

Abstract
Insulin receptor–related receptor (IRR) is a receptor tyrosine kinase of the insulin receptor family and functions as an extracellular alkali sensor that controls metabolic alkalosis in the regulation of the acid–base balance. In the present work, we sought to analyze structural features of IRR by comparing them with those of the insulin receptor, which is its closest homolog but does not respond to pH changes. Using small-angle X-ray scattering (SAXS) and atomic force microscopy (AFM), we investigated the overall conformation of the recombinant soluble IRR ectodomain (ectoIRR) at neutral and alkaline pH. In contrast to the well-known inverted U-shaped (or λ-shaped) conformation of the insulin receptor, the structural models reconstructed at different pH values revealed that the ectoIRR organization has a “droplike” shape with a shorter distance between the fibronectin domains of the disulfide-linked dimer subunits within ectoIRR. We detected no large-scale pH-dependent conformational changes of ectoIRR in both SAXS and AFM experiments, an observation that agreed well with previous biochemical and functional analyses of IRR. Our findings indicate that ectoIRR's sensing of alkaline conditions involves additional molecular mechanisms, for example engagement of receptor juxtamembrane regions or the surrounding lipid environment.
Keywords: insulin receptor, tyrosine-protein kinase (tyrosine kinase), atomic force microscopy (AFM), small-angle X-ray scattering (SAXS), protein structure, acid-base balance, alkali sensing, insulin receptor-related receptor, pH sensor, solution state
Introduction
Insulin receptor-related receptor (IRR),2 a receptor tyrosine kinase of the insulin receptor family, has attracted the attention of scientists due to its unusual properties. IRR gene was originally found in cloning experiments with the DNA of the insulin receptor (IR) as a probe (1). However, since this discovery in 1989, no endogenous ligands for IRR have been identified (2), and a physiological role of the IRR has remained uncertain for a long time. Recently, it was determined that IRR can be activated merely by an increase in the extracellular pH, and it was postulated that IRR is a regulator of the acid–base balance (3).
The activation of IRR by alkaline media has typical features of the ligand–receptor interaction (2, 4, 5). Because IR and type I insulin-like growth factor receptor (IGF-IR) cannot be activated by either alkali or acid, the IRR activation is quite specific. Upon alkali treatment, IRR shows a dose-dependent response with positive cooperativity that is quickly reversible when the media pH is neutralized. The IRR activation triggers intracellular signaling that starts with IRR autophosphorylation followed by phosphorylation of intracellular signaling adapters IRS-1 and AKT-1. The IRR activation leads to intense blebbing and actin stress fiber rearrangement in transfected CHO cells. Actin cytoskeleton rearrangement was also observed in beta pancreatic MIN6 cells where IRR is expressed endogenously (4, 6, 7). Finally, analyses of several chimeras of IR and IRR revealed the key importance of the IRR extracellular region in its pH sensitivity (5, 6).
The IRR tissue expression profile distinguishes it from its relatives of the IR minifamily (3). Whereas IR and IGF-IR are essentially ubiquitous, detectable concentrations of IRR were only found in some specialized cells of kidney, pancreatic, stomach, ovary, and nervous tissues. The physiological importance of IRR was also addressed by gene targeting. Unexpectedly, IRR-knockout mice appeared to be quite healthy under normal conditions, whereas IR and IGF-IR knockouts were not viable (8). However, upon experimental alkali challenge, the IRR-knockout mice showed defects in their renal function and behavioral abnormalities (9, 10).
Despite the obvious functional differences between the IR family members, their structural features are similar. Unlike other receptor tyrosine kinases that dimerize upon their ligand binding (a key step in their activation), IRR, IR, and IGF-IR are expressed as disulfide-linked dimers of two subunits, αβ half-receptors, which are derived from proteolytic cleavage but stay complexed due to covalent disulfide bonds. Thus, one can assume that transmembrane signaling by IRR and its homologs is not based on ligand-induced dimerization per se but should involve allosteric structural changes within the existing receptor dimer (4). This kind of structural transition can occur at both global and local levels. In the extracellular part, the local structural transition can possibly be limited by a local rotation of the domains due to flexible linkers between them (11).
The amino acid sequences of IRR, IR, and IGF-IR are highly homologous with about 50% identity and 65% similarity. They also have identical domain structures. Their large N-terminal extracellular regions contain two leucine-rich repeat domains, named L1 and L2, joined by the cysteine-rich C-domain and three C-terminal fibronectin type III repeats (FnIII-1, FnIII-2, and FnIII-3). The one transmembrane hydrophobic segment is followed by the C-terminal intracellular catalytic tyrosine kinase domain (12).
No structural data are available for IRR, whereas its homologs IR and IGF-IR have been thoroughly studied by X-ray crystallography, cryo-EM, and SAXS (11, 13–19). The analysis of the IR ectodomain cocrystallized with antibodies revealed its symmetrical λ-shaped structure with a head-to-tail complex of monomers (13). Major conformational changes of IR in the absence and presence of insulin were detected (20). It was shown that insulin binding to the dimeric receptor converts its ectodomain from the λ-shaped conformation to the T-shaped conformation (20). Single-particle cryo-EM investigations of the IR ectodomain–insulin complex described the structure of the complex with higher resolution (4–7 Å), revealed conformational changes upon ligand binding, and presented a number of conformations, the shape of which differed from those shown previously (11, 20). As of now, the mechanism of IRR homolog activation remains somewhat contradictory.
Although IR family members have highly related structures, fundamental differences that underlie their ligand specificities, in particular the pH sensitivity of IRR, must exist. The quantitative analysis of IRR/IR chimera activation by alkali showed the key importance of the ectodomain in IRR and the primary role of L1C, FnIII-2, and FnIII-3 domains (6, 21). As no structural information for IRR was available, the results of these studies were interpreted on the basis of the structure of the IR ectodomain obtained by crystallography (18).
This study is devoted to the structural analysis of the IRR ectodomain by two complementary methods of structural biology: small-angle X-ray scattering (SAXS) and atomic force microscopy (AFM). SAXS is a particularly useful tool for investigating the solution states of proteins that includes modeling the quaternary structure of multidomain proteins (22–27). SAXS was also used to analyze structural changes of IR and IGF-IR ectodomains induced by ligand binding with the conclusion that structural movements within the ectodomain upon ligand binding are small and limited to local rotation of domains (11). Another structural method, AFM, can provide direct real-space information about the size and shape of particles and electrochemical properties of a macromolecule based on its binding with surfaces. Complementarity between SAXS and AFM allows cross-validation of models, thereby increasing the reliability and confidence in the results (28, 29). On the basis of SAXS and AFM analyses of the IRR ectodomain, we propose its asymmetrical droplike model.
Results
Analysis of scattering curves
To obtain structural information, samples of recombinant ectoIRR were analyzed by SAXS. Because IRR is an alkali sensor, the measurements were made in parallel at pH 7.0 and 9.0. The processed experimental scattering patterns from ectoIRR are displayed in Fig. 1. Concentration-dependent effects on the measured scattering intensities were not observed for samples spanning 0.5–3 mg/ml. Fig. 1 shows scattering curves from 3 mg/ml protein solution.
Figure 1.

Experimental SAXS curves of ectoIRR and corresponding structural analysis. a, experimental SAXS curve at pH 7.0 (1), scattering patterns computed from the ab initio model DAMMIN at pH 7.0 (2), the transformed from p(r) and extrapolated to zero scattering angle intensity at pH 7.0 (3), experimental SAXS curve at pH 9.0 (4), scattering patterns computed from the ab initio model DAMMIN at pH 9.0 (5), and the transformed from p(r) and extrapolated to zero scattering angle intensity at pH 9.0 (6). The experimental curves for pH 7.0 (curve 1) and for pH 9.0 (curve 4) together with corresponding fits are shifted along the ordinate by 1 logarithmic unit for better visualization. Unshifted curves are presented in Fig. S2. The sample concentration was 3 mg/ml at both pH values. Inset, distance distribution functions, p(r), computed by GNOM at pH 7.0 (1) and at pH 9.0 (2). b, the Guinier plot at pH 7.0 (1) and at pH 9.0 (2); Guinier fits are presented by black lines. c, Kratky plots at pH 7.0 (1) and at pH 9.0 (2). d and e, typical structural models reconstructed by ab initio modeling program DAMMIN. d, DAMMIM shape reconstruction performed for the sample at pH 7.0. e, DAMMIN shape reconstruction performed for the sample at pH 9.0. P1 symmetry was used in both cases. The models d and e are shown in two different orientations.
The scattering profiles of ectoIRR, measured at pH 7.0 and 9.0, do not show dramatic differences and almost coincide when the data are scaled relative to each other (Figs. 1a, curves 1 and 4, and S2). Their similarity is emphasized by the superposition of the bell-shaped Kratky plots, which indicates that the protein is mostly folded (30), and by the corresponding p(r) distance distribution functions (Fig. 1, inset), which show little difference in the distribution of internal vector lengths or the respective maximum particle dimension (Dmax) of either sample (∼19 nm; Fig. 1c). The slope of the Guinier plots is also similar (Fig. 1b), yielding a very close radius of gyration from Guinier (Rg) of ectoIRR at pH 7.0 (Rg = 5.30 nm) and pH 9.0 (Rg = 5.25 nm). The experimental molecular mass (MM) estimates obtained from the data are consistent with the MM calculated from the electrophoresis of ectoIRR (Fig. S3) and indicate that ectoIRR forms a dimer in solution. A summary of the experimental structural parameters (Rg, Dmax, Porod volume (Vp) estimate, calculated molecular mass from the amino acid sequence without the contribution of glycosylation of the ectoIRR dimer (MMaa), molecular mass from Porod volume (MMPorod), and molecular mass by the Bayesian inference approach (MMBayesian)) extracted from the SAXS data are reported in Table 1.
Table 1.
SAXS-derived overall structural parameters of the ectoIRR dimer in solution
The associated errors for Rg represent standard deviations directly evaluated by the error propagation from the experimental data. For Dmax, the deviations were assessed by repetitive runs with varied analysis parameters.
| Sample | Rg | Dmax | Vp | MMaa | MMPorod | MMBayesian |
|---|---|---|---|---|---|---|
| nm | nm | nm3 | kDa | kDa | kDa | |
| ectoIRR, pH 7.0 | 5.30 ± 0.02 | 19.5 ± 0.3 | 460 ± 20 | 201.5 | 280 ± 15 | 243 credibility interval: 221–373 |
| ectoIRR, pH 9.0 | 5.25 ± 0.02 | 19.0 ± 0.2 | 455 ± 20 | 201.5 | 270 ± 15 | 243 credibility interval: 221–373 |
As revealed by the SAXS data analysis, the integral structural characteristics of the protein in solution remain the same despite changes in pH. These results are in some degree similar to the data obtained in the work of Whitten et al. (11). The authors suggest that structural changes of the IR and the homologous IGF-IR induced by ligand binding are small and possibly limited to local rotation of domains.
Typical ab initio shape reconstructions of the ectoIRR dimer at both pH values are presented in Fig. 1, d and e. At the given resolution, the ectodomain shape does not change significantly with a pH increase (Fig. S2b); the normalized spatial discrepancy (NSD) in this case is 0.50. The second-order symmetry was not used for DAMMIN shape reconstruction to obtain a model independent of artificially introduced symmetry because the use of any restrictions dramatically narrows the area of resulting models. Moreover, due to flexibility of the links between domains, the protein can achieve slightly different coexisting conformations in solution. Thus, ab initio reconstruction without symmetry more adequately reflects an average structure of the sample. Nevertheless, because the macromolecule of the protein is a dimer consisting of two identical monomers, P2 symmetry was also used for the shape reconstruction. However, a comparison of the obtained low-resolution models demonstrates their overall similarity. In contrast, reconstructions with P2 symmetry yield systematically worse fits to the experimental data through multiple runs of the program, whereas scattering patterns computed from the ab initio models obtained by DAMMIN without symmetry yield good fits to the experimental data: χ2 = 1.3 for pH 7.0 and 1.4 for pH 9.0 (Fig. 1a).
Therefore, at this stage of the SAXS data analysis, one can conclude that the overall low-resolution shape of the ectoIRR does not change at the alkalization. Given the availability of flexible links between protein domains, it should be assumed that pH changing leads to some local changes that occur at the level of rotation of the individual domains. This assumption is supported by small but detectable differences in the structural characteristics of the protein (Fig. 1 and Table 1). To obtain more detailed structural organization of the ectoIRR in solution, hybrid modeling was performed using CORAL. The available high-resolution X-ray crystal structure of the insulin receptor ectodomain (derived from Protein Data Bank (PDB) code 4ZXB) as the closest IRR homolog was split into individual subdomains. The splitting was done between the head and tail domains such that the first subdomain contained amino acids 1–465 and the second contained amino acids 472–910. Amino acids 466–471 constitute the flexible linker, as this loop contains only several contacts with the rest of the monomer structure (Fig. 2). Usage of only two intact domains per polypeptide chain was chosen to keep the number of free parameters low with a reasonable number of degrees of freedom to avoid overfitting.
Figure 2.

High-resolution X-ray crystal structure of the homodimer of the insulin receptor ectodomain (a) with flexible linkers (red) between the head and tail domains (b) that allowed hybrid modeling performed by program CORAL.
First, the modeling was performed for pH 9.0. The resulting model yields good fit to the experimental data with χ2 = 1.27 (Fig. 3a). Given the similarity of the SAXS experimental data at pH 7.0 and 9.0, further CORAL modeling (against pH 7.0 data) was performed starting from the rigid-body model obtained for pH 9.0. This refinement also yielded a good fit with χ2 = 1.30 (Fig. 3b). Structural comparison of these two CORAL models performed by the program SUPCOMB once again demonstrated their overall similarity with a normalized spatial discrepancy of 0.97 (Fig. 3c). When comparing CORAL models with the corresponding DAMMIN shapes, NSDs were found to be 1.3 for both models at pH 7.0 and 9.0 (Fig. 3, d and e).
Figure 3.

Rigid body modeling of the ectoIRR dimer structure. a, modeling at pH 7. 0. b, modeling at pH 9.0. Experimental scattering curves (1) and scattering patterns computed from the CORAL models (2) are shown. Insets, structural organizations of the ectoIRR dimer in solution obtained by CORAL. Red circles mark obtained configuration of amino acids 466–471 as a dummy-residue chain to fit the computed scattering patterns. c, comparison of the CORAL models obtained for pH 7.0 (red) and for pH 9.0 (green); NSD = 0.97. d and e, comparison of the CORAL models with the corresponding DAMMIN shapes at pH 7.0 and 9.0, respectively; NSD = 1.3. P1 symmetry was used for all shape reconstructions.
Thus, the hybrid modeling has demonstrated that the ectoIRR dimers achieve similar low-resolution shapes at two different pH values. It should be noted that due to the flexibility of interdomain links and the coexistence of slightly different protein conformations in solution, the resulting structures are averaged and correspond to the most probable constitution of the protein under given conditions. The overall structural characteristics for the CORAL shapes of the ectoIRR dimer calculated by the program CRYSOL from their atomic coordinates almost coincide: Rg = 5.3 and 5.4 nm, envelope diameter D = 21.9 and 21.8 nm, and average electron density ρ = 0.5205 and 0.5204, respectively. These parameters also correlate well with those obtained from the experimental small-angle scattering data (Table 1).
An attempt was also made to obtain a symmetrical conformation by imposing the 2-fold symmetry axis between the ectoIRR dimer subunits such that for each atom with the coordinates (x, y, z) in the first monomer a symmetry mate in the second monomer has the coordinates (−x, −y, z). Using the P2 symmetry, as in the case of ab initio shape reconstruction, the constraint, however, yielded worse fits to the experimental data with χ2 = 1.6 (Fig. S4).
AFM analysis and comparison with the SAXS results
The SAXS data were further verified by atomic force microscopy analysis (Fig. 4). AFM experiments of ectoIRR adsorbed on the surface of atomically flat mica revealed shapes of the homodimer similar to those observed by SAXS, without significant differences under conditions of pH 7.0 and 9.0. Fig. 4 shows that the typical structures observed by AFM overlapped with the SAXS models. The average sizes of the fitted homodimers are listed in Table 2. Thus, using AFM we were able to detect an ectoIRR conformation essentially similar to that observed by SAXS at both pH 7.0 and 9.0.
Figure 4.

Representative AFM topology images of the ectoIRR dimer absorbed on the mica surface from water solution (150 mm NaCl, 20 mm Tris) at pH 7.0 (a) and 9.0 (b). The full z-scale (height) is 6.0 nm in both images. The spatial structure of the ectoIRR dimer obtained by CORAL modeling is overlapped with the AFM topology images by the most suitable rotation.
Table 2.
AFM-derived spatial dimensions of the ectoIRR dimer absorbed on the mica surface
| pH 7.0 | pH 9.0 | |
|---|---|---|
| Height (nm) | 3.0 ± 0.6 (S.D., n = 28) | 3.2 ± 0.3 (S.D., n = 9) |
| Length (nm) | 20 ± 3 (S.D., n = 28) | 20 ± 2 (S.D., n = 9) |
| Width (nm) | 9 ± 3 (S.D., n = 28) | 10 ± 2 (S.D., n = 9) |
Discussion
The present work is the first reconstruction of the 3D structural model of the IRR extracellular ectodomain in solution at both neutral and alkaline pH by means of small-angle X-ray scattering and atomic force microscopy. The droplike structures obtained by ab initio reconstruction of the ectoIRR dimer under pH 7.0 and 9.0 in solution did not differ significantly; both SAXS and AFM gave the same integral structural (Tables 1 and 2).
More detailed structural study of ectoIRR by hybrid modeling using the available high-resolution X-ray crystal structure of the insulin receptor ectodomain demonstrated that the ectoIRR dimer can attain asymmetric droplike conformation, quite different from the λ shape of the IR ectodomain (Fig. 5). It was interesting to compare the experimental SAXS curve of ectoIRR and a theoretically calculated scattering curve based on the published three-dimensional structure of the insulin receptor ectodomain, which shows a λ-shaped symmetrical head-to-tail complex of two disulfide-linked monomers (18) (derived from PDB code 4ZXB). The comparison of the experimental and computed data confirmed that the solution organization of ectoIRR and the crystal structure of the IR ectodomain stabilized by complexing with antibodies are different (Fig. S5).
Figure 5.
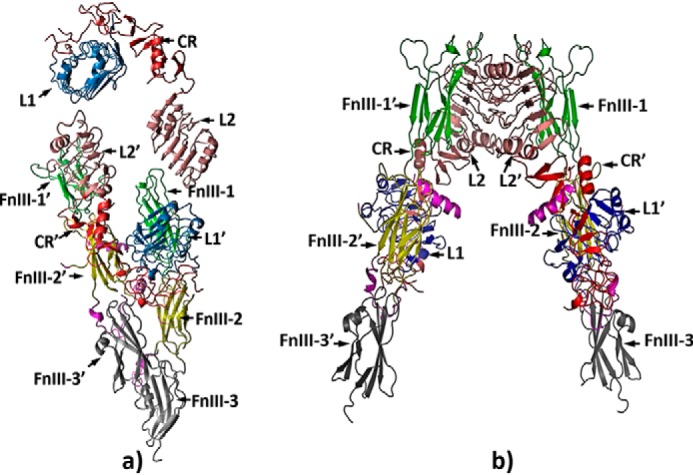
Structural organization of the droplike conformation of the ectoIRR in solution obtained by CORAL (a) and a comparison of this conformation with the λ shape of the IR ectodomain (b).
As reported recently, it is possible that two identical insulin receptor or epidermal growth factor receptor dimer subunits can be complexed symmetrically or asymmetrically (20, 31, 32). It is tempting to speculate that the asymmetrical shape of ectoIRR may reflect activation-induced conformational changes of the full-size IRR that involve a relative shift of the transmembrane segments with consequent repositioning of the catalytic domains resulting in receptor autophosphorylation. In a recent cryo-EM study of the IR ectodomain complexed with insulin and artificially connected juxtamembrane C termini by a leucine zipper, an asymmetrical dimeric structure was described (33). Remarkably, this structure of the IR ectodomain bound to one insulin molecule (33) resembles the shape of ectoIRR in solution. It is also worth noting that the proposed structural organization of ectoIRR is different from the major T-shaped dimer conformation (labeled as 1T) of IR with close proximity of fibronectin domains from different monomers, which was demonstrated by EM after insulin binding (20). However, it is similar to an alternative T-shaped dimer conformation of the full-length IR after its reconstitution into nanodiscs (20). This shape was indicated as a II-shaped conformation, which represents an asymmetric arrangement of the monomers with close positions of opposite fibronectin domains.
Another key feature of the found ectoIRR conformation is a small distance between the fibronectin domains of the two subunits (Fig. 5a). This structural feature agrees with the mutant analysis of ectoIRR indicating that FnIII-2 and FnIII-3 domains are most critical for IRR activity (5). Swapping FnIII-2 and FnIII-3 domains or all FnIII domains of IRR with the IR led to significant glycosylation of these chimeras and completely abolished the IRR pH-sensing property. Partial removal of glycosylation by point mutagenesis in IRR/IR chimeras resulted in a partial restoration of IRR pH-sensing activity (34), suggesting that a larger carbohydrate coat of FnIII domains provides a steric hindrance to IRR activation.
The published mutagenesis data also revealed the importance of five conserved amino acid residues (6) of the L1C domains. In the λ model, these residues are exposed to the aqueous phase, whereas in the droplike asymmetrical model five of them are located at the contact surface between L1C and fibronectin domains within the tethered subunits of the ectoIRR dimer (Fig. 6), which better explains their role in IRR function and identifies L1C domains of the tethered subunit together with the fibronectin domain of both subunits as the pH-sensing mechanism with one or two centers of activation.
Figure 6.

Distribution of the five amino acid residues playing a key role in alkali sensing (6) within the droplike conformation of the asymmetric ectoIRR dimer (a) (highlighted by green and red in the extended and tethered subunits of the dimer, respectively) and within the tethered dimer subunit zoomed in on (b) and with some rotation (c). The subdomain names of the extended and tethered subunits of the dimer are highlighted by green and pink, respectively.
Experimental procedures
Production of recombinant IRR ectodomain
The IRR ectodomain expression construct was obtained by inserting a fragment of cDNA coding for the first 918 amino acids of human IRR protein fused with c-myc tag to pEE6.HCMV expression vector for the GS expression system (Lonza). CHO-K1 cells were transfected by this plasmid, and a stable expression clone of CHO-K1 was obtained. The cells were grown in serum-free medium GibriC-1-CHO (PanEco), and the medium was collected and purified by anion-exchange, gel-filtration, and hydrophobic chromatography as described (35). The protein identity was verified by SDS-PAGE and Western blotting with antibodies against ectoIRR and c-myc as described (36). Purity was controlled by high-performance gel filtration on a Superose 6 Increase 10/300 GL column (Fig. S1).
Scattering experiments and data analysis
Synchrotron SAXS measurements were performed at the European Molecular Biology Laboratory (EMBL) on the EMBL-P12 BioSAXS beamline at the PETRAIII storage ring (Deutsches Elektronen-Synchrotron (DESY), Hamburg, Germany) equipped with a robotic sample changer and a 2D photon-counting pixel X-ray detector Pilatus 2M (DECTRIS, Switzerland). The scattering intensity, I(s), was recorded in the range of the momentum transfer 0.08 < s < 3.5 nm−1 where s = (4πsinθ)/λ, 2θ is the scattering angle, and λ = 0.124 nm, the X-ray wavelength (37). The measurements were carried out in either 150 mm NaCl, 20 mm Tris, pH 7.0, or 150 mm NaCl, 20 mm Tris, pH 9.0, buffer at 10 °C using continuous sample flow operation over a total exposure time of 1 s, collected as 20 × 50-ms individual frames to monitor for potential radiation damage (no radiation effects were detected (38)). The data were corrected for the solvent scattering and processed using standard procedures (39). To account for the interparticle interactions, solutions of ectoIRR at concentrations of 0.5 and 3 mg/ml were measured.
The values of the total forward scattering at zero angle, I(0), and radii of gyration, Rg, were calculated from the experimental SAXS profiles using the Guinier approximation, which is valid in the range of (sRg) approximately <1.3 (27, 39).
| (Eq. 1) |
These parameters and the Dmax were also computed from the probable distance distribution function, p(r), using the program GNOM (40).
| (Eq. 2) |
The MMs of each sample were calculated from the SAXS data using the value of I(0) combined with protein concentration relative to a BSA standard (41) as well as from the concentration-independent excluded Porod volume (MMPorod) (42). The latter was determined given that the empirical ratio between the Vp and MM of a protein is ∼1.65 (25). A Bayesian inference approach was also applied, affording an accuracy above that of the individual methods and reporting MM (MMBayesian) estimates together with a credibility interval (43).
The low-resolution shapes of ectodomain IRR were reconstructed ab initio using the program DAMMIN (44) that generates 3D dummy-atom (bead) model spatial representations of a particle. Starting from a random assembly, the program utilizes a simulated annealing algorithm to build models fitting the experimental data Iexp(s) that minimizes the discrepancy.
| (Eq. 3) |
i.e. the reduced χ2 test where N is the number of experimental points, c is a scaling factor, and Icalc(sj) and σ(sj) are the calculated intensity from the model and the experimental error on the intensities at the momentum transfer sj, respectively.
Hybrid rigid-body modeling was performed by the program CORAL (24), which refines the spatial positions of domains with known high-resolution structures combined with linkers connecting the domains that are represented as chains of dummy amino acids. The program searches for the optimal orientations of the domains and the possible conformation(s) of the linkers by minimizing the fit to the experimental SAXS data using the reduced χ2 test. The high-resolution structures used for each of the domains were obtained from PDB code 4ZXB (the first subdomain contained amino acids 1–465, the second subdomain contained amino acids 472–910, and amino acids 466–471 constituted the flexible linker). The modeling was performed using P1 and P2 symmetry. The program CRYSOL v.3.0 (24, 45) was used to calculate the theoretical scattering from the atomic coordinates of the insulin receptor crystal structure (PDB code 4ZXB) and from those of its domains to select ensembles of conformations whose combined weighted scattering best fit the experimental data.
To assess the convergence of the models obtained in individual simulated annealing runs and to average the models, an NSD (46) is employed. This approach reveals a dissimilarity measure between individual models containing, in general, a different number of scatterers, e.g. two ab initio models or a bead model and a rigid-body model. Superposition of distinct models is done by the program SUPCOMB (46). The averaging of ab initio models is performed by the program DAMAVER (47). First, the most typical model (having the lowest average NSD compared with all the others) is selected and used as a reference. Then, after the alignment of all the models with the reference model, the entire collection of beads is remapped onto a densely packed hexagonal grid, which is filtered to yield an average model with the volume equal to the averaged volume of the input models.
Atomic force microscopy
Atomic force microscopy of ectoIRR was carried out on the Multimode Nanoscope IV setup (Veeco Digital Instruments) equipped with a J-type scanner and an electrochemical fluid cell. Scanning was performed in tapping mode with the aid of SiN3 cantilevers with a nominal spring constant of 0.06 Newton/metre (type SNL, Bruker) and a tip radius of ∼2 nm. All experiments were conducted in the working buffer solution (150 mm NaCl, 20 mm Tris) with the corresponding pH value of 7.0 or 9.0. Samples were prepared by placing 200-μl droplets of the protein solution with a bulk concentration of 7.5 μg/ml on the surface of freshly cleaved mica. After 45 min of incubation, the sample was rinsed five times with the working buffer solution to remove any unbound protein. The sample was then placed into the AFM cell filled with the corresponding buffer solution for scanning. All images were processed using WSXM software (48). Dimensions of ectoIRR were obtained as the maximal size of the cross-section profile in the corresponding plane and corrected in the lateral plane by the tip radius of the cantilever.
Author contributions
E. V. S., I. E. D., E. V. B., O. V. B., and A. G. P. conceptualization; E. V. S., A. A. M., and A. G. P. resources; E. V. S., A. A. M., I. E. D., L. A. D., D. I. S., O. V. B., and A. G. P. data curation; E. V. S., M. V. P., A. A. M., I. E. D., L. A. D., E. V. B., D. I. S., O. V. B., and A. G. P. formal analysis; E. V. S., D. I. S., O. V. B., and A. G. P. supervision; E. V. S., D. I. S., O. V. B., and A. G. P. funding acquisition; E. V. S., M. V. P., A. A. M., I. E. D., C. M. J., D. I. S., O. V. B., and A. G. P. validation; E. V. S., M. V. P., L. A. D., N. A. L., A. S. G., C. M. J., and O. V. B. investigation; E. V. S., L. A. D., and O. V. B. visualization; E. V. S., M. V. P., D. I. S., and O. V. B. methodology; E. V. S., M. V. P., A. A. M., I. E. D., L. A. D., E. V. B., C. M. J., O. V. B., and A. G. P. writing-original draft; E. V. S., O. V. B., and A. G. P. project administration; E. V. S., I. E. D., E. V. B., D. I. S., O. V. B., and A. G. P. writing-review and editing; M. V. P. software.
Supplementary Material
Acknowledgments
AFM measurements were performed using equipment of CKP FMI of A. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences. We thank Drs. M. Lawrence and J. Menting (The Walter and Eliza Hall Institute of Medical Research) for collaboration and for making available the cell line expressing ectoIRR. We also thank Dr. L. Garber and V. Petrenko for careful editing of the manuscript.
The work was supported by the Russian Foundation for Basic Research (RFBR) Projects 17-00-00487-KOMFI (to E. V. S., M. V. P., and L. A. D.), 17-00-00488-KOMFI (to O. V. B. and N. A. L.), and 17-00-00489-KOMFI (to A. G. P., A. A. M., I. E. D., and E. V. B.) and by Infrastructure for NMR, EM and X-rays for Translational Research (iNEXT) Project 653706 funded by the Horizon 2020 program of the European Union (to M. V. P, A. A. M., and E. V. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
- IRR
- insulin receptor–related receptor
- SAXS
- small-angle X-ray scattering
- AFM
- atomic force microscopy
- ectoIRR
- IRR ectodomain
- FnIII
- fibronectin type III
- IR
- insulin receptor
- IGF-IR
- type I insulin-like growth factor receptor
- CHO
- Chinese hamster ovary
- MM
- molecular mass
- Rg
- radius of gyration from Guinier
- Vp
- Porod volume
- Dmax
- maximum particle dimension
- MMaa
- calculated molecular mass from the amino acid sequence without the contribution of glycosylation of the ectoIRR dimer
- MMPorod
- molecular mass from Porod volume
- MMBayesian
- molecular mass by the Bayesian inference approach
- NSD
- normalized spatial discrepancy
- PDB
- Protein Data Bank.
References
- 1. Shier P., and Watt V. M. (1989) Primary structure of a putative receptor for a ligand of the insulin family. J. Biol. Chem. 264, 14605–14608 [PubMed] [Google Scholar]
- 2. Dissen G. A., Garcia-Rudaz C., Tapia V., Parada L. F., Hsu S. Y., and Ojeda S. R. (2006) Expression of the insulin receptor-related receptor is induced by the preovulatory surge of luteinizing hormone in thecal-interstitial cells of the rat ovary. Endocrinology 147, 155–165 10.1210/en.2005-0386 [DOI] [PubMed] [Google Scholar]
- 3. Petrenko A. G., Zozulya S. A., Deyev I. E., and Eladari D. (2013) Insulin receptor-related receptor as an extracellular pH sensor involved in the regulation of acid-base balance. Biochim. Biophys. Acta 1834, 2170–2175 10.1016/j.bbapap.2012.11.011 [DOI] [PubMed] [Google Scholar]
- 4. Deyev I. E., Sohet F., Vassilenko K. P., Serova O. V., Popova N. V., Zozulya S. A., Burova E. B., Houillier P., Rzhevsky D. I., Berchatova A. A., Murashev A. N., Chugunov A. O., Efremov R. G., Nikol'sky N. N., Bertelli E., et al. (2011) Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 13, 679–689 10.1016/j.cmet.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deyev I. E., Popova N. V., and Petrenko A. G. (2015) Determination of alkali-sensing parts of the insulin receptor-related receptor using the bioinformatic approach. Acta Naturae 7, 80–86 [PMC free article] [PubMed] [Google Scholar]
- 6. Deyev I. E., Mitrofanova A. V., Zhevlenev E. S., Radionov N., Berchatova A. A., Popova N. V., Serova O. V., and Petrenko A. G. (2013) Structural determinants of the insulin receptor-related receptor activation by alkali. J. Biol. Chem. 288, 33884–33893 10.1074/jbc.M113.483172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deyev I. E., Popova N. V., Serova O. V., Zhenilo S. V., Regoli M., Bertelli E., and Petrenko A. G. (2017) Alkaline pH induces IRR-mediated phosphorylation of IRS-1 and actin cytoskeleton remodeling in a pancreatic β cell line. Biochimie 138, 62–69 10.1016/j.biochi.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 8. Kitamura T., Kido Y., Nef S., Merenmies J., Parada L. F., and Accili D. (2001) Preserved pancreatic β-cell development and function in mice lacking the insulin receptor-related receptor. Mol. Cell Biol. 21, 5624–5630 10.1128/MCB.21.16.5624-5630.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shayahmetova D. M., Zhevlenev E. S., Mozhaev A. A., Deyev I. E., and Petrenko A. G. (2016) Genetic link between IRR-receptor and Ly6/PLAUR protein. Russ. J. Bioorg. Chem. 42, 449–452 10.1134/S1068162016040166 [DOI] [Google Scholar]
- 10. Zubkov E. A., Morozova A. Y., Chachina N. A., Shayahmetova D. M., Mozhaev A. A., Deyev I. E., Chekhonin V. P., Petrenko A. G. (2017) Behavioral phenotype of mice with alkali sensor IRR gene knockout. Zh. Vyssh. Nerv. Deiat. Im. I. P. Pavlova. 67, 106–112 [PubMed] [Google Scholar]
- 11. Whitten A. E., Smith B. J., Menting J. G., Margetts M. B., McKern N. M., Lovrecz G. O., Adams T. E., Richards K., Bentley J. D., Trewhella J., Ward C. W., and Lawrence M. C. (2009) Solution structure of ectodomains of the insulin receptor family: the ectodomain of the type 1 insulin-like growth factor receptor displays asymmetry of ligand binding accompanied by limited conformational change. J. Mol. Biol. 394, 878–892 10.1016/j.jmb.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 12. De Meyts P. (2008) The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 33, 376–384 10.1016/j.tibs.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 13. McKern N. M., Lawrence M. C., Streltsov V. A., Lou M. Z., Adams T. E., Lovrecz G. O., Elleman T. C., Richards K. M., Bentley J. D., Pilling P. A., Hoyne P. A., Cartledge K. A., Pham T. M., Lewis J. L., Sankovich S. E., et al. (2006) Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 443, 218–221 10.1038/nature05106 [DOI] [PubMed] [Google Scholar]
- 14. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., et al. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 10.1038/nature11781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hubbard S. R., Wei L., Ellis L., and Hendrickson W. A. (1994) Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 372, 746–754 10.1038/372746a0 [DOI] [PubMed] [Google Scholar]
- 16. Li Q., Wong Y. L., and Kang C. (2014) Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta 1838, 1313–1321 10.1016/j.bbamem.2014.01.005 [DOI] [PubMed] [Google Scholar]
- 17. Cabail M. Z., Li S., Lemmon E., Bowen M. E., Hubbard S. R., and Miller W. T. (2015) The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 6, 6406 10.1038/ncomms7406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Croll T. I., Smith B. J., Margetts M. B., Whittaker J., Weiss M. A., Ward C. W., and Lawrence M. C. (2016) Higher-resolution structure of the human insulin receptor ectodomain: multi-modal inclusion of the insert domain. Structure 24, 469–476 10.1016/j.str.2015.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y., Kong G. K., Menting J. G., Margetts M. B., Delaine C. A., Jenkin L. M., Kiselyov V. V., De Meyts P., Forbes B. E., and Lawrence M. C. (2018) How ligand binds to the type 1 insulin-like growth factor receptor. Nat. Commun. 9, 821 10.1038/s41467-018-03219-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gutmann T., Kim K. H., Grzybek M., Walz T., and Coskun Ü. (2018) Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J. Cell Biol. 217, 1643–1649 10.1083/jcb.201711047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deyev I. E., Chachina N. A., Shayahmetova D. M., Serova O. V., and Petrenko A. G. (2015) Mapping of alkali-sensing sites of the insulin receptor-related receptor. The role of L2 and fibronectin domains. Biochimie 111, 1–9 10.1016/j.biochi.2014.12.014 [DOI] [PubMed] [Google Scholar]
- 22. Tria G., Mertens H. D., Kachala M., and Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 10.1107/S205225251500202X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 10.1021/ja069124n [DOI] [PubMed] [Google Scholar]
- 24. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Svergun D. I., Burkhardt N., Pedersen J. S., Koch M. H., Volkov V. V., Kozin M. B., Meerwink W., Stuhrmann H. B., Diedrich G., and Nierhaus K. H. (1997) Solution scattering structural analysis of the 70 S Escherichia coli ribosome by contrast variation. II. A model of the ribosome and its RNA at 3.5 nm resolution. J. Mol. Biol. 271, 602–618 10.1006/jmbi.1997.1191 [DOI] [PubMed] [Google Scholar]
- 26. Blanchet C. E., and Svergun D. I. (2013) Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 64, 37–54 10.1146/annurev-physchem-040412-110132 [DOI] [PubMed] [Google Scholar]
- 27. Svergun D. I., Koch M. H. J., Timmins P. A., and May R. P. (2013) Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules, pp. 93–151, Oxford University Press, Oxford [Google Scholar]
- 28. Shtykova E. V., Baratova L. A., Fedorova N. V., Radyukhin V. A., Ksenofontov A. L., Volkov V. V., Shishkov A. V., Dolgov A. A., Shilova L. A., Batishchev O. V., Jeffries C. M., and Svergun D. I. (2013) Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH. PLoS One 8, e82431 10.1371/journal.pone.0082431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shtykova E. V., Dadinova L. A., Fedorova N. V., Golanikov A. E., Bogacheva E. N., Ksenofontov A. L., Baratova L. A., Shilova L. A., Tashkin V. Y., Galimzyanov T. R., Jeffries C. M., Svergun D. I., and Batishchev O. V. (2017) Influenza virus matrix protein M1 preserves its conformation with pH, changing multimerization state at the priming stage due to electrostatics. Sci. Rep. 7, 16793 10.1038/s41598-017-16986-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mertens H. D., and Svergun D. I. (2010) Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141 10.1016/j.jsb.2010.06.012 [DOI] [PubMed] [Google Scholar]
- 31. Bocharov E. V., Sharonov G. V., Bocharova O. V., and Pavlov K. V. (2017) Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta Biomembr. 1859, 1417–1429 10.1016/j.bbamem.2017.01.025 [DOI] [PubMed] [Google Scholar]
- 32. Mi L. Z., Lu C., Li Z., Nishida N., Walz T., and Springer T. A. (2011) Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat. Struct. Mol. Biol. 18, 984–989 10.1038/nsmb.2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weis F., Menting J. G., Margetts M. B., Chan S. J., Xu Y., Tennagels N., Wohlfart P., Langer T., Müller C. W., Dreyer M. K., and Lawrence M. C. (2018) The signalling conformation of the insulin receptor ectodomain. Nat. Commun. 9, 4420 10.1038/s41467-018-06826-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deyev I. E., Chachina N. A., Zhevlenev E. S., and Petrenko A. G. (2017) Site-directed mutagenesis of the fibronectin domains in insulin receptor-related receptor. Int. J. Mol. Sci. 18, E2461 10.3390/ijms18112461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cosgrove L., Lovrecz G. O., Verkuylen A., Cavaleri L., Black L. A., Bentley J. D., Howlett G. J., Gray P. P., Ward C. W., and McKern N. M. (1995) Purification and properties of insulin receptor ectodomain from large-scale mammalian cell culture. Protein Expr. Purif. 6, 789–798 10.1006/prep.1995.0010 [DOI] [PubMed] [Google Scholar]
- 36. Krasnoperov V., Deyev I. E., Serova O. V., Xu C., Lu Y., Buryanovsky L., Gabibov A. G., Neubert T. A., and Petrenko A. G. (2009) Dissociation of the subunits of the calcium-independent receptor of α-latrotoxin as a result of two-step proteolysis. Biochemistry 48, 3230–3238 10.1021/bi802163p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanchet C. E., Spilotros A., Schwemmer F., Graewert M. A., Kikhney A., Jeffries C. M., Franke D., Mark D., Zengerle R., Cipriani F., Fiedler S., Roessle M., and Svergun D. I. (2015) Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 48, 431–443 10.1107/S160057671500254X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jeffries C. M., Graewert M. A., Svergun D. I., and Blanchet C. E. (2015) Limiting radiation damage for high-brilliance biological solution scattering: practical experience at the EMBL P12 beamline PETRAIII. J. Synchrotron Radiat. 22, 273–279 10.1107/S1600577515000375 [DOI] [PubMed] [Google Scholar]
- 39. Feigin L. A., and Svergun D. I. (1987) Structure Analysis by Small-Angle X-Ray and Neutron Scattering, pp. 59–90, Plenum Press, New York [Google Scholar]
- 40. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 41. Mylonas E., and Svergun D. I. (2007) Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 40, s245–s249 10.1107/S002188980700252X [DOI] [Google Scholar]
- 42. Porod G. (1982) General theory. In Small-Angle X-ray Scattering (Glatter O., and Kratky O., eds) pp. 17–51, Academic Press, London [Google Scholar]
- 43. Hajizadeh N. R., Franke D., Jeffries C. M., and Svergun D. I. (2018) Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 8, 7204 10.1038/s41598-018-25355-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 10.1016/S0006-3495(99)77443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Svergun D. I., Barberato C., and Koch M. H. J. (1995) CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 46. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 10.1107/S0021889800014126 [DOI] [Google Scholar]
- 47. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horcas I., Fernández R., Gómez-Rodríguez J. M., Colchero J., Gómez-Herrero J., and Baro A. M. (2007) WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 78, 013705 10.1063/1.2432410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.