

Abstract
Objective
High-density lipoprotein (HDL) infusion reduces atherosclerosis in animal models and is being evaluated as a treatment in humans. Studies have shown either anti- or pro-inflammatory effects of HDL in macrophages, and there is no consensus on the underlying mechanisms. Here, we interrogate the effects of HDL on inflammatory gene expression in macrophages.
Approach and Results
We cultured bone marrow-derived macrophages, treated them with reconstituted HDL or HDL isolated from APOA1Tg;Ldlr−/− mice, and challenged them with lipopolysaccharide (LPS). Transcriptional profiling showed that HDL exerts a broad anti-inflammatory effect on LPS-induced genes and pro-inflammatory effect in a subset of genes enriched for chemokines. Cholesterol removal by POPC-liposomes or β-methylcyclodextrin mimicked both pro- and anti-inflammatory effects of HDL, whereas cholesterol loading by POPC/cholesterol-liposomes or acetylated LDL prior to HDL attenuated these effects, indicating that these responses are mediated by cholesterol efflux. While early anti-inflammatory effects reflect reduced Toll-like receptor 4 levels, late anti-inflammatory effects are due to reduced interferon receptor signaling. Pro-inflammatory effects occur late and represent a modified ER stress response, mediated by IRE1a/ASK1/p38 MAPK signaling, that occurs under conditions of extreme cholesterol depletion. To investigate the effects of HDL on inflammatory gene expression in myeloid cells in atherosclerotic lesions, we injected rHDL into Apoe−/− or Ldlr−/− mice fed a Western-type diet. Reconstituted HDL infusions produced anti-inflammatory effects in lesion macrophages without any evidence of pro-inflammatory effects.
Conclusion
Reconstituted HDL infusions in hypercholesterolemic atherosclerotic mice produced anti-inflammatory effects in lesion macrophages suggesting a beneficial therapeutic effect of HDL in vivo.
Graphical Abstract
Introduction
Epidemiological studies 1 and experimental animal models 2, 3 suggest that high-density lipoprotein (HDL) protects against atherosclerosis. However, the causal relationship of HDL cholesterol levels to atherosclerosis has been challenged by Mendelian randomization studies 4, 5 and failure or mediocre success of HDL cholesterol elevating treatments in clinical trials of statin treated subjects 6, 7. To reconcile these findings, it has been suggested that evaluating the functionality of HDL, instead of the HDL cholesterol level, may provide a better biomarker of its ability to confer atheroprotection 8. HDL exerts a plethora of functions in vitro that are thought to be atheroprotective, such as cholesterol efflux, anti-inflammatory, anti-oxidant and anti-thrombotic effects 9. Amongst these, the cholesterol efflux capacity of HDL has been shown to be a better predictor of coronary heart disease risk compared to HDL cholesterol levels 10, 11.
The central role of inflammatory responses in human coronary heart disease has been clearly shown by the positive outcome of the CANTOS trial 12. Given the importance of macrophage inflammation in the initiation and progression of atherosclerosis 13, recent studies have directly evaluated the anti-inflammatory potential of HDL and its major apolipoprotein, APOA1 in macrophages 14-19, mainly in the context of lipopolysaccharide (LPS) stimulation and Toll-like receptor 4 (TLR4) activation 14-17. These studies reported conflicting results indicating either anti- or pro-inflammatory effects and suggested distinct mechanisms. For example, anti-inflammatory effects were attributed to induction of ATF3, an inhibitor of Toll like receptor (TLR)/nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling by HDL, 15 or defective induction of Type I interferons, downstream of TRAM/TRIF signaling 14. Another study showed anti-inflammatory effects of HDL in IFNγ-stimulated macrophages 20. In contrast, pro-inflammatory effects were associated with increased activity of Protein Kinase C (PKC) 17, enhancing TLR signaling, or direct stimulation of TLRs by APOA1 16. Notably, despite the relation of HDL to atherosclerosis, only two of these studies examined its effects on lesion macrophages 18, 19. Moreover, one of these HDL studies, showing pro-inflammatory effects, associated the observed changes with the ability of HDL to remove cholesterol 17. This contrasts with the notion that cholesterol accumulation in macrophages amplifies TLR signaling and thus cholesterol removal dampens this response 21, 22.
Given the discrepancies concerning the effects of HDL on macrophage inflammation, and the potential concern that reconstituted HDL infusion in humans could have adverse effects in plaque macrophages, we have carried out a systematic examination of the inflammatory potential of reconstituted human and native mouse HDL (rHDL and nHDL, respectively) in cultured macrophages and in plaque myeloid cells. Transcriptome analysis using RNA-seq showed anti- and pro-inflammatory effects of HDL; both effects are mediated by cholesterol removal. The anti-inflammatory effects are associated with reduced TLR4 expression and decreased IFNAR (interferon-α/β receptor) signaling, whereas the pro-inflammatory effects represent an ER stress response, involving activation of the IRE1a/ASK1/p38 MAPK signaling axis. Both pro- and anti-inflammatory effects of HDL were attenuated by prior macrophage cholesterol loading. Importantly, infusions of rHDL in hypercholesterolemic atherosclerotic mice led to reduced expression of inflammatory genes in aortic macrophages without any evidence of increased inflammatory gene expression.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure on reasonable request.
Animals
All animal studies were approved by the Animal Care and Use Committees of Columbia University (protocol AAAO6351) and University of Washington (protocol 3154-01). Wild type C57BL/6J (stock number 000664), Apoe−/− (stock number 002052), Ldlr−/− (stock number 002207), Ifnar1−/− (stock number 32045-JAX) and APOA1Tg (stock number 001927) mice were obtained from The Jackson Laboratory. The APOA1Tg mice were crossed with Ldlr−/− mice to generate APOA1Tg Ldlr−/− mice and Ldlr−/− littermates. All mice were on the C57BL/6J background and females were used aged 8-16 weeks. Female mice were selected based on their higher susceptibly to diet-induced atherosclerosis compared to male mice 23-27. This allowed us to isolate enough CD11b+ from the atherosclerotic plaques after a short-term Western-type diet to perform RNA analysis (see Supplemental Methods and Figures 6D, E). For consistency we used female mice in all experiments (e.g. bone marrow derived macrophage or peritoneal macrophage isolation). Animals were kept under specific pathogen-free conditions with ad libitum access to both food and water. Mice were fed irradiated normal laboratory diet (Purina Mills diet 5053) or Western-type diet (WTD; Harlan Teklad TD88137). Housing temperatures were kept within a range of 71-73°F (21.7-22.8°C). Water and cages were autoclaved, and cages were changed once weekly. The health status of the mice was monitored using a dirty bedding sentinel program and no health status issues or changes in immune status were identified.
Figure 6. In vivo effects of HDL on macrophage inflammatory gene expression.

(A) Wild type C57BL/6 mice fed a normal laboratory diet were injected intravenously with 80 mg/Kg rHDL (n=8) or PBS daily (n=7), for a total of 5 days before sacrificing the mice 2 hours after the last rHDL injection. Thioglycollate was injected intraperitoneally 3 days before sacrificing the mice and peritoneal macrophages were collected, stimulated with LPS (100 ng/ml, 2 h) and inflammatory gene expression was assessed. (B) Effect of LPS (10 ng/ml, 4 hours) on inflammatory genes in resident peritoneal macrophages isolated from normal laboratory diet-fed APOA1Tg;Ldlr−/− mice or Ldlr−/− (WT) mice (n=7-8 per group). (C) Neutral lipid staining with BODIPY in resident macrophages isolated from APOA1Tg;Ldlr−/− mice (n=4) or Ldlr−/− littermates (n=4). (D) Apoe−/− mice fed a western type diet for 4 weeks were injected intravenously with 80 mg/Kg rHDL or PBS daily (n=29 per group), for a total of 5 days before sacrificing the mice 2 hours after the last rHDL injection. CD11b+ cells from whole aorta were collected and assessed for inflammatory gene expression, Hmgcr and Abca1 expression. (E) Ldlr−/− mice fed a western type diet for 8 weeks were injected intravenously with 80 mg/Kg rHDL or PBS daily (n=12 per group), for a total of 5 days before sacrificing the mice 2 hours after the last rHDL injection. CD11b+ cells from whole aorta were collected and assessed for inflammatory gene expression, Hmgcr and Abca1 expression. The results are shown as mean ± SEM. Significance was determined by multiple t-tests using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, *p<0.05, ****p<0.0001 (A), one-way ANOVA with Tukey’s multiple comparisons test (B) or by unpaired t test with Welch’s correction for data not having equal variances or Mann-Whitney U test for data not passing normality *p<0.05 (D, E). See also Supplemental Figure VI.
Cell culture
Bone marrow-derived macrophages (BMDMs) were obtained by isolating bone marrow cells from femurs and tibias of mice and incubated in DMEM (Corning), 10% FBS (Thermo), 1% pen-strep (Thermo) supplemented with 20% L929-conditioned medium in tissue culture plates (Thermo). Fresh medium was added on day 3 and day 5. After 7 days, macrophages were fully differentiated and the culturing media were replaced with DMEM, 1% pen-strep supplemented with 4% L929-conditioned medium to normalize exposure to serum-derived lipoproteins. On day 8, macrophages were treated with reconstituted HDL CSL111 (CSL Behring) with an APOA1 to soybean phosphatidylcholine ratio of 1:150 28 for 20 hours. The BMDMs were washed twice with PBS, fresh DMEM, 1% pen-strep supplemented with 4% L929-conditioned medium was added with or without LPS (Cell signaling) or recombinant mouse IFNβ (R&D systems) for the indicated time. For thioglycollate-elicited macrophages isolation and culture, wild-type or Ldlr−/− mice were injected with thioglycollate (1 ml; 3% thioglycollate, BD, in PBS) i.p. and peritoneal macrophages were collected via PBS lavage 3 days after the injection and cultured in DMEM supplemented with 4% L929-conditioned medium for 24 hours. After stimulation with LPS cells were lysed in RNA lysis buffer for RNA isolation. In some experiments chemical inhibitors or ER stressors were added after rHDL treatment but prior to LPS stimulation and remained in the medium during the LPS stimulation. These included: p38 MAPK inhibitor (BIRB0796 1 μM, Axon Medchem), ASK1 inhibitor (Selonsertib 10 μM, Selleckchem), ER stress inhibitor Sodium phenylbutyrate (4PBA 5mM, Sigma), IRE1a kinase inhibitor (KIRA6 100 nM or 500 nM, EMD Millipore), PKC inhibitor (Ro31-8425 10 μM, Sigma), Tunicamycin (Tm 2.5 μg/ml, Sigma). The duration of the treatment is described in the Figure legend of each experiment. To assess TLR4 degradation lysosomal (Chloroquine 50 μM, Invivogen), proteasomal (MG132 10 μM, Sigma) or protease inhibitors (Pepstatin 10 μM, Leupeptin 20 μM, E64 20 μM, Calpepstin 10 μM, Sigma) were added during the last 4-6 hours of the rHDL treatment (Supplemental Figure IV-C). Human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by density gradient centrifugation using Ficoll-Pague Plus (GE Healthcare) and 2.5×106 PBMCs were plated into 12-well cell culture plates in RPMI-1640 medium with L-glutamine (Corning). After 3 hours, non-adherent cells were removed and adherent monocytes were differentiated into macrophages by culture in RPMI-1640 supplemented with 10% human serum (Sigma), and 20 ng/mL recombinant human macrophage colony-stimulating factor (M-CSF, Peprotech) for 7 days. PBMC-derived macrophages were then treated with reconstituted HDL CSL111 for 20 hours followed by LPS stimulation for 4 hours. For the nHDL experiments, BMDMs were incubated in RPMI1600 (Corning) supplemented with 30% L929-conditioned medium, 7% FBS (Thermo), 1% pen-strep (Thermo) in tissue culture plates (Thermo). After 7 days, BMDMs were treated with nHDL in RPMI 1600 media containing 2% FBS for 18 hours, followed by washing of cells and LPS stimulation (ultrapure LPS; 10 ng/ml; List Biological Laboratories Inc.; NC9633766) for 4 hours in the presence of 30% L929-conditioned medium. For some experiments, BMDMs were treated with the cholesterol-depleting agent, methyl-β-cyclodextrin (MβCD 10 μM, Sigma) or with MβCD complexed with cholesterol (100 μg/ml, Sigma) to prevent cholesterol depletion before stimulation of cells with LPS. The human THP-1 monocytic cell line was obtained from American Type Culture collection (Manassas, VA; ATCC® TIB-202™) and maintained in RPMI with 10% FBS, 2 mM L-glutamine and 1% penicillin/streptomycin. THP-1 monocytes were differentiated into macrophage-like cells by incubation in medium supplemented with phorbol 12-myristate 13-acetate (PMA, 50 ng/ml, Sigma). After 3 days of differentiation, cells were pre-treated with nHDL for 18 hours, followed by washing of the cells and stimulation with LPS (10 ng/ml, 4 hours).
Antisense oligonucleotides (ASO) transfection
BMDMs were cultured in non-tissue culture plates (Thermo) and cellstripper (Corning) was used to gently dislodge them from the plates on day 5. BMDMs were then plated on 24 well plates (Thermo) and received either control or Ask1 single-stranded antisense nucleotides (Qiagen, custom made Antisense LNA GapmeRs). Briefly, in a single well 2 ul Lipofectamine RNAiMAX Reagent (Thermo) in 50 μl opti-MEM (Thermo) was mixed with 100 nM of ASO in 50 μl opti-MEM and incubated in room temperature for 20 minutes. BMDMs in DMEM (Corning) 10% FBS (Thermo) supplemented with 20% L929-conditioned medium (no pen-strep was added) was added in the well (400 μl, total of 350000 cells per well) and incubated overnight. The next day cells were washed with PBS and treated with rHDL, followed by LPS stimulation.
Quantitative PCR
Macrophages were washed twice with cold PBS and lysed in RNA lysis buffer (Qiagen, Zymo Research or Takara Bio USA, Inc). RNA was isolated using RNeasy kits (Qiagen), RNA MiniPrep kits (Zymo Research) or the Nucleospin RNA plus (Takara Bio USA, Inc) kit according to the manufacturers’ protocol. cDNA was prepared using first strand synthesis kits (Thermo) and qPCR was performed on an ABI StepOnePlus machine with SYBR reagents (Thermo). Primers for qPCR assays are provided in Supplemental Table V. The housekeeping gene m36B4 RNA or Rn18s was used to account for variability in the initial quantities of mRNA.
RNA-Seq
For RNA-Seq, macrophages were washed twice with cold PBS and lysed in TRIzol reagent (Thermo). RNA was isolated from the aqueous phase using RNeasy kits (Qiagen). RNA with RIN > 8 was subjected to poly-dT pulldown using magnetic beads (NEB) before preparation for RNA-Seq using RNA Ultra kits (NEB). Libraries were sequenced on a NextSeq 500 (Illumina) and reads were aligned to the mm10 transcriptome using HISAT2 29 after adaptor trimming using cutadapt 30. Reads counts per gene for RefSeq genes were computed using featureCounts 31. Counts were normalized to reads per kilobase per million (RPKM) and processed for pairwise differential expression analysis of selected conditions using DESeq2 32 with an False Discovery Rate (FDR)-adjusted p-value cutoff of 0.05. Gene Ontology analysis was performed using the PANTHER database 33 and sequence motif analysis was performed using HOMER 34.
Immunoblot analysis
BMDMs were washed with PBS twice and lysed with RIPA buffer (Boston bioproducts) supplemented with protease and phosphatase inhibitors (Thermo). The protein quantity was measured with the Pierce BCA protein assay kit (Thermo), subjected to SDS-PAGE on 5-20% gradient gels (Biorad) with a protein ladder (Biorad) and transferred to nitrocellulose membrane (Biorad). The membranes were blocked in 5% non-fat dry milk (Biorad) in Tris-buffered saline with 0.1% Tween-20 (Thermo). The primary and secondary antibodies and the dilution which they were used are: IκΒα (Cell signaling, 1:1000 dilution), p-p38 MAPK (Cell signaling, 1:1000 dilution), p38 MAPK (Cell signaling, 1:2000 dilution), p-ERK1/2 (Cell signaling, 1:1000 dilution), ERK1/2 (Cell signaling, 1:2000 dilution), p-JNK1/2 (Cell signaling, 1:1000 dilution), JNK1/2 (Cell signaling, 1:1000 dilution), p-IRF3 (Cell signaling, 1:1000 dilution), IRF3 (Cell signaling, 1:2000 dilution), p-IRF7 (Cell signaling, 1:500 dilution), IRF7 (Abcam, 1:1000 dilution), p-STAT1 (Cell signaling, 1:1000 dilution), STAT1 (Cell signaling, 1:2000 dilution), p-STAT3 (Cell signaling, 1:1000 dilution), STAT3 (Cell signaling, 1:2000 dilution), Ire1a (Cell signaling, 1:500 dilution), TLR4 (Cell signaling, 1:1000 dilution), IFNAR1 (Santa Cruz Biotechnology, 1:500 dilution), IFIT3 (EMD Millipore, 1:500 dilution), β-actin (Sigma, 1:8000 dilution), anti-rabbit IgG HRP-linked antibody (cell signaling, 1:5000 dilution), anti-mouse IgG HRP-linked antibody (GE Healthcare, 1:10000 dilution). For detection of horseradish peroxidase (HRP) activity, the Pierce ECL Western Blotting Substrate (Thermo) was used.
Secretion analysis
TNFA and CCL2 secretion was measured by enzyme-linked immunosorbent assay (ELISA) (R&D systems).
Flow cytometry
BMDMs were cultured in non-tissue culture plates (Thermo) and cellstripper (Corning) was used to gently dislodge them from the plates to avoid damage to cell surface antigens. DMEM with 10% FBS and 1% PS was used to neutralize the cellstripper and the BMDMs were centrifuged, washed, and resuspended in HBSS (0.1% BSA, 5 mM EDTA). The cells were stained for 30 minutes with a cocktail of antibodies against mouse F4/80-Pacific Blue clone BM8 (Biolegend; 123124, 1:200 dilution) to identify macrophages and TLR4-PE/Cy7 clone SA15-21 (Biolegend; 145408, 1:200 dilution). For intracellular staining cell were fixed with a fixation/permeabilization solution for 10 minutes (BD Biosciences), washed with BD Perm/Wash buffer (BD Biosciences) and then stained with antibodies. All samples were analyzed on an LSRII (BD Biosciences) or LSRFORTESSA (BD Biosciences), running FACSDiVa software. Resident peritoneal macrophages were collected from APOA1Tg Ldlr−/− mice and Ldlr−/− littermates using PBS containing ultrapure 5 mM EDTA (Invitrogen). Cell suspensions were preincubated with anti-CD16/CD32 mAb (eBiosiences; 25-4801-82, 1:2000 dilution) to block FcγRII/III receptors and stained on ice. The cells were stained for 30 minutes with fluorochrome-conjugated mAb in the following color staining combination of antibodies: PE-Cy7-labeled F4/80 clone BM8 (eBiosiences; 25-4801-82, 1:2000 dilution), APC-labeled Ly-6C clone 1A8 (eBiosiences; 17-593-80, 1:2000 dilution), PE-labeled CD11b clone M1/70 (eBiosciences; 12-0112-82, 1:4000 dilution), FITC-labeled CD45 clone 30-F11 (eBiosciences; 54-0451-82, 1:2000 dilution). All samples were analyzed on a FACSCanto II (BD Biosciences), running FlowJo software.
Liposome preparation
Cholesterol-rich and cholesterol free liposome was prepared using a modified protocol 35-37. Liposomes were prepared using 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids) with or without adding cholesterol (Sigma) (referred as POPC/cholesterol-liposomes or POPC-liposomes). For POPC/cholesterol-liposomes, 40 mg of POPC and 80 mg of cholesterol were dissolved in chloroform and dried under a gentle stream of nitrogen gas. To ensure complete removal of chloroform the liposomes were placed in a Speed-vac (Thermo) for 1 hour. Multilamellar vesicles (>1 μM) were formed by addition of DMEM to the lipid film and then shaking the mixture vigorously to detach the lipid layer from the walls of the tube. The liposomes were reduced in size by probe sonication on ice for 20 minutes (5 times of 4 minutes sonication with 5 minutes break in-between to avoid excessive heat generation). The preparation was subjected to centrifugation at 10,000 g for 20 minutes. The supernatant was collected and stored at 4°C for no more than 24 hours. The cholesterol in the liposomes was assessed using an enzymatic kit from Wako (Cholesterol E). For the POPC liposomes preparation, the same procedure was followed without adding any cholesterol. To load macrophages with cholesterol, they were incubated with POPC/cholesterol-liposomes (~ 1 mg cholesterol/ml) for 20 hours. To remove cholesterol, macrophages received rHDL or POPC-liposomes (cholesterol-free), that similar to rHDL act as cholesterol acceptors, for 20 hours. In some experiments, BMDMs or thioglycollate-elicited macrophages were cholesterol loaded using AcLDL (50 μg/ml human acetylated LDL; Kalen Biomedical, 770201) for 48 hours before HDL and LPS treatments.
Preparation of nHDL and isolation of HDL subpopulations
HDL (density 1.063–1.21 g/ml) was isolated from plasma of APOA1Tg;Ldlr−/− mice by density gradient ultracentrifugation 38. Total HDL isolated by ultracentrifugation from plasma was used to further separate small, medium and large fractions of HDL by FPLC. In short, nHDL was separated by a Superdex 200 Increase 10/300 GL column at 4°C in 150 mM ammonium acetate buffer with an ÄKTA 10 fast protein liquid chromatography system (GE Healthcare Bio-Sciences, Pittsburgh, PA). Fractions containing APOA1 (200 μL) were collected and combined to represent different sizes of HDL. The size distribution of each class of particles before and after isolation, as determined by calibrated ion mobility analysis 39, are shown in Supplemental Figure II-C,D. HDL particle concentrations were determined by calibrated ion mobility analysis 39.
Cholesterol efflux assay
Macrophage cholesterol efflux capacity was assessed with J774 macrophages labeled with [3H]cholesterol and stimulated with a cAMP analog, as described by Rothblat and colleagues40. Efflux of [3H]cholesterol was measured after a 4 h incubation in medium with nHDL subpopulations (100 nM HDL particle concentration, equivalent to about 10 μg HDL protein per milliliter). Cholesterol efflux capacity was calculated as the percentage of total [3H]cholesterol (medium plus cell) released into the medium of J774 cells stimulated with cAMP after the value obtained with cells stimulated with medium alone was subtracted.
BODIPY staining
Lipid droplets were identified with the neutral lipid dye BODIPY 493/503 (10 μg/ml, Invitrogen) using confocal microscopy. Resident peritoneal macrophages from Ldlr−/− and APOA1Tg Ldlr−/− mice were plated on eight-chambered Lab-Tek borosilicate cover glasses (ThermoFisher Scientific, Nunc), after flow cytometric verification of similar cell populations in the mice (Supplemental Figure VI-B). Staining was performed after one hour of adhesion purification of macrophages using BODIPY 493/503. Pictures were taken using a Nikon A1R Confocal microscope at a 60X magnification.
Aortic digestion and CD11b+ cell isolation
Aortas were isolated from Apoe−/− mice or Ldlr−/− mice fed after 4 and 8 weeks of WTD respectively and rHDL infusions as described in the figure legend (Figure 6) of each experiment. Whole aortas were isolated, cut in small pieces and incubated for 1 h at 37°C with 1.25 mg/ml liberase TH (Sigma), 120 units/ml hyaluronidase (Sigma), and 160 units/ml DNase (Sigma). Samples were centrifuged and CD11b+ cells were isolated using CD11b+ coated beads (Miltenyi Biotec). RNA from aortic CD11b+ cells was isolated using a Qiagen RNeasy kit.
Statistics
All data are presented as mean ± SEM. In cell culture experiments, sample size (n) represents the number of individually differentiated primary macrophage cultures in each experiment. In aortic CD11b+ and thioglycollate-elicited peritoneal macrophage experiments from rHDL–infused mice (Figure 6), sample size (n) represents the number of individual mice in each experiment. The statistical parameters (n, mean, SEM, and statistical tests used) can be found within the figure legends. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. To define differences between 2 datasets, the unpaired Student’s t test was used or unpaired t test with Welch’s correction (for datasets without equal variance). When the criteria were not met for analyses by parametric test, the nonparametric Mann Whitney test was used. To assess differences between three groups or more, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used. When the criteria were not met for analyses by parametric test, the nonparametric Kruskal-Wallis test with Dunn’s multiple comparisons test was used. In the case of time-course data two-way ANOVA with Sidak’s post hoc test was used to determine significance. The criterion for significance was set at p<0.05. Statistical analyses were performed using GraphPad Prism Version 8.2.1 (San Diego, CA). For RNA-Seq, gene expression differences were evaluated by Wald test after linear model fitting using DESeq2 and genes significant at 5% FDR were considered to be differentially expressed.
Data availability
The NCBI GEO accession number for high throughput sequencing data reported in this paper is GSE129347.
Results
Transcriptome profiling of HDL-treated macrophages
Toll-like receptors have been suggested to bridge innate immunity to atherosclerosis 41 and danger signals in the atherosclerotic plaque likely activate TLRs, including TLR4 42. To study the effects of HDL on inflammatory gene expression in macrophages we cultured bone marrow-derived macrophages (BMDMs) from wild type mice, treated them with reconstituted HDL (rHDL, CSL111) for 20 hours, washed the cells thoroughly to remove any residual HDL, and then stimulated them with LPS. The rHDL consists of human APOA1 (the main protein component of HDL) complexed with phospholipids and we have shown that it can promote cholesterol efflux both through ABCA1/G1 transporters as well as in a transporter-independent passive fashion 43. LPS is a TLR4 ligand that upon binding activates the expression of many inflammatory genes in macrophages.
To obtain a global view of gene expression changes, we carried out RNA-seq transcriptomic analysis. LPS induced the expression of 1404 genes (fold change ≥1.5, false discovery rate (FDR)-corrected p ≤ 0.05) in BMDMs (Supplemental Figure I-A). Pre-incubation with rHDL (150 μg of human APOA1 protein/ml) repressed the expression of 728 genes whereas it induced the expression of 761 genes (Supplemental Figure I-A). Out of the 728 genes down-regulated by rHDL, 300 were LPS inducible, indicating a widespread anti-inflammatory effect. However, 85 out of 782 genes up-regulated by rHDL were also LPS inducible (Figure 1A and Supplemental Figure I-A). While this confirms a pro-inflammatory effect of rHDL, this effect was more circumscribed than reported by van der Vorst and colleagues who found that about 50% of all LPS-induced genes were up-regulated by HDL using microarray analysis 17. To identify biological functions that are related to the gene changes promoted by rHDL we used Gene Ontology (GO) analysis. The top GO categories for rHDL-induced genes were cholesterol biosynthetic process, followed by chemokine signaling pathway, autophagy and vacuole organization (Figure 1B and Supplemental Table I). In response to cholesterol removal by rHDL, macrophages induced several cholesterol biosynthetic genes, such as Hmgcr and Dhcr24. Interestingly, there was enrichment for the category macroautophagy consistent with the association of cholesterol depletion with cell stress/autophagy 44. The pro-inflammatory effect of rHDL involved chemokines and some of the most up-regulated genes were Ccl2 (Mcp-1), Cxcl1 and Cxcl2 (Figure 1D). GO analysis for the rHDL-repressed genes revealed the following categories: cytokine biosynthetic process, cellular response to IFNβ, defense response to virus and regulation of IL-1β production (Figure 1C, E and Supplemental Table II). We next performed HOMER analysis to identify transcriptional regulatory motifs associated with gene expression changes. Consistently, for rHDL-induced genes HOMER analysis revealed E-boxes likely associated with SREBP activation as well as TFE3 and MITF, associated with autophagy and lysosomal biogenesis 45, 46 (Supplemental Figure I-B) whereas rHDL-repressed genes were enriched for interferon response elements (ISREs and IRFs) (Supplemental Figure I-C).
Figure 1. Transcriptome profiling of rHDL-treated macrophages in response to LPS.

Wild type BMDMs were treated for 20 hours with 150 μg/ml rHDL (reconstituted HDL), washed with PBS and stimulated with 100 ng/mL LPS for 4 hours and harvested for RNA-Seq (n=3/condition). For cholesterol loading (CL) macrophages were incubated with POPC/cholesterol-liposomes (~ 1 mg cholesterol/ml) for 20 hours prior to rHDL treatment. (A) Heatmap of >1.5-fold induced or repressed genes at 5% FDR colored by row-normalized Z score, with extent of induction by LPS indicated on right (FC; Fold change). (B, C) PANTHER GO categories enriched in rHDL-induced or rHDL-repressed genes (Bonferroni-adjusted P < 0.05). Row-normalized z-score for (D) rHDL-induced genes in the GO category “Chemokine signaling pathway”, (E) rHDL-repressed genes in the GO category “Cellular response to interferon β” and (F) ER stress response genes affected by rHDL in both the cholesterol loaded and non-cholesterol loaded conditions. See also Supplemental Figure I.
To explore the role of cholesterol, we loaded BMDMs with cholesterol by treating them with POPC/cholesterol-liposomes, washed thoroughly, added rHDL then stimulated with LPS. Surprisingly, RNA-seq analysis showed that cholesterol loading attenuated rHDL induced differential gene expression: both the anti- and pro-inflammatory effect of rHDL were reduced (Figure 1A). In this setting, the only GO category that came up in the rHDL-induced genes was cholesterol biosynthetic process, but there were fewer cholesterol biosynthetic genes that were induced by rHDL compared to the number of these genes in the non-cholesterol loaded condition (Supplemental Table III and Supplemental Table IV). The GO analysis for the rHDL-repressed genes showed enrichment for ER stress and unfolded protein response (Supplemental Table III). Interestingly, the same set of ER stress related genes that were down regulated by rHDL in the cholesterol loaded condition were upregulated by rHDL in the non-cholesterol loaded condition (Figure 1F). This suggests that perturbation of ER cholesterol content in either direction can induce a stress response in LPS-treated macrophages. The impact of HDL-mediated cholesterol efflux may be either to induce or alleviate ER stress depending on cholesterol loading status of the macrophage.
Pro- and anti-inflammatory effects of HDL depend on cholesterol efflux
We sought to verify and extend the main findings suggested by RNA-seq by using different amounts and forms of HDL, by varying the cholesterol loading status of BMDMs and by assessing kinetics of responses. Further in vitro experiments in BMDMs using different doses of rHDL, ranging from 50-300 μg/ml, showed a robust pro-inflammatory effect only at the higher doses of rHDL, as indicated by the mRNA expression of Tnfa and Ccl2 and the respective protein secretion (Figure 2A and Supplemental Figure II-A). In contrast, the anti-inflammatory effect, as indicated by the mRNA and protein expression of Ifit3, was present at all rHDL doses (Figure 2A and Supplemental Figure II-A). Incubation of rHDL alone, without any LPS stimulation, did not have a pro-inflammatory effect (not shown). Since both anti- and pro-inflammatory effects were readily observed at 150 μg/ml rHDL, we chose to use this concentration in most of the rHDL experiments unless stated otherwise. To examine if similar effects are exerted by different forms of HDL, we isolated HDL (native HDL; nHDL) by ultracentrifugation from APOA1Tg;Ldlr−/− mice fed a normal laboratory diet, pretreated BMDMs with this nHDL and then stimulated them with LPS. These mice express a human APOA1 transgene, which results in a marked suppression of mouse APOA1 and a human-like HDL particle profile 47. Similar to rHDL, nHDL at higher doses exerted a pro-inflammatory effect in LPS-stimulated BMDMs but did not induce pro-inflammatory effects in the absence of LPS (Supplemental Figure II-B). Because nHDL is a mixture of HDL particles differing in size and shape, we fractionated nHDL in small HDL (7-8.5 nm diameter) medium HDL (8.5-10 nm diameter) and large HDL (10-12.5 nm) (Supplemental Figure II-C, D) and treated BMDMs. Small nHDL exhibited the highest capacity to promote cholesterol efflux from macrophages (Figure 2B) and increased inflammatory gene expression at a higher level than medium and large nHDL when compared at equal HDL particle concentrations (Figure 2C). A high dose of nHDL brought out pro-inflammatory effects in response to LPS in human THP1 cells as well (Supplemental Figure II-E). However, at a similar high dose rHDL exhibited only anti-inflammatory effects on LPS-induced inflammatory gene expression in human PBMC-derived macrophages (Supplemental Figure II-F).Together, these data demonstrate that different forms of HDL not only promoted anti-inflammatory effects on LPS-stimulated macrophages, but also a chemokine-specific pro-inflammatory effect. The magnitude of these effects correlates with the ability of HDL to promote cholesterol efflux from macrophages. In contrast to the results with BMDMs and THP1 cells, primary human monocyte-derived macrophages showed an anti-inflammatory effect at a relatively high dose of rHDL.
Figure 2. Pro- and anti-inflammatory effects of HDL on macrophages depend on cholesterol content.

Wild type BMDMs were treated for 20 hours with rHDL (reconstituted HDL) or nHDL (native HDL), washed with PBS and stimulated with LPS, as described below and harvested for RNA. For altering cholesterol content, macrophages were incubated with POPC/cholesterol-liposomes or AcLDL prior to rHDL or nHDL treatment to load them with cholesterol, and then washed before rHDL or nHDL treatment. To remove cholesterol, BMDMs received POPC-liposomes (cholesterol-free) instead of rHDL, that similar to rHDL act as cholesterol acceptors. (A) Dose effect of rHDL (50, 150 and 300 μg/ml) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (B) Effect of different nHDL subpopulations (S-Small, M-Medium, L-Large) (10 μg protein/ml HDL particles, corresponding to 0.1 μM HDL particles as measured by calibrated ion mobility analysis) on cholesterol efflux capacity in J774 cells. (C) Effect of different nHDL subpopulations (50 μg/ml, corresponding to 0.5 μM HDL particles) on inflammatory gene expression in LPS-stimulated (10 ng/ml, 4 hours) BMDMs. (D) Effect of rHDL (150 μg/ml) on LPS-induced (100 ng/ml, 4 hours) inflammatory gene expression, Hmgcr and Abca1 expression in BMDMs pre-treated with POPC/cholesterol-liposomes (~ 1 mg cholesterol/ml) for 20 hours, prior to rHDL treatment. (E) Effect of nHDL (100 μg/ml) on inflammatory gene expression in LPS-stimulated (10 ng/ml, 4 hours) BMDMs preloaded with AcLDL cholesterol (50 μg/ml, 48 hours). (F) Effect of rHDL (150 μg/ml), POPC-liposomes and POPC/cholesterol-liposomes (~ 1 mg cholesterol/ml) on LPS-induced (100 ng/ml, 4 hours) inflammatory gene expression, Hmgcr and Abca1 expression in BMDMs. mRNA expression was evaluated by qPCR and mean ± SEM is plotted (n=4). Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. Significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (A, C, E, F) or nonparametric Kruskal-Wallis with Dunn's multiple comparisons test (D), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data are representative of at least two independent experiments. See also Supplemental Figure II.
To confirm the RNA-seq data, we loaded either BMDMs or thioglycollate-elicited peritoneal macrophages using POPC/cholesterol-liposomes or acetylated LDL (AcLDL). As shown in Figure 2D, cholesterol loading using POPC/cholesterol-liposomes blocked the rHDL-induced expression of Tnfa and Ccl2 as well as the rHDL-repressed expression of the IFN-responsive genes, Ifit3 and Mx1. Similarly, cholesterol loading of BMDMs or thioglycollate-elicited peritoneal macrophages using AcLDL reduced the pro-inflammatory effects of nHDL (Figure 2E and Supplemental Figure II-G). To confirm the cholesterol loading status of the macrophages we measured the expression of Hmgcr and Abca1. As expected, rHDL increased the expression of Hmgcr and reduced the expression of Abca1, reflecting the depletion of cholesterol in the cells (Figure 2D). In contrast, cholesterol loading significantly reduced the expression of Hmgcr and increased the expression of Abca1. Incubation of rHDL in the cholesterol loaded condition did not affect Hmgcr expression, consistent with the RNA-seq results (Supplemental Table IV) and only modestly reduced Abca1 expression (Figure 2D). A similar effect was observed even when the cholesterol-loaded macrophages were incubated with the highest dose of rHDL (300 μg/ml). These results indicate that both pro- and anti-inflammatory effects of HDL are reduced by cholesterol loading of macrophages, paralleling a decreased ability of HDL to perturb cholesterol homeostasis as measured by expression of cholesterol biosynthesis and efflux genes.
To further assess whether the pro- and anti-inflammatory effects depend on cholesterol removal, we treated the BMDMs with POPC-liposomes that do not contain cholesterol and can absorb cholesterol from the cell membranes by aqueous diffusion. Similar to rHDL, the POPC-liposomes induced the expression of Tnfa and Ccl2 and repressed the expression of Ifit3 and Mx1 (Figure 2F). The impact of POPC-liposomes on cholesterol homeostatic gene expression was comparable to rHDL, as indicated by increased Hmgcr and reduced Abca1 expression (Figure 2F). Cholesterol pre-loading attenuated the POPC-liposomes effects, especially the increase of Tnfa and Ccl2, further supporting a cholesterol dependent effect on inflammatory gene expression (Supplemental Figure II-H). Similar to POPC-liposomes, depletion of cholesterol with β-methylcyclodextrin caused an induction of inflammatory gene expression, whereas β-methylcyclodextrin complexed with cholesterol did not significantly induce inflammatory gene expression (Supplemental Figure II-I). Overall, our experiments indicate that both the pro- and anti-inflammatory effects of HDL or liposomes are related to cholesterol efflux and are dependent on the macrophage cholesterol content.
To gain a more complete picture of how HDL affects TLR4 signaling, we stimulated BMDMs with LPS at time points ranging from 15 minutes to 4 hours. These experiments revealed a biphasic response involving an initial phase (0-60 min) when only anti-inflammatory effects were produced by rHDL, followed by a second phase (120-240 min) in which there was an increase in Tnfa, Ccl2, and Cxcl1/2 expression. The reduced expression of Type I interferon responsive genes (Ifit3, Mx1) also appeared to be biphasic with an initial suppression followed by a more sustained reduction (Figure 3A). In addition, the initial anti-inflammatory effects of rHDL were associated with decreased IκB degradation and decreased p38 MAPK, JNK and ERK phosphorylation (Figure 3B).
Figure 3. Kinetics of inflammatory responses to rHDL.

Wild type BMDMs were treated for 20 hours with rHDL, washed with PBS and stimulated with LPS, as described below and harvested for RNA. (A) Effect of rHDL (150 μg/ml) on inflammatory gene expression in LPS-stimulated (100 ng/ml, time points from 15-240 minutes) BMDMs. Results are expressed as the change of mRNA expression in rHDL treated versus non-treated cells. (B) Effect of rHDL (150 μg/ml) on IκBa protein expression and p38 MAPK/JNK/ERK phosphorylation under similar LPS conditions. Results are shown as mean ± SEM (n=4). Significance was determined by two-way ANOVA with Sidak’s post-hoc test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data are representative of at least two independent experiments.
The pro-inflammatory effect of HDL is triggered by activation of the IRE1a/ASK1/p38 MAPK axis
Studies by van der Vorst and colleagues suggested that HDL induces a pro-inflammatory effect in macrophages by increasing PKC activation 17. We treated BMDMs with the broad-spectrum PKC inhibitor Ro31-8425 at the same concentration used in the van der Vorst study. Although, PKC inhibition blocked the HDL pro-inflammatory effect, it also markedly reduced LPS signaling (over 85% percent), virtually blocking the LPS response (Supplemental Figure III-A). This suggests a general suppression of LPS responses rather than an HDL-specific effect. Thus, we sought evidence for an alternative pro-inflammatory mechanism.
HOMER motif analysis of the rHDL-induced genes indicated an enrichment for JunB and AP1 transcription factors (Supplemental Figure I-B). Activation of AP1 by MAPKs downstream of TLR signaling induces the expression of inflammatory genes, including many of the chemokine genes that are induced by rHDL, such as Cxcl1, Cxcl22, Ccl2 and Ccl7 48. Moreover, we observed that there was an increase in p38 MAPK phosphorylation by rHDL in the later LPS time points (Figure 3B), indicating that p38 MAPK may be involved in the pro-inflammatory effect of rHDL. We first confirmed in multiple experiments that rHDL increases p38 MAPK phosphorylation after prolonged LPS stimulation (Figure 4A). To assess if the pro-inflammatory effect is specific to p38 MAPK, we treated BMDM with the p38 inhibitor, BIRB0796. Indeed, p38 MAPK inhibition blocked the induction of Tnfa, Ccl2, Cxcl1 and Cxcl2 by rHDL but did not affect the reduction of Ifit3 and Mx1 (Figure 4B). Pre-treatment of the BMDMs with POPC/cholesterol-liposomes attenuated the effect of rHDL on p38 MAPK phosphorylation (Supplemental Figure III-B), in agreement with the gene expression results (Figure 2D). In contrast, POPC-liposomes increased p38 MAPK phosphorylation, similarly to rHDL (Supplemental Figure III-B). This indicates that cholesterol efflux-dependent late inflammatory responses are mediated through increased p38 MAPK signaling.
Figure 4. The pro-inflammatory effect of HDL is triggered by activation of the IRE1a/ASK1/p38 MAPK axis.

Wild type BMDMs were treated for 20 hours with rHDL, washed with PBS and stimulated with LPS, as described below. Macrophages were treated with the indicated inhibitors after removing rHDL and washing the cells, but prior to LPS stimulation and remained in the medium during the LPS stimulation. (A) Effect of rHDL (150 μg/ml) on p38 MAPK phosphorylation in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (B) Effect of rHDL (150 μg/ml) and p38 MAPK inhibitor (BIRB0796, 1 μM, added 1 hour prior to LPS) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (C) Effect of rHDL (150 μg/ml) and ASK1 inhibitor (Selonsertib, 10 μM, added 2 hours prior to LPS) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (D) Effect of rHDL (150 μg/ml) and Ask1 knock-down on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. BMDMs were treated for 24 hours with control or Ask1 ASO (100 nM), then treated with rHDL for 20 hours, followed by LPS stimulation (100 ng/ml, 4 hours). (E) Effect of rHDL (150 μg/ml) and the ER stress inhibitor Sodium phenylbutyrate (4PBA, 5 mM, added 2 hours prior to LPS) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (F) Effect of rHDL (300 μg/ml) and the ER stressor Tunicamycin (Tm, 2.5 μg/ml, added 2 hours prior to LPS) on IRE1a protein expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (G) Effects of cholesterol pre-loading followed by rHDL (300 μg/ml) or the ER stressor Tunicamycin (Tm, 2.5 μg/ml, added 2 hours prior to LPS) on IRE1a protein expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. (H) Effect of rHDL (300 μg/ml) and the IRE1a kinase inhibitor (KIRA6, 500 nM, added 1 hour prior to LPS) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 4 hours) BMDMs. The results are shown as mean ± SEM (n=4 for gene expression, n=3 for protein expression). Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. Significance was determined by unpaired t test (A) or one-way ANOVA with Tukey’s multiple comparisons test , *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (B, C, D, E, F, G, H). Data are representative of at least two independent experiments. See also Supplemental Figure III.
ASK1 enhances p38 MAPK phosphorylation in response to TLR4 activation in macrophages 49. ASK1 is a MAPKKK (MAPK kinase kinase) upstream of p38 MAPK that is activated in response to stress signals 50. Such signals include oxidative stress, calcium overload, endoplasmic reticulum stress and inflammatory stimuli, like TNFα and LPS49, 51, 52. Selonsertib is a selective ASK1 inhibitor that in early clinical trials showed promise against NASH by improving inflammation and fibrosis 53. Pre-treatment of BMDMs with Selonsertib blocked the rHDL-mediated induction of Tnfa, Ccl2 and Cxcl1 (Figure 4C) as well as p38 MAPK phosphorylation (Supplemental Figure III-C). Consistently, ASO-mediated knock-down of Ask1 also blocked the induction of these genes by rHDL (Figure 4D). rHDL pre-treatment did not increase ROS production more than LPS alone and N-acetyl-L-cysteine, an ROS scavenger, did not affect the pro-inflammatory effect of rHDL (not shown), indicating that ROS generation is not responsible for ASK1 activation by rHDL. ER stress can activate ASK1 and this involves the association of IRE1a, an ER membrane protein that acts as an ER stress signal transducer, with TRAF2 and ASK1 51, 54. The RNA-seq results showed that rHDL may cause cell stress as indicated by the GO categories of macroautophagy/vacuole organization and the induction of ER stress genes (Figure 1B, F). Although, rHDL did not increase the expression of typical ER stress targets like Ddit3 (Chop) and spliced Xbp1 (Supplemental Figure III-D), treatment of the BMDMs with 4-phenylbutyrate, commonly used to alleviate ER stress, blocked the pro-inflammatory effect of rHDL (Figure 4E). Moreover, rHDL increased IRE1a levels (both total and phosphorylated) similar to tunicamycin, a potent ER stressor (Figure 4F). Cholesterol loading attenuated the rHDL-mediated increase of Ire1a but did not affect Ire1a in response to tunicamycin, indicating a distinct mechanism of Ire1a activation (Figure 4G). While HDL-treatment alone was sufficient to increase IRE1a, the induction of p38 MAPK and inflammatory responses required an additional LPS stimulus. In turn, treating the BMDMs with Ire1a kinase inhibitors KIRA6 55 or 4μ8c 56, 57, attenuated the induction of Tnfa, Ccl2 and Cxcl1 by rHDL (Figure 4H and Supplemental Figure III-E). These results support that rHDL plus LPS stimulation exerts a pro-inflammatory effect through a modified ER stress response involving the IRE1a/ASK1/p38 MAPK signaling axis likely secondary to ER cholesterol depletion.
Early anti-inflammatory effects reflect reduced TLR4 levels and signaling
The reduction of IκB degradation and p38 MAPK, JNK and ERK phosphorylation in early LPS stimulation by rHDL (Figure 3B) suggests reduced TLR4 signaling upstream of ΙkB and MAPKs. Our previous studies suggested that membrane cholesterol accumulation in macrophages lacking both of the major cholesterol efflux transporters, ABCA1 and ABCG1 (Abca1−/−Abcg1−/− macrophages), increases TLR4 cell surface expression, thus augmenting LPS responses 21. We hypothesized that the initial rHDL anti-inflammatory effect may involve a reduction in TLR4 expression. Indeed, flow cytometry experiments showed that rHDL pre-incubation reduced both cell surface and intracellular TLR4 expression before and after LPS stimulation (Figure 5A and Supplemental Figure IV-A). A reduction of total TLR4 was also confirmed by Western blot (Supplemental Figure IV-B). This does not involve increased protein degradation through the endosomal/lysosomal pathway since treating the macrophages with lysosomal, proteasomal or protease inhibitors did not block the downregulation of TLR4 by rHDL (Supplemental Figure IV-C). In cholesterol loaded BMDMs, rHDL treatment failed to decrease TLR4 expression (Figure 5B), inhibit IκB degradation and reduce p38 MAPK phosphorylation (Figure 5C), consistent with an effect of cholesterol loading to block the initial reduction of inflammatory gene expression by rHDL (Supplemental Figure IV-D). In contrast, POPC-liposomes, similarly to rHDL, reduced TLR4 protein levels (Figure 5B), indicating that cholesterol removal drives the early reduction in TLR4 and the early anti-inflammatory effect. In contrast to an earlier report that proposed increased Atf3 expression as an anti-inflammatory effect of HDL 15, we found a reduction in Atf3 mRNA levels by rHDL in LPS-stimulated macrophages (Supplemental Figure IV-E). Since expression of Atf3 is increased upon TLR stimulation 15, 58, this is consistent with our finding of reduced TLR4 signaling.
Figure 5. Early anti-inflammatory effects reflect reduced TLR4 expression and late anti-inflammatory effects reflect reduction in STAT1 phosphorylation and Type 1 interferon signaling.

BMDMs were treated for 20 hours with rHDL, washed with PBS and stimulated with LPS or IFNb, as indicated, and harvested for RNA or protein. For altering cholesterol content macrophages were incubated with POPC/cholesterol-liposomes (to load them with cholesterol) prior to rHDL treatment or cholesterol-free POPC-liposomes (instead of rHDL, to remove cholesterol). (A) Effect of rHDL (150 μg/ml) on surface TLR4 expression in LPS-stimulated (100 ng/ml) BMDMs at the indicated time point. (B) Effect of rHDL (150 μg/ml) on TLR4 protein expression in BMDMs pre-treated with POPC/cholesterol-liposomes (~ 1 mg cholesterol/ml) for 20 hours or cells pre-treated with POPC-liposomes alone and stimulated with LPS (100 ng/ml) at the indicated time points. (C) Effect of rHDL (150 μg/ml) on IκBa protein expression and p38 MAPK phosphorylation in cholesterol loaded BMDM stimulated with LPS (100 ng/ml) at the indicated time point. (D) Effect of rHDL (150 μg/ml) on IRF7 phosphorylation in LPS-stimulated (100 ng/ml) BMDMs for the indicated time points. (E) Effect of rHDL (150 μg/ml) on inflammatory gene expression in LPS-stimulated (100 ng/ml, 2 hours) Ifnar−/− BMDMs. (F) Expression of Ifit3 and Mx1 in BMDMs treated with rHDL (150 μg/ml) followed by IFNb stimulation (5 ng/ml, 2 hours) (G). Effect of rHDL (150 μg/ml) on IFNAR1 expression and STAT1 phosphorylation in LPS-stimulated (100 ng/ml, 2 hours) or IFNb-stimulated (1 ng/ml, 2 hours) BMDMs. The results are shown as mean ± SEM (n=4 for gene expression, n=3 for protein expression). Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. Significance was determined by two-way ANOVA with Sidak’s post-hoc test (A, D), one-way ANOVA with Tukey’s multiple comparisons test (B, C), unpaired t test (G) or by unpaired t test with Welch’s correction (E, F), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data are representative of at least two independent experiments. See also Supplemental Figure IV and Supplemental Figure V.
Late reduction in Type I interferon responses reflects reduced interferon receptor signaling
Similar to our findings, Suzuki and colleagues 14 have shown in macrophages stimulated for 4 hours with LPS, that HDL selectively inhibits LPS-induced genes that are downstream of TRAM/TRIF signaling. However, rHDL produced no change in IRF3 phosphorylation which is immediately downstream of TRAM/TRIF (Supplemental Figure V-A), suggesting that a more distal signaling effect may be responsible for the sustained Type I Interferon anti-inflammatory effect. IFN-related genes, like Ifit3 and Mx1, show late induction in response to LPS downstream of IFNβ expression and subsequent IFNAR activation 59. IRF7 is an important mediator of interferon responses that acts downstream of the interferon receptor in a delayed manner 60. In contrast to the effects of IRF3, BMDMs pre-treated with rHDL exhibited reduced IRF7 phosphorylation (Figure 5D), consistent with reduced signaling downstream of the IFNAR. Moreover, in macrophages lacking IFNAR1, rHDL failed to repress Ifit3 and Mx1 expression but still increased Tnfa and Cxcl1 expression in response to LPS (Figure 5E). To further assess if rHDL exerts additional anti-inflammatory effects downstream of IFNAR activation, we treated wild type BMDMs with rHDL, followed by IFNβ stimulation. As in LPS-stimulated cells, rHDL repressed the expression of Ifit3 and Mx1 in response to IFNβ (Figure 5F). Consistently, rHDL reduced STAT1 and STAT3 phosphorylation, in response to LPS and IFNβ stimulation, without affecting IFNAR1 expression (Figure 5G and Supplemental Figure V-B). Our results show that rHDL reduces IFNAR signaling, explaining the sustained IFN-related anti-inflammatory effect that is observed in response to LPS.
In vivo effects of rHDL on macrophage inflammatory gene expression
To determine the in vivo relevance of findings in BMDMs, normal laboratory diet fed wild type mice were injected intravenously with 80 mg/Kg rHDL or PBS daily, for a total of 5 days before sacrificing the mice 2 hours after the last rHDL injection. The RNA from thioglycollate-elicited peritoneal macrophages stimulated with LPS ex vivo was extracted and assessed for inflammatory gene expression. Macrophages from the rHDL-infused mice showed significantly decreased expression of MYD88-dependent genes, such as Tnfa, Ccl2 and Il6 (Figure 6A), consistent with the early anti-inflammatory effect observed in vitro, but there was no difference in the expression of genes in the Type I interferon response. No pro-inflammatory effects were observed. In contrast, in a similar experiment in Ldlr−/− mice fed a Western-type diet (WTD) for 8 weeks, rHDL infusions failed to promote any changes in inflammatory gene expression (Supplemental Figure VI-A). Moreover, rHDL infusions in the wild type mice modestly increased Hmgcr expression, suggesting a lower macrophage cholesterol content in these mice (Figure 6A). Therefore, the anti-inflammatory effects observed in the macrophages of normal laboratory diet fed wild type mice likely reflect a better capability of rHDL to reduce macrophage cholesterol content compared to the WTD-fed Ldlr−/− mice.
In another approach to determine in vivo relevance, we isolated resident peritoneal macrophages from APOA1Tg;Ldlr−/− mice or Ldlr−/− littermate controls fed a normal laboratory diet (Supplemental Figure VI-B), adhesion purified the macrophages, and then stimulated them with LPS. The macrophages from APOA1Tg;Ldlr−/− mice exhibited enhanced inflammatory gene expression in response to LPS compared to the macrophages isolated from Ldlr−/− mice (Figure 6B). The sustained cholesterol efflux by high levels of circulating HDL in these mice was sufficient to cause a decrease in the lipid content of the macrophages as observed by BODIPY staining (Figure 6C) as well as a prominent increase in Hmgcr (Figure 6B). This supports the hypothesis that extensive cholesterol removal by HDL is necessary to significantly affect pro-inflammatory gene expression in vivo.
Reduced inflammatory gene expression in myeloid cells of atherosclerotic aortas
To determine if rHDL affects inflammatory gene expression in the macrophages of the atherosclerotic plaque, we fed a WTD to Apoe−/− mice (female, n=29 per group) for 4 weeks or Ldlr−/− mice (female, n=12 per group) for 8 weeks to promote the formation of early atherosclerotic lesions and then administered rHDL 80 mg/Kg or PBS intravenously once daily for 5 days. The thoracic aorta was digested and CD11b+ cells (comprising inflammatory macrophages, monocytes, and neutrophils) 61 were isolated and assessed for inflammatory gene expression. In the Apoe−/− mice, rHDL significantly reduced the expression of some, but not all, MYD88-dependent inflammatory genes including Cxcl1, Il6 and Il1b (Figure 6D). In contrast to the effects in BMDMs in vitro, but consistent with the peritoneal macrophage response in vivo, there was no effect on Type I interferon genes. In rHDL-infused Ldlr−/− mice there was also a trend towards reduced expression of the same genes, but it did not reach statistical significance possibly due to highly variable expression in the control group (Figure 6E). We did not detect any changes in Abca1 and Hmgcr expression in aortic CD11b+ cells (Figure 6D, E), suggesting that the amount of cholesterol efflux was insufficient to activate cholesterol homeostatic responses.
Discussion
The effects of HDL on inflammatory gene expression in macrophages are controversial. There are studies either supporting anti-inflammatory14, 15 or pro-inflammatory effects of APOA1/HDL 16, 17. Our studies may help to reconcile these discrepant results by showing that the pro-inflammatory response only occurs under conditions of marked cholesterol depletion. Importantly, we show an anti-inflammatory effect and no evidence of pro-inflammatory effects of rHDL infusion in myeloid cells isolated from atherosclerotic plaques and only anti-inflammatory effects in primary human/monocyte macrophage cultures.
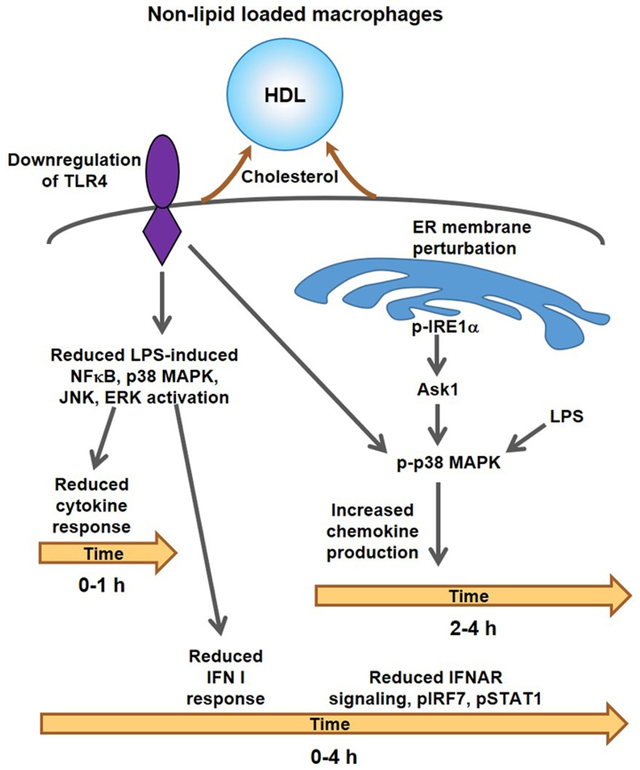
In contradistinction to many earlier studies 14-17, we show that both anti- and pro-inflammatory effects of HDL are primarily responses to cholesterol efflux and are attenuated by cholesterol loading of macrophages. Initial anti-inflammatory effects of rHDL reflect reduced TLR4 levels and signaling (Graphic abstract). This is followed by a sustained anti-inflammatory effect mediated downstream of IFNAR and involving suppression of Type I interferon responses. At the same time there is increased expression of Tnfa and some chemokine genes. The late induction of chemokine-enriched inflammatory genes represents an ER stress response mediated by IRE1a/ASK1/p38 MAPK signaling. Highlighting that these changes are mediated by cholesterol depletion, both pro- and anti-inflammatory responses to HDL could be simulated by cholesterol-poor, but not cholesterol-enriched PC liposomes, or β-methylcyclodextrin and were blunted or abolished in cholesterol loaded macrophages.
Our experiments show that the pro-inflammatory effect of HDL depends on p38 MAPK phosphorylation and entails the activation of the IRE1a/ASK1 axis. IRE1a and ASK1 are activated in response to ER stress signals 50, 51, 54 and subsequently can activate MAPKs 51, 52, 62. The RNA-seq results also pointed to a stress response induced by rHDL as evident by the GO categories enrichment of macroautophagy/vacuole organization and the induction of specific ER stress genes. Moreover, the pro-inflammatory effect was blocked in cholesterol loaded macrophages. Therefore, it is possible that severe cholesterol depletion by high levels of rHDL may cause perturbations in the ER membrane that activates IRE1a and together with prolonged LPS stimulation result in sustained p38 MAPK phosphorylation and inflammatory gene expression. Although, rHDL activates IRE1a, an ER stress transducer in mammalian cells, and induces the expression of some ER stress genes, other genes like Ddit3 (Chop) and spliced Xbp1 remain unaffected. Our data support primarily activation of the kinase activity of IRE1a rather than its endonuclease activity (that leads to Xbp1 splicing) and likely excludes the involvements of other ER stress transducers, like ATF6 and PERK. Notably, cholesterol loading reversed Ire1a activation by rHDL but not by tunicamycin. Thus, marked cholesterol depletion led to a rather specific stress response that was less general than that of strong ER stressors such as Tunicamycin or Thapsigargin. Interestingly, our RNA-seq results suggested induction of a circumscribed set of ER stress genes in cholesterol loaded macrophages that was reversed by HDL treatment. This is reminiscent of earlier studies showing that free cholesterol loading of macrophages (by treatment with AcLDL plus an ACAT inhibitor) induced a widespread ER stress response 63. This suggests that either too much or too little cholesterol in the ER can induce a stress response.
Previous animal studies have clearly shown that rHDL infusions reduce lesion macrophage content and the protein expression of inflammatory markers (MCP1, COX2, iNOS) in whole aorta 64-66 and APOA1 reduces macrophage chemotaxis in vitro and in vivo 67. In a model of atherosclerosis regression, where plaque-bearing aortic arches from Apoe−/− mice were transplanted into recipient APOA1Tg/Apoe−/− or Apoe−/− mice, plaque macrophages from hypercholesterolemic APOA1 transgenic mice also displayed reduced expression of inflammatory genes compared to hypercholesterolemic controls 18. Similarly, native APOA1 injections in Apoe−/− mice resulted in a decrease of Il1b and Ccl2 mRNA expression in plaque macrophages compared to control mice 19. In our study, infusions of rHDL elicited an anti-inflammatory response in plaque macrophages. However, we found no evidence that rHDL infusion led to reduced expression of Type I interferon genes in atherosclerotic plaques. This could reflect limited induction of Type I interferon genes in plaques, compared to in vitro treatment of macrophages with LPS. Notably, there was no evidence of a pro-inflammatory response to rHDL infusion in plaque myeloid cells. These cells did not show significant changes in Hmgcr or Abca1 mRNA, indicating limited disturbance of cholesterol homeostasis despite 5 infusions of rHDL. The dose of rHDL in our studies (80 mg/Kg) was chosen to simulate that being employed in phase 3 clinical 68. The studies by Feig and colleagues 18 and Hewing and colleagues 19 led to a more sustained cholesterol efflux as indicated by Oil Red O staining and Hmgcr expression. This is likely because Feig and colleagues 18 employed APOA1Tg mice whereas Hewing and colleagues 19 injected a total of 60 mg of APOA1, which are higher than 5 doses of 80 mg//Kg rHDL (total of 8 mg only). It is also likely that in our model, abundant sources of cholesterol within macrophages or derived from other cells prevented the dramatic depletion of ER cholesterol that led to a stress-induced inflammatory response in BMDMs. Consistent with the present findings including reduced expression of Il1b, rHDL infusion reduced inflammasome activation in Ldlr−/− mice transplanted with myeloid cell-specific Abca1/g1 deficient bone marrow 61. Additional anti-atherogenic mechanisms of rHDL infusions could include reduced platelet activation and reduced expression of monocyte integrins and their counter-receptors on endothelial cells 69, 70, leading to a decrease in macrophage content.
Infusions of rHDL (CSL111) in humans and mice were associated with ALT elevations, indicating hepatocyte toxicity 71, 72. A new formulation of rHDL (CSL112) containing less phospholipids relative to APOA1 (CSL112; APOA1:PC molar ratio, 1:55 vs CSL111; APOA1:PC molar ratio, 1:150) 71, 73 produced lower levels of cholesterol efflux and did not induce ALT elevations when infused into mice or humans 68, 72. Moreover, mice deficient in SR-B1, which has an important role in rHDL-mediated cholesterol efflux, did not show ALT elevations when infused with CSL111 72. Thus, hepatocyte toxicity related to rHDL infusions likely also reflected excessive depletion of cholesterol and could be mediated through an ER stress response similar to macrophages.
Recently, two rHDL preparations, namely MDCO-216 and CER-001, failed to show a reduction in plaque volume as determined by intravascular ultrasound 74, 75. Possible reasons for their failure are the low dose, short treatment and even their protein/lipid composition 76. Reconstituted HDL (CSL112) infusions are currently being assessed in a phase 3 clinical trial (AEGIS-II) at a much higher dose of 6 g APOA1 (approximately 80 mg/kg) 68, 77. Our rHDL infusion experiments in mice, at a dose comparable to the clinical trial, showed an anti-inflammatory effect in CD11b+ cells that could contribute in plaque stabilization and atheroprotection. Nevertheless, the anti-inflammatory effect was modest and observed in a limited number of genes. It is possible that higher doses or longer rHDL treatments could have had a greater impact. Moreover, rHDL exhibited strong substantial anti-inflammatory effects in human PBMC-derived macrophages. It remains to be seen if CSL112, at the dose and treatment duration of the AEGIS-II trial, succeeds in reducing atherosclerotic burden, inflammation and cardiovascular disease.
Supplementary Material
Highlights:
HDL exerts anti- and pro-inflammatory effects that are mediated by cholesterol removal in cultured macrophages.
Anti-inflammatory are associated with reduced TLR4 levels and reduced interferon receptor signaling.
Pro-inflammatory effects of rHDL represent a modified ER stress response that involves IRE1a/ASK1/p38 MAPK signaling and occurs under conditions of extreme cholesterol depletion.
Reconstituted HDL infusions in mice produced anti-inflammatory effects in lesion macrophages suggesting a beneficial therapeutic effect of HDL in vivo.
Acknowledgements
We would like to thank Wei Wang, Joanne Hsieh, Trevor Fidler, Liana Tascau, Wenli Liu, Kaori Umeda, Huijuan Dou and Kristin McCabe (all affiliated with Columbia University) for technical assistance and Maria Chronopoulou for proofreading the manuscript.
Sources of funding
This work was supported by grants from the National Institutes of Health HL107653 (to A.R.T.), P01HL092969, R01HL127694, R01HL126028, DP3DK108209, P30DK017047 (to K.E.B), a postdoctoral fellowship award from the American Diabetes Association #9-18-CVD1-002 (to V.K), VIDI grant 917.15.350 from the Netherlands Organization of Sciences (NOW) (to M.W.) and a Rosalind Franklin Fellowship from the University Medical Center Groningen (to M.W.). Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviation List
- HDL
High-density lipoprotein
- LPS
Lipopolysaccharide
- BMDM
Bone marrow derived macrophages
- PBMC
Peripheral blood mononuclear cell
- WTD
Western type diet
- TLR4
Toll-like receptor 4
- TNFa
Tumor necrosis factor-a
- CCL2
C-C Motif chemokine ligand 2
- IFIT3
Interferon-induced protein with tetratricopeptide repeats 3
- IRE1a
Inositol-requiring enzyme 1 a
- ASK1
Apoptosis signal-regulating kinase 1
- P38 MAPK
P38 mitogen-activated protein kinases
Footnotes
Disclosures
Dr. Tall reports being a consultant to Amgen, CSL, Staten Biotechnology, Fortico Biotech and Janssen Pharmaceuticals. Dr. Bornfeldt reports receiving research support from Novo Nordisk A/S on an unrelated project. The other authors report no conflicts.
References
- 1.Di AE, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein ai. Nature. 1991;353:265–267 [DOI] [PubMed] [Google Scholar]
- 3.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma hdl cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet. 2012;380:572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tall AR, Rader DJ. Trials and tribulations of cetp inhibitors. Circ Res. 2018;122:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267 [DOI] [PubMed] [Google Scholar]
- 8.Ronsein GE, Heinecke JW. Time to ditch hdl-c as a measure of hdl function? Curr Opin Lipidol. 2017;28:414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karathanasis SK, Freeman LA, Gordon SM, Remaley AT. The changing face of hdl and the best way to measure it. Clin Chem. 2017;63:196–210 [DOI] [PubMed] [Google Scholar]
- 10.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. Hdl cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shea S, Stein JH, Jorgensen NW, McClelland RL, Tascau L, Shrager S, Heinecke JW, Yvan-Charvet L, Tall AR. Cholesterol mass efflux capacity, incident cardiovascular disease, and progression of carotid plaque. Arterioscler Thromb Vasc Biol. 2019;39:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
- 13.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, de Beer FC, Miyake K, Oram JF, Heinecke JW. High-density lipoprotein suppresses the type i interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122:1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Nardo D, Labzin LI, Kono H, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator atf3. Nat Immunol. 2014;15:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, Cavigiolio G, Oda MN, Parks JS, Fessler MB. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 2010;11:493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Vorst EPC, Theodorou K, Wu Y, et al. High-density lipoproteins exert pro-inflammatory effects on macrophages via passive cholesterol depletion and pkc-nf-kappab/stat1-irf1 signaling. Cell Metab. 2017;25:197–207 [DOI] [PubMed] [Google Scholar]
- 18.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. Hdl promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hewing B, Parathath S, Barrett T, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-i on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanson M, Distel E, Fisher EA. Hdl induces the expression of the m2 macrophage markers arginase 1 and fizz-1 in a stat6-dependent process. PLoS One. 2013;8:e74676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in abc transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage abca1 reduces myd88-dependent toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and hdl-lipid levels in male, female, and testosterone-treated female mice from strains c57bl/6, balb/c, and c3h. Atherosclerosis. 1987;64:215–221 [DOI] [PubMed] [Google Scholar]
- 24.Warden CH, Hedrick CC, Qiao JH, Castellani LW, Lusis AJ. Atherosclerosis in transgenic mice overexpressing apolipoprotein a-ii. Science. 1993;261:469–472 [DOI] [PubMed] [Google Scholar]
- 25.Paszty C, Maeda N, Verstuyft J, Rubin EM. Apolipoprotein ai transgene corrects apolipoprotein e deficiency-induced atherosclerosis in mice. J Clin Invest. 1994;94:899–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda N, Johnson L, Kim S, Hagaman J, Friedman M, Reddick R. Anatomical differences and atherosclerosis in apolipoprotein e-deficient mice with 129/svev and c57bl/6 genetic backgrounds. Atherosclerosis. 2007;195:75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett BJ, Orozco L, Kostem E, Erbilgin A, Dallinga M, Neuhaus I, Guan B, Wang X, Eskin E, Lusis AJ. High-resolution association mapping of atherosclerosis loci in mice. Arterioscler Thromb Vasc Biol. 2012;32:1790–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lerch PG, Fortsch V, Hodler G, Bolli R. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sang. 1996;71:155–164 [DOI] [PubMed] [Google Scholar]
- 29.Kim D, Langmead B, Salzberg SL. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015;12:357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 2011;17:10–12 [Google Scholar]
- 31.Liao Y, Smyth GK, Shi W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930 [DOI] [PubMed] [Google Scholar]
- 32.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. Panther: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell 2010;38:576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorisky A, Kucera GL, Rittenhouse SE. Stimulated cholesterol-enriched platelets display increased cytosolic ca2+ and phospholipase a activity independent of changes in inositol trisphosphates and agonist/receptor binding. Biochem J. 1990;265:747–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shattil SJ, Anaya-Galindo R, Bennett J, Colman RW, Cooper RA. Platelet hypersensitivity induced by cholesterol incorporation. J Clin Invest. 1975;55:636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper RA, Leslie MH, Fischkoff S, Shinitzky M, Shattil SJ. Factors influencing the lipid composition and fluidity of red cell membranes in vitro: Production of red cells possessing more than two cholesterols per phospholipid. Biochemistry. 1978;17:327–331 [DOI] [PubMed] [Google Scholar]
- 38.Mendez AJ, Oram JF, Bierman EL. Protein kinase c as a mediator of high density lipoprotein receptor-dependent efflux of intracellular cholesterol. J Biol Chem. 1991;266:10104–10111 [PubMed] [Google Scholar]
- 39.Hutchins PM, Ronsein GE, Monette JS, Pamir N, Wimberger J, He Y, Anantharamaiah GM, Kim DS, Ranchalis JE, Jarvik GP, Vaisar T, Heinecke JW. Quantification of hdl particle concentration by calibrated ion mobility analysis. Clin Chem. 2014;60:1393–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via abca1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtiss LK, Tobias PS. Emerging role of toll-like receptors in atherosclerosis. J Lipid Res. 2009;50 Suppl:S340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. Cd36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Westerterp M, Murphy AJ, Wang M, et al. Deficiency of atp-binding cassette transporters a1 and g1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res 2013;112:1456–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A, Fujimoto T. Cholesterol depletion induces autophagy. Biochem Biophys Res Commun. 2006;351:246–252 [DOI] [PubMed] [Google Scholar]
- 45.Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor tfe3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7:ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila mitf regulates the v-atpase and the lysosomal-autophagic pathway. Autophagy. 2016;12:484–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein a-i in transgenic mice results in reduced plasma levels of murine apolipoprotein a-i and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell. 2016;165:165–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. Ros-dependent activation of the traf6-ask1-p38 pathway is selectively required for tlr4-mediated innate immunity. Nat Immunol. 2005;6:587–592 [DOI] [PubMed] [Google Scholar]
- 50.Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol. 2008;48:199–225 [DOI] [PubMed] [Google Scholar]
- 51.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. Ask1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. Ask1 is required for sustained activations of jnk/p38 map kinases and apoptosis. EMBO Rep. 2001;2:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loomba R, Lawitz E, Mantry PS, et al. The ask1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the er to activation of jnk protein kinases by transmembrane protein kinase ire1. Science. 2000;287:664–666 [DOI] [PubMed] [Google Scholar]
- 55.Ghosh R, Wang L, Wang ES, et al. Allosteric inhibition of the ire1alpha rnase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tufanli O, Telkoparan Akillilar P, Acosta-Alvear D, Kocaturk B, Onat UI, Hamid SM, Cimen I, Walter P, Weber C, Erbay E. Targeting ire1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci U S A. 2017;114:E1395–E1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mrna splicing by an ire1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify atf3 as a negative regulator of toll-like receptor 4. Nature. 2006;441:173–178 [DOI] [PubMed] [Google Scholar]
- 59.Sheikh F, Dickensheets H, Gamero AM, Vogel SN, Donnelly RP. An essential role for ifn-beta in the induction of ifn-stimulated gene expression by lps in macrophages. J Leukoc Biol. 2014;96:591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type i ifn genes by the ifn-inducible transcription factor irf-7. FEBS Lett. 1998;441:106–110 [DOI] [PubMed] [Google Scholar]
- 61.Westerterp M, Fotakis P, Ouimet M, et al. Cholesterol efflux pathways suppress inflammasome activation, netosis, and atherogenesis. Circulation. 2018;138:898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raciti M, Lotti LV, Valia S, Pulcinelli FM, Di Renzo L. Jnk2 is activated during er stress and promotes cell survival. Cell Death Dis. 2012;3:e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires er stress pathways and engagement of the type a scavenger receptor. J Cell Biol. 2005;171:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ibanez B, Giannarelli C, Cimmino G, Santos-Gallego CG, Alique M, Pinero A, Vilahur G, Fuster V, Badimon L, Badimon JJ. Recombinant hdl(milano) exerts greater anti-inflammatory and plaque stabilizing properties than hdl(wild-type). Atherosclerosis. 2012;220:72–77 [DOI] [PubMed] [Google Scholar]
- 65.Ibanez B, Vilahur G, Cimmino G, Speidl WS, Pinero A, Choi BG, Zafar MU, Santos-Gallego CG, Krause B, Badimon L, Fuster V, Badimon JJ. Rapid change in plaque size, composition, and molecular footprint after recombinant apolipoprotein a-i milano (etc-216) administration: Magnetic resonance imaging study in an experimental model of atherosclerosis. J Am Coll Cardiol. 2008;51:1104–1109 [DOI] [PubMed] [Google Scholar]
- 66.Tardy C, Goffinet M, Boubekeur N, Ackermann R, Sy G, Bluteau A, Cholez G, Keyserling C, Lalwani N, Paolini JF, Dasseux JL, Barbaras R, Baron R. Cer-001, a hdl-mimetic, stimulates the reverse lipid transport and atherosclerosis regression in high cholesterol diet-fed ldl-receptor deficient mice. Atherosclerosis. 2014;232:110–118 [DOI] [PubMed] [Google Scholar]
- 67.Iqbal AJ, Barrett TJ, Taylor L, et al. Acute exposure to apolipoprotein a1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. Elife. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michael Gibson C, Korjian S, Tricoci P, et al. Safety and tolerability of csl112, a reconstituted, infusible, plasma-derived apolipoprotein a-i, after acute myocardial infarction: The aegis-i trial (apoa-i event reducing in ischemic syndromes i). Circulation. 2016;134:1918–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calkin AC, Drew BG, Ono A, Duffy SJ, Gordon MV, Schoenwaelder SM, Sviridov D, Cooper ME, Kingwell BA, Jackson SP. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation. 2009;120:2095–2104 [DOI] [PubMed] [Google Scholar]
- 70.Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–971 [DOI] [PubMed] [Google Scholar]
- 71.Tardif JC, Gregoire J, L’Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J, Effect of r HDLoA-S, Efficacy I. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. JAMA. 2007;297:1675–1682 [DOI] [PubMed] [Google Scholar]
- 72.Herzog E, Pragst I, Waelchli M, Gille A, Schenk S, Mueller-Cohrs J, Diditchenko S, Zanoni P, Cuchel M, Seubert A, Rader DJ, Wright SD. Reconstituted high-density lipoprotein can elevate plasma alanine aminotransferase by transient depletion of hepatic cholesterol: Role of the phospholipid component. J Appl Toxicol. 2016;36:1038–1047 [DOI] [PubMed] [Google Scholar]
- 73.Diditchenko S, Gille A, Pragst I, Stadler D, Waelchli M, Hamilton R, Leis A, Wright SD. Novel formulation of a reconstituted high-density lipoprotein (csl112) dramatically enhances abca1-dependent cholesterol efflux. Arterioscler Thromb Vasc Biol. 2013;33:2202–2211 [DOI] [PubMed] [Google Scholar]
- 74.Nicholls SJ, Puri R, Ballantyne CM, Jukema JW, Kastelein JJP, Koenig W, Wright RS, Kallend D, Wijngaard P, Borgman M, Wolski K, Nissen SE. Effect of infusion of high-density lipoprotein mimetic containing recombinant apolipoprotein a-i milano on coronary disease in patients with an acute coronary syndrome in the milano-pilot trial: A randomized clinical trial. JAMA Cardiol. 2018;3:806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nicholls SJ, Andrews J, Kastelein JJP, et al. Effect of serial infusions of cer-001, a pre-beta high-density lipoprotein mimetic, on coronary atherosclerosis in patients following acute coronary syndromes in the cer-001 atherosclerosis regression acute coronary syndrome trial: A randomized clinical trial. JAMA Cardiol. 2018;3:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rader DJ. Apolipoprotein a-i infusion therapies for coronary disease: Two outs in the ninth inning and swinging for the fences. JAMA Cardiol. 2018;3:799–801 [DOI] [PubMed] [Google Scholar]
- 77.Gille A, D’Andrea D, Tortorici MA, Hartel G, Wright SD. Csl112 (apolipoprotein a-i [human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. 2018;38:953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NCBI GEO accession number for high throughput sequencing data reported in this paper is GSE129347.