

Abstract
Resistance to inhibitors of cholinesterase 8A (Ric8A) protein is an important G protein–coupled receptor (GPCR)-independent regulator of G protein α-subunits (Gα), acting as a guanine nucleotide exchange factor (GEF) and a chaperone. Insights into the complex between Ric8A and Gα hold the key to understanding the mechanisms underlying noncanonical activation of G-protein signaling as well as the folding of nascent Gα proteins. Here, we examined the structure of the complex of Ric8A with minimized Gαi (miniGαi) in solution by small-angle X-ray scattering (SAXS) and exploited the scattering profile in modeling of the Ric8A/miniGαi complex by steered molecular dynamics (SMD) simulations. A small set of models of the complex featured minimal clash scores, excellent agreement with the experimental SAXS data, and a large-scale rearrangement of the signal-transducing α5-helix of Gα away from its β-sheet core. The resulting interface involved the Gα α5-helix bound to the concave surface of Ric8A and the Gα β-sheet that wraps around the C-terminal part of the Ric8A armadillo domain, leading to a severe disruption of the GDP-binding site. Further modeling of the flexible C-terminal tail of Ric8A indicated that it interacts with the effector surface of Gα. This smaller interface may enable the Ric8A-bound Gα to interact with GTP. The two-interface interaction with Gα described here distinguishes Ric8A from GPCRs and non-GPCR regulators of G-protein signaling.
Keywords: G protein, G protein-coupled receptor (GPCR), guanine nucleotide exchange factor (GEF), chaperone, small-angle X-ray scattering (SAXS), molecular dynamics, signal transduction, Ric8
Introduction
The resistance to inhibitors of cholinesterase 8 (Ric8)2 proteins are guanine nucleotide exchange factors (GEFs) and chaperones for G protein α-subunits (Gα) (1–4). The Ric8A isoform regulates a diverse subset of Gα subunits, including Gαi, Gαq, and Gα12/13, and in this capacity it is essential to multiple cellular signaling pathways, including asymmetric cell division and synaptic transmission (5–7). Ric8 selectively interacts with the GDP-bound state of Gα and induces release of GDP. A stable intermediate complex of Ric8 and nucleotide-free Gα is formed and persists until Gα binds GTP and dissociates from Ric8 (2, 8). Although the mechanism of Ric8A GEF activity is thought to be very different from that of G protein–coupled receptor (GPCR)-dependent activation of heterotrimeric G proteins (Gαβγ), one striking parallel has emerged (i.e. both Ric8A and GPCRs interact with the C termini of Gα, and transmission of the GPCR-induced activation signal involves the Gα α5-helix) (9–13). In particular, the largest conformational change in Gα is an outward translation with rotation of the α5-helix that disrupts the guanine ring binding loop β6-α5 of Gα (11). The first structural clues to the mechanism of Gα activation by Ric8A have been provided by the recent crystal structure of the complex of Ric8A with the C-terminal fragment of Gα corresponding to the α5-helix (10). Based on this structure, we modeled the complex of Ric8A with miniGαi and the full-length Gαi subunit (10). The key premise for the model was the observation that the steric overlap between Ric8A and Gα is markedly reduced when a GPCR-bound conformation of Gα was used in the modeling that involved superimposition of the α5-helix (10). The remaining clashes in the model were resolved with an assumption that Ric8A adopts an open conformation to accommodate the Ras-like domain (RD) of Gα. Indeed, the steered molecular dynamics (SMD) simulations with force applied to the Ric8A region that clashed with Gα readily yield such an “open” conformation (10). In this study, we examined the solution structure of the Ric8A/miniGαi complex by small-angle X-ray scattering (SAXS) to evaluate and/or refine this model. Unexpectedly, the experimental SAXS profile of the Ric8A/miniGαi complex revealed a very poor agreement with the theoretical SAXS profile of the model, necessitating its revision. We explored the possibility that the complex formation leads to conformational changes in Gα with SMD simulations where force is applied to the miniGαi α5-helix. Thus, we obtained a group of similar conformations of miniGαi that show no significant clashes in modeling of the Ric8A/miniGαi complex. Importantly, the resulting models are in excellent agreement with the experimental SAXS profile, and they feature large rearrangement of the Gα α5-helix.
Results
Analysis of the Ric8A/miniGαi complex solution structure by SAXS
We utilized minimized Gαi lacking flexible parts of the protein, the helical domain (HD) and the N-terminal αN-helix (ΔαN-miniGαi), in SAXS experiments to limit conformational uncertainty. A highly purified sample of the Ric8A1–492/ΔαN-miniGαi complex was analyzed by size-exclusion chromatography (SEC)-SAXS (Fig. 1). Previously, we generated two models of the Ric8A1–492/miniGαi complex that differ in the position of the distal C-terminal tail of Ric8A (10). Comparison of the theoretical SAXS profiles of the two corresponding Ric8A1–492/ΔαN-miniGαi models 1 and 2 with the experimental SAXS profile revealed poor fits for both models (Fig. 2). We also evaluated the theoretical SAXS profile of the Ric8A1–452/ΔαN-miniGαi model (previous SMD model) lacking residues 453–492 of Ric8A, which served as a template for models 1 and 2 (Fig. 2A). The quality of the fit (χ2 value) remained poor, suggesting that the disagreement of the Ric8A1–492/ΔαN-miniGαi models with the experimental SAXS data was not due to the incorrectly modeled residues 453–492 of Ric8A. The above-mentioned models of Ric8A1–492/ΔαN-miniGαi were derived using the “open” conformation of Ric8A1–452 that resulted from an SMD simulation with force applied to the clashing part of the protein (10). In light of the disagreement of the model for the complex with the SAXS data, we re-examined the validity of “open” conformation of Ric8A by reverting to its conformation observed in the crystal structures (10, 14). The crystal structure conformation leads to an improbable model in which the two proteins extensively clash (“clash” model) (Fig. 2A). However, the theoretical SAXS profile of the clashing model of Ric8A1–452/ΔαN-miniGαi complex agreed with the experimental data somewhat better than the models with the “open” conformation of Ric8A (Fig. 2B). Thus, the “open” conformation of Ric8A is not supported by the experimental SAXS profile of the Ric8A1–492/ΔαN-miniGαi complex.
Figure 1.

Analysis of the Ric8A1–492/ΔαN-miniGαi complex by SAXS. A, SEC elution profile of the Ric8A1–492/ΔαN-miniGαi complex during purification. Peak 1, Ric8A1–492/ΔαN-miniGαi complex; peak 2, free ΔαN-miniGαi. Inset, Coomassie-stained gel. Arrows, positions of 25 and 50 kDa markers. B, elution profile of the Ric8A1–492/ΔαN-miniGαi complex during SEC-SAXS. C, time evolution of the integrated SAXS intensity and radius of gyration (Rg). Blue box, region for the average SAXS profile. D, experimental SAXS profile of Ric8A1–492-ΔαN-miniGαi. Inset, Guinier plot for the low q region (q·Rg < 1.3); Rg = 32.3 Å.
Figure 2.
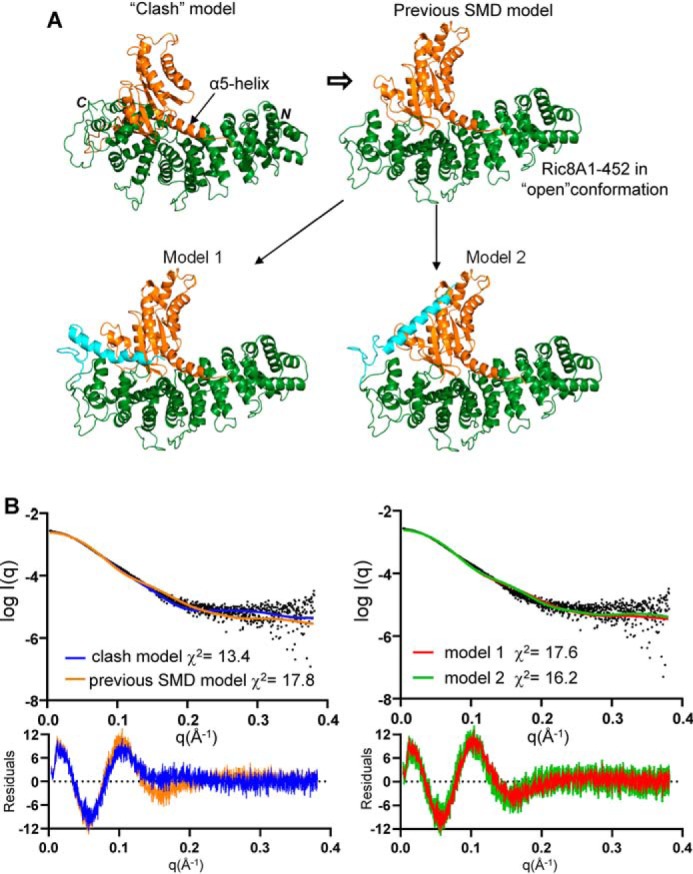
Models of the Ric8A complexes with ΔαN-miniGαi and their theoretical SAXS profiles. A, a “clash” model of the Ric8A1–452/ΔαN-miniGαi complex derived from the superimposition of the Gα α5-helix based on the Ric8A1–492/MBP-Gαt327–350 structure (PDB entry 6N85). The previous SMD model of the Ric8A1–452/ΔαN-miniGαi complex was based on the “open” conformation of Ric8A1–452 (10). Shown are FloppyTail models 1 and 2 of the Ric8A1–492/ΔαN-miniGαi complex (10). Green, Ric8A1–452; orange, ΔαN-miniGαi; cyan, modeled distal C-terminal tail of Ric8A (residues 453–492). B, fits of the theoretical SAXS profiles of the models in A to the experimental SAXS profile of the Ric8A1–492/ΔαN-miniGαi complex.
SAXS-directed modeling of the Ric8A/miniGαi complex indicates rearrangement of the Gα α5-helix
To avoid clashes, and barring the “open” conformation of Ric8A, conformational changes more extensive than the GPCR-induced changes would have to occur in Gα. To simulate the forces that act on the Gα α5-helix upon binding of Ric8A, we conducted an SMD simulation of ΔαN-miniGαi that was further truncated by 5 N-terminal residues with conformational ambiguity (ΔΔαN-miniGαi). The 12-ns SMD trajectory yielded 300 conformations of ΔΔαN-miniGαi (Fig. 3A). Models of the Ric8A1–452/ΔΔαN-miniGαi complex were constructed for each of the SMD conformations of ΔΔαN-miniGαi. Top SMD models of the Ric8A1–452/ΔΔαN-miniGαi complex were selected based on the combinations of low clash score and agreement with the experimental SAXS profile (low χ2 value) (Fig. 3, B and C). Compared with the starting “clashing” model (clash score 100.1, χ2 = 13.4), the top six SMD models displayed markedly improved clash scores (<25) and χ2 values (<2.0) (Fig. 3, E and F). All of these models were similar and featured a large hinge-like movement of the Gα α5-helix away from the β-sheet core of the RD (Fig. 3D). This movement allowed positioning the Gα β-sheet on top of the C-terminal portion of Ric8A1–452 and minimized clashes between the two proteins (Fig. 3E). Residual clashes in the top SMD models were readily eliminated with energy minimization of the model structures.
Figure 3.

Modeling with steered molecular dynamics simulation. A, root mean square deviation (RMSD) plot for the 12-ns SMD of ΔΔαN-miniGαi. B, clash scores for the models of the Ric8A1–452/ΔΔαN-miniGαi complex were calculated using MolProbity. C, χ2 values for the fits of theoretical SAXS profiles of models to the experimental SAXS data. Among the top six SMD models of ΔΔαN-miniGαi (D) and the corresponding models of the Ric8A1–452/ΔΔαN-miniGαi complex (clash score < 25, χ2 value < 2) (E), models a and b featured the lowest χ2 value (1.31) and clash score (13.5), respectively. F, fits of the theoretical SAXS profiles of the models a and b to the experimental SAXS profile of the Ric8A1–492/ΔαN-miniGαi complex.
Modeling of the C-terminal tail of Ric8A in the complex with Gα
Evidence suggests that the flexible C-terminal tail of Ric8A is important for its GEF and chaperone activities (9, 10, 15). We performed FloppyTail modeling of the C-terminal residues 453–492 using a top SMD Ric8A1–452/ΔΔαN-miniGαi model with the best fit to the SAXS data (χ2 = 1.31) as template (16). In total, 2446 FloppyTail models were generated using linear distance constraints of 30 Å for the previously identified cross-linked pairs Ric8A-K488/miniGαi-K122 and K462/miniGαi-K21 (10, 17). To allow use of the latter cross-link constraint in the modeling, the N terminus of ΔΔN-miniGαi was extended to that of ΔαN-miniGαi. More stringent surface distance constraints were then applied in the initial model selection. Of 2446 models, 966 were consistent with a surface distance constraint of 30 Å for the Ric8A-K488/ΔαN-miniGαi-K122 pair (distance d1). Considering a degree of uncertainty in position of ΔαN-miniGαi-K21, a more relaxed surface distance constraint of 35 Å was applied for the Ric8A-K462/ΔαN-miniGαi-K21 pair (distance d2), yielding 287 models. This pool of models was further narrowed to 129 models that satisfied a criterion of χ2 < 2. The top 50% of these models (65 models) in terms of energy score were clustered, yielding three main clusters, cluster 1 (23), cluster 2 (15), and cluster 3 (10) (Fig. 4). In cluster 1 models, the C-terminal tail of Ric8A wraps around ΔαN-miniGαi, with the C-terminal α-helix (residues 471–490) lying near the groove between Gα switch II and α3-helix (i.e. in the vicinity of the effector surface of Gα). In cluster 2 and 3 models, the C-terminal helix of Ric8A is positioned near the Gα α4-helix and the switch I/II regions, respectively (Fig. 4). The mean values for the key model parameters (χ2, energy score, d1, and d2) showed that cluster 1 models had the best average energy score, whereas cluster 2 models best fit the SAXS data (Fig. 4E). However, cluster 2 models had an average d2 exceeding 30 Å, indicating that these models are not probable. Therefore, we favor cluster 1 models, although cluster 3 models cannot be excluded. Both cluster 1 and cluster 3 models are consistent with an earlier analysis of the Ric8A/Gα complex using hydrogen-deuterium exchange–MS (15). Subsequently, we modeled the complex of Ric8A with the full-length Gαi (Fig. 4D). In this model, the HD of Gαi does not interact with Ric8A, and it can sample all conformations within the known HD/RD distance distribution (18).
Figure 4.

Models of the Ric8A1–492/ΔαN-miniGαi complex. Shown are three representative FloppyTail models from each of the clusters, cluster 1 (A), 2 (B), and 3 (C). The switch II region of Gαi is shown in cyan; the modeled distal C-terminal tail of Ric8A (residues 453–492) is shown in magenta, gray, or light green. D, model of the Ric8A1–492/Gαi complex. Arrows, HD (gray) and RD domains and the αN-helix of Gαi. The distal C-terminal tail of Ric8A in a representative conformation from cluster 1 is shown in magenta. E, parameters of the FloppyTail models of the Ric8A1–492/ΔαN-miniGαi complex from clusters 1–3 (mean ± S.D.).
As an additional tool to evaluate our models of the Ric8A1–492/ΔαN-miniGαi complex, an ab initio electron density map of the complex was reconstructed from the SAXS data using DENSS (19). This reconstruction yielded a 32 Å resolution map, which closely correlated (correlation ∼0.94) with the electron density maps obtained for the cluster 1 models and filtered to the same resolution (Fig. 5).
Figure 5.
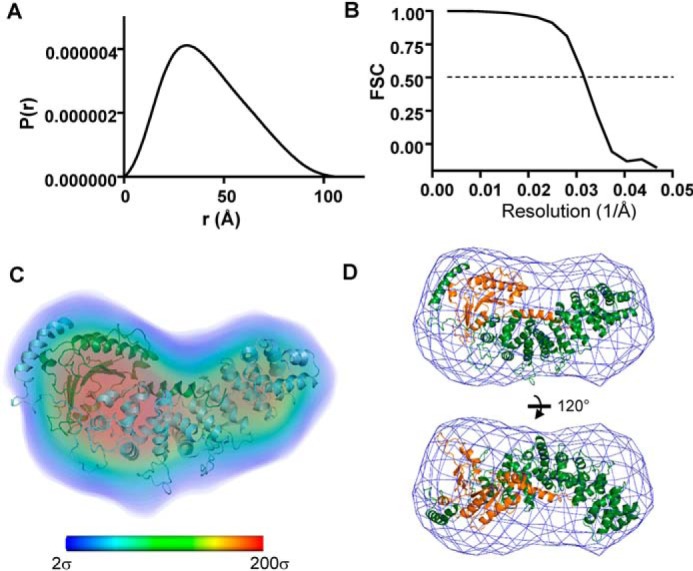
DENSS electron density map of the Ric8A1–492/ΔαN-miniGαi complex. A, the pairwise distance distribution function P(r) was calculated from the SAXS data using GNOM (Dmax = 107 Å) and served as an input into the DENSS reconstruction. B, Fourier shell correlation (FSC) curve for the DENSS reconstruction estimates resolution at 32 Å. C, electron density is shown as volume colored according to density (bar, electron density values in σ from 2 to 200 (blue to red)). Shown is a representative model of the Ric8A1–492/ΔαN-miniGαi complex from cluster 1 aligned with the DENSS map. D, DENSS electron density envelope contoured at 2σ and aligned with the model in C.
Discussion
The analysis of the structure of the Ric8A1–492/ΔαN-miniGαi complex in solution by SAXS reported in this study led us to propose a new model of this complex. The starting point in our modeling lies in the established location of the Gα α5-helix with respect to Ric8A (10). Although all known conformations of Gα produce extensive clashes with Ric8A when modeled according to the position of the α5-helix, the clashes are less severe using the GPCR-bound conformation. To avoid steric overlap, additional conformational changes must take place either in Ric8A, Gα, or both. The previously proposed model hypothesized conformational changes in Gα that are similar to those induced by agonist-bound GPCRs and additional changes in the armadillo core domain of Ric8A that lead to a more stretched “open” conformation (10). Such a model appeared to be the most parsimonious, as the transition of the Gα subunit to its GPCR-bound conformation is well understood, and conformational flexibility of the N- and C-terminal parts of armadillo-fold proteins has been described (11, 20–23). However, the experimental scattering profile of the Ric8A1–492/ΔαN-miniGαi complex revealed strong disagreement with the theoretical profile of the model based on the “open” conformation of Ric8A, thereby refuting it. Accordingly, we explored the possibility of further conformational changes in Gα that extend beyond those induced by GPCRs using the SMD simulation mimicking potential Ric8A-induced changes in Gα. Remarkably, low clash scores and low χ2 values converged in a small set of models with similar SMD conformations of ΔαN-miniGαi. The key feature of these Gα conformations is a large dislocation of the Gα α5-helix, whereby the latter becomes completely detached from the hydrophobic β-sheet core of Gα. The Gα β-sheet core is then stabilized by interactions with the C-terminal part of the Ric8A armadillo domain.
Two routes have been shown to transmit GPCR-induced conformational changes from the Gα α5-helix to the GDP binding site: disruption of the guanine nucleotide-binding loop β6-α5 and rearrangement of the interfaces between α5, α1, and β2-β3 that causes destabilization of the phosphate-binding β1-α1 loop (11, 21, 22). The model of the Ric8A/Gα complex suggests a major disruption of the β6-α5 loop, and a disordering of the β1-α1 loop can be predicted as well.
The modeling of the flexible C-terminal tail of Ric8A (residues 453–492) in complex with Gα provides further insight into the mechanism of Ric8A GEF and chaperone activities, as this region is critical for both activities of the protein (9, 10, 15). Interestingly, whereas our SAXS analyses argue against significant conformational changes in the core armadillo domain of Ric8A, they support a large conformational change in the flexible distal portion of the Ric8A C-terminal tail on binding of Gα. The SAXS analysis of the Ric8A/ΔαN-miniGαi complex reveals a smaller Rg value than that for the apo-Ric8A (10), which is indicative of immobilization of the extended floppy tail into a more compact conformation. Such immobilization occurs when, according to the favored cluster of models, the C-terminal α-helix of Ric8A interacts with the groove between the switch II region and α3-helix of Gα. This is a conformation-sensitive surface that Gα subunits utilize to bind multiple partners. At this surface, GαGTP binds effector molecules, whereas GαGDP interacts with GoLoco/GPR proteins or guanine-nucleotide exchange modulators, such as GIV/Girdin (24–29).
Our model and existing biochemical evidence suggest two not mutually exclusive roles for the distal C-terminal tail of Ric8A, Ric8A452–492. The C-tail of Ric8A may serve as a “hook” that helps to hold Gα while the α5-helix is being pulled away from the β-sheet core. Thus, it may facilitate transition to a stable intermediate complex of Ric8A with nucleotide-free Gα. Another role, and possibly the key role of the Ric8A C-tail is to promote binding of GTP to Gα. Compared with GPCRs, the interface of Ric8A with Gα is more extensive, and the disruption of the nucleotide-binding site is more severe. It then becomes imperative to stabilize the structural elements of Gα involved in the interaction with the γ-phosphate of GTP, which would enable GTP binding. By interacting with the conformation-sensitive switch II/α3-helix region, the C-tail of Ric8A may nudge the switch II region and its β3-α2 loop toward the GTP γ-phosphate binding position with cooperative changes in the switch I region, all of which would promote binding of GTP.
In summary, this study suggests a novel and unusual type of interface between Gα and its GPCR-independent GEF. The main interface involves the α5-helix and the β-sheet core of Gα that interact extensively with the concave surface of the Ric8A armadillo core and its C-terminal part, respectively. This interface is made possible by a large-scale dislocation the Gα α5-helix. A second smaller interface is between the distal C-terminal tail of Ric8A and the effector surface of Gα. The two interfaces on the opposite sides of Gα appear to be a unique feature of Ric8A as a GEF, and they may be critical to its function as a Gα chaperone.
Experimental procedures
Protein expression and purification
Bovine Ric8A1–492 was expressed and purified as described previously (10). Sequence encoding ΔαN-miniGαi (which starts with Gαi1 residue Glu-28 following Met) was amplified from the miniGαi vector (10). ΔαN-miniGαi was cloned into the modified pET21a vector containing an N-terminal His6 tag followed by the B1 domain of streptococcal protein G (GB1) tag and a tobacco etch virus cleavage site. The ΔαN-miniGαi construct was expressed and purified as described previously for miniGαi. The His6-GB1 tag was removed from ΔαN-miniGαi by adding tobacco etch virus protease in a 1:50 molar ratio to the protein eluted from cobalt-nitrilotriacetic acid resin. The sample was incubated overnight at 4 °C and purified by SEC using a HiLoad 16/600 Superdex 75-pg column equilibrated with 20 mm Tris-HCl (pH 8.0) buffer containing 150 mm KCl, 10% glycerol, 20 mm MgSO4, 10 μm GDP, and 1 mm TCEP.
For the small-angle X-ray scattering data collection, Ric8A1–492/ΔαN-miniGαi complex was prepared by mixing Ric8A with ΔαN-miniGαi at a 1:1.5 molar ratio. The complex was purified by SEC using a Superdex 200 10/300 GL (GE Healthcare) column equilibrated with 20 mm Tris-HCl (pH 8.0) buffer containing 150 mm KCl, 5% glycerol, and 1 mm TCEP. This procedure removed excess ΔαN-miniGαi, ensuring 1:1 stoichiometry of the complex.
SAXS
SAXS data were collected at the Bio-CAT beamline 18-ID-D at the Advanced Photon Source (Argonne, IL) using an in-line SEC-SAXS configuration (30) with a Superdex 200 column (GE Healthcare). A 250-μl volume of 10 mg/ml sample in 20 mm Tris, 150 mm KCl, 5% glycerol, 1 mm TCEP, pH 7.5, buffer was loaded onto the column at a flow rate of 0.9 ml/min. SAXS data were collected as described previously (10) and deposited in the Small Angle Scattering Biological Data Bank (ID SASDGB6). BioXTAS RAW and ATSAS 2.8 were used for SAXS data reduction and analysis (31, 32). The pair distribution function was calculated with GNOM, and an ab initio electron density map was calculated from the SAXS data with DENSS (19, 32).
SMD simulations
SMD simulations was performed on a homology model of miniGαi lacking conformationally ambiguous residues from the N-terminal αN-helix and αN-β1 loop (ΔΔαN-miniGαi, which starts with Gαi1 residue Glu-33). The structure of GPCR-bound miniGαi served as a template for the homology model (PDB entry 5G53) (33). The structure file for ΔΔαN-miniGαi was prepared using VMD (34) and the plugin QwikMD (35). The simulations employed the NAMD molecular dynamics package (36) and the CHARMM36 force field (37) as described previously (10). The SMD simulations (38) of constant velocity stretching (SMD-CV protocol) employing a pulling speed of 2.5 Å/ns and a harmonic constraint force of 7.0 kcal/mol/Å2 were performed for 12.0 ns. In this step, SMD was employed by harmonically restraining the positions of ΔΔαN-miniGαi corresponding to Glu-33 and Val-34 of Gαi1 and moving second restraint residues corresponding to Gαi1 residues 329–354. The force direction was defined by the axis between the center of mass of Ric8A atoms that clash with ΔΔαN-miniGαi and the center of mass of the Gαi α5-helix.
Modeling of the Ric8A1–452/ΔΔαN-miniGαi complex
The FloppyTail model of Ric8A1–452 was generated previously (10). The SMD trajectory yielded 300 conformations of ΔΔαN-miniGαi. Models of the Ric8A1–452/ΔΔαN-miniGαi complex were constructed for each of these conformations by superimposition of the α5-helix of ΔΔαN-miniGαi with the Gα α5-helical residues from the structure of the Ric8A1–492/MBP-Gαt327–350 complex (PDB entry 6N85). Clash scores for the complex models were calculated using MolProbity to identify models with the least steric clash (39). The Crysol program was used to generate and compare fits of theoretical SAXS profiles of the models to experimental SAXS data (χ2 values) (40). Top models of the Ric8A1–452/ΔΔαN-miniGαi complex were energy-minimized using YASARA Structure 18.2.7.
FloppyTail modeling
The model of the Ric8A1–452/ΔΔαN-miniGαi complex with the best fit to the experimental SAXS data was selected among the top six models with low clash scores and low χ2 values as a template for the FloppyTail simulations (16) of the Rosetta software suite. Residues Ric8A453–492 in extended conformation were added to the Ric8A1–452/ΔΔαN-miniGαi model. Residues 471–490 were kept helical, based on the secondary structure prediction (10). Also, the N terminus of ΔΔαN-miniGαi in the complex was extended by 5 residues to that of ΔαN-miniGαi using YASARA. The FloppyTail protocol was similar to that described previously (10). Two experimental linear distance constraints of 30 Å for the previously identified cross-linked pairs Ric8A-K488/miniGαi-K122, and Ric8A-K462/miniGαi-K21 were used during the simulation (10). In addition to energy scores from Rosetta and χ2 values, surface distances between cross-linked residues were used in model selection. The solvent-accessible surface distances were calculated using Jwalk as a more accurate estimate for the feasibility of cross-linking between two residues (41). Models of the Ric8A1–492/ΔαN-miniGαi complex were aligned with the DENSS ab initio electron density map using UCSF Chimera (42). The model of the Ric8A1–492/Gαi complex was generated by superimposition of the Gαi structure (PDB 6CMO) (43) lacking the α5-helix with ΔαN-miniGαi.
Author contributions
D. S. and N. O. A. formal analysis; D. S. and N. O. A. investigation; D. S. methodology; D. S. and N. O. A. writing-original draft; N. O. A. conceptualization; N. O. A. funding acquisition.
Acknowledgments
We thank Lokesh Gakhar for discussion and Srinivas Chakravarty (BioCAT facility, Advanced Photon Source) for help in SAXS data collection. This research used resources of the Advanced Photon Source, Argonne National Laboratory.
This work was supported by NEI, National Institutes of Health, Grant RO1 EY-12682 (to N. O. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SAXS data were deposited in the Small Angle Scattering Biological Data Bank (SASBDB) under accession number SASDGB6.
- Ric8
- resistance to inhibitors of cholinesterase 8
- GEF
- guanine nucleotide exchange factor
- GPCR
- G protein–coupled receptor
- miniGαi
- minimized Gαi
- RD
- Ras-like domain
- SMD
- steered molecular dynamics
- SAXS
- small-angle X-ray scattering
- HD
- helical domain
- TCEP
- tris(2-carboxyethyl)phosphine
- SEC
- size-exclusion chromatography.
References
- 1. Miller K. G., Emerson M. D., McManus J. R., and Rand J. B. (2000) RIC-8 (Synembryn): a novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron 27, 289–299 10.1016/S0896-6273(00)00037-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tall G. G., Krumins A. M., and Gilman A. G. (2003) Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 10.1074/jbc.M211862200 [DOI] [PubMed] [Google Scholar]
- 3. Tall G. G. (2013) Ric-8 regulation of heterotrimeric G proteins. J. Recept. Signal Transduct. Res. 33, 139–143 10.3109/10799893.2013.763828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan P., Thomas C. J., Sprang S. R., and Tall G. G. (2013) Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein α subunits. Proc. Natl. Acad. Sci. U.S.A. 110, 3794–3799 10.1073/pnas.1220943110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tall G. G., and Gilman A. G. (2005) Resistance to inhibitors of cholinesterase 8A catalyzes release of Gαi-GTP and nuclear mitotic apparatus protein (NuMA) from NuMA/LGN/Gαi-GDP complexes. Proc. Natl. Acad. Sci. U.S.A. 102, 16584–16589 10.1073/pnas.0508306102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuzaki F. (2005) Drosophila G-protein signalling: intricate roles for Ric-8? Nat. Cell Biol. 7, 1047–1049 10.1038/ncb1105-1047 [DOI] [PubMed] [Google Scholar]
- 7. Wilkie T. M., and Kinch L. (2005) New roles for Gα and RGS proteins: communication continues despite pulling sisters apart. Curr. Biol. 15, R843–R854 10.1016/j.cub.2005.10.008 [DOI] [PubMed] [Google Scholar]
- 8. Papasergi M. M., Patel B. R., and Tall G. G. (2015) The G protein α chaperone Ric-8 as a potential therapeutic target. Mol. Pharmacol. 87, 52–63 10.1124/mol.114.094664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas C. J., Briknarová K., Hilmer J. K., Movahed N., Bothner B., Sumida J. P., Tall G. G., and Sprang S. R. (2011) The nucleotide exchange factor Ric-8A is a chaperone for the conformationally dynamic nucleotide-free state of Gαi1. PLoS One 6, e23197 10.1371/journal.pone.0023197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Srivastava D., Gakhar L., and Artemyev N. O. (2019) Structural underpinnings of Ric8A function as a G-protein α-subunit chaperone and guanine-nucleotide exchange factor. Nat. Commun. 10, 3084 10.1038/s41467-019-11088-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oldham W. M., and Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 10.1038/nrm2299 [DOI] [PubMed] [Google Scholar]
- 13. Natochin M., Moussaif M., and Artemyev N. O. (2001) Probing the mechanism of rhodopsin-catalyzed transducin activation. J. Neurochem. 77, 202–210 10.1046/j.1471-4159.2001.t01-1-00221.x [DOI] [PubMed] [Google Scholar]
- 14. Zeng B., Mou T. C., Doukov T. I., Steiner A., Yu W., Papasergi-Scott M., Tall G. G., Hagn F., and Sprang S. R. (2019) Structure, function, and dynamics of the Gα binding domain of Ric-8A. Structure 27, 1137–1147.e5 10.1016/j.str.2019.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kant R., Zeng B., Thomas C. J., Bothner B., and Sprang S. R. (2016) Ric-8A, a G protein chaperone with nucleotide exchange activity induces long-range secondary structure changes in Gα. Elife 5, e19238 10.7554/eLife.19238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleiger G., Saha A., Lewis S., Kuhlman B., and Deshaies R. J. (2009) Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell 139, 957–968 10.1016/j.cell.2009.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Merkley E. D., Rysavy S., Kahraman A., Hafen R. P., Daggett V., and Adkins J. N. (2014) Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759 10.1002/pro.2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Eps N., Thomas C. J., Hubbell W. L., and Sprang S. R. (2015) The guanine nucleotide exchange factor Ric-8A induces domain separation and Ras domain plasticity in Gαi1. Proc. Natl. Acad. Sci. U.S.A. 112, 1404–1409 10.1073/pnas.1423878112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grant T. D. (2018) Ab initio electron density determination directly from solution scattering data. Nat. Methods 15, 191–193 10.1038/nmeth.4581 [DOI] [PubMed] [Google Scholar]
- 20. Chung K. Y., Rasmussen S. G., Liu T., Li S., DeVree B. T., Chae P. S., Calinski D., Kobilka B. K., Woods V. L. Jr., and Sunahara R. K. (2011) Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615 10.1038/nature10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dror R. O., Mildorf T. J., Hilger D., Manglik A., Borhani D. W., Arlow D. H., Philippsen A., Villanueva N., Yang Z., Lerch M. T., Hubbell W. L., Kobilka B. K., Sunahara R. K., and Shaw D. E. (2015) Signal transduction. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365 10.1126/science.aaa5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaya A. I., Lokits A. D., Gilbert J. A., Iverson T. M., Meiler J., and Hamm H. E. (2016) A conserved hydrophobic core in Gαi1 regulates G protein activation and release from activated receptor. J. Biol. Chem. 291, 19674–19686 10.1074/jbc.M116.745513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bujalowski P. J., Nicholls P., Barral J. M., and Oberhauser A. F. (2015) Thermally-induced structural changes in an armadillo repeat protein suggest a novel thermosensor mechanism in a molecular chaperone. FEBS Lett. 589, 123–130 10.1016/j.febslet.2014.11.034 [DOI] [PubMed] [Google Scholar]
- 24. Sprang S. R., Chen Z., and Du X. (2007) Structural basis of effector regulation and signal termination in heterotrimeric Gα proteins. Adv. Protein Chem. 74, 1–65 10.1016/S0065-3233(07)74001-9 [DOI] [PubMed] [Google Scholar]
- 25. Kimple R. J., Kimple M. E., Betts L., Sondek J., and Siderovski D. P. (2002) Structural determinants for GoLoco-induced inhibition of nucleotide release by Gα subunits. Nature 416, 878–881 10.1038/416878a [DOI] [PubMed] [Google Scholar]
- 26. Kalogriopoulos N. A., Rees S. D., Ngo T., Kopcho N. J., Ilatovskiy A. V., Sun N., Komives E. A., Chang G., Ghosh P., and Kufareva I. (2019) Structural basis for GPCR-independent activation of heterotrimeric Gi proteins. Proc. Natl. Acad. Sci. U.S.A. 116, 16394–16403 10.1073/pnas.1906658116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blumer J. B., and Lanier S. M. (2014) Activators of G protein signaling exhibit broad functionality and define a distinct core signaling triad. Mol. Pharmacol. 85, 388–396 10.1124/mol.113.090068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siderovski D. P., and Willard F. S. (2005) The GAPs, GEFs, and GDIs of heterotrimeric G-protein α subunits. Int. J. Biol. Sci. 1, 51–66 10.7150/ijbs.1.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aznar N., Kalogriopoulos N., Midde K. K., and Ghosh P. (2016) Heterotrimeric G protein signaling via GIV/Girdin: breaking the rules of engagement, space, and time. Bioessays 38, 379–393 10.1002/bies.201500133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathew E., Mirza A., and Menhart N. (2004) Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J. Synchrotron Radiat. 11, 314–318 10.1107/S0909049504014086 [DOI] [PubMed] [Google Scholar]
- 31. Hopkins J. B., Gillilan R. E., and Skou S. (2017) BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553 10.1107/S1600576717011438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carpenter B., Nehmé R., Warne T., Leslie A. G., and Tate C. G. (2016) Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536, 104–107 10.1038/nature18966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 35. Ribeiro J. V., Bernardi R. C., Rudack T., Stone J. E., Phillips J. C., Freddolino P. L., and Schulten K. (2016) QwikMD: integrative molecular dynamics toolkit for novices and experts. Sci. Rep. 6, 26536 10.1038/srep26536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Izrailev S., Stepaniants S., Balsera M., Oono Y., and Schulten K. (1997) Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72, 1568–1581 10.1016/S0006-3495(97)78804-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 41. Matthew Allen Bullock J., Schwab J., Thalassinos K., and Topf M. (2016) The importance of non-accessible crosslinks and solvent accessible surface distance in modeling proteins with restraints from crosslinking mass spectrometry. Mol. Cell Proteomics 15, 2491–2500 10.1074/mcp.M116.058560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 43. Kang Y., Kuybeda O., de Waal P. W., Mukherjee S., Van Eps N., Dutka P., Zhou X. E., Bartesaghi A., Erramilli S., Morizumi T., Gu X., Yin Y., Liu P., Jiang Y., Meng X., et al. (2018) Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 10.1038/s41586-018-0215-y [DOI] [PMC free article] [PubMed] [Google Scholar]