: 10.1089/jamp.2012.1001. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science SEA-PHAGES Program, Modlin RL, Hendrix RW, Hatfull GF. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434:187–201. doi: 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanjee Z, Amico KR, Li F, Mbolekwa K, Moll AP, Friedland GH. 2012. Tuberculosis infection control in a high drug-resistance setting in rural South Africa: information, motivation, and behavioral skills. J Infect Public Health 5:67–81. doi: 10.1016/j.jiph.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Roach DR, Leung CY, Henry M, Morello E, Singh D, di Santo JP, Weitz JS, Debarbieux L. 2017. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 22:38–47. doi: 10.1016/j.chom.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Bowen LE, Rivers K, Trombley JE, Bohannon JK, Li SX, Boydston JA, Eichelberger MC. 2012. Development of a murine nose-only inhalation model of influenza: comparison of disease caused by instilled and inhaled A/PR/8/34. Front Cell Infect Microbiol 2:74. doi: 10.3389/fcimb.2012.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler AJ, Ganji AR. 2010. Introduction to engineering experimentation, 3rd ed Pearson Higher Education, Upper Saddle River, NJ. [Google Scholar]
- 50.Ochs M, Nyengaard JR, Jung A, Knudsen L, Voigt M, Wahlers T, Richter J, Gundersen HJ. 2004. The number of alveoli in the human lung. Am J Respir Crit Care Med 169:120–124. doi: 10.1164/rccm.200308-1107OC. [DOI] [PubMed] [Google Scholar]
- 51.Guirado E, Mbawuike U, Keiser TL, Arcos J, Azad AK, Wang S-H, Schlesinger LS. 2015. Characterization of host and microbial determinants in individuals with latent tuberculosis infection using a human granuloma model. mBio 6:e02537-14. doi: 10.1128/mBio.02537-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Żaczek M, Łusiak-Szelachowska M, Jończyk-Matysiak E, Weber-Dąbrowska B, Międzybrodzki R, Owczarek B, Kopciuch A, Fortuna W, Rogóż P, Górski A. 2016. Antibody production in response to staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front Microbiol 7:1681. doi: 10.3389/fmicb.2016.01681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Belleghem JD, Dąbrowska K, Vaneechoutte M, Barr JJ, Bollyky PL. 2018. Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 11:10. doi: 10.3390/v11010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fauvart M, De Groote VN, Michiels J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60:699–709. doi: 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 55.Fairchild GA. 1972. Measurement of respiratory volume for virus retention studies in mice. Appl Microbiol 24:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin AR, Ang A, Katz IM, Häussermann S, Caillibotte G, Texereau J. 2011. An in vitro assessment of aerosol delivery through patient breathing circuits used with medical air or a helium-oxygen mixture. J Aerosol Med Pulm Drug Deliv 24:225–234. doi: 10.1089/jamp.2010.0871. [DOI] [PubMed] [Google Scholar]
- 57.Kuehl PJ, Anderson TL, Candelaria G, Gershman B, Harlin K, Hesterman JY, Holmes T, Hoppin J, Lackas C, Norenberg JP, Yu H, McDonald JD. 2012. Regional particle size dependent deposition of inhaled aerosol in rats and mice. Inhal Toxicol 24:27–35. doi: 10.3109/08958378.2011.632787. [DOI] [PubMed] [Google Scholar]
- 58.Soutiere SE, Tankersley CG, Mitzner W. 2004. Differences in alveolar size in inbred mouse strains. Respir Physiol Neurobiol 140:283–291. doi: 10.1016/j.resp.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Larsen S, Baldwin S, Orr M, Reese V, Pecor T, Granger B, Dubois Cauwelaert N, Podell B, Coler R. 2018. Enhanced anti-Mycobacterium tuberculosis immunity over time with combined drug and immunotherapy treatment. Vaccines (Basel) 6:30. doi: 10.3390/vaccines6020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertholet S, Ireton GC, Ordway DJ, Windish HP, Pine SO, Kahn M, Phan T, Orme IM, Vedvick TS, Baldwin SL, Coler RN, Reed SG. 2010. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci Transl Med 2:53ra74. doi: 10.1126/scitranslmed.3001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saini D, Hopkins GW, Seay SA, Chen CJ, Perley CC, Click EM, Frothingham R. 2012. Ultra-low dose of Mycobacterium tuberculosis aerosol creates partial infection in mice. Tuberculosis (Edinb) 92:160–165. doi: 10.1016/j.tube.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gern B, Plumlee C, Gerner M, Urdahl K. 2017. Investigating immune correlates of protection to tuberculosis using an ultra-low dose infection in a mouse model. Open Forum Infect Dis 4:S47–S48. doi: 10.1093/ofid/ofx162.112. [DOI] [Google Scholar]