

Abstract
Sleep has numerous advantages for aligning clinical and preclinical (basic neuroscience) studies of neuropsychiatric illness. Sleep has high translational relevance, because the same endpoints can be studied in humans and laboratory animals. In addition, sleep experiments are conducive to continuous data collection over long periods (hours/days/weeks) and can be based on highly objective neurophysiological measures. Here, we provide a translationally-oriented review on what is currently known about sleep in the context of autism spectrum disorder (ASD), including ASD-related conditions, thought to have genetic, environmental, or mixed etiologies. In humans, ASD is frequently associated with comorbid medical conditions including sleep disorders. Animal models used in the study of ASD frequently recapitulate dysregulation of sleep and biological (diurnal, circadian) rhythms, suggesting common pathophysiologies across species. As our understanding of the neurobiology of ASD and sleep each become more refined, it is conceivable that sleep-derived metrics may offer more powerful biomarkers of altered neurophysiology in ASD than the behavioral tests currently used in humans or lab animals. As such, the study of sleep in animal models for ASD may enable fundamentally new insights on the condition and represent a basis for strategies that enable the development of more effective therapeutics.
Subject terms: Sleep, Autism spectrum disorders
Introduction
A recent estimate suggests that as many as 2.5% of children have an autism spectrum disorder (ASD) [1]. Despite the high prevalence of ASD, its cause is unknown in most individuals. There is a strong genetic contribution to ASD, with an estimated genetic heritability of between 50–80% [2, 3]. The remaining risk is thought to be related to environmental risk factors: as examples, prenatal exposure to hypoxia, inflammation, or maternal obesity all increase the risk of developing ASD [4] or may be related to other forms of non-heritable risk such as de novo genetic mutations [5]. While genetic and environmental factors are often studied in isolation, ASD is likely the result of a complex interplay of both. ASD is defined by core features including (1) dysregulated social behavior, (2) deficits in communication, and (3) the presence of restricted and repetitive behavior. Behavioral signs of ASD often emerge within the first couple years of life and can be reliably diagnosed by 24 months of age [6]. Prior to the diagnostic threshold there may be prognostic features of ASD in the first years of life related to the underlying biology including low attention to eyes [7], atypical cries [8], and potentially abnormal electrophysiological signatures in the brain [9]. By necessity, the diagnosis of ASD is made on the basis of these core behavioral features, since definitive biomarkers that can readily identify ASD are yet to be discovered. As a result, ASD is not a unitary condition, rather a set of converging symptoms that likely stem from multiple biological etiologies. While the diagnosis of ASD is based on core behavioral features, there is a growing appreciation that medical conditions including epilepsy, gastrointestinal disturbances, sensory abnormalities, and sleep dysregulation are frequently comorbid and thus also represent important aspects of these conditions [10]. A better understanding of these comorbidities—including sleep dysregulation—may offer the opportunity to better understand and define the causes of ASD. Further, these comorbidities may also have potential as more objective and translationally-relevant endpoints in animal models used in the study of ASD, facilitating the development of effective interventions and therapeutics.
Sleep problems in ASD
Prevalence of sleep problems
Here we provide a brief overview of findings related to sleep disturbances in ASD, complementing more detailed reviews focused on findings in humans [11–14]. Sleep disturbances are more common in ASD than in the general population. A recent study of 522 children with ASD, 228 with ASD and other developmental delays, and 703 from the general population demonstrated that sleep problems are twice as prevalent in children with ASD (odds ratio: 2.12) or ASD and developmental delay (odds ratio: 2.37) [15]. This may be independent of environmental factors: in a study of siblings, children with ASD have higher rates of sleep problems (47%) than their typically developing (TD) sibling (16%) [16]. Such findings are broadly consistent with prevalence studies, which estimate sleep problems in 40–80% of children with ASD compared to 25–40% in TD children [11, 13]. The range of these estimates reflects the difficulty in determining an accurate prevalence of sleep problems in ASD, as factors including age, cognitive ability, and biological sex must be considered, all of which is further complicated by the lack of a clear consensus on the criteria and definitions of sleep disorders in pediatric populations [14]. Differences in methodology may also have an influence. Parental report is most commonly used to assess sleep in children and is the method of the majority of studies on children with ASD. However, actigraphy or polysomnography (PSG) studies—which offer more objective measures of sleep—are emerging and also suggest greater rates of sleep abnormalities in individuals with ASD compared to TD individuals [17]. Finally, considering that ASD is likely a cluster of disorders with multiple neurobiological etiologies, the prevalence and nature of sleep disorders may differ between these subtypes further adding variability. In spite of these complicating factors, there is a growing consensus that sleep disturbances are substantially more common in individuals with ASD.
Types of sleep problems
There is no singular form of sleep disruption in ASD. Symptoms of insomnia—difficulty initiating or maintaining sleep—are the most consistently reported sleep difficulty in ASD, including increases in sleep onset latency (time to fall asleep), poor sleep efficiency (proportion of time spent asleep while in bed), and less total sleep time [13]. Parental reports indicate that children with ASD are more likely to have bedtime resistance, increased awakenings, and decreases in the duration and continuity of sleep [18, 19]. Difficulties with sleep-onset insomnia (going to sleep) are more common than sleep-maintenance insomnia (staying asleep), although both frequently occur [11]. Parasomnias—undesirable physical or experiential events accompanying sleep—also occur at higher rates in ASD, according to some but not all studies [11, 20]. A PSG study of 23 children with ASD and 23 age-matched controls identified more frequent disorders of partial arousal, consistent with sleep terrors or confusional arousals, and more frequent spontaneous arousals [21]. Such findings are consistent with a growing number of studies in which actigraphy and PSG have been used to study sleep in individuals with ASD. While these studies are often restricted by smaller sample sizes, two recent meta-analyses have aggregated and summarized these findings [17, 22]. From studies involving actigraphy, an increase in sleep onset latency emerged as the sole significant parameter affected in ASD (5 studies, n = 276) [17]. From studies involving PSG, parameters significantly affected in ASD include a decrease in total sleep time (8 studies, n = 247), a reduction in sleep efficiency (7 studies, n = 238), an increase in sleep latency (7 studies, n = 211), an increase in wake time (7 studies, n = 211) and a decrease in REM sleep (9 studies, n = 273) [17]. Another meta-analysis of these PSG studies in ASD found significant differences in total sleep time (10 studies, mean decrease of 32.8 min in ASD), sleep latency (9 studies, mean increase of 10.9 min in ASD), and sleep efficiency (10 studies, mean decrease of 1.9%) [22]. Characterizing the nature of sleep problems in ASD has many of the same limitations as those seen with prevalence estimates, including small sample sizes, the heterogeneity of ASD, and differences in methodology. In addition, many of the commonly used survey instruments, such as the Children’s Sleep Habits Questionnaire (CSHQ), are designed primarily as screening tools to identify the presence of sleep problems, and have less power to discriminate between types of sleep problems [23].
A role for melatonin in ASD
Melatonin is an important regulator of the circadian sleep-wake rhythm, and there is evidence that dysregulation of this hormone might have a role in ASD-related sleep problems. Under normal physiological conditions in humans, plasma concentrations of melatonin follow a regular circadian pattern with low levels during the day and high levels at night [24]. The production of melatonin mainly occurs in the pineal gland, where release is entrained by ambient light via connections with the central circadian clock in the suprachiasmatic nucleus of the hypothalamus (SCN). The biosynthesis of melatonin occurs through an enzymatic process starting with the conversion of tryptophan to serotonin, which is then converted to melatonin through actions of two enzymes arylalkylamine-N-acetyltransferase (AA-NAT) and acetylserotonin O-methyltransferase (ASMT) [24]. Melatonin is of particular interest in ASD because of its role in neurodevelopment and sleep-wake rhythms, but also because abnormalities in the serotonin system are frequently identified in ASD [25]. The most consistent of these findings in ASD is a reduction in peripheral melatonin levels [26, 27]. The cause of this reduction in melatonin is thought to be an impairment of the serotonin-melatonin biosynthetic pathway, as there is evidence for reduced activity of the ASMT enzyme [27, 28]. Genetic variants within the promotor region of ASMT are more frequent in individuals with ASD and are associated with a decrease in the number of ASMT transcripts [27]. Consistent with these findings, it has been reported that 40% of those with ASD had abnormally high levels of serotonin in blood and 51% had abnormally low levels of melatonin, which would be expected if ASMT activity is reduced [28]. Additional evidence for a role for melatonin comes from the efficacy of supplemental melatonin in treating sleep problems in ASD, especially with improving sleep duration and sleep onset latency [29]. However, providing supplemental melatonin is not directly associated with changes in levels of melatonin, suggesting a more complex mechanism than simply replacing low levels of melatonin [30].
Relationships between sleep and behavioral dysregulation
The relationship between sleep dysregulation and behavioral dysregulation has received considerable attention in studies of ASD. In children and adolescents, sleep disturbances are associated with a wide variety of emotional and behavioral difficulties [31]. Numerous studies support the possibility that insufficient sleep exacerbates the behavioral symptoms of ASD [reviewed in ref. [13]]. In children with ASD, fewer hours of sleep per night predict greater severity of core ASD behaviors, such as deficits in social skills [32]. Additionally, higher levels of sleep problems are associated with higher levels of externalizing behavioral problems (e.g., aggression, impulsivity) and internalizing behavioral problems (e.g., withdrawal, anxiety, depression) [33]. Multivariate analyses suggest that sleep problems account for between 22–32% of the variance of behavioral problems in children with ASD [34]. While sleep problems are thought to worsen behavior, the inverse might also be true, with behavioral problems worsening sleep problems. Each of the core behavioral features of ASD may play roles: deficits in communication can interfere with social-related circadian cues, whereas difficulty understanding parental communication can interfere with putting children to bed, and repetitive and/or ritualistic behaviors can lead to difficulties with sleep when bedtime routine is disrupted [35]. Additionally, abnormal sensory functioning is common in ASD and over-responsivity to environmental stimuli and difficulties in regulating arousal can lead to problems with sleep-onset and insomnia [36]. In a population of hospitalized children with ASD, higher scores on the Aberrant Behavior Checklist-Community scale (ABC-C) (Irritability, Stereotypy, and Hyperactivity subscales) at admission were correlated with significantly fewer minutes slept during the last five nights of hospitalization [37]. These findings have led to the hypothesis that sleep and behavioral dysregulation may have a bidirectional relationship in ASD (Fig. 1, Condition B.). While this is a prevalent hypothesis, the vast majority of support is based on cross-sectional studies in which the relationship between sleep and behavior is only captured at one point in time, making both directionality and causality difficult to establish. A recent longitudinal study of sleep in 5151 children with ASD has challenged the assumption of a bidirectional relationship between sleep and autistic behavior [38]. Evaluating sleep and behavioral problems in children with ASD between 1.5 and 9 years of age revealed that even though higher levels of sleep problems are more likely to be associated with higher autistic traits, neither precedes or predicts the other over time [38]. These findings raise the possibility that sleep dysregulation may instead be a separate construct directly related to the underlying etiology of ASD (Fig. 1, Condition A).
Fig. 1.
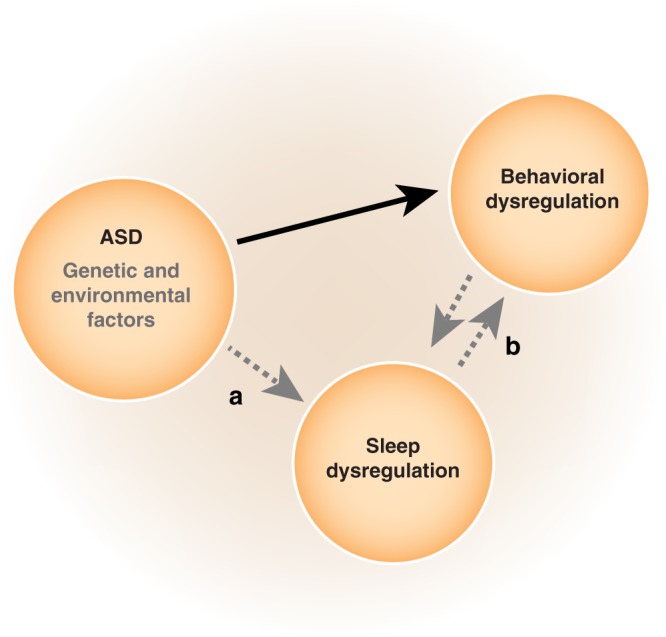
Schematic of hypothesized etiology of sleep dysregulation in ASD. Sleep dysregulation may stem directly from the underlying pathology of ASD (a) and/or indirectly as the consequence of a bidirectional relationship with behavioral dysregulation (b)
The cause of sleep problems in ASD
The cause of sleep problems in ASD is still unknown. Mazzone, Postorino [35] have proposed three possible etiological explanations for sleep problems in ASD. Possibility 1 is that sleep problems arise as a direct consequence of the underlying biology and genetics of ASD. This explanation would suggest that underlying changes in the neural process and circuits controlling the sleep-wake cycle is disrupted in ASD, for example, physiological changes in the melatonin, GABAergic circuits, or overall synaptic balance. Possibility 2 is that sleep problems arise from the clinical phenotype and core symptoms of ASD. As discussed above, several of the core behavioral features of ASD communication and social deficits and the presence of repetitive/stereotyped behaviors could directly lead to difficulties with sleep onset and insomnia. Possibility 3 is that sleep problems reflect a co-occurring condition that is completely independent from ASD and are related to psychiatric comorbidities. Numerous common psychiatric comorbidities of ASD such as attention deficit hyperactivity disorder (ADHD) or anxiety and mood disorders are also associated with sleep difficulties and as such sleep may be an intermediate phenotype of these conditions. Given the complex and heterogenous nature of ASD, the etiology is likely to be multifactorial and a single explanation is unlikely to be true for all individuals with ASD. Regardless, understanding the etiology of sleep problems in ASD is crucial as it can have direct implications for care and treatment. The growing understanding of the neurobiology of both ASD and sleep fueled by discoveries in animal models may present opportunities to begin to address these questions.
A role for animal models in the study of ASD
Types of animal models for ASD
Most fundamentally, animal models for ASD can be categorized as focusing on genetic (G) factors, environmental (E) factors, or G × E factors. Genetic models involve deletions or mutations that are associated with ASD—most commonly mice—but also in rats, drosophila, zebrafish, and recently non-human primates [39]. They often imitate monogenic syndromic ASD, which are forms of ASD due to chromosomal abnormalities or mutations that affect a single gene. These are powerful models due to being able to directly model a known cause for select forms of ASD (e.g., Fragile X syndrome), but are limited in scope as such conditions represent a small portion (~5%) of ASD prevalence [40]. Other genetic models alter the function of ASD associated risk genes or introduce identified copy number variations (CNVs) identified in humans with ASD, or use various inbred mice that exhibit behaviors resembling ASD. Explanations for the genetic basis of ASD are becoming increasingly complex, with more recent suggestions that the majority of genetic risk for ASD may be the result of numerous common genetic variants [2]. Each of these genes individually tend to have small effects, but impacts can be synergistic when combined. Unfortunately, modeling such a complex genetic landscape in laboratory animals is currently not possible and represents a substantial hurdle for genetic models used to study ASD.
In contrast, environmental models mimic identified risk factors for ASD during early development. Two of the most commonly used models are maternal exposure to valproic acid (e.g., refs [41–43]), which is based on the identified risk of valproate during fetal development and ASD, and maternal immune activation, which is based on epidemiological findings of viral and bacterial infection during pregnancy increasing the offspring’s risk of developing ASD (e.g., refs [44, 45]). A variety of other environmental risk factors have also been examined in the context of ASD such as exposure to diesel exhaust, maternal autoantibodies, and hypoxia [46–48]. A limitation of these approaches is that these epidemiological risk factors are often not unique to ASD; for instance, maternal inflammation has been associated with a wide range of psychiatric conditions including schizophrenia, bipolar disorder, and ADHD [49–52]. Furthermore, whereas certain genetic models reflect direct causal relationships between genes and illness that have been established in humans, in environmental models the causal links in humans are based on an associational studies, making assessment of construct validity (i.e., how accurately the model reflects the underlying biology) more complicated.
Uncovering the relationship between ASD and sleep
Animals models offer unique insights into the relationship between sleep dysregulation and ASD. Use of animal models enables exquisite control over numerous potentially confounding variables while utilizing objective measures of sleep (electroencephalography [EEG], electromyography [EMG], and actigraphy) that are analogous to human measurements. Whereas studies in humans are challenging because of the broad inclusiveness of the ASD diagnosis, animal models enable the study sleep in relation to known or hypothesized etiologies and pathophysiologies. By their very nature, animal models eliminate extraneous environmental factors that often complicate and/or confound studies in humans. For instance, impaired social ability in ASD might make parental interactions more difficult and complicate bedtime behaviors, whereas in animal models such concerns are mitigated as sleep can be studied in isolated subjects. Considering that there are many ways in which the core ASD behavioral phenotype (deficits in social behavior and communication and repetitive/stereotyped behaviors) might directly contribute to sleep dysregulation, animal models can offer key evidence for uncovering the relationship between the dysregulation of behavior and sleep in ASD. Findings of sleep dysregulation in animal models more directly support that sleep difficulties emerge as a construct (core feature) of ASD (Fig. 1, Condition A) rather than as a secondary consequence of behavioral dysregulation (Fig. 1, Condition B).
Sleep as a translational endpoint for understanding ASD
Studying sleep may provide opportunities to improve the animal models for ASD by enhancing their translational value. Modelling neuropsychiatric disorders in animals has been a significant challenge due to both the complexity of these conditions and the unique human attributes of many of the symptoms, as well as a lack of objective endpoints that are homologous across species. These problems are especially salient within the study of ASD, considering that recently there have been several prominent failures of drugs for Fragile X syndrome that were based heavily upon their initial and promising efficacy in genetic animal models of this condition [53]. Broad failures of drug candidates that were based on promising findings in animals models to translate to the human condition have contributed to a massive divestment of industry development of neuropsychiatric drugs and led to fundamental questions of the utility of animal models in studying psychiatric conditions [54]. Animal models for ASD often use classical behavioral tests related to core ASD symptoms [55]. For example, social behaviors are often assessed by examining the duration with which a mouse interacts with a novel age- and sex-matched partner mouse. Communication-related behaviors are often inferred from changes in the quantity of ultrasonic vocalizations. To test repetitive or stereotyped behaviors, the degree of self-grooming or performance on a rotarod (which can be enhanced) are often used as endpoints. However, these classical behavior tests have significant limitations. First, these tests are based on a face validity that is ultimately derived from anthropomorphic evidence as they require inferences of the cognitive and emotional states of an animal in a given situation. Second, they often only capture brief “snapshots” of time, with most tests occurring only over 5–10 min during a brief (and often arbitrary) portion of the day, missing any abnormalities that are not occurring during this period. Finally, they often lack tests or endpoints that are directly analogous in humans, adding more complexity to bench-to-bedside translation. There is increasing interest in using measures that overcome these limitations [56]. In this regard, studying sleep/wake physiology may have numerous advantages. Sleep is thought to be a critical biological process and is based on neurophysiological states that can be readily determined in both animals and humans using the same types of objective measures. Additionally, sleep and actigraphy studies are able to examine function over longer time frames, from days to weeks, instead of minutes in classical behavioral studies. Prospectively, the emergence of digital technologies and devices (e.g. fitness trackers, smartphones) are making studies of sleep/wake physiology more easily accessible and may provide new opportunities to align basic and clinical research [57].
Considerations in using animal models for sleep studies
To effectively study sleep in laboratory animals, one must consider the known similarities and differences of sleep across species. Most—if not all—vertebrate animals display the characteristic signs of sleep and there is evidence that sleep may serve an essential biological function [58, 59]. Many of the molecular mechanisms that subserve sleep are conserved between diverse species, from flies to humans [60]. Sleep can be identified through behavioral, physiological, and electrophysiological measures. In mammals and birds, sleep/wake states are often determined from EEG and EMG recordings. Sleep can be divided into rapid-eye-movement (REM) sleep (also referred to as paradoxical sleep [PS]) or non-rapid-eye-movement (NREM) sleep (also referred to as slow-wave sleep [SWS]). NREM sleep is characterized by low-frequency, high amplitude delta EEG oscillations, low muscle tone, and sleep spindles. In humans, NREM can be further divided into different stages. REM sleep is characterized by pronounced theta oscillations and a complete loss of muscle tone (muscle atonia). Wake is characterized by muscle activity and higher frequency EEG oscillations. EEG features can vary across species; for example, sleep involves only a single brain hemisphere in some species, whereas others lack the characteristic features of REM sleep [61]. Sleep can also be determined behaviorally, characterized by a quiescence of motor activity, elevated thresholds for arousal, and through homeostatic need [60]. There is variation in the daily duration, cyclical organization, and diurnal timing of sleep among different species and even different strains of inbred laboratory mice [62]. In contrast to humans, mice and rats predominantly sleep during the light phase and are awake during the dark phase. Sleep in humans is normally consolidated and monophasic (one daily sleep/wake cycle), whereas sleep in mice and rats is more fragmented and polyphasic (multiple daily sleep/wake cycles) [61]. As a result, sleep in animal models must always be considered in the context of the species being studied. However, despite these differences, the common and conserved features of sleep and the ways in which its architecture is defined across species makes it an endpoint of great potential relevance for translational studies.
Considerations in using animal models for ASD
Animal models are increasingly being generated to assess underlying mechanisms of ASD. However, numerous technical and interpretive issues may limit the reproducibility of findings in these models. With genetic models, the ways in which a particular mutation is created and maintained can be critical for accurate characterization of how a gene relates to a particular phenotype [reviewed in refs [63–65]. An unfortunately common issue relates to use of mixed genetic backgrounds, where a mutation is produced in one strain of mouse and bred to a second strain. The mixed genetic background of the progeny may inadvertently influence the phenotype if not properly controlled, as the chromosomal regions linked or flanked by the target gene will contain more of the genomic materials from the strain of the mutant founder. Several strategies to reduce this bias include using congenic mice in which backcrossing for 10 generations or more is performed, using co-isogenic mice in which the cells used to generate the mutant mouse originate from the same strain, or using an F1 hybrid in which the first generation cross is evaluated [64]. Additionally, it is becoming clear that environmental factors can have a strong influence on phenotypes of rodent models, such as rearing environment or the caging system used and should be carefully controlled and reported [66].
Incorporating sex differences
Identifying and understanding sex differences have recently gained considerable interest, driven by changing policies by funding agencies, an increasing awareness of their impact, and a growing consensus that continuing to neglect sex differences will be detrimental to brain- and health-related sciences [67]. ASD is a heavily sex-biased disease with a 4:1 male to female sex ratio in prevalence [68]. ASD may manifest differently within females, and there is a hypothesis that females may be protected against some symptoms of ASD—the so-called “female protective effect” [69, 70]. Alternatively, the “extreme male brain theory of autism” proposes that there are differences in male and female brains, and in ASD there is an extreme or hypermasculinized version of the male brain [71]. As such, sex differences have been influential in the conceptualization of ASD and may provide insights into the underlying mechanisms of ASD. Sex differences also exist within sleep. While large-scale studies are sparse, there are reports that women have greater total sleep time and have a higher percentage of slow-wave activity both at baseline and during recovery sleep following sleep deprivation [72]. Paradoxically, women also have higher rates of a wide range of sleep problems including poorer overall sleep quality, disrupted sleep, and insomnia [72]. Studies in rodents also indicate sex differences in sleep and suggest a crucial role for gonadal hormones. At baseline, female mice spend more time awake and decreased NREM compared to male mice, but following sleep deprivation show a more robust NREM recovery response [73]. The majority of these differences disappear following gonadectomy [73]. Furthermore, administration of estradiol alone or in combination with progesterone to rats promotes arousal by consolidating wake and fragmenting sleep in the dark phase, but not light phase [74]. These results are consistent with a well-established effect of the estrous cycle on sleep and activity, which in rats and mice follows a 4–5-day cycle consisting of multiple hormonal and behavioral phenomenon that often—but not always—coincide. The estrous cycle can be observed in the regular pattern of body temperature oscillations in female mice [45, 75]. Whereas male mice have consistent body temperature oscillations, female mice show a polyphasic profile (multiple peaks) during most of the cycle, but show a pronounced monophasic profile (one peak) during the middle of the dark phase at the start of estrus [75] (Fig. 2). Coinciding with this peak is a selective increase in locomotor activity 6–9 h following start of the dark phase. Both the increase in temperature and locomotor activity are dependent on gonadal hormones, and are lost following ovariectomy [75]. While readily modulated by environmental factors, typically this period corresponds to behavioral estrus (spontaneous activity and sexual receptivity) that is followed shortly by the start of ovulation in the same night [76]. Elevations in locomotor activity, body temperature, and sexual receptivity are inherent to the term “estrus”, which is derived from the Greek word for “gadfly” or “frenzy”; indeed, a colloquial (and increasingly anachronistic) euphemism for this period is “in heat” [77]. Coinciding with these alterations, during the dark phase of proestrus rats show significantly less NREM and REM sleep and spend greater time awake [78, 79]. Whether or not humans with a menstrual cycle have a corresponding behavioral estrus period is debated [80]; thorough investigation of sex differences may require examination of potential differences both across time-of-day and stage of cycle. In a mouse model of prenatal immune system activation, we recently identified a phenotype that is “silent” in females except within the small portion of the dark phase where the activity is increased during estrus [45]. While identifying sex-related differences in sleep behavior may require complex experimental designs, understanding the factors that contribute to them may enable unique new insights on the pathophysiology of ASD.
Fig. 2.

Sex differences and influence of body temperature and locomotor activity. Males have regular monophasic temperature oscillations, seen on example recordings from eight days (a), and averaged temperatures (b), that correspond to rhythms of locomotor activity (c). Due to the estrous cycle females have a four-day pattern of polyphasic oscillations during anestrus and a monophasic peak (E) that readily distinguishes the night of estrus (d, e). Locomotor activity is also selectively increased in estrus during the middle of the dark phase (f). Data from 5–6 C57BL/6J mice. **p < 0.01
Sleep in animal models for ASD
Environmental models for ASD
Prenatal valproic acid (VPA) exposure
Valproate is a medication used for the treatment of epilepsy and bipolar disorder. However, it has been discovered that prenatal (in utero) exposure to valproate significantly increases the risk of developing ASD [81]. The mechanism of these effects is not understood, but may be related to general toxicities of the drug or its actions as a histone deacetylase inhibitor. In rodent (rat or mouse) studies, prenatal valproic acid (VPA) exposure induces numerous sex-dependent behavioral abnormalities later in life that resemble core features of ASD [41–43]. Prenatal VPA exposure also induces sleep dysregulation. For example, rats treated prenatally with VPA exhibit frequent arousals during the light/sleep phase, as measured by increased levels of locomotor activity and increased bouts of feeding [82]. Consistent with these findings, another report indicated that prenatal VPA-treated rats spend more time in wake and less in NREM sleep during the light phase [83]. These changes coincide with an increase in high-frequency EEG spectral power during wake and REM sleep, and a decrease in theta power across all vigilance states [83]. Two potential mechanisms have been proposed for these alterations in sleep. First, prenatal VPA treatment increases basal levels of serotonin in the brain [82], which is consistent with findings of dysfunction in the serotonin-melatonin biosynthetic pathway in ASD. Second, prenatal VPA decreases cortical levels of GAD65 and GAD67—markers of GABAergic synapses—suggesting potential disruption of GABAergic signaling that might lead to imbalances in overall inhibitory tone [83]. These possibilities are not mutually exclusive: either mechanism, or both, could be contributing to the effects of VPA on sleep.
Prenatal immune activation
There is accumulating evidence for a potential immune involvement in ASD [84]. There are epidemiological associations between various forms of inflammation during pregnancy—in the form of bacterial or viral infection, or a parental autoimmune disorder—with an increased risk for the offspring to develop ASD [49, 85, 86]. Prenatal (or maternal) immune activation models have been developed where immune-stimulating agents are administered to animals (mice, rats, monkeys) during pregnancy to assess the consequences to neurodevelopment of the offspring. Across these species, maternal immune activation can lead to disruptions in behaviors that resemble aspects of both ASD and schizophrenia [87, 88]. Two studies have included evaluations of sleep in these models. In one study, lipopolysaccharides (LPS) administered near the end of gestation in rats led to no significant changes in sleep at baseline or following an acute stressor [89]. In contrast, a study from our group examined sleep in a “two-hit” immune activation model in mice, where polyinosinic-polycytidylic acid (Poly I:C) is administered during mid-gestation and a second “hit” (LPS) is administered on postnatal day 9 (roughly equivalent to the end of the third trimester in humans based on neurodevelopmental milestones) [90]. This regimen produces offspring with reductions in social behavior and increases in anxiety-like behavior, changes in synaptic strength of prefrontal to amygdala neural circuits and a persistent increase in microglia and neuroinflammatory markers in the brain [44]. The combination of prenatal and early postnatal immune activation also altered sleep with a selective increase in SWS, a shift in EEG spectral power, and spontaneous epileptiform activity (spike-wave discharges) [90]. Effects on sleep were strongest in mice that received the combined treatment; however, qualitatively similar effects were seen in mice that received only the postnatal (second) “hit”, whereas prenatal Poly I:C had no effects. These studies raise the possibility that either the timing of the immune system activation and/or the specific receptor target of LPS (in this case, toll-like receptor-4 [TLR4]) may play critical roles in regulating the effects on sleep. Many of these findings resemble the effects in the first several hours following acute administration of cytokines or LPS either peripherally or directly into the brain of rodents [91, 92]. As such, alterations in sleep in this model may reflect a state of elevated inflammatory signaling in the brain. The two different findings with prenatal Poly I:C and postnatal LPS are not surprising as animal models of perinatal immune activation have often had variable results. Emerging research is suggesting that environmental factors, the gut microbiome, the strain and vendor of mice, and the timing and type of immune activation used can all lead to highly divergent phenotypes [45, 66, 93].
Genetic models for ASD
Fragile X syndrome (FXS)
Fragile X syndrome (FXS) is the most common heritable cause of intellectual disability and one of the most thoroughly studied of the monogenic syndromes associated with ASD [94]. In FXS, the fragile X mental retardation gene (FMR1) is silenced, which leads to nonfunctional fragile X mental retardation protein (FMRP), an RNA-binding protein involved in the development of synaptic connections [95]. There are reports of sleep dysfunction in FXS, with parental reports indicating that 32% of children with the condition have sleep difficulties [96]. Additionally, a PSG study indicates reduced time in bed, lower sleep efficiency, and decreased NREM sleep [97]. Sleep and circadian disturbances are found in multiple models of FXS. Deletions of the Drosophila homolog to FMR1 (dFmr1) leads to arrhythmic locomotor activity in conditions of constant darkness (DD), but not under normal light:dark conditions (LD), whereas overexpression of dFmr1 leads to a lengthened circadian period implicating the dFmr1 gene in regulation of circadian rhythms [98, 99]. Furthermore, dFmr1 directly regulates daily sleep need with a loss of dFmr1 increasing sleep duration and overexpression of dFmr1 decreasing sleep duration [100]. In mammalian species, models are more complicated as two paralogs to Fmr1 exist—Fxr1 and Fxr2—that can compensate for loss of Fmr1. In mice, loss of either Fmr1 or Fxr2 decreases activity rhythms in DD conditions only, but combined deletion of Fmr1 and Fxr2 decreases activity rhythms in DD and LD conditions [101]. Interestingly, sleep disruption may follow a developmental time-course, as Fmr1 deletion leads to significant decreases in sleep during the light phase at later stages in development (postnatal day 70 and 180) but not at early stages (postnatal day 21), and sleep loss is worsened by incorporating the heterozygous deletion of Fxr2 [102]. While the mechanisms of these effects are unknown, the Fmr1 gene may directly modulate the cortical networks that regulate sleep. In vivo Ca2+ imaging in mice lacking Fmr1 revealed hypersynchronous neural activity in cerebral cortex, with increased neuronal involvement during the neuronal “up states” that occur every 1–2 s and are characteristic of SWS and quiet wakefulness [103]. Dysregulation of melatonin secretion is another potential mechanism, as chronic melatonin treatment is capable of reducing the learning deficits and enhanced anxiety-like behaviors of mice lacking Fmr1 [104].
Rett syndrome (RTT)
Rett syndrome (RTT) is an X-linked neurological disorder caused by mutations in the MECP2 gene encoding for methyl-CpG-binding protein 2 (MeCP2) [105]. RTT syndrome occurs almost exclusively in females, and individuals with RTT often meet criteria for ASD based on behavioral features during childhood [106]. RTT was historically classified as an ASD, but was removed from Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM 5) due to its known etiology reclassifying it as a medical condition [107]. Sleep problems occur in more than 80% of patients with RTT, with daytime napping, night-time laughing, teeth grinding, night screaming, and seizures being the most common problems [108, 109]. Animal models of RTT involve different strategies to alter MeCP2 expression, such as conditional deletion, cell-type-specific deletion, or introduction of various mutations in Mecp2, which can all recreate many of the behavioral ASD-like features [110–112]. Sleep disruption is found in many of these models. Mecp2-deficient mice have substantially reduced locomotor activity levels, highly fragmented wakefulness, and alterations in the timing but not quantity of sleep [113–115]. Additionally, the frequency of hippocampal theta rhythms is decreased and there is spontaneous abnormal rhythmic EEG cortical activity in these mice [116]. The advent of new gene-editing technologies (e.g., CRISPR-Cas9) has enabled the development of genetic models of syndromic ASD in non-human primates [39]. Expressing loss-of-function mutations within the human MeCP2 gene in cynomolgus monkeys creates a model that strongly resembles RTT on multiple behavioral and physiological measures [117]. For example, eye-tracking of MeCP2-deficient monkeys suggests that they show less interest in emotionally charged monkey faces, but prefer neutral monkey faces as compared to wildtype monkeys. This model also exhibits altered sleep patterns. Using video recording and experimenter rating, MeCP2-deficient monkeys spend significantly more time awake and have more fragmented sleep, showing more bouts of wake and sleep in comparison to wildtype monkeys [117]. Studies in drosophila also demonstrate that cell-type-specific changes in MeCP2 expression can affect sleep. Expression of human MeCP2 in astrocytes leads to a reduction in total sleep time, whereas expression in octopamine (homolog of mammalian noradrenaline) neurons causes phase-specific sleep deficits, increases in sleep latency, and sleep fragmentation [118, 119]. Mechanistically, MeCP2—which functionally is involved in DNA methylation—can directly modulate the central circadian system. MeCP2 is expressed in the SCN, where it becomes phosphorylated following light stimulation [120] and transcriptionally activates the circadian clock genes Per1 and Per2 [121]. SCN neurons of Mecp2-deficient mice have reduced spontaneous firing rates and decreased rhythmic expression of PER2 [113, 114].
Prader-Willi syndrome (PWS)
Prader-Willi syndrome (PWS)—together with Angelman syndrome (AS) (see below)—is a disorder caused by imprinting deficits in chromosomal region 15q11-q13. The 15q11-q13 region contains (1) a set of non-imprinted genes, (2) imprinted genes that are paternally expressed only including the bistronic SNURF-SNRPN gene, (3) preferentially maternally expressed genes, including the E3 ubiquitin ligase (UBE3A) gene, and (4) non-imprinted genes with paternally biased expression [122]. AS is due to loss of expression from the maternally inherited portion of this region, with alterations in UBE3A being critical, whereas PWS is due to loss expression of paternally inherited genes in this region. PWS is associated with excessive appetite with a gradual development of obesity, short stature, intellectual disability, and behavioral problems [122]. In those with PWS due to uniparental disomy, an estimated 35% met criteria for ASD, and in those with PWS due to deletion, 18% met criteria for ASD [123]. In an elegant cross-species comparison, Lassi, Priano [124] examined the role of the PWS gene candidate small nucleolar ribonucleic acid-116 (SNORD116) comparing sleep in paternal heterozygous deletion of Snord116 in mice, with sleep in two human PWS analogous cases characterized by deletions of SNORD116. Mutant mice had increased REM and decreased NREM, with an increase in the number of REM sleep episodes, and specific increases in EEG theta power during REM. The individuals with PWS with SNORD116 deletions had remarkably similar changes with increased REM and decreased NREM, an increased number of REM sleep episodes, REM sleep intrusions, and a selective increase in theta power during REM [124].
Angelman syndrome (AS)
Angelman syndrome (AS) is a due to the disruption of the maternally inherited UBE3A gene. AS typically presents with motor dysfunction, severe intellectual disability, seizures, significant language impairment, happy demeanor, and is commonly comorbid with ASD [125]. Besides the loss-of-function in AS, multiple mutations of UBE3A are associated with ASD including duplications, triplications, and other gain-of-function mutations [126]. Sleep problems are common in AS with reductions in total sleep time, increases in sleep onset latency, disrupted sleep architecture, and reductions in REM sleep [127, 128]. Maternal-specific deletion of Ube3a in mice increases the number, but not duration, of SWS and wake episodes, decreases the number and duration of REM episodes, and leads to characteristic paroxysmal EEG discharges [129]. Furthermore, loss of maternal Ube3a drastically disrupts sleep homeostasis. Following 4 h of sleep restriction, these mutant mice fail to show a rebound increase in SWS duration or an increase in delta power, as well as a smaller increase in REM sleep [129, 130]. In one study, maternal Ube3a deletion altered circadian rhythms, causing disruptions in circadian locomotor activity patterns in free-running conditions and in response to entrainment, as well as circadian changes in metabolic homeostasis and functional circadian periodicity that are restored in neurons by unsilencing of the paternal Ube3a gene [131]. However, in another study circadian rhythms were mostly intact, although maternal Ube3a deletion eliminated the siesta (timed rest period) during the dark phase, which may be the consequence of an inability to accumulate sleep pressure [130]. The mechanisms for sleep dysregulation in AS could be diverse, since UBE3A is a ubiquitin ligase involved in targeting a wide range of proteins for degradation. UBE3A is important in synaptic plasticity, and deficits in Ube3a impairs experience-dependent maturation of cortical circuits and Ube3a ubiquitination of the protein Arc is critical for synaptic plasticity [132, 133].
Tuberous sclerosis complex (TSC)
Tuberous sclerosis complex (TSC) is multiple organ system disorder caused by a loss of function in one of two genes: TSC1 or TSC2. The clinical features of the disease include tumors in multiple organs, seizures, cognitive disability, and ASD [134]. TSC1 and TSC2 form a protein complex that acts as an inhibitor of the mechanistic target of rapamycin (mTOR), a key cell growth and regulation pathway that regulates the translation of thousands of genes [135]. Disruptions of sleep and circadian rhythm are frequently reported in TSC patients including early arousals, sleep-maintenance insomnia, and fragmented sleep architecture especially in the presence of seizures [136, 137]. In Drosophila, heterozygous loss of homologs to TSC1 or TSC2 results in significantly weaker circadian rhythms and longer circadian periods [138]. Recently, a conditional deletion of TSC1/2 mouse model revealed that a direct interaction of the TSC protein complex with the central clock component BMAL1 could be the mechanism explaining these circadian abnormalities [139]. Deletion of TSC1 or TSC2 in post-mitotic neurons led to abnormalities in circadian rhythms with a loss of rhythmicity and shorter circadian periods under DD conditions, but were capable of entrainment on LD conditions. The mTOR inhibitor rapamycin—which is currently under clinical trials for treatment of TSC—was able to normalize the shortened period phenotype. Importantly, mTOR can regulate the proteostasis of BMAL1, and BMAL1 function is increased when Tsc gene function is lost. By genetically lowering BMAL1 expression, the abnormal circadian phenotype of Tsc2 deficient mice can be rescued [139].
Smith–Magenis syndrome (SMS) and Potocki–Lupski syndrome (PTLS)
Smith–Magenis syndrome (SMS) and Potocki–Lupski syndrome (PTLS) are neurodevelopmental conditions caused by either deletion of chromosomal region 17p11.2 (in SMS) or duplication of 17p11.2 (in PTLS) [140]. While not typically considered ASDs, both frequently involve ASD behavioral features, with ~90% of individuals with SMS and ~80% of individuals with PTLS meeting the criteria for ASD [141, 142]. Aberrant function of the Retinoic Acid Inducible 1 (RAI1) gene, which is within the genomic interval for both SMS and PTLS, seems critical to both of these disorders: point-mutations within RAI1 in SMS recapitulate almost all of the phenotype of larger deletions [143] and RAI1 is the smallest overlapping genomic region in PTLS duplications [144]. SMS and to a lesser extent PTLS are characterized by severe sleep disruptions and circadian abnormalities [141, 145]. Strikingly, the circadian rhythm of melatonin secretion is essentially inverted in SMS [146]. Mouse models of these conditions recapitulate many of the phenotypes of these disorders, including changes in body weight (increased in SMS and decreased in PTLS), altered activity (hypoactivity in SMS and hyperactivity in PTLS), learning and memory deficits, disrupted social behavior, and abnormal circadian period [142, 147]. Rai1 deletion in specific neuronal populations is sufficient to recapitulate learning deficits and obesity characteristic of the human conditions [148]. Together, these disorders represent an example of two neurodevelopmental disorders that have severe disruptions in sleep and circadian rhythms that are also present with ASD, raising the possibility of common pathophysiologies.
Timothy syndrome
Timothy syndrome is a neurodevelopmental condition caused by mutations in the CACNAC1C gene encoding for Cav1.2, that lead to multiorgan dysfunction, including lethal heart arrhythmias, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and ASD in ~80% of cases [149]. The most common mutation, a G406R missense mutation, alters the negative feedback mechanisms of voltage and calcium-dependent inactivation in L-type calcium channels [150]. As yet, sleep has not been in evaluated in an animal model that fully recapitulates the type of alteration in L-type channels of this mutation. Nonetheless, heterozygous deletion of Cacnac1c in mice suggest its involvement, as this results in normal total sleep amounts per day but more sleep in the dark period, a reduction in REM sleep recovery following sleep deprivation or restraint stress, and decreased EEG gamma power compared to wildtype mice [151].
Cortical dysplasia-focal epilepsy syndrome (CDFE)
First described in an Amish family in 2006, Cortical dysplasia-focal epilepsy syndrome (CDFE) is characterized by epileptic seizures, language regression, hyperactivity, aggressive behavior, and intellectually disability, of which ~66% of cases meet the criteria for ASD [152]. In this family a recessive mutation was identified within the gene connactin associated protein-like 2 (CNTNAP2) also known as (CASPR2), that encodes for a protein within the neurexin family [152, 153]. Numerous groups have now demonstrated that common and rare variants in CNTNAP2 conferred risk to both ASD and ASD-related endophenotypes including language delay [153, 154]. In mice, deletion of Cntnap2 produces an ASD-like phenotype, including deficits in social interaction, communication-like behavior, and repetitive behaviors-which are mitigated by risperidone treatment—as well as hyperactivity and epileptic seizures [155]. Abnormal sleep-wake physiology was recently described following the deletion of Cntnap2 in both mice and rats, although there were important species differences [156]. In mice, deletion of Cntnap2 induces absence seizure-like events, decreases in activity, and wake fragmentation. In rats, deletion of Cntnap2 induces severe motor seizures, increases in activity, and a consolidation of wakefulness. In both species, Cntnap2 deletion reduces spectral power in the alpha range during wake.
16p11.2 hemideletion
Chromosomal copy number variations (CNVs) are associated with ASD [157], including hemideletion of the chromosomal region 16p11.2, which greatly increases the risk for both ASD and ADHD [158]. Mice with 16p11.2 hemideletion have numerous behavioral phenotypes reminiscent of the human condition, with alterations in brain structure coinciding with cognitive and communication-related behavioral deficits [159, 160]. Angelakos, Watson [161] characterized sleep and circadian alterations in mice with 16p11.2 hemideletion. In both male and female mice, 16p11.2 hemideletion leads to robust hyperactivity, with no overt changes in circadian rhythm. In males, but not females time awake is increased while NREM is decreased. Furthermore, male 16p11.2 hemideletion mice have a greater proportion of prolonged bouts of wakefulness [161].
Neuroligins (NLG)
Neuroligins (NLG) are a family of cell adhesion molecules found on the surface of the postsynaptic membrane that facilitate the connection of neurons at synapses [162]. Some studies have identified rare single gene variations of NLGs in ASD [163, 164], whereas other studies failed to demonstrate an association [165]. There are five identified members of the neuroligin family in humans and four in rodents [166]. The distribution of the NLGs is isoform specific, with NLG1 being found predominantly at excitatory synapses, NLG2 exclusively on inhibitory synapses, and NLG3 at both excitatory and inhibitory synapses [166]. Nlg1 deletion in mice causes deficits in spatial learning and memory, along with repetitive, stereotyped grooming behavior—a potentially autism-related abnormality [167]. Several of the NLGs may be involved in sleep-wake regulation. Nlg1 deletion in mice decreases wake and increases NREM sleep [168]. Additionally, sleep deprivation can either increase or decrease expression of Nlg1 depending on the specific variant [168]. Nlg4 deletion in drosophila leads to longer sleep onset and less total sleep time and impairs GABAergic synaptic transmission [169]. The NLG3R341C point mutation is found in rare cases in ASD [163]. Introducing the Nlg3R451C mutation into mice leads to impaired social interaction, enhanced spatial learning abilities, and an increase in inhibitory synaptic transmission, effects that do not occur following complete deletion of Nlg3 [170]. Nlg3R451C mice have a normal duration and distribution of sleep states, but have differences in their EEG power spectrum, with increases in sigma and beta power during wake and decreases in delta power during NREM sleep [171].
Phosphatase and tensin homolog (PTEN)
Cowden Syndrome and several related disorders caused by mutations in phosphatase and tensin homolog (PTEN) are associated with multiple tumor-like growths, macroencephaly, and neurobehavioral features that often meet criteria for ASD [172, 173]. Conditional deletions of Pten in the CNS of mice can mimic behavioral traits of several of the phenotypes of ASD [174, 175]. Results suggesting circadian abnormalities in these mice have been mixed and may be dependent on the type of deletion. For example, conditional deletion of Pten in cortical and hippocampal neurons results in spontaneous epileptiform activity and a constant, lengthened circadian free-running period [176]. However, while haploinsufficiency of Pten reproduces many of the behavioral features of ASD in mice, including sex-specific changes in locomotor activity, no changes in the length of the free-running period have been observed [175].
Synthesis and future directions
There is now substantial evidence for a diverse array of sleep abnormalities in animal models for ASD. In general, animal models for ASD exhibit some form of alteration in sleep or circadian rhythm (Table 1). The diversity of these changes is not surprising given the heterogeneity of ASD, but in the few instances when specific models are compared to the human condition of the same etiology, the types of alterations observed are consistent across species. These findings support the concept that sleep dysfunction is inherent to many forms of ASD—a frequently co-occuring medical condition rather than a diagnostic criterion. Additionally, findings in animal models suggest that sleep problems derive from the underlying neurobiological pathophysiology of ASD, as opposed to being a non-specific (secondary) biproduct of behavioral problems.
Table 1.
Summary of studies of sleep in animal models for ASD
| Study | Relevant human condition | Manipulation | Species | Sleep and circadian-related phenotypes |
|---|---|---|---|---|
| Tsujino et al, 2007 | ASD | Prenatal VPA | Rat | More frequent arousals during the light phase |
| Cusmano and Mong 2014 | Prenatal VPA | Rat | Increased wake and decreased NREM during the light phase | |
| Zager et al. 2014 | ASD and SCZ | Prenatal Immune Activation | Rat | No changes in sleep at baseline or following stressor |
| Missig et al. 2018 | Perinatal Immune Activation | Mouse | Increased SWS and epileptiform activity | |
| Dockendorff et al. 2002 | Fragile X syndrome | dFMR1 loss-of-function | Drosophila | Arrhythmic circadian activity and erratic locomotor activity |
| Inoue et al. 2002 | dFMR1 loss-of-function | Drosophila | Arrhythmic circadian activity in DD | |
| Zhang et al 2008 | FMR1 and FXR2 deletion | Mouse | Shortened circadian period in DD and decreased rhythmicity in LD | |
| Bushey et al. 2009 | dFMR1 deletion | Drosophila | Deletion of dFMR1 increased- and overexpression decreased sleep time | |
| Wither et al. 2012 | Rett syndrome | MeCP2 deletion | Mouse | Disrupted circadian body temperature and activity rhythms |
| Tsuchiya et al. 2015 | MeCP2 deletion | Mouse | Decreased circadian rhythms in DD and altered central clock gene expression | |
| Li et al. 2015 | MeCP2 deletion | Mouse | Fragmented sleep, decreased strength and precision of circadian rhythms | |
| Chen et al. 2017 | MeCP2 deletion | Cynomolgus Monkey | Increased wake and fragmented sleep | |
| Lassi et al. 2016 | Prader-Willi syndrome | SNORD116 deletion | Mouse | Increased REM and decreased NREM duration, and increased REM episodes |
| Colas et al. 2005 | Angelman syndrome | Ube3a deletion | Mouse | Increased number of SWS and wake episodes |
| Shi et al. 2015 | Ube3a deletion | Mouse | Weakened circadian rhythms and disrupted metabolic homeostasis | |
| Ehlen et al. 2015 | Ube3a deletion | Mouse | Blunted recovery following sleep deprivation | |
| Zheng and Sehgal 2010 | Tuberous Sclerosis Complex | TSC1/TSC2 deletion | Drosophila | Weaker circadian rhythms and longer circadian period |
| Lipton et al. 2017 | TSC1/TSC2 deletion in neurons | Mouse | Loss of rhythmicity and shorter circadian period in DD | |
| Kumar et al. 2015 | Timothy syndrome | Cacna1c (Cav1.2) deletion | Mouse | Reduced REM sleep recovery |
| Thomas et al. 2017 | CDFE | Cntnap2 deletion | Mouse and Rat |
Rats: hyperactivity and increased consolidation of wake and REM, Mice: hypoactivity and fragmented wake |
| Angelakos et al. 2016 | ASD and ADHD | 16p11.2 −/+deletion | Mouse | Hyperactivity, increased and consolidated wake in males |
| El Helou et al. 2013 | ASD | NLG1 deletion | Mouse | Decreased wake and increased NREM |
| Li et al. 2013 | ASD | NLG4 deletion | Drosophila | Delayed sleep onset and decreased sleep time |
ADHD attention deficit hyperactivity disorder, ASD autism spectrum disorder, CDFE cortical dysplasia-focal epilepsy syndrome, DD dark/dark conditions or constant darkness, LD light/dark conditions, NREM non-rapid eye movement sleep, REM rapid eye movement sleep, SCZ schizophrenia, SWS slow-wave sleep
One intriguing possibility is that sleep dysregulation in ASD may reflect imbalances in synaptic homeostasis. In ASD, a dysregulation in synaptic homeostasis has been proposed as one of the central pathophysiological mechanisms [177]. Many of the monogenic syndromes can be considered “developmental synaptopathies” and converge on processes critical to synaptic signaling, plasticity, and development [178]. As examples, FXS, TSC, and AS are all characterized as disorders of transcriptional regulation, local translation, and protein quality control [178]. Many of the risk genes for ASD identified in genome-wide association studies are involved in synaptic plasticity, synthesis, or degradation, and transcriptome analysis from post-mortem brain tissue from ASD suggest a dysregulation cluster of synaptic and neurotransmitter signaling genes [177, 179]. Concurrently, there is substantial evidence that sleep may be crucial for synaptic homeostasis [180]. The highly influential sleep homeostasis hypothesis (SHY) proposes that the function of sleep is to regulate synaptic strength [181]. SHY suggests that during wake there is a net increase in overall synaptic strength, and that sleep globally downscales synaptic strength to restore cellular homeostasis [182]. While phenomena potentially contrary to SHY have been observed, such as synaptic strengthening during sleep, the crucial role of sleep in synaptic homeostasis has consistently been supported [183, 184]. Together, this may suggest that in ASD an intrinsic imbalance in synaptic homeostasis may manifest in abnormalities in sleep. This may suggest that altered sleep would be a direct consequence of an underlying pathophysiology of ASD. As such, measurements of sleep would provide an accessible neurophysiological metric of impairment in ASD that may be an ideal target in mechanistic studies aiming to treat or reverse the condition.
While a substantial body of work indicates that sleep is disrupted in ASD and in animal models for ASD, how this relationship changes over development has been far less studied. Sleep is highly developmentally regulated, changing dramatically during brain development, and the coincident maturation of sleep and synaptic physiology has been speculated to play a role in sleep disruption in neurodevelopmental disorders [reviewed in ref.: [185]]. In one of the few longitudinal studies, children with ASD followed a different trajectory of sleep problems compared to TD children. In TD children, sleep problems tended to decrease over time, whereas in children with ASD sleep problems worsened [38]. Additionally, alterations in sleep may have predictive value for the development of ASD. In one study infants with more sleep problems by 12 months had an increase in ASD symptoms a year later [186]. Future studies are needed to understand how the relationship between sleep and ASD evolves over the course of development and to determine whether altered sleep could be a factor that contributes to the pathophysiology ASD. Parallel studies and consistent results in animal models would increase confidence in the translational relevance of sleep-related endpoints. Furthermore, while not discussed in detail in this review, robust electrophysiological differences often exist in humans with ASD and animal models for ASD, including changes in baseline levels of spectral power and the presence of epileptiform activity [90, 187–189]. While these types of physiological measures have features that could make them valuable as biomarkers of ASD, alterations in baselines can complicate the interpretation of any putative changes in sleep architecture. Additionally, the majority of studies to date have focused on establishing that sleep differences exist while thus far offering limited mechanistic insights. With the advent of new methods (e.g., wireless EEG) in neuroscience there have been considerable advances in the understanding of the underlying neural circuits regulating sleep/wake physiology [190, 191] under increasingly ethologically-relevant conditions (e.g., freely moving/untethered) and time periods (e.g., weeks to months). How these specific neural circuits are affected in ASD is still unknown, but many of the technologies now exist that have the ability to uncover the mechanisms governing sleep disruptions in ASD.
Conclusions
Sleep is commonly disrupted in ASD. Animal models of ASD frequently recapitulate these sleep disruptions, suggesting that altered sleep/wake physiology appear to be well preserved across species. Studies in laboratory animals also support the interpretation that sleep disruptions likely derive directly from the underlying pathophysiology of ASD, as opposed to reflecting non-specific signs of more generalized behavioral problems. Beyond these implications, examining sleep may have particular utility in animal models for ASD in translating to humans. Sleep has several advantages compared to classical behavioral approaches, including being highly objective, having common definitions and analogous metrics that can be measured across species, and being conducive to collection of continuous data sets over long periods of time. As theories of ASD pathophysiology and the function of sleep become more refined and focused on key roles for synaptic homeostasis, sleep may offer a more direct measure of altered neurophysiology in ASD compared to traditional behavioral tests in humans or lab animals. As such, the study of sleep in animal models for ASD may enable fundamentally new insights on the condition and strategies for more effective therapeutics.
Funding and disclosure
Authorship of this article was supported by awards from the Robert and Donna Landreth Family Fund (to C.J.M., W.A.C.), the Teamsters Local 25 Autism Fund (to W.A.C.), MH063266 (to W.A.C.), the Andrew P. Merrill Memorial Research Fellowship (to G.M.), and the Brain & Behavior Research Foundation (to G.M.). Within the past 2 years, W.A.C. has served as a paid consultant for Psy Therapeutics. The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xu G, Strathearn L, Liu B, Bao W. Prevalence of autism spectrum disorder among US children and adolescents, 2014–2016. JAMA. 2018;319:81–82. doi: 10.1001/jama.2017.17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–5. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The Heritability of Autism Spectrum Disorder. JAMA. 2017;318:1182–4. doi: 10.1001/jama.2017.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism. 2017;8:13. doi: 10.1186/s13229-017-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson CP, Myers SM, American Academy of Pediatrics Council on Children With Disabilities. identification and evaluation of children with autism spectrum disorders. Pediatrics. 2007;120:1183–215. doi: 10.1542/peds.2007-2361. [DOI] [PubMed] [Google Scholar]
- 7.Jones W, Klin A. Attention to eyes is present but in decline in 2-6-month-old infants later diagnosed with autism. Nature. 2013;504:427–31. doi: 10.1038/nature12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esposito Gianluca, Hiroi Noboru, Scattoni Maria Luisa. Cry, Baby, Cry: Expression of Distress As a Biomarker and Modulator in Autism Spectrum Disorder. International Journal of Neuropsychopharmacology. 2017;20(6):498–503. doi: 10.1093/ijnp/pyx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosl WJ, Tager-Flusberg H, Nelson CA. EEG analytics for early detection of autism spectrum disorder: A data-driven approach. Sci Rep. 2018;8:6828. doi: 10.1038/s41598-018-24318-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argyropoulos A, Gilby KL, Hill-Yardin EL. Studying autism in rodent models: reconciling endophenotypes with comorbidities. Front Hum Neurosci. 2013;7:417. doi: 10.3389/fnhum.2013.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds AM, Malow BA. Sleep and autism spectrum disorders. Pedia Clin North Am. 2011;58:685–98. doi: 10.1016/j.pcl.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Devnani PA, Hegde AU. Autism and sleep disorders. J Pedia Neurosci. 2015;10:304–7. doi: 10.4103/1817-1745.174438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen S, Conduit R, Lockley SW, Rajaratnam SM, Cornish KM. The relationship between sleep and behavior in autism spectrum disorder (ASD): a review. J Neurodev Disord. 2014;6:44. doi: 10.1186/1866-1955-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richdale AL, Schreck KA. Sleep problems in autism spectrum disorders: prevalence, nature, & possible biopsychosocial aetiologies. Sleep Med Rev. 2009;13:403–11. doi: 10.1016/j.smrv.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds Ann M., Soke Gnakub N., Sabourin Katherine R., Hepburn Susan, Katz Terry, Wiggins Lisa D., Schieve Laura A., Levy Susan E. Sleep Problems in 2- to 5-Year-Olds With Autism Spectrum Disorder and Other Developmental Delays. Pediatrics. 2019;143(3):e20180492. doi: 10.1542/peds.2018-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park Subin, Cho Soo-Churl, Cho In Hee, Kim Boong-Nyun, Kim Jae-Won, Shin Min-Sup, Chung Un-Sun, Park Tae-Won, Son Jung-Woo, Yoo Hee Jeong. Sleep problems and their correlates and comorbid psychopathology of children with autism spectrum disorders. Research in Autism Spectrum Disorders. 2012;6(3):1068–1072. [Google Scholar]
- 17.Diaz-Roman A, Zhang J, Delorme R, Beggiato A, Cortese S. Sleep in youth with autism spectrum disorders: systematic review and meta-analysis of subjective and objective studies. Evid Based Ment Health. 2018;21:146–54. doi: 10.1136/ebmental-2018-300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couturier JL, Speechley KN, Steele M, Norman R, Stringer B, Nicolson R. Parental perception of sleep problems in children of normal intelligence with pervasive developmental disorders: prevalence, severity, and pattern. J Am Acad Child Adolesc Psychiatry. 2005;44:815–22. doi: 10.1097/01.chi.0000166377.22651.87. [DOI] [PubMed] [Google Scholar]
- 19.Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, Croen LA, Hansen RL. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res. 2008;17:197–206. doi: 10.1111/j.1365-2869.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreck KA, Mulick JA. Parental report of sleep problems in children with autism. J Autism Dev Disord. 2000;30:127–35. doi: 10.1023/a:1005407622050. [DOI] [PubMed] [Google Scholar]
- 21.Ming X, Sun YM, Nachajon RV, Brimacombe M, Walters AS. Prevalence of parasomnia in autistic children with sleep disorders. Clin Med Pedia. 2009;3:1–10. doi: 10.4137/cmped.s1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elrod MG, Hood BS. Sleep differences among children with autism spectrum disorders and typically developing peers: a meta-analysis. J Dev Behav Pedia. 2015;36:166–77. doi: 10.1097/DBP.0000000000000140. [DOI] [PubMed] [Google Scholar]
- 23.Owens JA, Spirito A, McGuinn M. The Children’s Sleep Habits Questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep. 2000;23:1043–51. [PubMed] [Google Scholar]
- 24.Zawilska JB, Skene DJ, Arendt J. Physiology and pharmacology of melatonin in relation to biological rhythms. Pharm Rep. 2009;61:383–410. doi: 10.1016/s1734-1140(09)70081-7. [DOI] [PubMed] [Google Scholar]
- 25.Muller CL, Anacker AMJ, Veenstra-VanderWeele J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience. 2016;321:24–41. doi: 10.1016/j.neuroscience.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y. Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol Psychiatry. 2005;57:134–8. doi: 10.1016/j.biopsych.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Melke J, Goubran Botros H, Chaste P, Betancur C, Nygren G, Anckarsater H, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry. 2008;13:90–8. doi: 10.1038/sj.mp.4002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagan C, Delorme R, Callebert J, Goubran-Botros H, Amsellem F, Drouot X, et al. The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl Psychiatry. 2014;4:e479. doi: 10.1038/tp.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossignol DA, Frye RE. Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev Med Child Neurol. 2011;53:783–92. doi: 10.1111/j.1469-8749.2011.03980.x. [DOI] [PubMed] [Google Scholar]
- 30.Goldman SE, Adkins KW, Calcutt MW, Carter MD, Goodpaster RL, Wang L, et al. Melatonin in children with autism spectrum disorders: endogenous and pharmacokinetic profiles in relation to sleep. J Autism Dev Disord. 2014;44:2525–35. doi: 10.1007/s10803-014-2123-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gregory AM, Sadeh A. Sleep, emotional and behavioral difficulties in children and adolescents. Sleep Med Rev. 2012;16:129–36. doi: 10.1016/j.smrv.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Schreck KA, Mulick JA, Smith AF. Sleep problems as possible predictors of intensified symptoms of autism. Res Dev Disabil. 2004;25:57–66. doi: 10.1016/j.ridd.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Sikora DM, Johnson K, Clemons T, Katz T. The relationship between sleep problems and daytime behavior in children of different ages with autism spectrum disorders. Pediatrics. 2012;130(Suppl 2):S83–90. doi: 10.1542/peds.2012-0900F. [DOI] [PubMed] [Google Scholar]
- 34.Mazurek MO, Sohl K. Sleep and Behavioral Problems in Children with Autism Spectrum Disorder. J Autism Dev Disord. 2016;46:1906–15. doi: 10.1007/s10803-016-2723-7. [DOI] [PubMed] [Google Scholar]
- 35.Mazzone Luigi, Postorino Valentina, Siracusano Martina, Riccioni Assia, Curatolo Paolo. The Relationship between Sleep Problems, Neurobiological Alterations, Core Symptoms of Autism Spectrum Disorder, and Psychiatric Comorbidities. Journal of Clinical Medicine. 2018;7(5):102. doi: 10.3390/jcm7050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazen EP, Stornelli JL, O’Rourke JA, Koesterer K, McDougle CJ. Sensory symptoms in autism spectrum disorders. Harv Rev Psychiatry. 2014;22:112–24. doi: 10.1097/01.HRP.0000445143.08773.58. [DOI] [PubMed] [Google Scholar]
- 37.Sannar EM, Palka T, Beresford C, Peura C, Kaplan D, Verdi M, et al. Sleep problems and their relationship to maladaptive behavior severity in psychiatrically hospitalized children with autism spectrum disorder (ASD) J Autism Dev Disord. 2018;48:3720–6. doi: 10.1007/s10803-017-3362-3. [DOI] [PubMed] [Google Scholar]
- 38.Verhoeff ME, Blanken LME, Kocevska D, Mileva-Seitz VR, Jaddoe VWV, White T, et al. The bidirectional association between sleep problems and autism spectrum disorder: a population-based cohort study. Mol Autism. 2018;9:8. doi: 10.1186/s13229-018-0194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Jiang YH, Zhang YQ. Modeling autism in non-human primates: Opportunities and challenges. Autism Res. 2018;11:686–94. doi: 10.1002/aur.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sztainberg Y, Zoghbi HY. Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci. 2016;19:1408–17. doi: 10.1038/nn.4420. [DOI] [PubMed] [Google Scholar]
- 41.Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30:80–9. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- 42.Roullet FI, Wollaston L, Decatanzaro D, Foster JA. Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience. 2010;170:514–22. doi: 10.1016/j.neuroscience.2010.06.069. [DOI] [PubMed] [Google Scholar]
- 43.Schneider T, Roman A, Basta-Kaim A, Kubera M, Budziszewska B, Schneider K, et al. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology. 2008;33:728–40. doi: 10.1016/j.psyneuen.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Missig G, Finger BC, Landino SM, Alexander AJ, Mokler E, et al. Maternal and Early Postnatal Immune Activation Produce Dissociable Effects on Neurotransmission in mPFC-Amygdala Circuits. J Neurosci. 2018;38:3358–72. doi: 10.1523/JNEUROSCI.3642-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Missig G, Robbins JO, Mokler EL, McCullough KM, Bilbo SD, McDougle CJ, et al. Sex-dependent neurobiological features of prenatal immune activation via TLR7. Mol Psychiatry, 2019. 10.1038/s41380-018-0346-4. [DOI] [PMC free article] [PubMed]
- 46.Thirtamara Rajamani K, Doherty-Lyons S, Bolden C, Willis D, Hoffman C, Zelikoff J, et al. Prenatal and early-life exposure to high-level diesel exhaust particles leads to increased locomotor activity and repetitive behaviors in mice. Autism Res. 2013;6:248–57. doi: 10.1002/aur.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones KL, Pride MC, Edmiston E, Yang M, Silverman JL, Crawley JN, et al. Autism-specific maternal autoantibodies produce behavioral abnormalities in an endogenous antigen-driven mouse model of autism. Mol Psychiatry, 2018. [DOI] [PMC free article] [PubMed]
- 48.Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, Klein PM, et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One. 2012;7:e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav Immun. 2016;58:165–72. doi: 10.1016/j.bbi.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–21. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 51.Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M, et al. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry. 2014;171:557–63. doi: 10.1176/appi.ajp.2013.13070943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Instanes JT, Halmoy A, Engeland A, Haavik J, Furu K, Klungsoyr K. Attention-Deficit/Hyperactivity Disorder in Offspring of Mothers With Inflammatory and Immune System Diseases. Biol Psychiatry. 2017;81:452–9. doi: 10.1016/j.biopsych.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 53.Berry-Kravis EM, Lindemann L, Jonch AE, Apostol G, Bear MF, Carpenter RL, et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Disco. 2018;17:280–99. doi: 10.1038/nrd.2017.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hyman SE. Revolution stalled. Sci Transl Med. 2012;4:155cm11. doi: 10.1126/scitranslmed.3003142. [DOI] [PubMed] [Google Scholar]
- 55.Crawley JN. Translational animal models of autism and neurodevelopmental disorders. Dialog- Clin Neurosci. 2012;14:293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lezak KR, Missig GM, Carlezon WAJ. Behavioral methods to study anxiety in rodents. Dialog- Clin Neurosci. 2017;19:181–91. doi: 10.31887/DCNS.2017.19.2/wcarlezon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker JT, Germine LT, Ressler KJ, Rauch SL, Carlezon WA., Jr. Digital devices and continuous telemetry: opportunities for aligning psychiatry and neuroscience. Neuropsychopharmacology. 2018;43:2499–503. doi: 10.1038/s41386-018-0172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cirelli C, Tononi G. Is sleep essential? PLoS Biol. 2008;6:e216. doi: 10.1371/journal.pbio.0060216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siegel JM. Do all animals sleep? Trends Neurosci. 2008;31:208–13. doi: 10.1016/j.tins.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allada R, Siegel JM. Unearthing the phylogenetic roots of sleep. Curr Biol. 2008;18:R670–R679. doi: 10.1016/j.cub.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toth LA, Bhargava P. Animal models of sleep disorders. Comp Med. 2013;63:91–104. [PMC free article] [PubMed] [Google Scholar]
- 62.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–69. [PubMed] [Google Scholar]
- 63.Zoghbi HY, Warren ST. Neurogenetics: advancing the “next-generation” of brain research. Neuron. 2010;68:165–73. doi: 10.1016/j.neuron.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiroi N. Critical reappraisal of mechanistic links of copy number variants to dimensional constructs of neuropsychiatric disorders in mouse models. Psychiatry Clin Neurosci. 2018;72:301–21. doi: 10.1111/pcn.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crusio WE, Goldowitz D, Holmes A, Wolfer D. Standards for the publication of mouse mutant studies. Genes Brain Behav. 2009;8:1–4. doi: 10.1111/j.1601-183X.2008.00438.x. [DOI] [PubMed] [Google Scholar]
- 66.Kentner Amanda C., Bilbo Staci D., Brown Alan S., Hsiao Elaine Y., McAllister A. Kimberley, Meyer Urs, Pearce Brad D., Pletnikov Mikhail V., Yolken Robert H., Bauman Melissa D. Maternal immune activation: reporting guidelines to improve the rigor, reproducibility, and transparency of the model. Neuropsychopharmacology. 2018;44(2):245–258. doi: 10.1038/s41386-018-0185-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bale TL. Sex matters. Neuropsychopharmacology. 2019;44:1–3. doi: 10.1038/s41386-018-0239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fombonne E. Epidemiology of pervasive developmental disorders. Pedia Res. 2009;65:591–8. doi: 10.1203/PDR.0b013e31819e7203. [DOI] [PubMed] [Google Scholar]
- 69.Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, et al. Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism. 2015;6:36. doi: 10.1186/s13229-015-0019-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferri SL, Abel T, Brodkin ES. Sex differences in autism spectrum disorder: a review. Curr Psychiatry Rep. 2018;20:9. doi: 10.1007/s11920-018-0874-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci. 2002;6:248–54. doi: 10.1016/s1364-6613(02)01904-6. [DOI] [PubMed] [Google Scholar]
- 72.Mong JA, Cusmano DM. Sex differences in sleep: impact of biological sex and sex steroids. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150110. doi: 10.1098/rstb.2015.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paul KN, Dugovic C, Turek FW, Laposky AD. Diurnal sex differences in the sleep-wake cycle of mice are dependent on gonadal function. Sleep. 2006;29:1211–23. doi: 10.1093/sleep/29.9.1211. [DOI] [PubMed] [Google Scholar]
- 74.Deurveilher S, Rusak B, Semba K. Female reproductive hormones alter sleep architecture in ovariectomized rats. Sleep. 2011;34:519–30. doi: 10.1093/sleep/34.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanchez-Alavez M, Alboni S, Conti B. Sex- and age-specific differences in core body temperature of C57Bl/6 mice. Age (Dordr) 2011;33:89–99. doi: 10.1007/s11357-010-9164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roscoe B. Jackson Memorial Laboratory. and E.L. Green, Biology of the laboratory mouse. 2d ed. 1975, New York: Dover Publications. x, 706 p., 3 leaves of plates.
- 77.Westwood FR. The female rat reproductive cycle: a practical histological guide to staging. Toxicol Pathol. 2008;36:375–84. doi: 10.1177/0192623308315665. [DOI] [PubMed] [Google Scholar]
- 78.Kleinlogel H. The female rat’s sleep during oestrous cycle. Neuropsychobiology. 1983;10:228–37. doi: 10.1159/000118016. [DOI] [PubMed] [Google Scholar]
- 79.Schwierin B, Borbely AA, Tobler I. Sleep homeostasis in the female rat during the estrous cycle. Brain Res. 1998;811:96–104. doi: 10.1016/s0006-8993(98)00991-3. [DOI] [PubMed] [Google Scholar]
- 80.Gangestad SW, Thornhill R. Human oestrus. Proc Biol Sci. 2008;275:991–1000. doi: 10.1098/rspb.2007.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christensen J, Gronborg TK, Sorensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA. 2013;309:1696–703. doi: 10.1001/jama.2013.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsujino N, Nakatani Y, Seki Y, Nakasato A, Nakamura M, Sugawara M, et al. Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci Res. 2007;57:289–95. doi: 10.1016/j.neures.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 83.Cusmano DM, Mong JA. In utero exposure to valproic acid changes sleep in juvenile rats: a model for sleep disturbances in autism. Sleep. 2014;37:1489–99. doi: 10.5665/sleep.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDougle CJ, Landino SM, Vahabzadeh A, O’Rourke J, Zurcher NR, Finger BC, et al. Toward an immune-mediated subtype of autism spectrum disorder. Brain Res. 2015;1617:72–92. doi: 10.1016/j.brainres.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 85.Wu S, Ding Y, Wu F, Li R, Xie G, Hou J, et al. Family history of autoimmune diseases is associated with an increased risk of autism in children: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2015;55:322–32. doi: 10.1016/j.neubiorev.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 86.Vinet E, Pineau CA, Clarke AE, Scott S, Fombonne E, Joseph L, et al. Increased risk of autism spectrum disorders in children born to women with systemic lupus erythematosus: results from a large population-based cohort. Arthritis Rheuma. 2015;67:3201–8. doi: 10.1002/art.39320. [DOI] [PubMed] [Google Scholar]
- 87.Careaga M, Murai T, Bauman MD. MateRnal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. 2017;81:391–401. doi: 10.1016/j.biopsych.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016;353:772–7. doi: 10.1126/science.aag3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zager A, Andersen ML, Tufik S, Palermo-Neto J. Maternal immune activation increases the corticosterone response to acute stress without affecting the hypothalamic monoamine content and sleep patterns in male mice offspring. Neuroimmunomodulation. 2014;21:37–44. doi: 10.1159/000355466. [DOI] [PubMed] [Google Scholar]
- 90.Missig Galen, Mokler Emery L, Robbins James O, Alexander Abigail J, McDougle Christopher J, Carlezon William A. Perinatal Immune Activation Produces Persistent Sleep Alterations and Epileptiform Activity in Male Mice. Neuropsychopharmacology. 2017;43(3):482–491. doi: 10.1038/npp.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, et al. Involvement of cytokines in slow wave sleep. Prog Brain Res. 2011;193:39–47. doi: 10.1016/B978-0-444-53839-0.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kovacs Z, Czurko A, Kekesi KA, Juhasz G. Intracerebroventricularly administered lipopolysaccharide enhances spike-wave discharges in freely moving WAG/Rij rats. Brain Res Bull. 2011;85:410–6. doi: 10.1016/j.brainresbull.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 93.Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–32. doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaufmann WE, Kidd SA, Andrews HF, Budimirovic DB, Esler A, Haas-Givler B, et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics. 2017;139(Suppl 3):S194–S206. doi: 10.1542/peds.2016-1159F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–5. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- 96.Kronk R, Bishop EE, Raspa M, Bickel JO, Mandel DA, Bailey DB, et al. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep. 2010;33:679–87. doi: 10.1093/sleep/33.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miano S, Bruni O, Elia M, Scifo L, Smerieri A, Trovato A, et al. Sleep phenotypes of intellectual disability: a polysomnographic evaluation in subjects with Down syndrome and Fragile-X syndrome. Clin Neurophysiol. 2008;119:1242–7. doi: 10.1016/j.clinph.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 98.Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–84. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 99.Inoue S, Shimoda M, Nishinokubi I, Siomi MC, Okamura M, Nakamura A, et al. A role for the Drosophila fragile X-related gene in circadian output. Curr Biol. 2002;12:1331–5. doi: 10.1016/s0960-9822(02)01036-9. [DOI] [PubMed] [Google Scholar]
- 100.Bushey D, Tononi G, Cirelli C. The Drosophila fragile X mental retardation gene regulates sleep need. J Neurosci. 2009;29:1948–61. doi: 10.1523/JNEUROSCI.4830-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, et al. Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am J Hum Genet. 2008;83:43–52. doi: 10.1016/j.ajhg.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sare RM, Harkless L, Levine M, Torossian A, Sheeler CA, Smith CB. Deficient Sleep in Mouse Models of Fragile X Syndrome. Front Mol Neurosci. 2017;10:280. doi: 10.3389/fnmol.2017.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goncalves JT, Anstey JE, Golshani P, Portera-Cailliau C. Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci. 2013;16:903–9. doi: 10.1038/nn.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Romero-Zerbo Y, Decara J, el Bekay R, Sanchez-Salido L, Del Arco-Herrera I, de Fonseca FR, et al. Protective effects of melatonin against oxidative stress in Fmr1 knockout mice: a therapeutic research model for the fragile X syndrome. J Pineal Res. 2009;46:224–34. doi: 10.1111/j.1600-079X.2008.00653.x. [DOI] [PubMed] [Google Scholar]
- 105.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 106.Neul JL. The relationship of Rett syndrome and MECP2 disorders to autism. Dialog- Clin Neurosci. 2012;14:253–62. doi: 10.31887/DCNS.2012.14.3/jneul. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Volkmar FR, McPartland JC. From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu Rev Clin Psychol. 2014;10:193–212. doi: 10.1146/annurev-clinpsy-032813-153710. [DOI] [PubMed] [Google Scholar]
- 108.Young D, Nagarajan L, de Klerk N, Jacoby P, Ellaway C, Leonard H. Sleep problems in Rett syndrome. Brain Dev. 2007;29:609–16. doi: 10.1016/j.braindev.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wong K, Leonard H, Jacoby P, Ellaway C, Downs J. The trajectories of sleep disturbances in Rett syndrome. J Sleep Res. 2015;24:223–33. doi: 10.1111/jsr.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–9. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–6. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 112.Ezeonwuka CD, Rastegar M. MeCP2-Related Diseases and Animal Models. Diseases. 2014;2:45–70. doi: 10.3390/diseases2010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tsuchiya Y, Minami Y, Umemura Y, Watanabe H, Ono D, Nakamura W, et al. Disruption of MeCP2 attenuates circadian rhythm in CRISPR/Cas9-based Rett syndrome model mouse. Genes Cells. 2015;20:992–1005. doi: 10.1111/gtc.12305. [DOI] [PubMed] [Google Scholar]
- 114.Li Q, Loh DH, Kudo T, Truong D, Derakhshesh M, Kaswan ZM, et al. Circadian rhythm disruption in a mouse model of Rett syndrome circadian disruption in RTT. Neurobiol Dis. 2015;77:155–64. doi: 10.1016/j.nbd.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 115.Wither RG, Colic S, Wu C, Bardakjian BL, Zhang L, Eubanks JH. Daily rhythmic behaviors and thermoregulatory patterns are disrupted in adult female MeCP2-deficient mice. PLoS One. 2012;7:e35396. doi: 10.1371/journal.pone.0035396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.D’Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol Dis. 2010;38:8–16. doi: 10.1016/j.nbd.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 117.Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G, et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell. 2017;169:945–55 e10. doi: 10.1016/j.cell.2017.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hess-Homeier DL, Fan CY, Gupta T, Chiang AS, Certel SJ. Astrocyte-specific regulation of hMeCP2 expression in Drosophila. Biol Open. 2014;3:1011–9. doi: 10.1242/bio.20149092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gupta T, Morgan HR, Bailey JA, Certel SJ. Functional conservation of MBD proteins: MeCP2 and Drosophila MBD proteins alter sleep. Genes Brain Behav. 2016;15:757–74. doi: 10.1111/gbb.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–69. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alvarez-Saavedra M, Antoun G, Yanagiya A, Oliva-Hernandez R, Cornejo-Palma D, Perez-Iratxeta C, et al. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum Mol Genet. 2011;20:731–51. doi: 10.1093/hmg/ddq519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38:1249–63. doi: 10.1007/s40618-015-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bennett JA, Germani T, Haqq AM, Zwaigenbaum L. Autism spectrum disorder in Prader-Willi syndrome: A systematic review. Am J Med Genet A. 2015;167A:2936–44. doi: 10.1002/ajmg.a.37286. [DOI] [PubMed] [Google Scholar]
- 124.Lassi G, Priano L, Maggi S, Garcia-Garcia C, Balzani E, El-Assawy N, et al. Deletion of the Snord116/SNORD116 Alters Sleep in Mice and Patients with Prader-Willi Syndrome. Sleep. 2016;39:637–44. doi: 10.5665/sleep.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman Syndrome. Neurotherapeutics. 2015;12:641–50. doi: 10.1007/s13311-015-0361-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vatsa N, Jana NR. UBE3A and Its Link With Autism. Front Mol Neurosci. 2018;11:448. doi: 10.3389/fnmol.2018.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9:434–41. doi: 10.1016/j.sleep.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 128.Spruyt K, Braam W, Curfs LM. Sleep in Angelman syndrome: A review of evidence. Sleep Med Rev. 2018;37:69–84. doi: 10.1016/j.smrv.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 129.Colas D, Wagstaff J, Fort P, Salvert D, Sarda N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol Dis. 2005;20:471–8. doi: 10.1016/j.nbd.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 130.Ehlen JC, Jones KA, Pinckney L, Gray CL, Burette S, Weinberg RJ, et al. Maternal Ube3a Loss Disrupts Sleep Homeostasis But Leaves Circadian Rhythmicity Largely Intact. J Neurosci. 2015;35:13587–98. doi: 10.1523/JNEUROSCI.2194-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shi SQ, Bichell TJ, Ihrie RA, Johnson CH. Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Curr Biol. 2015;25:537–45. doi: 10.1016/j.cub.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–16. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12:777–83. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Prim. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- 135.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–13. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bruni O, Cortesi F, Giannotti F, Curatolo P. Sleep disorders in tuberous sclerosis: a polysomnographic study. Brain Dev. 1995;17:52–6. doi: 10.1016/0387-7604(94)00118-h. [DOI] [PubMed] [Google Scholar]
- 137.van Eeghen AM, Numis AI, Staley BA, Therrien SE, Thibert RL, Thiele EA. Characterizing sleep disorders of adults with tuberous sclerosis complex: a questionnaire-based study and review. Epilepsy Behav. 2011;20:68–74. doi: 10.1016/j.yebeh.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zheng X, Sehgal A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr Biol. 2010;20:1203–8. doi: 10.1016/j.cub.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lipton JO, Boyle LM, Yuan ED, Hochstrasser KJ, Chifamba FF, Nathan A, et al. Aberrant Proteostasis of BMAL1 Underlies Circadian Abnormalities in a Paradigmatic mTOR-opathy. Cell Rep. 2017;20:868–80. doi: 10.1016/j.celrep.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, et al. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000;24:84–7. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- 141.Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633–49. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Molina J, Carmona-Mora P, Chrast J, Krall PM, Canales CP, Lupski JR, et al. Abnormal social behaviors and altered gene expression rates in a mouse model for Potocki-Lupski syndrome. Hum Mol Genet. 2008;17:2486–95. doi: 10.1093/hmg/ddn148. [DOI] [PubMed] [Google Scholar]
- 143.Dubourg C, Bonnet-Brilhault F, Toutain A, Mignot C, Jacquette A, Dieux A, et al. Identification of nine new RAI1-truncating mutations in Smith-Magenis syndrome patients without 17p11.2 deletions. Mol Syndr. 2014;5:57–64. doi: 10.1159/000357359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang F, Potocki L, Sampson JB, Liu P, Sanchez-Valle A, Robbins-Furman P, et al. Identification of uncommon recurrent Potocki-Lupski syndrome-associated duplications and the distribution of rearrangement types and mechanisms in PTLS. Am J Hum Genet. 2010;86:462–70. doi: 10.1016/j.ajhg.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smith AC, Dykens E, Greenberg F. Sleep disturbance in Smith-Magenis syndrome (del 17p11.2) Am J Med Genet. 1998;81:186–91. [PubMed] [Google Scholar]
- 146.De Leersnyder H. Inverted rhythm of melatonin secretion in Smith-Magenis syndrome: from symptoms to treatment. Trends Endocrinol Metab. 2006;17:291–8. doi: 10.1016/j.tem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 147.Carmona-Mora P, Molina J, Encina CA, Walz K. Mouse models of genomic syndromes as tools for understanding the basis of complex traits: an example with the smith-magenis and the potocki-lupski syndromes. Curr Genom. 2009;10:259–68. doi: 10.2174/138920209788488508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang WH, Guenthner CJ, Xu J, Nguyen T, Schwarz LA, Wilkinson AW, et al. Molecular and Neural Functions of Rai1, the Causal Gene for Smith-Magenis Syndrome. Neuron. 2016;92:392–406. doi: 10.1016/j.neuron.2016.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 150.Barrett CF, Tsien RW. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc Natl Acad Sci USA. 2008;105:2157–62. doi: 10.1073/pnas.0710501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kumar D, Dedic N, Flachskamm C, Voule S, Deussing JM, Kimura M. Cacna1c (Cav1.2) Modulates Electroencephalographic Rhythm and Rapid Eye Movement Sleep Recovery. Sleep. 2015;38:1371–80. doi: 10.5665/sleep.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–7. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 153.Penagarikano O, Geschwind DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med. 2012;18:156–63. doi: 10.1016/j.molmed.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–9. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–46. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Thomas, AM, Schwartz, MD, Saxe, MD, Kilduff, TS. Cntnap2 knockout rats and mice exhibit epileptiform activity and abnormal sleep-wake physiology. Sleep, 2017;40. 10.1093/sleep/zsw026. [DOI] [PubMed]
- 157.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry. 2015;77:785–93. doi: 10.1016/j.biopsych.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Arbogast T, Ouagazzal AM, Chevalier C, Kopanitsa M, Afinowi N, Migliavacca E, et al. Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet. 2016;12:e1005709. doi: 10.1371/journal.pgen.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar VJ, Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, et al. Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl Psychiatry. 2018;8:109. doi: 10.1038/s41398-018-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Angelakos CC, Watson AJ, O’Brien WT, Krainock KS, Nickl-Jockschat T, Abel T. Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res. 2017;10:572–84. doi: 10.1002/aur.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–11. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–9. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yu J, He X, Yao D, Li Z, Li H, Zhao Z. A sex-specific association of common variants of neuroligin genes (NLGN3 and NLGN4X) with autism spectrum disorders in a Chinese Han cohort. Behav Brain Funct. 2011;7:13. doi: 10.1186/1744-9081-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vincent JB, Kolozsvari D, Roberts WS, Bolton PF, Gurling HM, Scherer SW. Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:82–4. doi: 10.1002/ajmg.b.30069. [DOI] [PubMed] [Google Scholar]
- 166.Hu X, Luo JH, Xu J. The interplay between synaptic activity and neuroligin function in the CNS. Biomed Res Int. 2015;2015:498957. doi: 10.1155/2015/498957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30:2115–29. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.El Helou J, Belanger-Nelson E, Freyburger M, Dorsaz S, Curie T. F. La Spada, et al., Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc Natl Acad Sci USA. 2013;110:9974–9. doi: 10.1073/pnas.1221381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li Y, Zhou Z, Zhang X, Tong H, Li P, Zhang ZC, et al. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J Neurosci. 2013;33:15545–54. doi: 10.1523/JNEUROSCI.0819-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–6. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu JJ, Grace KP, Horner RL, Cortez MA, Shao Y, Jia Z. Neuroligin 3 R451C mutation alters electroencephalography spectral activity in an animal model of autism spectrum disorders. Mol Brain. 2017;10:10. doi: 10.1186/s13041-017-0290-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–4. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 173.Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–21. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–88. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Clipperton-Allen AE, Page DT. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum Mol Genet. 2014;23:3490–505. doi: 10.1093/hmg/ddu057. [DOI] [PubMed] [Google Scholar]
- 176.Ogawa S, Kwon CH, Zhou J, Koovakkattu D, Parada LF, Sinton CM. A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res. 2007;1168:112–23. doi: 10.1016/j.brainres.2007.06.074. [DOI] [PubMed] [Google Scholar]
- 177.Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–63. doi: 10.1038/nrn3992. [DOI] [PubMed] [Google Scholar]
- 178.Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. 2015;28:91–102. doi: 10.1097/WCO.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 179.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci. 2011;34:452–63. doi: 10.1016/j.tins.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull. 2003;62:143–50. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 182.Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Puentes-Mestril C, Aton SJ. Linking Network Activity to Synaptic Plasticity during Sleep: Hypotheses and Recent Data. Front Neural Circuits. 2017;11:61. doi: 10.3389/fncir.2017.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23:R774–88. doi: 10.1016/j.cub.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Barone Ilaria, Hawks-Mayer Hannah, Lipton Jonathan O. Mechanisms of sleep and circadian ontogeny through the lens of neurodevelopmental disorders. Neurobiology of Learning and Memory. 2019;160:160–172. doi: 10.1016/j.nlm.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 186.Nguyen A. K. Danny, Murphy Laura E., Kocak Mehmet, Tylavsky Frances A., Pagani Linda S. Prospective Associations Between Infant Sleep at 12 Months and Autism Spectrum Disorder Screening Scores at 24 Months in a Community-Based Birth Cohort. The Journal of Clinical Psychiatry. 2018;79(1):16m11127. doi: 10.4088/JCP.16m11127. [DOI] [PubMed] [Google Scholar]
- 187.Wang J, Barstein J, Ethridge LE, Mosconi MW, Takarae Y, Sweeney JA. Resting state EEG abnormalities in autism spectrum disorders. J Neurodev Disord. 2013;5:24. doi: 10.1186/1866-1955-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pedia Res. 2009;65:599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chez MG, Chang M, Krasne V, Coughlan C, Kominsky M, Schwartz A. Frequency of epileptiform EEG abnormalities in a sequential screening of autistic patients with no known clinical epilepsy from 1996 to 2005. Epilepsy Behav. 2006;8:267–71. doi: 10.1016/j.yebeh.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 190.Eban-Rothschild A, Appelbaum L, de Lecea L. Neuronal mechanisms for sleep/wake regulation and modulatory drive. Neuropsychopharmacology. 2018;43:937–52. doi: 10.1038/npp.2017.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Scammell TE, Arrigoni E, Lipton JO. Neural circuitry of wakefulness and sleep. Neuron. 2017;93:747–65. doi: 10.1016/j.neuron.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]