

Abstract
The association of plasma membrane (PM)-localized voltage-gated potassium (Kv2) channels with endoplasmic reticulum (ER)-localized vesicle-associated membrane protein-associated proteins VAPA and VAPB defines ER–PM junctions in mammalian brain neurons. Here, we used proteomics to identify proteins associated with Kv2/VAP-containing ER–PM junctions. We found that the VAP-interacting membrane-associated phosphatidylinositol (PtdIns) transfer proteins PYK2 N-terminal domain-interacting receptor 2 (Nir2) and Nir3 specifically associate with Kv2.1 complexes. When coexpressed with Kv2.1 and VAPA in HEK293T cells, Nir2 colocalized with cell-surface–conducting and -nonconducting Kv2.1 isoforms. This was enhanced by muscarinic-mediated PtdIns(4,5)P2 hydrolysis, leading to dynamic recruitment of Nir2 to Kv2.1 clusters. In cultured rat hippocampal neurons, exogenously expressed Nir2 did not strongly colocalize with Kv2.1, unless exogenous VAPA was also expressed, supporting the notion that VAPA mediates the spatial association of Kv2.1 and Nir2. Immunolabeling signals of endogenous Kv2.1, Nir2, and VAP puncta were spatially correlated in cultured neurons. Fluorescence-recovery-after-photobleaching experiments revealed that Kv2.1, VAPA, and Nir2 have comparable turnover rates at ER–PM junctions, suggesting that they form complexes at these sites. Exogenous Kv2.1 expression in HEK293T cells resulted in significant differences in the kinetics of PtdIns(4,5)P2 recovery following repetitive muscarinic stimulation, with no apparent impact on resting PtdIns(4,5)P2 or PtdIns(4)P levels. Finally, the brains of Kv2.1-knockout mice had altered composition of PtdIns lipids, suggesting a crucial role for native Kv2.1-containing ER–PM junctions in regulating PtdIns lipid metabolism in brain neurons. These results suggest that ER–PM junctions formed by Kv2 channel–VAP pairing regulate PtdIns lipid homeostasis via VAP-associated PtdIns transfer proteins.
Keywords: phosphatidylinositol signaling, plasma membrane, potassium channel, membrane protein, organelle, endoplasmic reticulum (ER), brain, neuron, lipids, membrane contact site (MCS), membrane-associated phosphatidylinositol transfer proteins, plasma membrane, plasma membrane junction
Introduction
Inter-organelle membrane contact sites (MCS)2 are a ubiquitously-conserved feature of eukaryotic cells. Of these, junctions between the plasma membrane (PM) and endoplasmic reticulum (ER) or ER–PM junctions serve as a vital platform for information transduction between these two membranes (1–4). These MCS were initially discovered in EM studies in mammalian muscle cells (5) and soon after in mammalian brain neurons (6–8), and they can engage >10% of the somatic neuronal PM (9). Since their initial discovery, ER–PM junctions have been described in a diverse array of mammalian cell types, and they are typically organized by various ER-resident tethers that bind to PM inner-leaflet phospholipids (10–15).
Much of what is known about the cellular physiological role(s) of ER–PM junctions is derived from studies performed in non-neuronal cells. ER–PM junctions act as hubs for specialized cellular Ca2+-signaling events (16–18). ER–PM junctions also serve as microdomains that mediate crucial aspects of lipid transfer and homeostasis, including dynamic recruitment of cytoplasmic membrane-associated phosphatidylinositol (PtdIns) transfer proteins (PITPs) to ER–PM junctions. These proteins act to homeostatically replenish PM PtdIns(4,5)-bisphosphate (PtdIns(4,5)P2) following G-protein–coupled receptor–mediated phospholipase C activation and PtdIns(4,5)P2 depletion (19–22). However, relatively little is known at the molecular level of the components underlying lipid trafficking and homeostasis at the ER–PM junctions that are prominent in mammalian brain neurons.
The PM Kv2.1 voltage-gated potassium (Kv) channel is a critical determinant of electrical excitability and underlies the majority of the delayed rectifier current in mammalian brain neurons (23–27). Genetic ablation of Kv2.1 in mice yields animals with enhanced behavioral and electrical excitability (28), and recently identified de novo mutations in Kv2.1 are associated with debilitating neurological disorders in children harboring these mutations (29–32). In neurons, Kv2.1 is expressed as PM clusters on the soma, proximal dendrites, and axon initial segment (33–36) specifically at ER–PM junctions (34, 37–39). Kv2.1 plays a role in organizing neuronal ER–PM junctions (40–42) as a nonconducting function (42), and knockout mice lacking expression of Kv2.1 and its paralog Kv2.2 have altered neuronal ER–PM junctions (42).
We previously used a mass spectrometry (MS)-based proteomics approach to identify interactions between ER-localized vesicle-associated membrane protein-associated proteins VAPA/B and Kv2.1 at ER–PM junctions in the mouse brain (43). Kv2.1 interacts with VAP proteins via a “cryptic,” phosphorylation-dependent FFAT motif within its cytoplasmic C-terminal proximal restriction and clustering or PRC domain (43, 44). The PRC motif is necessary and sufficient for Kv2.1 clustering at ER–PM junctions (45), and ER (42) and VAP (43) recruitment to ER–PM junctions.
Numerous proteins that play diverse roles in cellular physiology are localized to specific subcellular compartments through their interaction with VAPs (46–48). This raises the possibility that the Kv2–VAP interaction at neuronal ER–PM junctions could impact the localization and function of other VAP-interacting proteins at neuronal ER–PM junctions. Here, we show that the phosphatidylinositol transfer proteins isoforms 1 (Nir2) and 2 (Nir3) are membrane-associated components of mouse brain Kv2.1–VAP complexes. We find that Kv2.1 impacts Nir2 localization and PtdIns homeostasis in a VAP-dependent manner, suggesting that the Kv2–VAP interaction can impact neuronal lipid signaling and homeostasis.
Results
Kv2 channels and membrane-associated PITPs Nir2 and Nir3 associate in mouse brain
We previously employed immunoaffinity capture of Kv2-containing complexes from mouse brain to identify VAPA and VAPB as components of neuronal Kv2.1 complexes (43) by employing detergent (1% Triton X-100, 0.5% deoxycholate, and 0.1% SDS) extractions of mouse whole-brain homogenates subjected to DSP-mediated chemical cross-linking (49) during homogenization. Here, we modified this approach by using on-bead trypsin digestion instead of in-gel digestion prior to liquid chromatography–tandem mass spectrometry (LC-MS/MS). We used two parallel comparative immunopurification (IP) approaches: the first approach employs antibodies against either Kv2.1 or a related Kv channel Kv1.2 in IP reactions from WT mouse brain samples, and the second approach utilizes anti-Kv2.1 IPs against samples prepared from the brains of WT and Kv2.1 knockout (KO) mice. The specificity of our IP reactions and subsequent analyses was demonstrated by the presence of numerous tryptic peptide spectra for Kv2.1 itself in the Kv2.1 IP sample, their absence in the parallel samples from Kv1.2 IPs, and vice versa (Fig. 1A). Immunoblot analyses further confirmed the effective and specific capture of the target proteins in the respective IPs (Fig. 1B). We also found enriched in the Kv2.1 IPs (and absent from the Kv1.2 IPs) a number of previously reported Kv2.1-interacting proteins, including Kv2.2 (39, 43, 50) and VAPA and VAPB (43, 44). We note the nonspecific presence of GRP75, ROA2, and other proteins commonly found on the CRAPome database (http://crapome.org) in both Kv2.1 and Kv1.2 IPs. Importantly, the LC-MS/MS analyses of the Kv2.1 IPs exclusively reported the presence of the membrane-associated PITP isoforms Nir2 (PITPNM1, UniProt O35954) and Nir3 (PITPNM2, UniProt Q6ZPQ6) (Fig. 1A, red box). We also observed enriched recovery of Kv2.2, and VAPB, and Nir proteins in Kv2.1 IPs from WT but not Kv2.1 KO mouse brains (Fig. 1A). We also include here data for the Kv2.1 auxiliary subunit AMIGO-1, which exhibits an extensive association and colocalization with Kv2.1 in brain (38, 51), yet for which peptide spectra are present at relatively low abundance in the Kv2.1 IP samples relative to Kv2.2, VAPA, VAPBB, and the Nir proteins, and for TMEM24/C2CD2L, recently reported to colocalize with Kv2.1 at neuronal ER–PM junctions (52), and which we specifically recovered in Kv2.1 IPs from WT brain samples (Fig. 1A). These data support that in mouse brain the Nir proteins associate with protein complexes containing Kv2 channels and VAPs.
Figure 1.
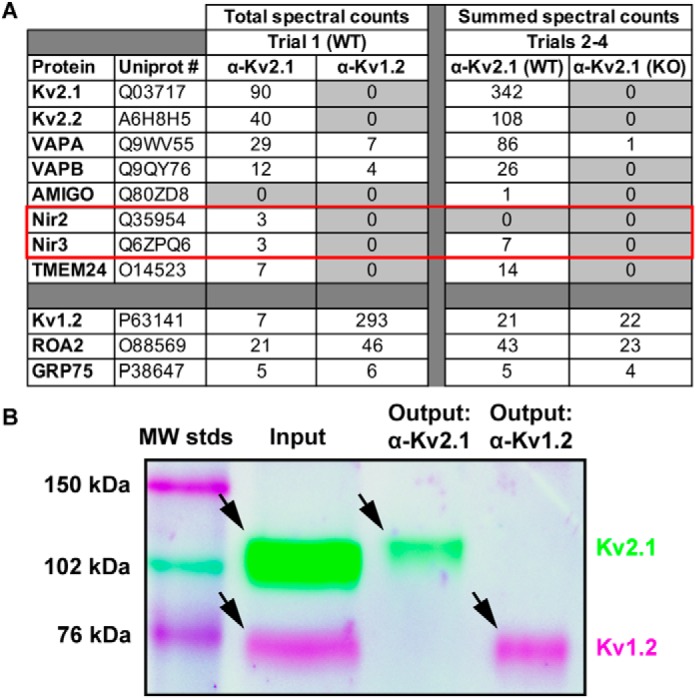
Membrane-associated phosphatidylinositol transfer proteins biochemically associate with Kv2.1 in mouse brain. A, total spectra counts of proteins recovered from a single trial of Kv2.1 or Kv1.2 IPs from WT mouse brain and summed spectra counts from three separate trials of Kv2.1 IPs from WT or Kv2.1 KO mouse brain. Note the enriched recovery of Kv2.2, VAPA and VAPB, and Nir2/3 in the Kv2.1 IPs relative to the Kv1.2 IPs. Note the nonspecific recovery of ROA2 and GRP75 in all samples. B, multiplexed immunoblot analysis of input and output fractions from a single trial of Kv2.1 or Kv1.2 IPs performed using WT mouse brain. Note the presence of both Kv2.1 (cyan band, ≈110 kDa) and Kv1.2 (magenta band, ≈70 kDa) in the input fraction and the exclusive presence of Kv2.1 in the Kv2.1 IP output fraction, and Kv1.2 in the Kv1.2 IP output fraction.
Muscarinic stimulation induces a reversible loss of PM PtdIns(4,5)P2 and accumulation of Nir2 at ER–PM junctions
Membrane-associated PITPs (Nir2 and Nir3) are components of the homeostatic machinery that mammalian cells employ to maintain and/or replenish PM PtdIns(4,5)P2. Depletion of PtdIns(4,5)P2 induces an accumulation of Nir2/3 at ER–PM junctions and a subsequent replenishment of PM PtdIns(4,5)P2 via retrieval of phosphatidic acid from, and insertion of PtdIns into, the PM (19–22). To begin exploring the role of Nir2 association with Kv2-containing ER–PM junctions in this process, we first validated the presence of these homeostatic mechanisms in HEK293T, on the basis that HEK293T cells recapitulate certain aspects of neuronal gene expression (53) but do not endogenously express either Kv2.1 or Kv2.2 (54). HEK293T cells that express exogenous type-1 muscarinic receptor (M1R) following transient transfection demonstrated robust PM-associated expression of YFP-tagged M1R (YFP–M1R), evident in spinning disk confocal (SDC) optical sections taken through the center of live cells (Fig. 2A). Importantly, the PM expression of YFP–M1R was functionally coupled to PM PtdIns(4,5)P2 depletion, such that acute muscarinic stimulation (10 μm oxotremorine M (OxoM) treatment for 40 s) was sufficient to deplete PM PtdIns(4,5)P2 as reported by displacement of the PtdIns(4,5)P2 biosensor, mCherry-tagged PHPLCδ1 (mCherry–PHPLCδ1), from the PM into the cytoplasm (Fig. 2, B and C). The depletion of PM PtdIns(4,5)P2 was transient as washout of OxoM enabled mCherry–PHPLCδ1 to re-accumulate at the PM following resynthesis of its lipid substrate (Fig. 2, B and C).
Figure 2.

Acute muscarinic stimulation induces a recoverable loss of PtdIns(4,5)P2 from the PM, and Nir2 recruitment to ER–PM junctions, in HEK293T cells expressing the M1 receptor. A, confocal optical section taken through the center of a single resting HEK293T cell cotransfected with YFP–M1R (shown in A) and mCherry–PHPLCδ (measured in B and shown in C). Note the efficient expression of YFP–M1R at the PM. The scale bar is 5 μm. B, time course of cytoplasmic mCherry–PHPLCδ intensity values, measured from the cell shown in A and C, prior to and during acute (40 s) stimulation (indicated by gray bar) with 10 μm OxoM and following washout. C, kymograph of PM and cytoplasmic mCherry–PHPLCδ expression taken from the same cell as in A and B. Note the loss of PM mCherry–PHPLCδ during 10 μm OxoM stimulation, and the recovery of PM mCherry–PHPLCδ following washout. Representative confocal optical sections of mCherry–PHPLCδ, taken through the center of the cell during rest, stimulation, and washout, are shown below the kymograph. The scale bar is 5 μm. Selection for kymograph is indicated by a red dashed line in these images. D, representative TIRF images of a pair of HEK293T cells cotransfected with GFP–VAPA (not shown), YFP–M1R (not shown), mCherry–Nir2 (magenta), and BFP–SEC61β (green) before (left) and after (right) acute stimulation with 10 μm OxoM. Merged image is shown at bottom. The scale bar in the large image is 10 μm. The PCC (mean ± S.D.) between mCherry–Nir2 and BFP–SEC61β following muscarinic stimulation is reported in the right merged image (n = 4 cells). E, line-scan analyses of mCherry–Nir2 (magenta) and BFP–SEC61β (green) intensity, following stimulation with 10 μm OxoM, from region indicated in the right merged image of D.
We next utilized total internal reflection fluorescence (TIRF) imaging of live cells to examine whether muscarinic stimulation was also sufficient to induce Nir2 accumulation at ER–PM junctions. We cotransfected HEK293T cells with YFP–M1R, GFP-tagged VAPA (GFP–VAPA), and BFP-tagged SEC61β (BFP–SEC61β), as a marker of cortical ER within the TIRF footprint (43, 55), and mCherry-tagged Nir2 (mCherry–Nir2). Acute stimulation with OxoM induced accumulation of the cytoplasmic mCherry–Nir2 protein into puncta localized to ER–PM junctions as marked by BFP–SEC61β in the TIRF field (Fig. 2D), such that enhanced colocalization between mCherry–Nir2 and BFP–SEC61β was present following stimulation (Fig. 2E). In line with these observations, measurements of Pearson's correlation coefficient (PCC) values between mCherry–Nir2 and BFP–SEC61B from these cells following muscarinic stimulation returned relatively high values (mean PCC ± S.D.: 0.76 ± 0.11, n = 4 cells). These experiments demonstrate that muscarinic stimulation of HEK293T cells is sufficient to induce a recoverable depletion of PM PtdIns(4,5)P2 and accumulation of the exogenously-expressed cytoplasmic Nir2 protein at ER–PM junctions, as seen previously in other cell types (19, 21).
Nir2 is recruited to ER–PM junctions mediated by cell-surface Kv2.1
We next examined the impact of Kv2.1 expression on Nir2 accumulation at ER–PM junctions by transiently transfecting HEK293T cells with GFP–VAPA, YFP–M1R, mCherry–Nir2, and CFP-tagged Kv2.1 (CFP–Kv2.1) and imaging live cells using TIRF microscopy. As shown in Fig. 3B, in ≈40% of unstimulated HEK293T cells coexpressing CFP–Kv2.1, GFP–VAPA, YFP–M1R, and mCherry–Nir2, the near-membrane signal of cytoplasmic mCherry–Nir2 (as imaged with TIRF) was present in large puncta that colocalized with clusters of CFP–Kv2.1, as evidenced by the white puncta in the merged panels in Fig. 3B, and by the overlap analysis in Fig. 3B. These large mCherry–Nir2 puncta were not present in unstimulated cells lacking CFP–Kv2.1 expression (see the representative image of diffuse mCherry–Nir2 signal observed in unstimulated cells lacking Kv2.1 in Fig. 2D) or in cells cotransfected with mCherry–Nir2, YFP–M1R, and CFP–Kv2.1 but lacking exogenous VAPA (data not shown). This demonstrates that unstimulated HEK293T cells expressing exogenous VAPA, exogenous coexpression of Kv2.1, that results in more robust ER–PM junctions (41, 42) via Kv2.1-mediated recruitment of VAPs (43, 44) is sufficient to impact Nir2 localization.
Figure 3.

Muscarinic stimulation triggers Nir2 recruitment to Kv2.1/VAP-mediated ER–PM junctions. A, TIRF image of a HEK293T cell cotransfected with YFP–M1R (not shown), GFP–VAPA (not shown), and mCherry–Nir2 (shown in inverted contrast) stimulated with 10 μm OxoM. The scale bar is 5 μm and holds for B. B, TIRF images of a resting HEK293T cell cotransfected with YFP–M1R (not shown), GFP–VAPA (not shown), mCherry–Nir2 (magenta), and CFP–Kv2.1 (green). Pixel overlap analysis of CFP–Kv2.1 and mCherry–Nir2 is shown to the right of the merged image. Note the robust colocalization of mCherry–Nir2 with CFP–Kv2.1. C, TIRF images of another resting HEK293T cell cotransfected with YFP–M1R (not shown), GFP–VAPA (not shown), mCherry–Nir2 (magenta), and CFP–Kv2.1 (green) prior to acute stimulation with 10 μm OxoM. The scale bar is 5 μm and holds for D. Line scan analysis of mCherry–Nir2 and CFP–Kv2.1 intensity, from selection indicated in merged image, is shown to right of merged image. D, TIRF images of same cell shown in C following acute stimulation with 10 μm OxoM. Line scan analysis of mCherry–Nir2 and CFP–Kv2.1 intensity, from selection indicated in merged image, shown to right of merged image. Note the overlap of Kv2.1 and Nir2 intensity profiles following stimulation with OxoM. E, summary graph of mean Nir2 puncta sizes, measured from 10 μm OxoM-stimulated HEK293T cells expressing mCherry–Nir2, GFP–VAPA, YFP–M1R (control), or coexpressing CFP–Kv2.1 (+Kv2.1). Bars are mean ± S.D. (****, p value = 6.87 × 10−7, n = 15–16 cells, two-tailed unpaired t test). F, cumulative frequency distributions of Nir2 puncta sizes measured from cells summarized in E.
In these experiments, other coexpressing/unstimulated cells lacked mCherry–Nir2 puncta (Fig. 3C). We next examined the impact of muscarinic stimulation on Nir2 accumulation at ER–PM junctions in these cells. Acute stimulation with 10 μm OxoM induced both a rapid accumulation of the cytoplasmic mCherry–Nir2 signal to the near-membrane TIRF field but also its presence in puncta that for the most part colocalized with clustered CFP–Kv2.1 (Fig. 3D). This extent and nature of mCherry–Nir2 clustering was not observed in stimulated cells lacking CFP–Kv2.1 expression (see the representative image of the smaller mCherry–Nir2 puncta observed in stimulated cells lacking Kv2.1 in Fig. 3A). To quantify these observations, we measured the size of mCherry–Nir2 puncta from stimulated HEK293T cells lacking or coexpressing Kv2.1. Consistent with our observations, there was a significant increase in mean Nir2 puncta size in cells coexpressing Kv2.1 (mean ± S.D.: 0.89 ± 0.35 μm2), relative to control cells lacking Kv2.1 expression (mean ± S.D.: 0.30 ± 0.11 μm2), as well as a rightward shift in the cumulative frequency distribution of Nir2 puncta sizes in cells coexpressing Kv2.1, relative to control cells (Fig. 3, E and F). These data demonstrate that Nir2 accumulates at Kv2.1/VAPA-containing ER–PM junctions following PM PtdIns(4,5)P2 depletion and that the presence of Kv2.1/VAPA-mediated ER–PM junctions can enhance Nir2 puncta at these sites, in both resting/unstimulated and muscarinic-stimulated cells.
Finally, as we have recently demonstrated that K+ conduction is not necessary for either cell-surface expression of Kv2.1 or organization of ER–PM junctions by Kv2.1/VAPs (42, 43), we compared the ability of a nonconducting mutant Kv2.1 isoform (Kv2.1 P404W (42, 43, 56)) to recruit mCherry–Nir2 puncta in unstimulated HEK293T cells (coexpressing exogenous mCherry–Nir2, GFP–VAPA, and YFP–M1R) relative to WT Kv2.1. In these experiments we labeled cell-surface Kv2.1 with GxTX-633, the membrane-impermeant and Kv2-specific tarantula toxin guanxgitoxin-1E (57) conjugated to DyLight 633 (42, 58) and imaged near-membrane signals using TIRF microscopy. No apparent differences were observed between WT Kv2.1 or Kv2.1 P404W in their ability to either traffic to the cell surface (as reported previously (42)) or induce large clusters of mCherry–Nir2 in unstimulated HEK293T cells (Fig. 4, A and B). Line-scan analyses of fluorescence intensity from these cells yielded comparable intensity profiles as well as R2 values for cells expressing conducting or nonconducting Kv2.1 (Fig. 4). These data taken together demonstrate the following: 1) Nir2 accumulates at Kv2.1 and VAPA-containing ER–PM junctions; 2) the presence of Kv2.1/VAPA-mediated ER–PM junctions can enhance the size of Nir2 puncta at these sites; and 3) Kv2.1 K+ conduction is not necessary for this impact on Nir2 localization.
Figure 4.
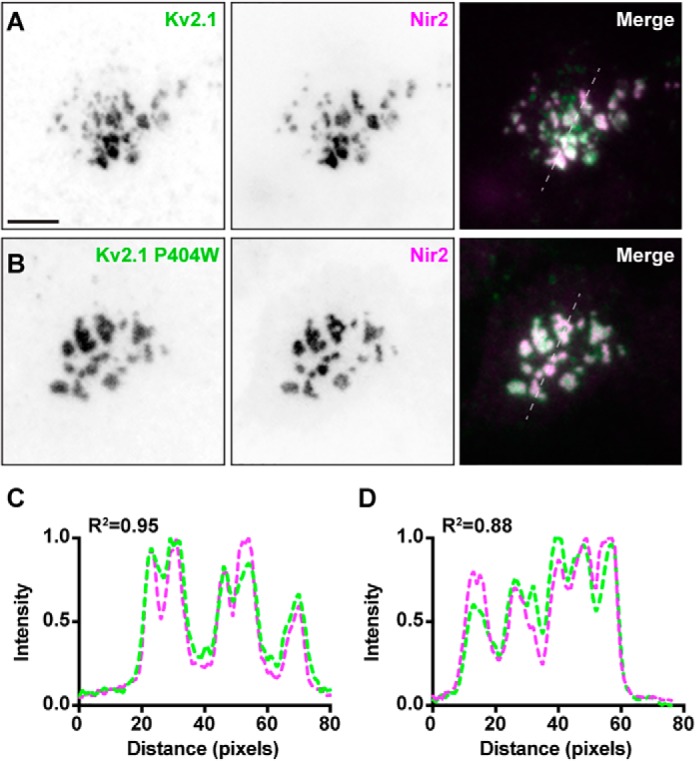
Conducting and nonconducting cell surface Kv2.1 colocalizes with Nir2 in resting HEK293T cells. A, TIRF images of a HEK293T cell cotransfected with CFP–Kv2.1 (surface labeled with GxTX-633, green), GFP–VAPA (not shown), YFP–M1R (not shown), and mCherry–Nir2 (magenta). The scale bar is 10 μm and holds for all images. B, TIRF image of another HEK293T cell cotransfected with Kv2.1 P404W (surface-labeled with GxTX-633, green) GFP–VAPA (not shown), YFP–M1R (not shown), and mCherry–Nir2 (magenta). C and D, line-scan analyses of GxTX-633 surface labeling and mCherry–Nir2 intensity from selections indicated in merged images of A and B, respectively.
Exogenous VAPA expression enhances colocalization of Kv2.1 and Nir2 at ER–PM junctions in cultured hippocampal neurons
We next investigated the spatial relationship between Kv2.1, VAPA, and Nir2 in live-cultured rat hippocampal neurons (CHNs) that endogenously express Kv2.1 and VAPs (43). In our initial experiments employing widefield live-cell imaging of CHNs, we observed weak colocalization of exogenously expressed CFP–Kv2.1 (or GFP-tagged Kv2.2 (GFP–Kv2.2)) and mCherry–Nir2 (data not shown). We hypothesized that endogenous VAPA levels in CHNs may be insufficient to mediate the effective association of exogenous Nir2 with Kv2.1 clusters or that endogenous VAP proteins are pre-associated with other binding partners or otherwise unavailable to mediate this association between exogenously expressed Kv2.1 and Nir2. As such, we examined the impact of exogenous VAPA coexpression on Kv2.1 and Nir2 colocalization. We evaluated subcellular localization using SDC microscopy of live CHNs transfected at 7–10 DIV and imaged 48 h later. We found that CHNs expressing mCherry–Nir2 alone produced a mostly-diffuse cytoplasmic signal with small puncta on the soma and processes (Fig. 5A). Coexpressing GFP–VAPA generally resulted in puncta of colocalized GFP–VAPA and mCherry–Nir2 on the soma and processes (Fig. 5B). In contrast, coexpressing CFP–Kv2.1 did not appear to substantially impact mCherry–Nir2 localization in most cells, despite the expression of large CFP–Kv2.1 clusters on the soma and processes (Fig. 5C). Coexpressing CFP–Kv2.1, GFP–VAPA, and mCherry–Nir2 resulted in colocalization of all three proteins in large clusters on the soma and processes (Fig. 5D). In each case, we noted the presence of mCherry–Nir2 puncta, which did not appear to colocalize with either CFP–Kv2.1 or GFP–VAPA, potentially indicating the association of mCherry–Nir2 with endogenous VAPs at these sites. To quantify these observations, we measured PCC values and found a significant increase in PCC measurements between CFP-Kv2.1 and mCherry–Nir2 upon GFP–VAPA coexpression (Fig. 5E). These experiments demonstrate that in CHNs exogenous VAPA expression is required for colocalization of exogenously expressed Kv2.1 and Nir.
Figure 5.

VAP coexpression triggers Nir2 recruitment to ER–PM junctions mediated by Kv2.1 in live cultured rat hippocampal neurons. A, SDC optical sections taken at the basal surface of a cultured rat hippocampal neuron transfected with mCherry–Nir2. The scale bar is 5 μm and holds for all images. B, SDC optical sections taken at the basal surface of a cultured rat hippocampal neuron cotransfected with mCherry–Nir2 (red) and GFP–VAPA (green). Note the robust colocalization of GFP–VAPA with mCherry–Nir2. Line scan analysis of mCherry–Nir2 and GFP–VAPA intensity from selection indicated in merged image of B is shown to the right of B. C, SDC optical sections taken at the basal surface of a cultured rat hippocampal neuron cotransfected with mCherry–Nir2 (red) and CFP–Kv2.1 (blue). Note the lack of minimal colocalization of mCherry–Nir2 with CFP–Kv2.1. Line scan analysis of mCherry–Nir2 and CFP–Kv2.1 intensity from selection indicated in merged image of C is shown to the right of C. D, SDC optical sections taken at the basal surface of a cultured rat hippocampal neuron cotransfected with mCherry–Nir2 (red), GFP–VAPA (green), and CFP–Kv2.1 (blue). Note the robust colocalization of mCherry–Nir2 with GFP–VAPA and CFP–Kv2.1. Line scan analysis of mCherry–Nir2, GFP–VAPA, and CFP–Kv2.1 intensity from selection indicated in merged image of D is shown to the right of D. E, enlarged images of mCherry–Nir2 (red), GFP–VAPA (green), and CFP–Kv2.1 (blue) from selection indicated in merged image of D. F, summary graph of Pearson's correlation coefficient values between mCherry–Nir2 and GFP–VAPA or CFP–Kv2.1, measured from cultured rat hippocampal neurons transfected with mCherry–Nir2 and CFP–Kv2.1 (circles) or mCherry–Nir2, CFP–Kv2.1, and GFP–VAPA (triangles). Bars are mean ± S.D. (VAPA: ****, p value = 0.0001, n = 16 cells; Kv2.1: ****, p value = 0.0001, n = 17 cells; ordinary one-way ANOVA followed by Dunnett's multiple comparisons test).
Spatial correlation of endogenous immunolabeled Nir2, Kv2.1, and VAPs in resting cultured rat hippocampal neurons
Given the biochemical association of Kv2.1 and Nir proteins detected in our proteomics experiments, and the colocalization of exogenous Nir2, Kv2.1, and VAPA in HEK293T cells and CHNs, we next investigated the subcellular localization of endogenous immunolabeled Nir2, Kv2.1, and VAPs. Until this point, we have largely utilized conventional TIRF imaging, under the rationale that TIRF microscopy, while diffraction-limited, provides the highest signal–to–noise for studying dynamic ER–PM junction-associated processes in live cells. Recent studies have demonstrated that the size of native ER–PM junctions in cultured mammalian cells (59) and mouse brain neurons (9) is below the diffraction limit, and as such, conventional light microscopy is not optimal for interrogating the spatial distribution of endogenous ER-PM junction components at native ER–PM junctions. Toward this end, we chose to employ super-resolution imaging (N-SIM) of fixed and immunolabeled but otherwise native CHNs, as we have done previously in similar studies (36, 37, 42, 43, 60). We immunolabeled CHNs that were cultured under resting conditions prior to fixation, followed by N-SIM imaging, similar to the approach that we utilized previously (43). We employed two distinct commercially available anti-Nir2 polyclonal antibodies raised against distinct regions of human Nir2 (amino acids 365–444 or 589–651) that each exhibit extensive (>80%) sequence identity with rat Nir2. For both anti-Nir2 antibodies, we observed robust immunolabeling of Nir2 puncta on the soma and proximal processes of these neurons (Fig. 6, A–C). There was no significant difference in the mean area (≈0.06 μm2) of Nir2 puncta labeled with these two different anti-Nir2 antibodies (Fig. S1, A and B). A subset of Nir2 puncta immunolabeling (≈20–25% of the total endogenous immunolabeled Nir2 signal) overlapped with immunolabeling for endogenous Kv2.1 (Fig. S1C), with additional Nir2 immunolabeling near but not directly overlapping with immunolabeled Kv2.1 clusters. The amount of overlap between Nir2 and Kv2.1 immunolabeling was not significantly different when either anti-Nir2 antibody was employed (Fig. S1D). We also immunolabeled for endogenous VAPs using a mAb recognizing both VAPA and VAPB (43), and we observed that immunolabeling of VAPs also overlapped with the immunolabeling of Kv2.1 and Nir2 (Fig. 6A).
Figure 6.

Spatial distributions of Kv2.1, Nir2, and VAPA/B immunolabeling in cultured hippocampal neurons. A, super-resolution (N-SIM) optical sections taken at the basal membrane of a cultured rat hippocampal neuron immunolabeled for endogenous Kv2.1 (shown in green), Nir2 (shown in red), and VAPA/B (shown in blue). Merged image shown to the right. Bottom panels show magnified selection from merged image in top panel. The scale bar in the upper left Kv2.1 panel is 5 μm and holds for all panels in that row. The scale bar in the magnified Kv2.1 panel is 1.25 μm and holds for all panels in the bottom two rows. B, super-resolution (N-SIM) optical sections taken at the basal membrane of a cultured rat hippocampal neuron immunolabeled for endogenous Kv2.1 and Nir2. The merged image is shown to the right, with Kv2.1 shown in green and Nir2 shown in magenta. The top row shows the original image, and the bottom row the same image after randomization of the Kv2.1 immunolabeling signal. Arrows point at spatially correlated signals in the original images that are lost in the randomized images. The scale bar is 5 μm and also holds for D. C, summary graph of percent overlap of Nir2 signal with Kv2.1 measured from the original images and from the same images after randomization of the Kv2.1 signal (randomized). Percent overlap values for Kv2.1 and Nir2 are original: 23.9 ± 5.97%, and randomized: 19.4 ± 4.56% (mean ± S.D.; ****, p value = 0.000594, n = 11 cells; two-tailed paired t test). D, super-resolution (N-SIM) optical sections taken at the basal membrane of a cultured rat hippocampal neuron immunolabeled for endogenous Kv2.1 and RyRs. The merged image is shown to the right, with Kv2.1 shown in green and RyRs shown in magenta. The top row shows the original image, and the bottom row the same image after randomization of the Kv2.1 immunolabeling signal. Arrows point at spatially correlated signals in the original images that are lost in the randomized images. E, summary graph of percent overlap of RyR signal with Kv2.1 measured from the original images and from the same images after randomization of the Kv2.1 signal (randomized). Percent overlap values for Kv2.1 and RyRs are original: 62.6 ± 9.96%, and randomized: 16.5 ± 7.00% (mean ± S.D.; ****, p value = 0.00000195, n = 8 cells; two-tailed paired t test).
Using these N-SIM images, we next performed two independent spatial interaction statistical analyses, FIJI-MOSAIC (61) and Icy-GcoPS (62), on immunolabeled puncta of endogenous Nir2, Kv2.1, and VAPA/B proteins within a region of interest outlining the neuronal soma and proximal processes. In both analyses, we found that the spatial distributions of immunolabeled Kv2.1 and Nir2 puncta were significantly correlated (versus the null hypotheses of independence) and could not be recapitulated in images in which their relative positions had been iteratively randomized in silico (63, 64). Furthermore, the spatial distributions of the immunolabeled Nir2 and Kv2.1 puncta were significantly correlated regardless of which anti-Nir2 antibody was used (p values listed in Table 1). The spatial distributions of Nir2 and VAPA/B puncta were also similarly correlated (p values listed in Table 1).
Table 1.
p values for spatial interaction statistical analyses of N-SIM images in Fig. 6
Finally, given the relatively low amount of signal overlap between Nir2 and Kv2.1 immunolabeling (≈20–25% of the total endogenous immunolabeled Nir2 signal) that we observed in these N-SIM images, we further examined whether the amount of overlap between Nir2 and Kv2.1 immunolabeling could be recapitulated in images in which the localization of Kv2.1 signal contained in the soma and proximal processes had been randomized. Toward this end, we segmented the Kv2.1 immunolabeling signal into blocks sized at 10 × 10 pixels (i.e. 313.4 × 313.4 nm), randomized the position of these blocks, and measured the amount of overlap between Nir2 with Kv2.1 in these images. We found that randomization of the Kv2.1 signal using this approach resulted in a significant decrease in the overlap of Kv2.1 and Nir2 immunolabeling (Fig. 6, B–D). We also found that randomization of the Kv2.1 signal similarly resulted in a significant decrease in the overlap of Kv2.1 with immunolabeled ryanodine receptors (RyRs, Fig. 6, E–G); a family of ER–PM junction-associated ER Ca2+-release channels previously found to colocalize with Kv2.1 (37, 40, 42, 43). We also found that Kv2.1 and RyRs were spatially correlated using the same two independent spatial interaction statistical analyses that we had employed on Kv2.1 and Nir2. Finally, we further examined whether altering the size of the blocks used to segment and randomize the Kv2.1 signal had any impact on these results. We found that various block sizes produced a similar significant decrease in Kv2.1 overlap with either Nir2 or RyRs (Fig. S2; Tables 2 and 3, respectively). Taken together these findings demonstrate that the subcellular distributions of endogenous immunolabeled Kv2.1, VAPs, and Nir2 are spatially correlated in native CHNs.
Table 2.
Quantification of the impact of Kv2.1 signal segmentation and randomization using various block sizes on overlap with Nir2
NA means not applicable.
| Sample | Percent of total Nir2 signal overlapping with Kv2.1 | S.D. | p value (two-tailed, paired t-test vs. percent overlap in original image) |
|---|---|---|---|
| Original image | 23.9 | 5.97 | NA |
| Kv2.1 randomized (1 × 1 pixel block size) | 1.99 | 0.437 | 0.000119 |
| Kv2.1 randomized (5 × 5 pixel block size) | 1.95 | 0.426 | 0.0000303 |
| Kv2.1 randomized (10 × 10 pixel block size) | 1.93 | 0.458 | 0.0000174 |
| Kv2.1 randomized (20 × 20 pixel block size) | 1.84 | 0.398 | 0.0000176 |
| Kv2.1 randomized (30 × 30 pixel block size) | 1.74 | 0.399 | 0.0000267 |
| Kv2.1 randomized (40 × 40 pixel block size) | 1.68 | 0.380 | 0.0000222 |
| Kv2.1 randomized (50 × 50 pixel block size) | 1.65 | 0.405 | 3.76E-06 |
Table 3.
Quantification of the impact of Kv2.1 signal segmentation and randomization using various block sizes on overlap with RyRs
NA means not applicable.
| Sample | Percent of total RyR signal overlapping with Kv2.1 | S.D. | p value (two-tailed, paired t-test vs. percent overlap in original image) |
|---|---|---|---|
| Original image | 62.6 | 9.96 | NA |
| Kv2.1 randomized (1 × 1 pixel block size) | 16.4 | 6.16 | 8.267E-07 |
| Kv2.1 randomized (5 × 5 pixel block size) | 16.0 | 5.85 | 7.43E-07 |
| Kv2.1 randomized (10 × 10 pixel block size) | 16.0 | 5.93 | 6.79E-07 |
| Kv2.1 randomized (20 × 20 pixel block size) | 16.8 | 7.41 | 8.55E-07 |
| Kv2.1 randomized (30 × 30 pixel block size) | 14.7 | 5.46 | 1.06E-06 |
| Kv2.1 randomized (40 × 40 pixel block size) | 15.4 | 5.42 | 7.22E-07 |
| Kv2.1 randomized (50 × 50 pixel block size) | 14.8 | 4.57 | 2.523E-07 |
Nir2 is preferentially recruited to ER–PM junctions organized by Kv2.1 compared with those formed via a rapamycin-mediated heterodimerization strategy
To test whether bulk formation of ER–PM junctions would result in recruitment of Nir2 as occurs upon formation of Kv2.1/VAPA-containing junctions, we increased the overall abundance of ER–PM junctions formed using a rapamycin-mediated heterodimerization strategy (60, 65). This entails coexpression of ER-localized CFP–CB5–FKBP (CB5) and PM-localized lyn11–FRB (lyn11), whose association is induced by treatment with 5 μm rapamycin to trigger enhanced ER–PM junctions stabilized by the rapamycin-dependent interaction of CB5 and lyn11. We first investigated the impact of bulk induction of ER–PM junctions on the subcellular distribution of GFP–VAPA, which in confocal images is widely distributed throughout the ER where it colocalizes with the bulk ER marker DsRed2–ER5 (Fig. 7A) in otherwise naïve HEK293T cells. Coexpression with CB5/lyn11 and triggered ER–PM junction formation via treatment with 5 μm rapamycin did not result in recruitment of GFP–VAPA out of bulk ER (marked by DsRed2–ER5) to these induced ER–PM junctions marked by CB5 (Fig. 7B), as indicated by the lack of overlap of the blue CB5 signal with the yellow signal resulting from overlap of GFP–VAPA and DsRed2–ER5 signals. This is in sharp contrast to the robust recruitment of GFP–VAPA to junctions formed by expression of (and that contain) CFP–Kv2.1 in the place of CB5/lyn11 (Fig. 6C), in which all three signals (CFP–Kv2.1, GFP–VAPA, and DsRed2–ER5) overlap to yield a white signal at ER–PM junctions at the cell periphery, consistent with previous reports (43, 44).
Figure 7.

Nir2 is preferentially recruited to ER–PM junctions enhanced by Kv2.1 expression, relative to those formed via a rapamycin-mediated heterodimerization strategy. A, SDC optical sections taken through the center of a HEK293T cell cotransfected with GFP–VAPA (green) and DsRed2–ER5 (red). The scale bar is 5 μm and holds for all images in A–C. Note the broad distribution of VAPA throughout bulk ER. B, confocal optical sections taken through the center of a HEK293T cell cotransfected with CFP–CB5–FKBP (blue), lyn11–FRB (not shown), GFP–VAPA (green), and DsRed2–ER5 (red) and treated with 5 μm rapamycin. Note the broad distribution of VAPA throughout bulk ER, and the lack of an enrichment of VAPA at ER–PM junctions induced by CFP–CB5–FKBP/lyn11–FRB heterodimerization. C, confocal optical sections taken through the center of a HEK293T cell cotransfected with CFP–Kv2.1 (blue), GFP–VAPA (green), and DsRed2–ER5 (red). Note the reduction of VAPA in bulk ER and the enrichment of GFP–VAPA at ER–PM junctions induced by CFP–Kv2.1 expression. D, summary graph of peripheral VAPA intensity measured from HEK293T cells cotransfected with GFP–VAPA and CFP–Kv2.1 or CFP–CB5–FKBP/lyn11–FRB and treated with 5 μm rapamycin (****, p value = 3.58 × 10−13, n = 27–31 cells, two-tailed unpaired t test). E and F, TIRF image of a HEK293T cell cotransfected with DsRed–Kv2.1 (green), CFP–CB5–FKBP (magenta), and lyn11–FRB (not shown) following 5 μm rapamycin treatment (E) and following 10 μm latrunculin A treatment (F). The scale bar is 10 μm and holds for E and F. G, summary graph of PCC measurements between DsRed–Kv2.1 and CFP–CB5–FKBP measured from HEK293T cells cotransfected with DsRed–Kv2.1, CFP–CB5–FKBP, and lyn11–FRB and treated with 5 μm rapamycin and further treated with 10 μm latrunculin A (ns, p value = 0.8092, n = 15 cells, two-tailed unpaired t test). H and I, TIRF images of a HEK293T cell cotransfected with YFP–M1R (not shown), GFP–VAPA (not shown), mCherry–Nir2 (magenta), CFP–CB5–FKBP (green), and lyn11–FRB (not shown) and treated with 5 μm rapamycin, at rest (H) and following (I) acute stimulation with 10 μm OxoM. The scale bar is 5 μm and holds for all images in H and I. J, line scan analysis of CFP–CB5–FKBP and mCherry–Nir2 intensity from selection indicated in merged image of I. Note the interdigitation of CFP–CB5–FKBP and mCherry–Nir2 intensity profiles. K, summary graph of PCC values between mCherry–Nir2 and CFP–Kv2.1 or CFP–CB5–FKBP measured from HEK293T cells cotransfected with mCherry–Nir2, GFP–VAPA, YFP–M1R, and CFP–Kv2.1 or CFP–CB5–FKBP/lyn11–FRB and treated with 10 μm OxoM (****, p value = 4.856 × 10−8, n = 17–18 cells, two-tailed unpaired t test).
To quantify the differential impacts on VAPA distribution mediated by Kv2.1 and CB5/lyn11 expression, we measured the intensity of VAPA fluorescence at the cell periphery associated with CB5/lyn11 or Kv2.1-mediated ER–PM junctions. Consistent with our visual observations, we quantified a significant increase in peripheral VAPA intensity in cells coexpressing Kv2.1 relative to cells coexpressing CB5/lyn11 (Fig. 6D). In addition, coexpression of DsRed-tagged Kv2.1 (DsRed–Kv2.1) with the components of the CB5/lyn11 system followed by rapamycin treatment yielded clusters of DsRed–Kv2.1 and CB5 that generally did not colocalize (Fig. 6E). Disruption of the actin cytoskeleton via latrunculin A treatment yielded the expected (42, 66) fusion of Kv2.1 clusters (Fig. 7F) but did not alter the mutual exclusion of Kv2.1-mediated ER–PM junctions with those formed by CB5/lyn11 coupling (Fig. 7F). PCC measurements taken from these cells prior to, or after, latrunculin A treatment were not significantly altered (Fig. 7G), further supporting the notion that ER–PM junctions mediated by Kv2.1/VAP association are distinct from those mediated by CB5/lyn11 interaction. Given the observed differences between Kv2.1- and CB5/lyn11-mediated ER–PM junctions, we addressed whether Nir2 would be recruited to ER–PM junctions mediated by CB5/lyn11. We coexpressed CB5/lyn-11, YFP–M1R, GFP–VAPA, and mCherry–Nir2 in HEK293T cells, treated with 5 μm rapamycin to induce coupling of CB5/lyn11 and formation of ER–PM junctions, and further stimulated cells with 10 μm OxoM to induce PtdIns(4,5)P2 depletion. We found that mCherry–Nir2 was not recruited to ER–PM junctions mediated by CB5/lyn11 to the same extent as those mediated by Kv2.1, such that PCC measurements of colocalization of mCherry–Nir2 and CB5 were significantly lower than those between mCherry–Nir2 and CFP–Kv2.1 in these experiments (Fig. 7J). This was particularly evident in a subset of cells in which mCherry–Nir2 appeared to be recruited to areas generally lacking CB5/lyn11 (Fig. 7, H and I), as indicated by the lack of overlap between their respective signals and interdigitated signal intensity line scans (Fig. 7J). We note that in contrast to the effects of Kv2.1 expression (Fig. 3), the formation of enhanced ER–PM junctions induced by CB5/lyn11 coupling did not lead to formation of Nir2 puncta in resting cells (i.e. prior to treatment with 10 μm OxoM). Taken together, these results demonstrate that ER–PM junctions mediated by Kv2.1 and VAP are distinct in their ability to recruit Nir2 compared with those formed upon CB5/lyn11 coupling.
Kv2.1, VAPA, and Nir2 colocalized at ER–PM junctions have comparable recovery rates after photobleaching
We next compared the turnover of Kv2.1, VAPA, and Nir2 present at Kv2.1–VAPA-mediated ER–PM junctions by performing fluorescence recovery after photobleaching (FRAP) experiments. We measured the recovery rates of CFP–Kv2.1, mCherry–Nir2, and GFP–VAPA fluorescence in the photobleached region of interest that occurs upon diffusion of these fluorescent proteins from unbleached areas into this region. We photobleached a large area of the basal surface of each cell, which often contained multiple large ER–PM junction puncta (Fig. 8, A–C). Following photobleaching, we monitored the recovery of CFP–Kv2.1, GFP–VAPA, or mCherry–Nir2 fluorescence over the next 360 s using SDC imaging, and we then used this to calculate the turnover rate (t½) for each protein. The turnover rates of CFP–Kv2.1, GFP–VAPA, and mCherry–Nir2 were all relatively low and not significantly different from each other (Fig. 8D) suggesting that Kv2.1, VAPA, and Nir2 not only colocalize but are comparably stable within ER–PM junctions.
Figure 8.

Exogenously expressed Kv2.1, VAPA, and Nir2 display comparable turnover rates at ER–PM junctions when coexpressed in HEK293T cells. A, confocal optical section of CFP–Kv2.1 expression taken from the basal surface of a resting HEK293T cell cotransfected with CFP–Kv2.1, GFP–VAPA (not shown), YFP–M1R (not shown), and mCherry–Nir2 (not shown) at rest (left), following photobleaching of area indicated by red circle (middle) and following recovery (right). The scale bar is 2.5 μm and holds for all images. B, confocal optical section of mCherry–Nir2 expression taken from the basal surface of a resting HEK293T cell cotransfected with CFP–Kv2.1 (not shown), GFP–VAPA (not shown), YFP–M1R (not shown), and mCherry–Nir2 at rest (left), following photobleaching of area indicated by red circle (middle) and following recovery (right). C, confocal optical section of GFP–VAPA expression taken from the basal surface of a resting HEK293T cell cotransfected with CFP–Kv2.1 (not shown), GFP–VAPA, and mCherry–Nir2 (not shown) at rest (left), following photobleaching of area indicated by red circle (middle), and following recovery (right). D, summary graph of t½ values of CFP–Kv2.1, mCherry–Nir2, and GFP–VAPA measured from FRAP experiments presented in A–C. Bars are mean ± S.D. Note the lack of a significant difference between t½ values of CFP–Kv2.1, GFP–VAPA, and mCherry–Nir2. (p value = 0.0923, n = 18–20 cells; ordinary one-way ANOVA).
Kv2.1-mediated ER–PM junction formation alters the extent of PtdIns(4,5)P2 depletion following repetitive muscarinic stimulation, but not resting PtdIns(4,5)P2, or PtdIns(4)P levels
Given the impact of expressing Kv2.1 on the enhanced levels of Nir2 associated with Kv2.1 and VAP containing ER–PM junctions, we examined whether Kv2.1 expression affected phosphoinositide homeostasis. We first examined whether Kv2.1 expression impacted steady-state PtdIns(4,5)P2 by labeling unstimulated HEK293T cells with the PtdIns(4,5)P2 reporter mCherry–PHPLCδ1 (67). In cells coexpressing mCherry–PHPLCδ and BFP–SEC61β, mCherry–PHPLCδ1 had a fairly homogeneous expression profile as visualized in TIRF microscopy (Fig. 9A); line-scan analyses suggested a subtle enrichment of mCherry–PHPLCδ at or near ER–PM junctions, as visualized by BFP–SEC61β TIRF signals (Fig. 8). Coexpression of GFP-tagged Kv2.1 (GFP–Kv2.1) led to the expected increase in the size of BFP–SEC61β puncta in the TIRF field yet did not lead to an appreciable impact on the overall distribution or intensity of mCherry–PHPLCδ1 (Fig. 9B). Line-scan analyses of these cells did not reveal an appreciable change of mCherry–PHPLCδ1 labeling at ER–PM junctions demarcated by BFP–SEC61β and GFP-Kv2.1 TIRF signals (Fig. 8). Similarly, PM targeting of mCherry–PHPLCδ1 visualized in SDC optical sections appeared unaltered in cells coexpressing CFP–Kv2.1 relative to control cells lacking Kv2.1 expression (Fig. 9, C and D), and ratiometric measurements of PM versus cytoplasmic mCherry–PHPLCδ1 signal intensity (measured from these SDC optical sections) showed Kv2.1 expression yielded no significant impact (Fig. 8E). Similarly, we did not observe a difference in the expression of PtdIns(4,5)P2 (a precursor of PtdIns(4,5)P2), as reported by the fluorescent PtdIns(4,5)P2 biosensor mCherry–P4Mx1 (68), in control cells relative to cells coexpressing CFP–Kv2.1 (data not shown). Ratiometric measurements of PM (TIRF) and cytoplasmic (SDC optical sections) mCherry–P4Mx1 expression yielded no significant differences in control cells relative to cells coexpressing CFP–Kv2.1 (Fig. 9F). From these results, we concluded that Kv2.1 expression does not significantly impact the steady-state levels of PtdIns(4,5)P2 or its precursor PtdIns(4)P in unstimulated HEK293T cells.
Figure 9.

Kv2.1 expression alters the kinetics of PtdIns(4,5)P2 recovery, following repetitive muscarinic stimulation, but does not alter the steady-state distributions of PtdIns(4,5)P2 or PtdIns(4)P. A, TIRF image of a resting HEK293T cell cotransfected with mCherry–PHPLCδ (red, shown left) and BFP–SEC61β (blue, shown right). Line scan analysis of selection indicated in merged image of A shown to right of A. The scale bar is 5 μm and holds for A and B. B, TIRF image of a resting HEK293T cell cotransfected with mCherry–PHPLCδ (red, shown left), GFP–Kv2.1 (shown middle), and BFP–SEC61β (middle right). Line scan analysis of selection indicated in merged image of B shown to right of B. C, SDC optical section taken through the center of a resting HEK293T cell transfected with mCherry–PHPLCδ alone (shown left). The scale bar is 5 μm and holds for C and D. D, SDC optical sections taken through the center of a resting HEK293T cell cotransfected with mCherry–PHPLCδ (red, shown left) and CFP–Kv2.1 (green, center). E, summary graph of the ratio of PM to cytoplasmic mCherry–PHPLCδ intensity values measured from HEK293T cells transfected with mCherry–PHPLCδ alone (control) or cotransfected with CFP–Kv2.1 (Kv2.1). ns, p value = 0.8654, n = 31 cells; two-tailed unpaired t test. F, summary graph of the ratio of PM (TIRF) to cytoplasmic (SDC) mCherry–P4Mx1 intensity values measured from HEK293T cells transfected with mCherry–P4Mx1 alone (control) or cotransfected with CFP–Kv2.1 (Kv2.1). (ns, p value = 0.1097, n = 25–31 cells; two-tailed unpaired t test). G, time course of cytoplasmic mCherry–PHPLCδ intensity values, measured from cells transfected with YFP–M1R and mCherry–PHPLCδ (control, black trace) or cotransfected with CFP–Kv2.1 (+Kv2.1, red trace) during two acute (40 s, indicated by gray bars) stimulations with 0.5 μm OxoM. Bars are mean ± S.D. Note that following the initial stimulation at 100 s, time points ranging from 110 to 165 s and 390 to 445 s are significantly different (0.0004186 ≤ p value ≤0.047207, n = 20 cells; two-tailed unpaired t test).
Finally, as we failed to observe differences in PtdIns(4,5)P2 levels in resting cells, we examined whether Kv2.1 expression would affect the kinetics of PtdIns(4,5)P2 hydrolysis and/or recovery following muscarinic stimulation. Toward this end, we employed a similar experimental paradigm as we used above (Fig. 2B) to measure cytoplasmic mCherry–PHPLCδ1 signal intensity in HEK293T cells expressing YFP–M1R alone (control) or coexpressing CFP–Kv2.1, during repetitive muscarinic stimulations with 0.5 μm OxoM. Interestingly, in these experiments we found that there were significant differences in cytoplasmic mCherry–PHPLCδ1 intensity in Kv2.1-expressing cells relative to control cells during these stimulations, such that Kv2.1-expressing cells exhibited a significantly-reduced response to OxoM stimulation relative to control cells (Fig. 9G). These data, taken together, demonstrate that whereas Kv2.1 expression has no apparent impact on resting or steady-state PtdIns(4,5)P2 or phosphatidylinositol 4-phosphate levels, Kv2.1 expression is sufficient to impact the extent of PtdIns(4,5)P2 metabolism and/or recovery during repetitive muscarinic stimulations in heterologous HEK293T cells.
Kv2.1 knockout mouse brains have altered phosphoinositide and phosphatidic acid levels
To this point, we have tested a role for Kv2.1 in regulating phosphoinositide metabolism in HEK293T cells. To examine whether Kv2.1 participates in tuning phosphoinositide levels in intact brain, we used a combination of high-performance LC (HPLC) with electrospray ionization (ESI) and tandem MS with multiple reaction monitoring (HPLC–ESI–MS/MS) (69) to quantitatively determine phosphoinositide levels from brains of WT mice and Kv2.1-deficient (Kv2.1 KO) littermates. Lipid analysis revealed that Kv2.1 KO brains had significantly less total PtdIns(4)P, PtdIns(4,5)P2, and phosphatidic acid, relative to WT controls (Fig. 10A), with the most abundant isoforms (38:4, 36:1; determined by their fatty-acyl chains) depleted by an equivalent extent across each phosphoinositide species. We also investigated whether these differences arose from differential expression of Nir2 in CHNs isolated from Kv2.1 KO and WT mice. However, in these immunocytochemistry and widefield-imaging experiments we did not detect a significant difference in Nir2 labeling intensity between CHNs cultured from neurons in Kv2.1 KO and WT mice (data not shown). From these results, we conclude that Kv2.1 makes significant contributions to the total abundance of several key phosphoinositide-signaling lipids in intact mouse brain.
Figure 10.
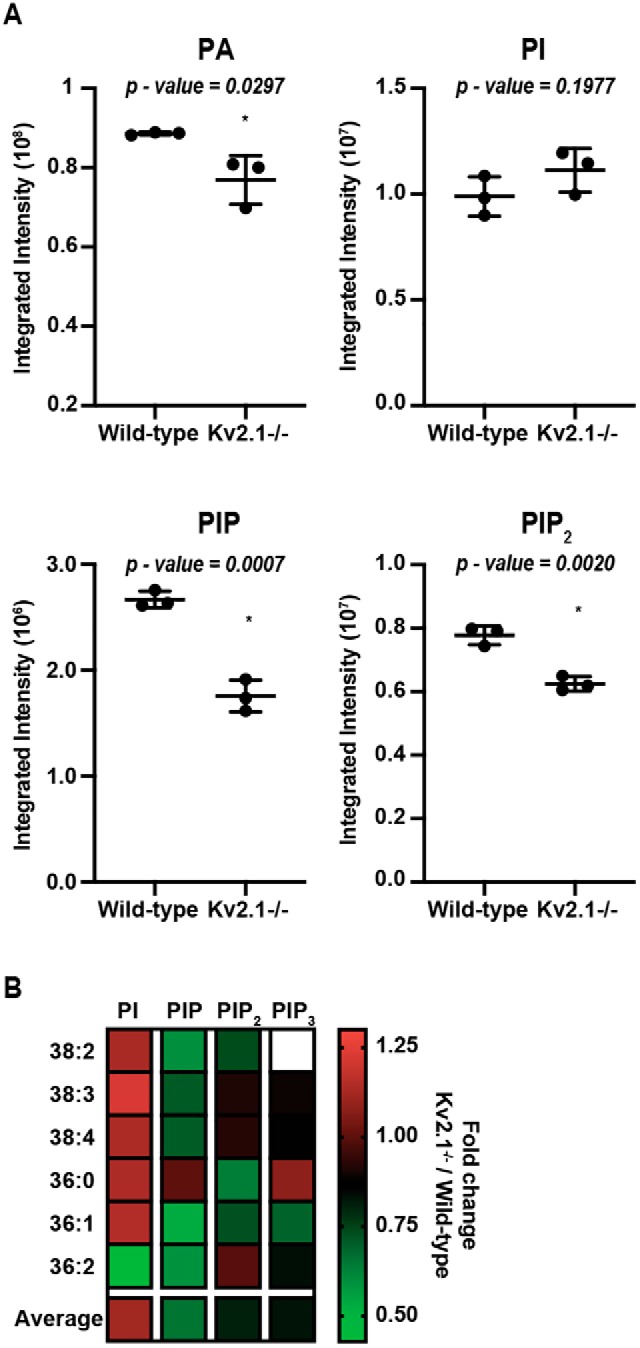
Kv2.1 KO alters phosphoinositide levels in mouse brains. A, histograms representing UPLC-MS/MS measurements of total PtdIns, PtdIns(4)P, PtdIns(4,5)P2, and phosphatidic acid levels from WT and Kv2.1 KO mouse brains (n = 3). Numbers above each histogram reflect p values. B, heat map profiling the fold change of each phosphoinositide species from Kv2.1 KO relative to WT brains. Each colored box is an average of measurements from three independent experiments (n = 3 mice each). White box denotes absence of lipid from analysis.
Discussion
Our previous findings identified the association of PM-localized Kv2 α-subunits and ER-localized VAPs as defining a subset of ER–PM junctions in mammalian brain neurons (43). The unbiased proteomic approach that we undertook in this study revealed the biochemical association of membrane-associated phosphatidylinositol transfer proteins isoforms 1 (Nir2) and 2 (Nir3) with Kv2.1- and VAP-containing protein complexes. We note that Nir2 and Nir3 were not present in the previous proteomic data set used to identify VAPA and VAPB as proteins associated with Kv2.1 (43). Here, we modified our approach to use on-bead tryptic digestion to elute tryptic peptides from the immunopurified Kv2.1-containing complexes, as opposed to performing SDS-gel electrophoresis and in-gel digests as we had done previously (43). This not only enhanced the overall peptide recovery (contained in our spectral counts) of Kv2 α-subunits and VAPs relative to our previous data set, but also revealed the presence of Nir2 and Nir3, albeit at relatively low abundance. Our findings are that Nir proteins biochemically copurify with Kv2.1, and that the subcellular distribution of Nir2 is spatially correlated with Kv2.1 and VAPs in HEK293T cells and cultured hippocampal neurons. These experiments support that a subpopulation of these Nir proteins expressed in brain neurons are spatially associated with Kv2.1 and VAP-containing ER–PM junctions.
A number of factors may explain the low abundance of Nir proteins in our proteomic data set. Although among all tissues examined both Nir2 (70–72) and Nir3 (73) have relatively high levels of expression at the mRNA level in brain tissue, their regional and cellular expression is more restricted than that of the widely-expressed Kv2 α-subunits and VAPs (e.g. in mouse brain (http://mouse.brain-map.org)). For example, Nir3 mRNA expression in brain is high only in granule cells within the dentate gyrus (73), which also express Kv2.1 (74, 75) and Kv2.2 (28), as well as VAPA and VAPB (43). However, even in these neurons their subcellular localization is only partial, as Nir3 immunoreactivity is found throughout the cell bodies, axons, and dendrites (73), whereas that for Kv2 α-subunits and VAPs is primarily present on the cell body and proximal dendrites (43). Nir2 is more broadly expressed than Nir3 at the regional and cellular level (e.g. in mouse brain (http://mouse.brain-map.org)), but its expression is still relatively limited compared with Kv2 α-subunits and VAPs.
Another factor potentially limiting the overall abundance of Nir proteins associated with immunopurified Kv2.1 complexes from brain is that their association with ER–PM junctions is conditional and is substantially enhanced after depletion of PtdIns(4,5)P2 in the PM (19–22). We also observed conditionality in Nir2 and Kv2.1 colocalization in our imaging experiments in HEK293T cells, such that while a subset of HEK293T cells had colocalized Kv2.1, VAPA, and Nir2 prior to muscarinic stimulation, this was substantially enhanced by such stimulation. Our immunopurification of Kv2.1 was performed using mouse brain samples that had not been exposed to any specific stimulus to deplete PM PtdIns(4,5)P2 prior to homogenization, although the brain undoubtedly contains substantial populations of neurons in which the Nir proteins were present at ER–PM junctions in response to a recent PLC-mediated signaling event. Nir3 is more sensitive and can detect subtle changes in PtdIns(4,5)P2, whereas Nir2 requires extensive PtdIns(4,5)P2 depletion to be recruited (19), perhaps consistent with Nir3 being more highly-associated with Kv2.1-containing ER–PM junctions in unstimulated brain samples, and as such more substantially represented in immunopurified Kv2.1-containing complexes than was Nir2. Additionally, our findings in HEK293T cells and CHNs support that the colocalization of Nir2 and Kv2.1 requires exogenous VAPA expression, arguing against a direct protein–protein interaction between Kv2.1 and Nir2. As such, the cross-linking of both Nir proteins to Kv2.1 may be less efficient than for the directly-associated VAP proteins.
We observed in our super-resolution imaging experiments that only a subset (≈20–25%) of endogenous Nir2 immunolabeling was spatially associated with that for Kv2.1 in CHNs. However, the extent of spatial overlap was significantly decreased in images in which the Kv2.1 immunolabeling signal had been randomized, and the spatial distributions of immunolabeled Kv2.1, Nir2, and VAPA were significantly correlated with one another, such that their positions relative to one another could not be recapitulated due to random chance. We note that a major inherent limitation to these analyses is our use of indirect immunocytochemistry (i.e. employing a primary antibody to recognize a target protein, and a fluorescently labeled secondary antibody to recognize the primary antibody) in conjunction with super-resolution microscopy. This limits our ability to accurately assess the precise position and/or size of the immunolabeled target, as the distance between the immunolabeled epitope and the fluorescence-emitting label on the secondary antibody in indirect immunocytochemistry likely reaches ≈30–35 nm, as recently reviewed (76). This technical limitation may also contribute to the relatively low level of spatially-correlated signals for endogenous Kv2.1 and Nir2 observed in our experiments.
That both Kv2 α-subunits (43, 44) and Nir proteins (77) bind VAP proteins via their FFAT motifs further support a model whereby VAPs act as intermediaries in a Kv2–VAP–Nir ternary complex. This is supported by our findings that the turnover rates measured from our FRAP experiments in HEK293T cells for colocalized Kv2.1, VAPA, and Nir2 at ER–PM junctions were not significantly different. That VAPA could form a ternary complex containing Kv2 α-subunits and Nir proteins is intriguing given that both bind to VAPs via FFAT motifs. Structural (78) and biochemical (77, 79, 80) studies support that VAPs can exist as dimers that simultaneously bind two FFAT motifs (78, 81), such that a single VAP protein could in principle simultaneously bind a Kv2.1 and Nir protein FFAT motif. Although the structures of the VAP-binding domains of Kv2 α-subunits and Nir proteins have not been elucidated, it seems plausible that such heterotypic binding would be impeded by steric hindrance between the two large cytoplasmic domains of Kv2.1 and Nir2. As Kv2.1 channels are tetramers, it seems likely that the VAP dimer would be occupied by two Kv2.1-derived FFAT motifs and unable to accommodate binding an FFAT motif from Nir2. However, the ER VAP proteins also form higher-order oligomers via their juxtamembrane coiled-coil domains (80). As such, it is possible that VAPs with free FFAT-binding domains could exist within VAP oligomers at a Kv2.1-mediated neuronal ER–PM junction that could serve as sites for Nir2 recruitment following muscarinic activation. Further experiments are needed to clarify this mechanism and to examine the contribution of other signals previously reported to impact Nir2 activity at ER–PM junctions (e.g. PM PtdIns(4,5)P2 and phosphatidic acid levels, extended synaptotagmin-1 activation, and ER–PM junctional distance (19–22)) relative to the Kv2–VAP–Nir association reported here.
Our findings suggest that Kv2.1 and VAP-mediated ER–PM junctions in brain neurons could play a role in phosphoinositide homeostasis by acting as sites for Nir protein recruitment. Kv2.1 expression is limited to neurons, striated and smooth muscle, and pancreatic cells, such that other cell types (including HEK293 cells) are able to maintain lipid homeostasis, including recruitment of Nir proteins in its absence. That Kv2.1 complexes contain Nir proteins and that recruitment of Nir2 to ER–PM junctions is enhanced by Kv2.1 expression suggest that Kv2.1-containing ER–PM junctions, which are abundant in brain neurons, could act as specialized sites of PM PtdIns(4,5)P2 replenishment. Nir2 is recruited to other classes of ER–PM junctions, for example those formed by the resident ER-extended synaptotagmin proteins. However, that enhanced ER–PM junction formation triggered by the expression and dimerization of CFP–CB5–FKBP and lyn11–FRB does not recruit Nir2 argues against a promiscuous association of Nir proteins with any ER–PM junctions. We note that the ER–PM junctions formed via CB5/lyn11 coupling have a narrower (≈4–6 nm) junctional gap (82) than those formed upon Kv2.1 expression in HEK293 cells (≈15 nm (41)) or those present in brain neurons (9), a substantial subset of which presumably contain Kv2.1.
That Kv2.1-mediated ER–PM junctions could play a role in lipid homeostasis is supported by two lines of evidence. First, we found that PtdIns(4,5)P2 metabolism and/or recovery are altered in HEK293T cells exogenously expressing Kv2.1 relative to control cells lacking Kv2.1 expression, following repetitive muscarinic stimulation. We note, however, that these experiments do not define whether the impact of Kv2.1 expression on PtdIns(4,5)P2 metabolism and/or recovery is mediated through effects on Nir2. Second, our lipidomic analyses reveal that the levels of two derivatives of Nir-mediated PtdIns transfer, phosphatidylinositol and PIP2, are altered in the brains of Kv2.1 KO mice. We note that the lipidomics analyses were performed on lipids extracted from whole mouse brains. Within the mouse brain, Kv2.1 is detectably expressed only in neurons (28, 39, 83), which comprise 65% of the cells in adult mouse brain (84, 85). However, lipids from non-neuronal brain cell types (predominantly astrocytes, microglia, oligodendrocytes, and endothelial cells (86)), which would not be affected by knockout of Kv2.1 expression, at least in a cell autonomous manner, would contribute to the lipid profiles in our analyses. As such, it remains possible that the differences that we found may be even more pronounced in brain neurons that express Kv2.1. Through their impact on diverse ion channels and transports, PtdIns lipids are important modulators of neuronal excitability (4, 87). Kv2.1 KO mice exhibit neuronal hyperexcitability, behavioral hyperactivity, and enhanced susceptibility to induced seizures (28). Given the complex effects of PtdIns lipids on excitability, and the lack of specific details as to changes in PtdIns lipids in specific classes of neurons, it is not possible to make a specific link between altered lipids in the brains of Kv2.1 KO mice and the observed phenotypes, although the results presented here will need to be considered in interpreting the molecular basis of their phenotype.
That we did not observe an apparent impact of Kv2.1 expression on the subcellular distribution of the fluorescent PtdIns(4,5)P2 biosensor, mCherry–PHPLCδ1, in HEK293T cells would appear to be in conflict with the results of our quantitative lipidomic analysis of absolute PI(4,5)P2 levels in brain samples from WT versus Kv2.1 KO brains. The basis for this discrepancy is as yet unclear, but we speculate that the heterologous HEK293T cell system does not recapitulate the entirety of Kv2.1-mediated regulation of lipid levels across the mammalian brain. It is also possible that the effects of transient expression of exogenous Kv2.1 expression on PI(4,5)P2 levels in resting HEK293T cells were not sufficiently robust in this particular fluorescence assay.
The differences in PtdIns seen in lipidomic analyses of samples from Kv2.1 KO versus WT mouse brains presented here have certain parallels to those obtained in a comparison with OxoM-treated cultured sympathetic neurons (88), in which reductions in PtdIns(4)P and PtdIns(4,5)P2 levels were also observed. Our lipidomic analyses of Kv2.1 KO brains also revealed a reduction in steady-state phosphatidic acid levels. Phosphatidic acid can be generated by the catalytic actions of DAG-kinase acting on DAG; therefore, reduced steady-state phosphatidic acid levels may be expected if the reduction in brain PIP2 observed in Kv2.1 KO animals translates to a decrease in available substrate for PM PLC, which is responsible for the generation of DAG and inositol 1,4,5-trisphosphate. Consequences of decreases in phosphatidic acid levels may be a reduced ability of Nir2 to associate with the PM at non-Kv2.1 ER–PM contact sites, further compromising the ability of Nir2 to be recruited to ER–PM MCS during periods of PIP2 depletion. Collectively, the reduced steady-state levels of these lipids may be consistent with a model whereby loss of Kv2.1 expression impacts the Nir-mediated recovery of PtdIns lipids in the neuronal PM as mediated by PM-associated Kv2.1–VAP–Nir complexes as well as PLC-mediated signaling events.
Together, these results support that Kv2.1- and VAP-mediated ER–PM junctions participate in phosphoinositide homeostasis, potentially via recruitment of Nir proteins. Further experiments are needed to define the underlying mechanisms and to rule out other interpretations, for example that Kv2.1 expression directly alters M1R activity and/or downstream signaling with subsequent effects on the kinetics of PM PtdIns(4,5)P2 metabolism/replenishment. However, we note that in our experiments we did not observe an appreciable difference in the subcellular localization of YFP–M1R upon Kv2.1 expression (data not shown), which perhaps argues against a direct interaction between Kv2.1 and M1R.
Although Kv2.1 is prominently expressed at ER–PM junctions in mammalian brain neurons (33–36, 75), its restricted localization at these sites is dynamically regulated by activity-dependent dephosphorylation of Kv2.1 that results in diffuse Kv2.1 localization (39, 89) and loss of Kv2.1 from ER–PM junctions (43). Recent studies demonstrating activity-dependent decreases in the extent of neuronal ER–PM junctions in hippocampal neurons (90) and altered levels of ER-derived cisternal stacks (91) may be consistent with a role for Kv2.1 in activity-dependent regulation of at least a subset of ER–PM junctions in brain neurons. That this could be further coupled to the conditional presence of Nir proteins at Kv2.1-associated ER–PM junctions suggests the possibility of a complex regulation of the Kv2–VAP–Nir ternary complex in response to neuronal activity and receptor-mediated phospholipase C activation. The contribution of these complexes to those present at other classes of neuronal ER–PM junctions in lipid signaling and homeostasis remains an important question for future research.
Materials and methods
Animals
All procedures involving mice and rats were approved by the University of California Davis Institutional Animal Care and Use Committee (IACUC) and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All mice and rats were maintained under standard light–dark cycles and allowed to feed and drink ad libitum. Age-matched adult (over 12 weeks old) male and female mice were used in proteomic experiments. Kv2.1 KO mice (RRID:IMSR_MGI:3806050) have been described previously (28, 43, 92) and were generated from breeding of Kv2.1+/− mice that had been back-crossed on the C57/BL6J background (RRID:IMSR_JAX:000664). Pregnant female Sprague-Dawley rats were used as a source of hippocampal neurons for primary CHN cultures.
Antibodies
All primary antibodies used in this study are described in Table 4. Validation in KO mice has been previously demonstrated for the anti-Kv2.1 mouse monoclonal K89/34 and rabbit polyclonal KC antibodies (28, 37–39). The rabbit Kv1.2C polyclonal antibody (75) and anti-VAPA/B mouse mAb N479/107 have also been previously described (43).
Table 4.
Antibody information
| Antigen and antibody name | Immunogen | Manufacturer's information | Concentration used | Figs. |
|---|---|---|---|---|
| Kv2.1 (KC) | Synthetic peptide aa 837–853 of rat Kv2.1 | Rabbit pAb, in-house (Trimmer Laboratory), RRID:AB_2315767 | Affinity-purified, 1:100 | 1 (immunopurifications) |
| Kv2.1 (K89/34) | Synthetic peptide aa 837–853 of rat Kv2.1 | Mouse IgG1 mAb, NeuroMab. RRID: AB_10672253 | Purified, 10 μg/ml | 1 (immunoblot) |
| Kv1.2 (Kv1.2C) | Synthetic peptide aa 463–480 of rat Kv1.2 | Rabbit pAb, in-house (Trimmer Laboratory), RRID:AB_2756300 | Affinity-purified, 1:100 | 1 (immunopurifications, immunoblot) |
| VAPA/B (N479/107) | Fusion protein aa 1–219 of rat VAPA | Mouse IgG2b mAb, NeuroMab. RRID:AB_2722711 | Tissue cultured supernatant (1:5) | 6 |
| Nir2 | Recombinant fragment of human Nir2 aa 589–651 | Rabbit pAb, Abcam catalog no. ab221381, RRID:AB_2801563 | 10 μg/ml (Fig. 6A), 1 μg/ml (Fig. 6E) | S1 |
| Nir2 | Recombinant peptide of human Nir2 aa 365–444 | Rabbit pAb, Novus Biologicals catalog no. NBP2–34132, RRID: AB_2801564 | 10 μg/ml (Fig. 6A) | 6, S1 |
| RyRs (34C) | Partially purified chicken pectoral muscle ryanodine receptor | Mouse IgG1 mAb, Developmental Studies Hybridoma Bank, RRID: AB_528457 | 1 μg/ml | 6 |
Immunopurification and proteomic analyses
Preparation of cross-linked mouse brain samples for immunopurification was as described previously (43). Briefly, mice were acutely decapitated in the absence of anesthesia, and brains were rapidly (dissection time ≤1 min) excised and mechanically homogenized over ice in a Dounce homogenizer containing 5 ml of ice-cold homogenization buffer containing 1 mm DSP (Lomant's reagent; Thermo Fisher Scientific catalog no. 22585), 320 mm sucrose (Sigma catalog no. S0389), 5 mm Na phosphate, pH 7.4, 0.1 m sodium fluoride, 1 mm PMSF, and protease inhibitors. Following a 1-h incubation on ice, DSP was quenched using 1 m Tris, pH 7.4 (J.T. Baker catalog no. 4109-01, Tris base, and catalog no. 4103-01, Tris-HCl), added to a final concentration of 0.02 m. 2 ml of this brain homogenate was added to an equal volume of ice-cold 2× radioimmunoprecipitation assay (RIPA) buffer (final concentrations: 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 150 mm NaCl, 50 mm Tris, pH 8.0) and incubated for 30 min on a tube rotator at 4 °C. Following this incubation, insoluble material was pelleted by centrifugation at 12,000 × g for 10 min at 4 °C. The supernatant was incubated with antibodies overnight at 4 °C, employing either the anti-Kv2.1 rabbit polyclonal antibody KC (33), directed against the Kv2.1 C terminus (see Refs. 28, 93 for KO validation), or the anti-Kv1.2 rabbit polyclonal antibody Kv1.2C (75), directed against the Kv1.2 C terminus. This was followed by the addition of 100 μl of magnetic protein G beads (Thermo Fisher Scientific) for 1 h on a tube rotator at 4 °C. The beads were then washed six times in 1× RIPA buffer and four times in 50 mm ammonium bicarbonate (pH 7.4). Tryptic peptide fragments of immunopurified proteins captured on magnetic beads were eluted by digestion with 1.5 mg/ml trypsin (Promega catalog no. V5111) in 50 mm ammonium bicarbonate overnight at 37 °C. The extracts were lyophilized and resuspended in 0.1% TFA in 60% acetonitrile.
Proteomic profiling was performed at the University of California, Davis Proteomics Facility. Digested peptides were analyzed by LC-MS/MS on a Thermo Fisher Scientific Q Exactive Plus Orbitrap mass spectrometer in conjunction with a Proxeon Easy-nLC II HPLC (Thermo Fisher Scientific) and Proxeon nanospray source. The digested peptides were loaded a 100 μm × 25 mm Magic C18 100 Å 5-unit reverse-phase trap where they were desalted online before being separated using a 75 μm × 150 mm Magic C18 200Å 3-unit reverse-phase column. Peptides were eluted using a 60-min gradient with a flow rate of 300 nl/min. An MS survey scan was obtained for the m/z range 350–1600; MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the MS spectra were subjected to high-energy collisional dissociation. An isolation mass window of 1.6 m/z was for the precursor ion selection, and normalized collision energy of 27% was used for fragmentation. A 15-s duration was used for the dynamic exclusion.
Tandem mass spectra were extracted and charge state–deconvoluted by Proteome Discoverer (Thermo Fisher Scientific). All MS/MS samples were analyzed using X! Tandem (The GPM, thegpm.org; version Alanine (2017. 2. 1.4)). X! Tandem was set up to search the UniProt Mouse database (May, 2017, 103,089 entries), the cRAP database of common proteomic contaminants (www.thegpm.org/crap; 114 entries), and the ADAR2 catalytic domain sequence plus an equal number of reverse protein sequences assuming the digestion enzyme trypsin. X! Tandem was searched with a fragment ion mass tolerance of 20 ppm and a parent ion tolerance of 20 ppm. Deamidation of asparagine and glutamine, oxidation of methionine and tryptophan, sulfone of methionine, tryptophan oxidation to formylkynurenin of tryptophan, and acetylation of the N terminus were specified in X! Tandem as variable modifications.
Scaffold (version Scaffold_4.8.4, Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they exceeded specific database search engine thresholds. X! Tandem identifications required at least −log scores (expected scores) of greater than 2.0 with a mass accuracy of 5 ppm. Protein identifications were accepted if they contained at least two identified peptides. Our threshold for peptide acceptance was greater than 95% probability.
Lipidomic analysis of phosphoinositide species
Kv2.1 KO and WT age- and sex-matched mice were acutely decapitated in the absence of anesthesia and brains were rapidly (dissection time ≤1 min) excised and mechanically homogenized over ice in a Dounce homogenizer containing 5 ml of ice-cold Hanks' balanced saline solution (HBSS, Gibco catalog no. 14025092) with 1 mm PMSF and protease inhibitors. Lipids were then extracted, derivatized, and analyzed by LC and MS as described previously (69). Peak areas for lipid species were quantified by integrating curves using Waters Quanlynx software. For comparisons of different samples, peak areas were corrected for extraction efficiencies by normalizing to synthetic 17:0, 20:4 (37:4) PtdIns(4,5)P2, PtdIns(4)P, and PtdIns internal standards and corrected for cell number variations using a Bradford assay.
Immunoblotting
Protein samples were boiled for 5 min in a reducing sample buffer containing SDS (lauryl sulfate, Sigma catalog no. L-5750) and 2-mercaptoethanol prior to size-fractionation on a 7.5% SDS-polyacrylamide gel alongside fluorescent molecular weight standards (Sigma catalog no. GERPN800E) diluted in the same reducing sample buffer. Size-fractionated proteins were then transferred to a nitrocellulose membrane for immunoblotting. Membranes were blocked for 1 h at room temperature in TBS containing 4% nonfat milk powder (BLOTTO), incubated for 1 h at room temperature with primary antibodies in blocking solution, washed three times for 5 min each in BLOTTO, and further incubated for 1 h at room temperature with mouse subclass-specific and anti-rabbit heavy- and light-chain–specific fluorescent secondary antibodies (all secondary antibodies from Thermo Fisher Scientific) diluted 1:1500 in BLOTTO. Membranes were washed three times for 5 min each in TBS and imaged using a fluorescent imager (Protein Simple).
Culture and transfection of rat hippocampal neurons
Hippocampi were dissected from embryonic day 18 rat embryos and dissociated enzymatically for 20 min at 37 °C in 0.25% (w/v) trypsin (Thermo Fisher Scientific catalog no. 15050065) in HBSS and further dissociated mechanically by triturating with flame-polished glass Pasteur pipettes. Dissociated cells were suspended in plating medium containing Neurobasal (Thermo Fisher Scientific catalog no. 21103049) supplemented with 10% fetal bovine serum (Invitrogen catalog no. 16140071), 2% B27 (Invitrogen catalog no. 17504044), 2% GlutaMAX (Thermo Fisher Scientific catalog no. 35050061), and 0.001% gentamycin (Gibco catalog no. 15710064) and plated at 60,000 cells per dish in glass bottom dishes (MatTek catalog no. P35G-1.5-14-C) or no. 1.5 glass coverslips coated with poly-l-lysine (Sigma catalog no. P2636). At 7 DIV, cytosine-d-arabinofuranoside (Millipore catalog no. 251010) was added to inhibit non-neuronal cell growth. Neurons were transiently transfected at DIV 7–10 using Lipofectamine 2000 (Invitrogen catalog no. 11668019) for 1.5 h as described previously (42, 43, 45). Neurons were imaged 40–48 h post-transfection.
Immunocytochemistry and super-resolution imaging of cultured rat hippocampal neurons
Immunocytochemistry of CHNs was performed as described previously (43). Briefly, CHNs were fixed in a solution of 4% formaldehyde, prepared by dissolving paraformaldehyde powder (Sigma catalog no. 158127) in PBS, pH 7.4, for 15 min at 4 °C. All subsequent steps were performed at room temperature. CHNs were subsequently washed three times for 5 min each in PBS and blocked in BLOTTO-T (4% milk powder, 0.1% Triton X-100 in TBS at pH 7.4) for 1 h. CHNs were immunolabeled for 1 h with primary antibodies diluted in BLOTTO-T (concentrations used for primary antibodies are listed in Table 4), and subsequently washed three times for 5 min each in BLOTTO-T. CHNs were immunolabeled with mouse subclass-specific and anti-rabbit heavy- and light-chain–specific fluorescent secondary antibodies (all secondary antibodies from Thermo Fisher Scientific) diluted at 1:1500 in BLOTTO-T for 1 h. After washing three times for 5 min each in PBS, coverslips were mounted on microscope slides with Prolong Gold mounting media (Thermo Fisher Scientific catalog no. P36930) and allowed to cure overnight before sealing. N-SIM images were acquired with a Hamamatsu ORCA-ERCCD camera on a SIM/wide-field–capable Nikon Eclipse Ti microscope with an EXFO X-Cite metal halide light source and a ×100 PlanApo TIRF/1.49 objective controlled by NIS Elements software (Nikon). Images were collected within NIS Elements as ND2 images and processed within NIS Elements. Images were subsequently exported as TIFFs, linearly scaled for min/max intensity, and flattened as RBG TIFFs in Photoshop (Adobe) prior to presentation and analysis.
Culture and transfection of HEK293T cells
HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% Fetal Clone III (HyClone catalog no. SH30109.03), 1% penicillin/streptomycin, and 1× GlutaMAX (Thermo Fisher Scientific catalog no. 35050061) in a humidified incubator at 37 °C and 5% CO2. HEK293T cells were transfected as described previously (38, 42, 43). Briefly, cells were split to 15% confluence on glass bottom dishes (MatTek catalog no. P35G-1.5-14-C; used in Figs. 2D, 3, 4, 6, E–H, 7, and 8, A and B) or no. 1.5 glass coverslips (used in Figs. 2, A–C, 6, A–C, and 8, C–G), coated with poly-l-lysine, then transiently transfected using Lipofectamine 2000 following the manufacturer's protocol. Cells were transiently transfected in DMEM without supplements and then returned to regular growth media 4 h after transfection. Cells were imaged 40–48 h post-transfection.
Plasmid constructs
CFP-tagged Kv2.1 (CFP–Kv2.1) has been previously described (94). GFP-tagged Kv2.2 (GFP–Kv2.2) has been previously described (42). BFP-tagged SEC61β (BFP–SEC61β) was a generous gift from Dr. Jodi Nunnari (University of California, Davis) and has been used previously in similar experiments to report Kv2-mediated ER–PM junctions (38, 42, 43). The following investigators were generous in providing the following gifts: Kv2.1 P404W, Dr. Jon Sack (University of California, Davis); YFP-tagged M1R (YFP-M1R), Dr. Bertil Hille (University of Washington); mCherry-tagged PHPLCδ (mCherry–PHPLCδ), Dr. Tamas Balla (National Institutes of Health); CFP-tagged CB5-FKBP (CFP-CB5-FKBP), Dr. Takanari Inoue (Johns Hopkins University); lyn11–FRB, Dr. Tobias Meyer (Stanford University); and mCherry-tagged Nir2, Dr. Jen Liou (University of Texas Southwestern); GFP-tagged VAPA (GFP–VAPA, Addgene plasmid no. 18874), Dr. Axel Brunger (Stanford University); DsRed2-tagged ER5 (DsRed2–ER5, Addgene plasmid no. 55836), Dr. Michael Davidson (Florida State University); mCherry-tagged P4Mx1 (mCherry-P4Mx1, Addgene plasmid no. 108143), Dr. Gerry Hammond (Pittsburgh University).
Live cell guangxitoxin labeling
The GxTX peptide used in surface labeling was synthesized at the Molecular Foundry of the Lawrence Berkeley National Laboratory under United States Department of Energy contract no. DE-AC02-05CH11231. HEK293T cells were surface-labeled with 1 μm GxTX, as described previously (42, 58), and imaged by TIRF microscopy in physiological saline solution (4.7 mm KCl, 146 mm NaCl, 2.5 mm CaCl2, 0.6 mm MgSO4, 1.6 mm NaHCO3. 0.15 mm NaH2PO4, 20 mm HEPES, pH 7.4) containing 8 mm glucose and 0.1% BSA.
TIRF microscopy imaging
All TIRF imaging of live HEK293T cells (excluding the data summarized in Fig. 8F) was performed at the University of California Davis MCB Imaging Facility. Live transfected HEK293T cells cultured on glass bottom dishes were imaged in a physiological saline solution (4.7 mm KCl, 146 mm NaCl, 2.5 mm CaCl2, 0.6 mm MgSO4, 1.6 mm NaHCO3. 0.15 mm NaH2PO4, 20 mm HEPES, pH 7.4) containing 8 mm glucose as described previously (38, 42, 43). Cells were maintained at 37 °C during imaging with a heated stage and objective heater. Images were obtained with an Andor iXon EMCCD camera installed on a TIRF/widefield-equipped Nikon Eclipse Ti microscope using a Nikon LUA4 laser launch with 405-, 488-, 561-, and 647-nm lasers and a ×100 PlanApo TIRF, 1.49 NA objective run with NIS Elements software. Images were collected within NIS Elements as ND2 images. For TIRF (and spinning disk confocal) data summarized in Fig. 8F, TIRF and spinning disk confocal images were acquired using a ×60 NA 1.49 objective mounted on an Olympus W-1 (IX71) microscope doubly equipped with a Yokugawa spinning disk unit and a TIRF apparatus.
Drug treatments
For experiments presented in Figs. 2D, 3C, 3D, and 7, H and I, HEK293T cells cultured on MatTek dishes were washed twice in physiological saline (4.7 mm KCl, 146 mm NaCl, 2.5 mm CaCl2, 0.6 mm MgSO4, 1.6 mm NaHCO3. 0.15 mm NaH2PO4, 20 mm HEPES, pH 7.4) containing 8 mm glucose, prior to treatment and imaging. Oxotremorine M (Millipore, catalog no. 1067) was added as a 2× solution in physiological saline, by pipette to MatTek dishes already containing physiological saline to a final concentration of 10 μm. For the experiment presented in Fig. 7F, latrunculin A (ThermoFisher Scientific, catalog no. 428021100UG) was similarly added as a 2× solution in physiological saline, by pipette to prewashed HEK293T cells cultured on MatTek dishes already containing physiological saline to a final concentration of 10 μm. For experiments presented in Fig. 7, B and E, rapamycin (Sigma, catalog no. R8781-200UL) was diluted in imaging saline to a final concentration of 5 μm and added to cells. For experiments presented in Figs. 2, A–C and 9G, HEK293T cells cultured on round glass no. 1.5 coverslips were mounted on a magnetic round coverslip holder (Live Cell Instruments) connected to a homemade gravity-based perfusion system allowing for a continuous exchange of Ringer's solution (160 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES pH 7.4, 8 mm glucose) over cells. Following perfusion of Ringer's solution for 100 s, oxotremorine M (10 μm as used in Fig. 2, A–C, or 0.5 μm as used in Fig. 8G) was added in the superfusate during a 40-s interval. A single interval was used in Fig. 2, A–C, and two intervals interspersed by 240 s of Ringer's solution were used in Fig. 8G.
Fluorescence recovery after photobleaching
For experiments summarized in Fig. 8, HEK293T cells cultured on MATTEK dishes were imaged at room temperature using a spinning disc module of a Marianas SDC real-time confocal microscope (Intelligent Imaging Innovations, 3i) equipped with a Yokogawa spinning disk unit, a ×100 1.46 NA objective (Olympus), and EMCCD camera controlled by Slidebook software (Intelligent Imaging Innovations, 3i). We focused on a single optical section that captured the basal surface of a cell. Following 36 s at rest, a single region containing CFP–Kv2.1, GFP–VAPA, or mCherry–Nir2 puncta was photobleached, and recovery was monitored out to 360 s. t½ values were calculated directly in Slidebook using the FRAP analysis plugin.
Spinning disk confocal imaging
For experiments summarized in Figs. 2, A–C, 5, 7, A–C, and 9, C–G, HEK293T cells, or dissociated rat hippocampal neurons, cultured on no. 1.5 round glass coverslips were imaged at room temperature using a ×60 NA 1.49 objective mounted on an Olympus W-1 (IX71) microscope doubly-equipped with a Yokugawa spinning disk unit and a TIRF apparatus.
Experimental design and statistical analysis
For all data sets presented in this study for which statistical analysis was performed, measurements were imported into GraphPad Prism for presentation and statistical analysis as noted in each figure legend. Exact p values are also noted in each figure legend (Figs. 3–9 and Fig. S1), within the figure itself (Fig. 10), or in Tables 1–3 for Fig. S2. Proteomics on brain samples were collected from a single WT female mouse (trial 1) and from three independent sets of age-matched male WT and Kv2.1 KO adult mice (trials 2–4). Lipidomics on brain samples were similarly collected from three independent sets of sex- and age-matched WT and Kv2.1 KO adult mice. For experiments involving HEK293T cells and mixed sex cultured rat hippocampal neurons, the number of samples (n) indicates the number of cells analyzed and is noted in each figure legend.
The colocalization analyses reported in Fig. 7 were performed within Nikon NIS Elements using ND2 files. A region of interest was drawn within a cell and PCC values were collected. The colocalization analysis reported in Figs. 2 and 5 was performed using FIJI (National Institutes of Health). A region of interest was drawn around the soma of a neuron, and PCC values were collected using the Coloc2 plugin. All intensity measurements were collected using FIJI. All intensity measurements reported in line scans were normalized to the maximum intensity measurement. Measurements of puncta sizes (employed in Figs. 3, E and F, and 6C) were performed essentially as described previously (42, 43, 60). Briefly, within FIJI, TIRF images of Nir2 puncta collected from control cells, or from cells coexpressing Kv2.1, were background subtracted and subsequently converted into a binary mask by thresholding. Puncta sizes were measured automatically using the “analyze particles” feature of FIJI. For size analyses of diffraction-limited TIRF imaging, puncta smaller than 0.0256 μm2 (essentially, the area of a single pixel in our camera) were considered smaller than the diffraction limit and excluded from our analysis.
Quantification of the percent of total Nir2 signal overlapping with Kv2.1 (Fig. 6D) was performed using a custom automated pipeline written in Python 3.7.1. Nir2 and Kv2.1 images were first background-subtracted by defining a threshold between signal and background pixels using the Li and Lee (95) minimum cross-entropy method. Masks defining the neuron area, containing the soma and proximal processes, were defined manually in FIJI. Randomized Kv2.1 images were computationally generated by decomposing the neuron area (defined by the mask) into pixel blocks (the size of the pixel block used is noted in the corresponding figure legend and table; a single pixel is sized at 31.34 nm in our images), which were then placed randomly within the neuron mask area to create a new randomized image. To calculate percent overlap, a binary mask was generated for both Nir2 and Kv2.1 from the background-subtracted images to define the spatial location of Nir2 and Kv2.1 signal. Using these binary mask images, percent overlap was calculated as the number of pixels containing both Nir2 and Kv2.1 signal divided by the total number of pixels containing Nir2 signal. The percent overlap analysis was performed identically on randomized and original images. The difference in overlap between the original and randomized image sets was evaluated using a two-tailed, paired t test. The image of pixel overlap presented in Fig. 3B was generated using the plugin “RBG2 colocalization” in FIJI. Analyses of the spatial distribution of endogenous Kv2.1, Nir2, and VAPs, in N-SIM images of CHNs, were performed in FIJI using the “Interaction analysis” plugin from the MOSAIC suite of plugins (63) and in Icy (96) using the GcoPS plugin (62).
Author contributions
M. K., E. J. D., and J. S. T. conceptualization; M. K., E. J. D., and J. S. T. resources; M. K., E. J. D., and J. S. T. data curation; M. K., T. E. G., E. J. D., and J. S. T. formal analysis; M. K., E. J. D., and J. S. T. supervision; M. K. and J. S. T. funding acquisition; M. K., T. E. G., E. J. D., and J. S. T. validation; M. K., E. J. D., and J. S. T. investigation; M. K., T. E. G., E. J. D., and J. S. T. visualization; M. K., T. E. G., E. J. D., and J. S. T. methodology; M. K., E. J. D., and J. S. T. writing-original draft; M. K., E. J. D., and J. S. T. project administration; M. K., T. E. G., E. J. D., and J. S. T. writing-review and editing; T. E. G. software.
Supplementary Material
Acknowledgments
We thank Michelle Salemi and the UC Davis Proteomics Core for expert advice on proteomics and Dr. Michael Paddy and the UC Davis MCB imaging facility for expert advice on imaging. We thank Grace Or Mizuno and Kimberly Nguyen for help in preparation of cultured rat hippocampal neurons.
This work was supported by National Institutes of Health Research Grants R01 HL144071 and R21 NS101648 (to J. S. T.), Grant R01 GM127513 (to E. J. D.), and Grant T32 GM007377 (to M. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- MCS
- membrane contact site
- PtdIns(4,5)P2
- PtdIns 4,5-bisphosphate
- PM
- plasma membrane
- ER
- endoplasmic reticulum
- PtdIns
- phosphatidylinositol
- PITP
- phosphatidylinositol transfer protein
- IP
- immunopurification
- PCC
- Pearson correlation coefficient
- OxoM
- oxotremorine M
- SDC
- spinning disk confocal
- ANOVA
- analysis of variance
- M1R
- type-1 muscarinic receptor
- CHN
- cultured rat hippocampal neuron
- PMSF
- phenylmethylsulfonyl fluoride
- DSP
- dithiobis(succinimidyl propionate)
- PtdIns(4)P
- phosphatidylinositol 4-phosphate
- TIRF
- total internal reflection fluorescence
- BFP
- blue fluorescent protein
- CFP
- cyan fluorescent protein
- DIV
- days in vitro
- RyR
- ryanodine receptor
- FRAP
- fluorescence recovery after photobleaching
- ESI
- electrospray ionization
- DAG
- diacylglycerol
- PLC
- phospholipase C
- HBSS
- Hanks' balanced saline solution
- DMEM
- Dulbecco's modified Eagle's medium
- GxTX
- guangxitoxin
- PIP2
- phosphatidylinositol 4,5-bisphosphate.
References
- 1. Henne W. M., Liou J., and Emr S. D. (2015) Molecular mechanisms of inter-organelle ER–PM contact sites. Curr. Opin. Cell Biol. 35, 123–130 10.1016/j.ceb.2015.05.001 [DOI] [PubMed] [Google Scholar]
- 2. Prakriya M., and Lewis R. S. (2015) Store-operated calcium channels. Physiol. Rev. 95, 1383–1436 10.1152/physrev.00020.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gallo A., Vannier C., and Galli T. (2016) Endoplasmic reticulum-plasma membrane associations: structures and functions. Annu. Rev. Cell Dev. Biol. 32, 279–301 10.1146/annurev-cellbio-111315-125024 [DOI] [PubMed] [Google Scholar]
- 4. Dickson E. J. (2017) Endoplasmic reticulum–plasma membrane contacts regulate cellular excitability. Adv. Exp. Med. Biol. 997, 95–109 10.1007/978-981-10-4567-7_7 [DOI] [PubMed] [Google Scholar]
- 5. Porter K. R., and Palade G. E. (1957) Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J. Biophys. Biochem. Cytol. 3, 269–300 10.1083/jcb.3.2.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenbluth J. (1962) Subsurface cisterns and their relationship to the neuronal plasma membrane. J. Cell Biol. 13, 405–421 10.1083/jcb.13.3.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henkart M., Landis D. M., and Reese T. S. (1976) Similarity of junctions between plasma membranes and endoplasmic reticulum in muscle and neurons. J. Cell Biol. 70, 338–347 10.1083/jcb.70.2.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peters A., Proskauer C. C., and Kaiserman-Abramof I. R. (1968) The small pyramidal neuron of the rat cerebral cortex. The axon hillock and initial segment. J. Cell Biol. 39, 604–619 10.1083/jcb.39.3.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu Y., Whiteus C., Xu C. S., Hayworth K. J., Weinberg R. J., Hess H. F., and De Camilli P. (2017) Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. U.S.A. 114, E4859–E4867 10.1073/pnas.1701078114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giordano F., Saheki Y., Idevall-Hagren O., Colombo S. F., Pirruccello M., Milosevic I., Gracheva E. O., Bagriantsev S. N., Borgese N., and De Camilli P. (2013) PI(4,5)P2-dependent and Ca2+-regulated ER–PM interactions mediated by the extended synaptotagmins. Cell 153, 1494–1509 10.1016/j.cell.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chung J., Torta F., Masai K., Lucast L., Czapla H., Tanner L. B., Narayanaswamy P., Wenk M. R., Nakatsu F., and De Camilli P. (2015) PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432 10.1126/science.aab1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lees J. A., Messa M., Sun E. W., Wheeler H., Torta F., Wenk M. R., De Camilli P., and Reinisch K. M. (2017) Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion. Science 355, eaah6171 10.1126/science.aah6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luik R. M., Wu M. M., Buchanan J., and Lewis R. S. (2006) The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 174, 815–825 10.1083/jcb.200604015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., and Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 10.1016/j.cell.2009.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takeshima H., Komazaki S., Nishi M., Iino M., and Kangawa K. (2000) Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22 10.1016/S1097-2765(00)00003-4 [DOI] [PubMed] [Google Scholar]
- 16. Hogan P. G., Lewis R. S., and Rao A. (2010) Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533 10.1146/annurev.immunol.021908.132550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 18. Calderón J. C., Bolaños P., and Caputo C. (2014) The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 6, 133–160 10.1007/s12551-013-0135-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang C. L., and Liou J. (2015) Phosphatidylinositol 4,5-bisphosphate homeostasis regulated by Nir2 and Nir3 proteins at endoplasmic reticulum–plasma membrane junctions. J. Biol. Chem. 290, 14289–14301 10.1074/jbc.M114.621375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang C. L., Hsieh T. S., Yang T. T., Rothberg K. G., Azizoglu D. B., Volk E., Liao J. C., and Liou J. (2013) Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum–plasma membrane junctions. Cell Rep. 5, 813–825 10.1016/j.celrep.2013.09.038 [DOI] [PubMed] [Google Scholar]
- 21. Kim Y. J., Guzman-Hernandez M. L., Wisniewski E., and Balla T. (2015) Phosphatidylinositol-phosphatidic acid exchange by Nir2 at ER–PM contact sites maintains phosphoinositide signaling competence. Dev. Cell 33, 549–561 10.1016/j.devcel.2015.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim Y. J., Guzman-Hernandez M. L., Wisniewski E., Echeverria N., and Balla T. (2016) Phosphatidylinositol and phosphatidic acid transport between the ER and plasma membrane during PLC activation requires the Nir2 protein. Biochem. Soc. Trans. 44, 197–201 10.1042/BST20150187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murakoshi H., and Trimmer J. S. (1999) Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J. Neurosci. 19, 1728–1735 10.1523/JNEUROSCI.19-05-01728.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guan D., Tkatch T., Surmeier D. J., Armstrong W. E., and Foehring R. C. (2007) Kv2 subunits underlie slowly inactivating potassium current in rat neocortical pyramidal neurons. J. Physiol. 581, 941–960 10.1113/jphysiol.2007.128454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu P. W., and Bean B. P. (2014) Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci. 34, 4991–5002 10.1523/JNEUROSCI.1925-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hönigsperger C., Nigro M. J., and Storm J. F. (2017) Physiological roles of Kv2 channels in entorhinal cortex layer II stellate cells revealed by Guangxitoxin-1E. J. Physiol. 595, 739–757 10.1113/JP273024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palacio S., Chevaleyre V., Brann D. H., Murray K. D., Piskorowski R. A., and Trimmer J. S. (2017) Heterogeneity in Kv2 channel expression shapes action potential characteristics and firing patterns in CA1 versus CA2 hippocampal pyramidal neurons. eNeuro 4, ENEURO.0267–17.2017 10.1523/ENEURO.0267-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Speca D. J., Ogata G., Mandikian D., Bishop H. I., Wiler S. W., Eum K., Wenzel H. J., Doisy E. T., Matt L., Campi K. L., Golub M. S., Nerbonne J. M., Hell J. W., Trainor B. C., Sack J. T., et al. (2014) Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav. 13, 394–408 10.1111/gbb.12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Torkamani A., Bersell K., Jorge B. S., Bjork R. L. Jr, Friedman J. R., Bloss C. S., Cohen J., Gupta S., Naidu S., Vanoye C. G., George A. L. Jr., and Kearney J. A. (2014) De novo KCNB1 mutations in epileptic encephalopathy. Ann. Neurol. 76, 529–540 10.1002/ana.24263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Srivastava S., Cohen J. S., Vernon H., Barañano K., McClellan R., Jamal L., Naidu S., and Fatemi A. (2014) Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 76, 473–483 10.1002/ana.24251 [DOI] [PubMed] [Google Scholar]
- 31. Thiffault I., Speca D. J., Austin D. C., Cobb M. M., Eum K. S., Safina N. P., Grote L., Farrow E. G., Miller N., Soden S., Kingsmore S. F., Trimmer J. S., Saunders C. J., and Sack J. T. (2015) A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J. Gen. Physiol. 146, 399–410 10.1085/jgp.201511444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Kovel C. G. F., Syrbe S., Brilstra E. H., Verbeek N., Kerr B., Dubbs H., Bayat A., Desai S., Naidu S., Srivastava S., Cagaylan H., Yis U., Saunders C., Rook M., Plugge S., Muhle H., et al. (2017) Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 74, 1228–1236 10.1001/jamaneurol.2017.1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trimmer J. S. (1991) Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 10764–10768 10.1073/pnas.88.23.10764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du J., Tao-Cheng J. H., Zerfas P., and McBain C. J. (1998) The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84, 37–48 10.1016/S0306-4522(97)00519-8 [DOI] [PubMed] [Google Scholar]
- 35. Sarmiere P. D., Weigle C. M., and Tamkun M. M. (2008) The Kv2.1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci. 9, 112 10.1186/1471-2202-9-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. King A. N., Manning C. F., and Trimmer J. S. (2014) A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J. Comp. Neurol. 522, 2594–2608 10.1002/cne.23551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mandikian D., Bocksteins E., Parajuli L. K., Bishop H. I., Cerda O., Shigemoto R., and Trimmer J. S. (2014) Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol. 522, 3555–3574 10.1002/cne.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bishop H. I., Cobb M. M., Kirmiz M., Parajuli L. K., Mandikian D., Philp A. M., Melnik M., Kuja-Panula J., Rauvala H., Shigemoto R., Murray K. D., and Trimmer J. S. (2018) Kv2 ion channels determine the expression and localization of the associated AMIGO-1 cell adhesion molecule in adult brain neurons. Front. Mol. Neurosci. 11, 1 10.3389/fnmol.2018.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bishop H. I., Guan D., Bocksteins E., Parajuli L. K., Murray K. D., Cobb M. M., Misonou H., Zito K., Foehring R. C., and Trimmer J. S. (2015) Distinct cell- and layer-specific expression patterns and independent regulation of Kv2 channel subtypes in cortical pyramidal neurons. J. Neurosci. 35, 14922–14942 10.1523/JNEUROSCI.1897-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Antonucci D. E., Lim S. T., Vassanelli S., and Trimmer J. S. (2001) Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108, 69–81 10.1016/S0306-4522(01)00476-6 [DOI] [PubMed] [Google Scholar]
- 41. Fox P. D., Haberkorn C. J., Akin E. J., Seel P. J., Krapf D., and Tamkun M. M. (2015) Induction of stable ER–plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 128, 2096–2105 10.1242/jcs.166009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kirmiz M., Palacio S., Thapa P., King A. N., Sack J. T., and Trimmer J. S. (2018) Remodeling neuronal ER–PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol. Biol. Cell 29, 2410–2432 10.1091/mbc.E18-05-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kirmiz M., Vierra N. C., Palacio S., and Trimmer J. S. (2018) Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum–plasma membrane junctions in mammalian brain neurons. J. Neurosci. 38, 7562–7584 10.1523/JNEUROSCI.0893-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson B., Leek A. N., Solé L., Maverick E. E., Levine T. P., and Tamkun M. M. (2018) Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. U.S.A. 115, E7331–E7340 10.1073/pnas.1805757115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim S. T., Antonucci D. E., Scannevin R. H., and Trimmer J. S. (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25, 385–397 10.1016/S0896-6273(00)80902-2 [DOI] [PubMed] [Google Scholar]
- 46. Lev S., Ben Halevy D., Peretti D., and Dahan N. (2008) The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol. 18, 282–290 10.1016/j.tcb.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 47. Rocha N., Kuijl C., van der Kant R., Janssen L., Houben D., Janssen H., Zwart W., and Neefjes J. (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 glued and late endosome positioning. J. Cell Biol. 185, 1209–1225 10.1083/jcb.200811005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mesmin B., Bigay J., Moser von Filseck J., Lacas-Gervais S., Drin G., and Antonny B. (2013) A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER–Golgi tether OSBP. Cell 155, 830–843 10.1016/j.cell.2013.09.056 [DOI] [PubMed] [Google Scholar]
- 49. Lomant A. J., and Fairbanks G. (1976) Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J. Mol. Biol. 104, 243–261 10.1016/0022-2836(76)90011-5 [DOI] [PubMed] [Google Scholar]
- 50. Kihira Y., Hermanstyne T. O., and Misonou H. (2010) Formation of heteromeric Kv2 channels in mammalian brain neurons. J. Biol. Chem. 285, 15048–15055 10.1074/jbc.M109.074260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peltola M. A., Kuja-Panula J., Lauri S. E., Taira T., and Rauvala H. (2011) AMIGO is an auxiliary subunit of the Kv2.1 potassium channel. EMBO Rep. 12, 1293–1299 10.1038/embor.2011.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun E. W., Guillén-Samander A., Bian X., Wu Y., Cai Y., Messa M., and De Camilli P. (2019) Lipid transporter TMEM24/C2CD2L is a Ca2+-regulated component of ER–plasma membrane contacts in mammalian neurons. Proc. Natl. Acad. Sci. U.S.A. 116, 5775–5784 10.1073/pnas.1820156116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shaw G., Morse S., Ararat M., and Graham F. L. (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16, 869–871 10.1096/fj.01-0995fje [DOI] [PubMed] [Google Scholar]
- 54. Yu S. P., and Kerchner G. A. (1998) Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J. Neurosci. Res. 52, 612–617 10.1002/(SICI)1097-4547(19980601)52:5<612::AID-JNR13>3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 55. Besprozvannaya M., Dickson E., Li H., Ginburg K. S., Bers D. M., Auwerx J., and Nunnari J. (2018) GRAM domain proteins specialize functionally distinct ER–PM contact sites in human cells. Elife 7, e31019 10.7554/eLife.31019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee H. C., Wang J. M., and Swartz K. J. (2003) Interaction between extracellular Hanatoxin and the resting conformation of the voltage–sensor paddle in Kv channels. Neuron 40, 527–536 10.1016/S0896-6273(03)00636-6 [DOI] [PubMed] [Google Scholar]
- 57. Herrington J., Zhou Y. P., Bugianesi R. M., Dulski P. M., Feng Y., Warren V. A., Smith M. M., Kohler M. G., Garsky V. M., Sanchez M., Wagner M., Raphaelli K., Banerjee P., Ahaghotu C., Wunderler D., et al. (2006) Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes 55, 1034–1042 10.2337/diabetes.55.04.06.db05-0788 [DOI] [PubMed] [Google Scholar]
- 58. Tilley D. C., Eum K. S., Fletcher-Taylor S., Austin D. C., Dupré C., Patrón L. A., Garcia R. L., Lam K., Yarov-Yarovoy V., Cohen B. E., and Sack J. T. (2014) Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc. Natl. Acad. Sci. U.S.A. 111, E4789–E4796 10.1073/pnas.1406876111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsieh T. S., Chen Y. J., Chang C. L., Lee W. R., and Liou J. (2017) Cortical actin contributes to spatial organization of ER–PM junctions. Mol. Biol. Cell 28, 3171–3180 10.1091/mbc.e17-06-0377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dickson E. J., Jensen J. B., Vivas O., Kruse M., Traynor-Kaplan A. E., and Hille B. (2016) Dynamic formation of ER–PM junctions presents a lipid phosphatase to regulate phosphoinositides. J. Cell Biol. 213, 33–48 10.1083/jcb.201508106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Helmuth J. A., Paul G., and Sbalzarini I. F. (2010) Beyond colocalization: inferring spatial interactions between sub-cellular structures from microscopy images. BMC Bioinformatics 11, 372 10.1186/1471-2105-11-372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lavancier F., Pecot T., Zengzhen L., and Kervrann C. (2019) Testing independence between two random sets for the analysis of colocalization in bio-imaging. Biometrics 2019 10.1111/biom.13115 [DOI] [PubMed] [Google Scholar]
- 63. Shivanandan A., Radenovic A., and Sbalzarini I. F. (2013) MosaicIA: an ImageJ/Fiji plugin for spatial pattern and interaction analysis. BMC Bioinformatics 14, 349 10.1186/1471-2105-14-349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., and Lockett S. (2004) Automatic and quantitative measurement of protein–protein colocalization in live cells. Biophys. J. 86, 3993–4003 10.1529/biophysj.103.038422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Inoue T., Heo W. D., Grimley J. S., Wandless T. J., and Meyer T. (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 10.1038/nmeth763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Connell K. M., Rolig A. S., Whitesell J. D., and Tamkun M. M. (2006) Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J. Neurosci. 26, 9609–9618 10.1523/JNEUROSCI.1825-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stauffer T. P., Ahn S., and Meyer T. (1998) Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8, 343–346 10.1016/S0960-9822(98)70135-6 [DOI] [PubMed] [Google Scholar]
- 68. Hammond G. R., Machner M. P., and Balla T. (2014) A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 205, 113–126 10.1083/jcb.201312072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Traynor-Kaplan A., Kruse M., Dickson E. J., Dai G., Vivas O., Yu H., Whittington D., and Hille B. (2017) Fatty-acyl chain profiles of cellular phosphoinositides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 513–522 10.1016/j.bbalip.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Aikawa Y., Hara H., and Watanabe T. (1997) Molecular cloning and characterization of mammalian homologues of the Drosophila retinal degeneration B gene. Biochem. Biophys. Res. Commun. 236, 559–564 10.1006/bbrc.1997.7009 [DOI] [PubMed] [Google Scholar]
- 71. Chang J. T., Milligan S., Li Y., Chew C. E., Wiggs J., Copeland N. G., Jenkins N. A., Campochiaro P. A., Hyde D. R., and Zack D. J. (1997) Mammalian homolog of Drosophila retinal degeneration B rescues the mutant fly phenotype. J. Neurosci. 17, 5881–5890 10.1523/JNEUROSCI.17-15-05881.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lev S., Hernandez J., Martinez R., Chen A., Plowman G., and Schlessinger J. (1999) Identification of a novel family of targets of PYK2 related to Drosophila retinal degeneration B (rdgB) protein. Mol. Cell. Biol. 19, 2278–2288 10.1128/MCB.19.3.2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lu C., Vihtelic T. S., Hyde D. R., and Li T. (1999) A neuronal-specific mammalian homolog of the Drosophila retinal degeneration B gene with expression restricted to the retina and dentate gyrus. J. Neurosci. 19, 7317–7325 10.1523/JNEUROSCI.19-17-07317.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maletic-Savatic M., Lenn N. J., and Trimmer J. S. (1995) Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J. Neurosci. 15, 3840–3851 10.1523/JNEUROSCI.15-05-03840.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rhodes K. J., Keilbaugh S. A., Barrezueta N. X., Lopez K. L., and Trimmer J. S. (1995) Association and colocalization of K+ channel α- and β-subunit polypeptides in rat brain. J. Neurosci. 15, 5360–5371 10.1523/JNEUROSCI.15-07-05360.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Huang B., Bates M., and Zhuang X. (2009) Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 78, 993–1016 10.1146/annurev.biochem.77.061906.092014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Amarilio R., Ramachandran S., Sabanay H., and Lev S. (2005) Differential regulation of endoplasmic reticulum structure through VAP–Nir protein interaction. J. Biol. Chem. 280, 5934–5944 10.1074/jbc.M409566200 [DOI] [PubMed] [Google Scholar]
- 78. Kaiser S. E., Brickner J. H., Reilein A. R., Fenn T. D., Walter P., and Brunger A. T. (2005) Structural basis of FFAT motif-mediated ER targeting. Structure 13, 1035–1045 10.1016/j.str.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 79. Nishimura Y., Hayashi M., Inada H., and Tanaka T. (1999) Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem. Biophys. Res. Commun. 254, 21–26 10.1006/bbrc.1998.9876 [DOI] [PubMed] [Google Scholar]
- 80. Kim S., Leal S. S., Ben Halevy D., Gomes C. M., and Lev S. (2010) Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 285, 13839–13849 10.1074/jbc.M109.097345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Furuita K., Jee J., Fukada H., Mishima M., and Kojima C. (2010) Electrostatic interaction between oxysterol-binding protein and VAMP-associated protein A revealed by NMR and mutagenesis studies. J. Biol. Chem. 285, 12961–12970 10.1074/jbc.M109.082602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Várnai P., Tóth B., Tóth D. J., Hunyady L., and Balla T. (2007) Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1–Orai1 Complex. J. Biol. Chem. 282, 29678–29690 10.1074/jbc.M704339200 [DOI] [PubMed] [Google Scholar]
- 83. Drewe J. A., Verma S., Frech G., and Joho R. H. (1992) Distinct spatial and temporal expression patterns of K+ channel mRNAs from different subfamilies. J. Neurosci. 12, 538–548 10.1523/JNEUROSCI.12-02-00538.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Herculano-Houzel S., Mota B., and Lent R. (2006) Cellular scaling rules for rodent brains. Proc. Natl. Acad. Sci. U.S.A. 103, 12138–12143 10.1073/pnas.0604911103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Herculano-Houzel S. (2014) The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62, 1377–1391 10.1002/glia.22683 [DOI] [PubMed] [Google Scholar]
- 86. McKenzie A. T., Wang M., Hauberg M. E., Fullard J. F., Kozlenkov A., Keenan A., Hurd Y. L., Dracheva S., Casaccia P., Roussos P., and Zhang B. (2018) Brain cell type specific gene expression and coexpression network architectures. Sci. Rep. 8, 8868 10.1038/s41598-018-27293-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dickson E. J., and Hille B. (2019) Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1–23 10.1042/BCJ20180022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kruse M., Vivas O., Traynor-Kaplan A., and Hille B. (2016) Dynamics of phosphoinositide-dependent signaling in sympathetic neurons. J. Neurosci. 36, 1386–1400 10.1523/JNEUROSCI.3535-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Misonou H., Mohapatra D. P., Park E. W., Leung V., Zhen D., Misonou K., Anderson A. E., and Trimmer J. S. (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718 10.1038/nn1260 [DOI] [PubMed] [Google Scholar]
- 90. Tao-Cheng J. H. (2018) Activity-dependent decrease in contact areas between subsurface cisterns and plasma membrane of hippocampal neurons. Mol. Brain 11, 23 10.1186/s13041-018-0366-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tao-Cheng J. H. (2018) Stimulation-induced structural changes at the nucleus, endoplasmic reticulum and mitochondria of hippocampal neurons. Mol. Brain 11, 44 10.1186/s13041-018-0387-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jacobson D. A., Kuznetsov A., Lopez J. P., Kash S., Ammälä C. E., and Philipson L. H. (2007) Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 6, 229–235 10.1016/j.cmet.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Misonou H., Menegola M., Mohapatra D. P., Guy L. K., Park K. S., and Trimmer J. S. (2006) Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J. Neurosci. 26, 13505–13514 10.1523/JNEUROSCI.3970-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kobrinsky E., Stevens L., Kazmi Y., Wray D., and Soldatov N. M. (2006) Molecular rearrangements of the Kv2.1 potassium channel termini associated with voltage gating. J. Biol. Chem. 281, 19233–19240 10.1074/jbc.M601231200 [DOI] [PubMed] [Google Scholar]
- 95. Li C. H., and Lee C. K. (1993) Minimum cross entropy thresholding. Pattern Recognition 26, 617–625 10.1016/0031-3203(93)90115-D [DOI] [Google Scholar]
- 96. de Chaumont F., Dallongeville S., Chenouard N., Hervé N., Pop S., Provoost T., Meas-Yedid V., Pankajakshan P., Lecomte T., Le Montagner Y., Lagache T., Dufour A., and Olivo-Marin J. C. (2012) Icy: an open bioimage informatics platform for extended reproducible research. Nat. Methods 9, 690–696 10.1038/nmeth.2075 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.