

Abstract
How cells utilize nutrients to produce the ATP needed for bioenergetic homeostasis has been well-characterized. What is less well-studied is how resting cells metabolically shift from an ATP-producing catabolic metabolism to a metabolism that supports anabolic growth. In metazoan organisms, the discovery of growth factors and the ability of their receptors to induce new transcription and translation led to the hypothesis that the bioenergetic and synthetic demands of cell growth were primarily met through the replacement of nutrients consumed during net macromolecular synthesis, a demand-based system of nutrient uptake. Recent data have challenged this hypothesis. Instead, there is increasing evidence that cellular nutrient uptake is a push system. Growth factor signaling has been linked to direct stimulation of nutrient uptake. The ability of growth factor signaling to increase the uptake of glucose, lipids, and amino acids to levels that exceed a cell's bioenergetic and synthetic needs has been documented in a wide variety of settings. In some tissues, this leads to the storage of the excess nutrients in the form of glycogen or fat. In others, the excess is secreted as lactate and certain nonessential amino acids. When growth factor signaling stimulates nutrient uptake to levels that exceed a cell's bioenergetic needs, adaptive changes in intermediate metabolism lead to the production of anabolic precursors that fuel the net synthesis of protein, lipids, and nucleic acids. Through the increased production of these precursors, growth factor signaling provides a supply-side stimulation of cell growth and proliferation.
Keywords: cell metabolism, growth factor, cell growth, bioenergetics, cell proliferation
Introduction
Most unicellular eukaryotes forage for food to maintain their survival. In contrast, most metazoan cells are surrounded by sufficient extracellular nutrients that the cells should never have to face a bioenergetic compromise. The traditional view was that mammalian cells take up glucose in response to the depletion of ATP and a rise in ADP (Fig. 1A). However, this model did not adequately explain how proliferating cells increased their nutrient uptake to not only maintain bioenergetics, but also to fuel net anabolic synthesis (Fig. 1B). This paradox received relatively little attention until it was discovered that mammalian cells required growth factor signaling to maintain sufficient nutrient uptake to sustain ATP production and cell survival (1, 2). When mammalian cells were isolated and cultured in the absence of endocrine, paracrine, and growth factor signaling normally present in vivo, the cells lost the ability to take up extracellular nutrients and could not maintain their bioenergetic homeostasis (1, 3). To survive the loss of signaling, cells turned to the degradation of internal proteins and lipids through autophagy to maintain mitochondrial ATP production (2). Only when extracellular signaling through lineage-specific survival/growth factors was reestablished would cells reuptake glucose, lipids, and amino acids from the environment and utilize these to sustain bioenergetics and engage in replacement macromolecular synthesis.
Figure 1.

The fate of nutrients taken up is different between resting and proliferating cells. A, in resting cells, the primary bioenergetic substrate taken up by the cell is glucose. Through combined activities of glycolysis and oxidative phosphorylation, glucose is oxidized to carbon dioxide (CO2) and water (H2O), converting ADP to ATP primarily through the electrochemical potential created by the electron transport chain (ETC) in the inner mitochondrial membrane. B, in proliferating cells, more glucose is taken up than the cells can utilize for either ATP production or macromolecular synthesis. Carbon in excess of those needs is secreted as lactate. Depletion of TCA cycle intermediates is prevented by increases in the uptake of amino acids, primarily glutamine.
G0 phase of the cell cycle
The above findings led us to hypothesize that G0 is the most dynamic cell state (4). Consistent with this, we found that primary parenchymal cells do not have a defined cell size in G0. Instead, their resting cell size is determined by the net input of extracellular growth factors directing nutrient uptake (5). When extracellular growth factors are low, cells in G0 shrink in size through the progressive consumption of intracellular proteins and lipids via autophagy (Fig. 2) (2). In contrast, when growth factors are in excess, signaling increases glucose and amino acid uptake and reprograms metabolism to support anabolic growth (5). Using fibroblasts, we were able to show that the length of time it takes a G0 fibroblast to reenter S phase upon serum stimulation can vary by up to 11 days, and the entire variation correlates directly with the time needed to regrow and reverse the effects of autophagy (2).
Figure 2.

The size of cells not actively engaged in the cell cycle is determined by the magnitude of growth factor–directed nutrient uptake. A, in noncycling (G0) cells that do not receive any growth factor signaling, G0 cells are unable to sustain the cell surface expression of nutrient transporters. The absence of growth factor–induced mTORC1 activity leads to activation of autophagy through Ulk 1/2. The autophagic degradation of intracellular contents supplies the mitochondria with substrates that can be oxidized to maintain ATP production and cell survival. As a result of the degradation of intracellular contents, cells deficient in basal growth factor signaling get smaller over time. B, when G0 cells receive more than basal levels of growth factor signaling, they increase their uptake of glucose and amino acids (AA) through enhanced cell surface expression of nutrient transporters. Glucose is captured intracellularly by phosphorylation, and amino acids (AA) are captured by increased tRNA charging and translation. As a result, cells grow larger in size.
Growth factor regulation of glucose and amino acid uptake
Glucose is the major metabolite used to support bioenergetics in mammalian cells and its uptake is primarily regulated by lineage-specific growth factor receptors (6). Many of the receptors that can support cell survival and growth share a common signaling pathway that involves the activation of phosphatidylinositol 3-kinase (PI3K)2 (7, 8) and leads to the downstream activation of the downstream effectors, AKT and mTOR (9, 10). Together, these effectors are responsible for increasing the cell surface expression of glucose and amino acid transporters and for programming intermediate metabolism to support the production of anabolic precursors that fuel protein and lipid production. In every species that has been examined to date, activation of the PI3K/AKT/mTOR pathway is sufficient to increase the cell size of G0 cells in their resting state (G0) (for a review, see Ref. 11). Thus, the basal parenchymal cell size is determined by the level of growth factor signaling the cell receives (Fig. 2).
Cells engage in anabolic processes in response to intracellular nutrient levels
The fact that metazoan cells have lost the cell-autonomous ability to take up nutrients suggested that cell growth and proliferation can be limited by intracellular nutrient supplies. Growth factors stimulate growth, at least in part, through stimulating an increased uptake of extracellular nutrients (6, 12). The growth factor–induced accumulation of nutrients is accompanied by an intracellular reprogramming of intermediate metabolism that supports the production of macromolecular precursors (13, 14). Excess glucose taken up in response to growth factor stimulation can be diverted from the glycolytic pathway to support ribose and nonessential amino acid production. Excess glycolysis-derived pyruvate entering the mitochondria is diverted from oxidative phosphorylation into the production of precursors that support fatty acid, nonessential amino acid, and pyrimidine biosynthesis. Both fatty acid and essential amino acid oxidation are suppressed as a result of PI3K/AKT/mTOR activation (8), and the nonessential amino acid glutamine becomes the major anapleurotic substrate that supports TCA cycle replenishment as TCA cycle intermediates are diverted into production of synthetic precursors (15).
Growth factor–dependent nutrient uptake regulates homeostatic proliferation
The above model has interesting implications for so-called homeostatic proliferation, where aging or apoptotic parenchymal cells within an organ are replaced through proliferation (5). As the number of parenchymal cells of an organ declines, the basal levels of paracrine, endocrine, and growth factors in the extracellular environment will increase on a per cell basis, directing more nutrient uptake to support growth/proliferation of the remaining cells. Thus, by being directly coupled to nutrient uptake, net growth factor stimulation can homeostatically regulate cell numbers within adult organs and lineages.
Many cancer cells display cell autonomous nutrient uptake
If cell growth in metazoan organisms is limited by nutrient uptake, then diseases associated with dysregulated growth should be associated with mutations that render a cell autonomous for the ability to take up nutrients. Cancer represents exactly such a disease, where transformed cells become dysregulated and grow autonomously. Thus, one would expect cancer cells to preferentially acquire mutations that render them cell-autonomous for nutrient uptake. In recent years, cancer genome sequencing efforts have revealed that mutations in growth factor receptors along with the PI3K/AKT/mTOR pathway components represent together the most commonly mutated pathway in human malignancy. Mutations in this pathway have been shown to correlate with the ability to image human tumors in vivo using 18-fluorodeoxyglucose (FDG) (for a review, see Ref. 16). Collectively, between 70 and 75% of human cancers exhibit a mutation in one of the components of this signaling pathway. This is close to the percentage of human tumors that display excess glucose uptake using FDG-positron emission tomography (PET). Thus, there is a strong association between in vivo glucose uptake and mutations that activate the PI3K/AKT/mTOR signaling pathway.
The Warburg effect
The close association of FDG-positive PET scans and mutations that activate the PI3K/AKT/mTOR pathway suggests a simple explanation for the so-called Warburg effect. In the 1920s, Otto Warburg and his colleagues noticed that cancer cells preferentially metabolized glucose through glycolysis rather than through respiration (17). He suggested that this was due to a defect in mitochondrial function (18). However, to date, no human cancers have been described that lack the ability to carry out oxidative phosphorylation. An alternative explanation is that signaling through the PI3K/AKT/mTOR pathway directs more glucose uptake than can be utilized by the cell and the cell must deal with the extra carbon by secreting it as lactate (19). The flux of nutrients created by activation of the PI3K/AKT/mTOR pathway also pushes glycolytic intermediates into alternative pathways, such as the pentose-phosphate shunt and the serine/glycine synthetic pathway, and into glycerol production (14, 16). The excess pyruvate available to the mitochondria also leads to excess TCA cycle intermediates that support lipid, nonessential amino acid, and pyrimidine biosynthesis (13, 15).
Cell proliferation when extracellular nutrients are limiting
Although the above associations are clear when cancer cells are growing in tissue culture, the fact remains that not all growing cancers exist within nutrient-rich environments. In vivo, many tumors do not exhibit excess glucose uptake based on FDG imaging. This suggests that there must be alternate mechanisms by which cancer cells take up sufficient nutrients to engage in growth. Even for nontransformed cells, there are forms of cellular growth and proliferation that occur in nutrient-depleted environments. During organ repair following injury, cell proliferation must occur to replace lost tissue. Often the vascular supply of homeostatic nutrients is compromised under such circumstances. Similarly, cuts and abrasions must heal through epithelial cell proliferation even though the underlying vascularity is compromised.
During tissue repair, relatively little of the replacement biomass comes as a direct product of glucose metabolism; 60% of cellular mass is proteins. In vivo, amino acids are the major nutrients that contribute to cell mass during growth (20). Furthermore, many amino acids required for growth are essential and cannot be synthesized from either other amino acids or glucose. Essential amino acids are among the lowest in absolute concentration in the extracellular fluid. Thus, in the absence of constant replenishment, essential amino acids will be rapidly depleted in vascularly compromised tissues.
Metazoan cells can use extracellular proteins as nutrients
A potential explanation for how cell growth and proliferation might be supported in the absence of mutations in growth factor receptors or the PI3K/AKT/mTOR pathway came from trying to understand a paradox observed in the extracellular environment of vascularly compromised tumors. Multiple studies have shown that as tumors outgrow their vasculature, among the nutrients that become depleted are nonessential amino acids (21–23). In contrast, essential amino acids are often found at higher levels in the tumor extracellular environment than in the surrounding normal tissue. How vascularly compromised tumors can accumulate excess essential amino acids while depleting nonessential amino acids has been a puzzle. This led to the discovery that tumor cells in vascularly compromised tissue could resort to an alternative and potentially more evolutionarily ancient nutrient uptake pathway known as macropinocytosis (24). In macropinocytosis, rather than engage in nutrient uptake through nutrient-specific transporters, cells produce evaginations that encircle extracellular fluid. The resulting macropinosomes then traffic through the endosomal pathway to the lysosome, where their intercellular contents are degraded by lipases and proteases to supply the cell with amino acids and lipids that can sustain bioenergetics and macromolecular synthesis (Fig. 3).
Figure 3.
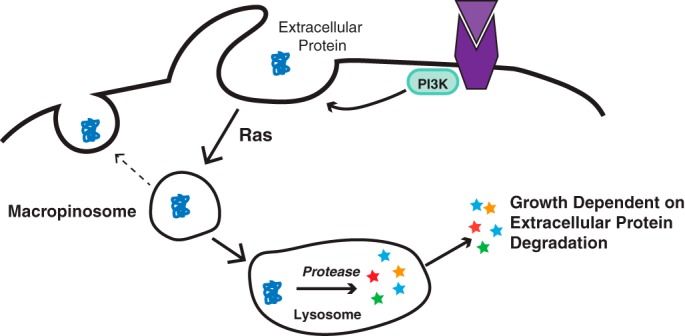
Growth factor–initiated PI3K activation leads to induction of macropinocytosis that can be amplified by Ras activation. When cells are depleted of free amino acids, macropinosomes traffic preferentially to the lysosome, where their engulfed contents are degraded, releasing amino acids and lipids to maintain bioenergetics and macromolecular synthesis.
Macropinocytosis can supply cells with the building blocks to support cell growth
A notable consequence of the use of macropinocytosis is that, as cells engulf and degrade extracellular protein to support cell growth, the cells preferentially accumulate essential amino acids. In contrast, nonessential amino acids become depleted as they are used during growth to support not only protein production but also nucleotide and lipid synthesis.
The stimulation of macropinocytosis can occur as a direct consequence of activating mutations of the Ras family of oncogenes (25). This finding may explain how certain cancers, such as pancreatic cancer and lung cancer, can be highly aggressive despite being vascularly compromised. However, whereas Ras increases the gain on the rate at which cells undertake macropinocytosis, the ability of a cell to engage in macropinocytosis is initiated by growth factor–induced PI3K signaling (26). In cancer cells bearing Ras mutations, there appears to be a sufficient increase of macropinocytosis at physiologic levels of PI3K activation to sustain cell growth. Thus, in cells that have a Ras mutation without a growth factor–signaling or PI3K mutation, the cells do not take up sufficient glucose or amino acids through amino acid transporters to fuel anabolic growth, but they are able to take up the proteins and lipids they need to grow as a result of an increased rate of macropinocytosis.
mTORC1 regulates the cellular nutrient uptake strategy
Recently, cross-talk between these two ancient mechanisms of nutrient uptake, macropinocytosis and selective nutrient transport, has been uncovered. The ability to maintain amino acid transporters on the cell surface depends on P13K/AKT-induced activation of mTORC1 (10). When amino acid transporters are able to take up sufficient amino acids to maintain mTORC1 activity, mTORC1 suppresses the trafficking of macropinosomes to the lysosome (27) (Fig. 4). Cells with active mTORC1 are unable to survive and grow using macropinocytosis as a source of nutrient uptake.
Figure 4.

mTORC1 suppresses macropinocytosis in nutrient-replete environments. Growth factor–initiated PI3K/AKT/mTORC1 signaling leads to surface expression of amino acid transporters. If this results in sufficient amino acid uptake to further activate the mTOR-containing mTORC1 complex, then the mTORC1 complex suppresses the trafficking of macropinosomes to the lysosome.
The fact that mTORC1 provides feedback inhibition of macropinocytosis suggests a potential explanation for the other major criticism concerning the association of PI3K/AKT/mTOR1 mutations with deregulated nutrient uptake. If the key effect of this pathway was to increase nutrient uptake, it would be expected that the closer the mutation in the pathway was to the critical effect, the more frequently mutations would be observed in that effector. Human tumor sequencing efforts have demonstrated that in naturally occurring tumors, whereas growth factor receptor and PI3K mutations are quite frequent, mutations in AKT and mTORC1 are relatively rare. Because growth factor activation of PI3K initiates both macropinocytosis and transporter-mediated nutrient uptake, mutations in either of these effectors would make the cell self-autonomous for the ability to take up nutrients. Similarly, a mutation in Ras would allow the tumor cells to grow in all nutritional environments, including vascularly depleted environments. However, mutations in mTORC1 would be predicted to permit cell growth only in a highly vascularized environment. Experiments are ongoing to test this hypothesis.
Concluding remarks
The idea that extracellular nutrient uptake is a bottleneck that regulates the growth and proliferation in multicellular organisms is still evolving. Although growth factor signaling does more than regulate nutrient uptake, it is clear that the role of growth factors in stimulating supply-side nutrient uptake and metabolism has important implications for our understanding of organismal homeostasis, tissue integrity, and disease states including cancer and degenerative diseases.
Acknowledgments
We thank past and present members of the Thompson Laboratory for allowing us to summarize their work. In addition, this review is a reflection on work carried out in the laboratory over the past 20 years, but it depends on the large body of work carried out by others in the fields of biochemistry, molecular biology, signal transmission, and bioenergetics and by our colleagues who have helped renew the field of cellular metabolism. We realize this reflection overemphasizes the work undertaken in this laboratory. It is meant to be primarily a summary of the work that led to the recognition bestowed by the ASBMB 2019 Bert and Natalie Vallee Award in Biomedical Sciences. Funding support for the work summarized here can be found in the referenced articles.
Dr. Thompson has been a past member of the Board of Directors and stockholder in Merck and Charles River Laboratories and is a founder and member of the Scientific Advisory Board of Agios Pharmaceuticals. He is a named inventor on a number of patents in the area of cellular metabolism.
- PI3K
- phosphatidylinositol 3-kinase
- mTOR
- mammalian target of rapamycin
- TCA
- tricarboxylic acid
- FDG
- 18-fluorodeoxyglucose
- PET
- positron emission tomography
- mTORC1
- mammalian target of rapamycin complex 1.
References
- 1. Rathmell J. C., Vander Heiden M. G., Harris M. H., Frauwirth K. A., and Thompson C. B. (2000) In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 6, 683–692 10.1016/S1097-2765(00)00066-6 [DOI] [PubMed] [Google Scholar]
- 2. Lum J. J., Bauer D. E., Kong M., Harris M. H., Li C., Lindsten T., and Thompson C. B. (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 10.1016/j.cell.2004.11.046 [DOI] [PubMed] [Google Scholar]
- 3. Lindsten T., Golden J. A., Zong W. X., Minarcik J., Harris M. H., and Thompson C. B. (2003) The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J. Neurosci. 23, 11112–11119 10.1523/JNEUROSCI.23-35-11112.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., and Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 5. Rathmell J. C., Farkash E. A., Gao W., and Thompson C. B. (2001) IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167, 6869–6876 10.4049/jimmunol.167.12.6869 [DOI] [PubMed] [Google Scholar]
- 6. Vander Heiden M. G., Plas D. R., Rathmell J. C., Fox C. J., Harris M. H., and Thompson C. B. (2001) Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 21, 5899–5912 10.1128/MCB.21.17.5899-5912.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frauwirth K. A., Riley J. L., Harris M. H., Parry R. V., Rathmell J. C., Plas D. R., Elstrom R. L., June C. H., and Thompson C. B. (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 10.1016/S1074-7613(02)00323-0 [DOI] [PubMed] [Google Scholar]
- 8. Deberardinis R. J., Lum J. J., and Thompson C. B. (2006) Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J. Biol. Chem. 281, 37372–37380 10.1074/jbc.M608372200 [DOI] [PubMed] [Google Scholar]
- 9. Elstrom R. L., Bauer D. E., Buzzai M., Karnauskas R., Harris M. H., Plas D. R., Zhuang H., Cinalli R. M., Alavi A., Rudin C. M., and Thompson C. B. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 64, 3892–3899 10.1158/0008-5472.CAN-03-2904 [DOI] [PubMed] [Google Scholar]
- 10. Edinger A. L., and Thompson C. B. (2002) Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 13, 2276–2288 10.1091/mbc.01-12-0584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wullschleger S., Loewith R., and Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 12. Bauer D. E., Harris M. H., Plas D. R., Lum J. J., Hammerman P. S., Rathmell J. C., Riley J. L., and Thompson C. B. (2004) Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. FASEB J. 18, 1303–1305 10.1096/fj.03-1001fje [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatzivassiliou G., Zhao F., Bauer D. E., Andreadis C., Shaw A. N., Dhanak D., Hingorani S. R., Tuveson D. A., and Thompson C. B. (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8, 311–321 10.1016/j.ccr.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 14. Ye J., Mancuso A., Tong X., Ward P. S., Fan J., Rabinowitz J. D., and Thompson C. B. (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 109, 6904–6909 10.1073/pnas.1204176109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., and Thompson C. B. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350 10.1073/pnas.0709747104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warburg O., Wind F., and Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 19. Lum J. J., Bui T., Gruber M., Gordan J. D., DeBerardinis R. J., Covello K. L., Simon M. C., and Thompson C. B. (2007) The transcription factor HIF-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 21, 1037–1049 10.1101/gad.1529107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hosios A. M., Hecht V. C., Danai L. V., Johnson M. O., Rathmell J. C., Steinhauser M. L., Manalis S. R., and Vander Heiden M. G. (2016) Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell 36, 540–549 10.1016/j.devcel.2016.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaupel P., Kallinowski F., and Okunieff P. (1989) Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465 [PubMed] [Google Scholar]
- 22. Kamphorst J. J., Nofal M., Commisso C., Hackett S. R., Lu W., Grabocka E., Vander Heiden M. G., Miller G., Drebin J. A., Bar-Sagi D., Thompson C. B., and Rabinowitz J. D. (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 10.1158/0008-5472.CAN-14-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sullivan M. R., Danai L. V., Lewis C. A., Chan S. H., Gui D. Y., Kunchok T., Dennstedt E. A., Vander Heiden M. G., and Muir A. (2019) Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235 10.7554/eLife.44235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Commisso C., Davidson S. M., Soydaner-Azeloglu R. G., Parker S. J., Kamphorst J. J., Hackett S., Grabocka E., Nofal M., Drebin J. A., Thompson C. B., Rabinowitz J. D., Metallo C. M., Vander Heiden M. G., and Bar-Sagi D. (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 10.1038/nature12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bar-Sagi D., and Feramisco J. R. (1986) Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233, 1061–1068 10.1126/science.3090687 [DOI] [PubMed] [Google Scholar]
- 26. Palm W., Araki J., King B., DeMatteo R. G., and Thompson C. B. (2017) Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proc. Natl. Acad. Sci. U.S.A. 114, E8628–E8636 10.1073/pnas.1712726114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palm W., Park Y., Wright K., Pavlova N. N., Tuveson D. A., and Thompson C. B. (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270 10.1016/j.cell.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]