

Abstract
The choroid plexus (CP), composed of capillaries surrounded by a barrier epithelium, is the main producer of cerebrospinal fluid (CSF). The CP epithelium regulates the transport of ions and water between the blood and the ventricles, contributing to CSF production and composition. Several studies suggest a connection between the cation channel transient receptor potential vanilloid-4 (TRPV4) and transepithelial ion movement. TRPV4 is a nonselective, calcium-permeable cation channel present in CP epithelia reported to be activated by cytokines and inflammatory mediators. Utilizing the PCP-R (porcine choroid plexus-Riems) cell line, we investigated the effects of various cytokines and inflammatory mediators on TRPV4-mediated activity. Select proinflammatory cytokines (TNF-α, IL-1β, TGF-β1) had inhibitory effects on TRPV4-stimulated transepithelial ion flux and permeability changes, whereas anti-inflammatory cytokines (IL-10, IL-4, and IL-6) had none. Quantitative mRNA analysis showed that these cytokines had no effect on TRPV4 transcription levels. Inhibition of the transcription factor NF-κB, involved in the production and regulation of several inflammatory cytokines, inhibited TRPV4-mediated activity, suggesting a link between TRPV4 and cytokine production. Contrary to published studies, the proinflammatory mediator arachidonic acid (AA) had inhibitory rather than stimulatory effects on TRPV4-mediated responses. However, inhibition of AA metabolism also caused inhibitory effects on TRPV4, suggesting a complex interaction of AA and its metabolites in the regulation of TRPV4 activity. Together these data imply that TRPV4 activity is involved in the inflammatory response; it is negatively affected by proinflammatory mediators. Furthermore, arachidonic acid metabolites, but not arachidonic acid itself, are positive regulators of TRPV4.
Keywords: arachidonic acid, blood-choroid plexus barrier, epoxyeicosatrienoic acid, nuclear factor-κB
INTRODUCTION
Neurological and neurodegenerative conditions in the brain can result in an increase in the release of inflammatory mediators into the injured areas. These conditions can include head trauma, intracranial hemorrhage, infection, brain tumor, genetic defect, or neurodegenerative disease (11, 13, 16, 18, 20, 21, 27, 29, 45, 46). In many of these cases, the development of transient or chronic enlargement of the brain ventricles and an increase in pressure due to the accumulation of cerebrospinal fluid (CSF) occurs. This accrual of fluid could be caused by increases in inflammation affecting the production and/or absorption of the CSF.
The main production of CSF comes from the choroid plexus (CP), present in the lateral, third, and fourth ventricles (10). Composed of a barrier epithelium that surrounds a network of capillaries, the CP epithelial cells regulate the transport of ions and water between the ventricles and capillaries via electrolyte transporters, thus controlling the production and composition of CSF. Aberrant regulation of these transporters could be a causative agent leading to the emergence of fluid/electrolyte imbalances in the brain.
Previous data suggest a link between transepithelial ion movement and the cation channel transient receptor potential vanilloid-4 (TRPV4) (29, 37, 44). This ion channel is found in choroid plexus epithelium (10, 37). TRPV4 is a mechano-, osmo-, and temperature-sensitive, nonselective, calcium-permeable cation channel that can serve as a hub protein for the activation of other transporters (26, 32, 37, 42). For example, in the porcine choroid plexus-Riems (PCP-R) cell line, TRPV4 activation can lead to an influx of calcium into the cells, which subsequently stimulates calcium-activated ion channels such as the intermediate conductance potassium channels (37). Once these channels are activated, there is the potential for significant ion flux across the epithelia, resulting in compensatory water movement and a change in CSF production. TRPV4 can also be modulated through a number of stimuli, including chemical activators such as cytokines and inflammatory mediators (26, 32, 37, 42). Indeed, it has been established that the inflammatory lipid endocannabinoid anandamide (AEA) and its metabolites, including arachidonic acid (AA), are capable of activating the channel (32). In addition, cytochrome P450 epoxygenases, cyclooxygenases, and lipoxygenases can metabolize AA into both inflammatory and anti-inflammatory factors epoxyeicosatrienoic acids (EETs), prostaglandins, and hydroperoxides, respectively, which are also believed to stimulate TRPV4 activity (32, 33).
Microglia, the macrophage-like cells of the central nervous system (CNS), can be triggered to release proinflammatory cytokines in response to abnormal protein accumulation or neuronal injury from chemical or physical means (7, 50). It is possible that such inflammation is associated with increases in CSF production associated with neurological disease (4, 17, 18, 35, 46). Summarized in Table 1, proinflammatory cytokines found upregulated in neurodegenerative states include interleukin (IL)-1β and tumor necrosis factor-α (TNF-α). IL-10 and IL-4 are well-documented anti-inflammatory cytokines also found in various neurological diseases. TGF-β and IL-6 have both been implicated in pro- as well as anti-inflammatory effects (2, 7, 8, 14, 15, 24, 38, 41, 48). These cytokines can, therefore, potentially play a role in altering TRPV4 activity.
Table 1.
Reported cytokines function in the inflammatory pathway
As an in vitro model, the PCP-R cell line can be utilized as a surrogate for in vivo CP tissue. This cell line exhibits many of the characteristics of the native epithelium, including the formation of a barrier epithelial monolayer, expression of tight junctional proteins claudin-1 and -3, zona occludins-1, and occludin as well as the polarization of transporters and channels similar to those found in vivo (37, 39). A previous study has used this line to monitor changes in transepithelial ion flux and barrier permeability in response to factors that modulate choroid plexus electrolyte transporters in vivo (37). The polarity and barrier function exhibited by this cell line allows for experimentation modeling changes in the barrier function of choroid plexus in vivo (37, 39). Using a TRPV4 agonist, GSK1016790A, we are able to stimulate TRPV4 activation and observe changes in ion flux and transepithelial conductance in the presence of various cytokines and inflammatory mediators.
In this paper, we show that specific cytokines and inflammatory mediators can alter the function of TRPV4 in PCP-R cell epithelia. This provides more insight into the role of the cation channel in neuroinflammatory states and suggests this hub protein as a potential effector in CSF production.
MATERIALS AND METHODS
Cell culture.
PCP-R cells were grown on 0.4-µm pore diameter filter inserts (no. PIHP03050; Millipore Sigma, Burlington, MA) until the cultures developed a high transepithelial electrical resistance (TER; 10–12 days) (37). Only cultures with resistances >500 Ωcm2 were utilized. During experimental protocols that result in a change in resistance, cells that drop below 100 Ωcm2 were also removed from analysis, as their junctional complexes appear irreversibly altered. Cells were fed three times weekly with DMEM media (no. 12100-046; Gibco, Gaithersburg, MD) containing 4.5 g/L glucose, 3.7 g/L NaHCO3, 5.71 g/L HEPES, 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 100 U/mL penicillin, 100 mg/mL streptomycin, and 5 µg/mL insulin. Each plate contained six filters in six different wells. Filtered inserts were bathed in 2 mL feeding media on the basolateral (bottom) side of the membrane and 1.5 mL feeding media on the apical (top) side of the membrane.
Reverse transcriptase-PCR.
For the reverse transcriptase (RT)‐PCR, cells were grown in 75 cm2 flasks until confluent. The monolayers were trypsinized and collected, and RNA from the cells was extracted, following the manufacturer’s instructions for the New Monarch Total RNA Miniprep Kit (no. T2010S; New England BioLabs, Ipswich, MA). No template controls and cDNA were prepared by following manufacturer’s protocol for the New England BioLabs LunaScript RT SuperMix Kit (no. E3010S). Three primer sets for each gene were generated using Primer3Plus with mRNA sequences and tested on a temperature gradient. Only those primers used in the final images were included in Table 2. Sus scrofa mRNA sequences were obtained from Ensembl and additionally confirmed with sequences from the NCBI database. Forward and reverse primers (IDT) were combined with cDNA (∼500 ng) and GoTaq Green Master Mix (no. M7122; Promega, Madison, WI) to perform gradients of PCR to determine the optimal annealing temperature. PCR products were then run on 1.5% agarose gels with ethidium bromide and 100bp flanking ladders. Gels were imaged using a ChemiDoc XRS imager (Bio-Rad, Hercules, CA). Following confirmation of single-band amplification, PCR products were Sanger sequenced by Eton Bioscience (Union, NJ). Amplicons were confirmed to be the predicted transcript for each gene, validated using NCBI BLAST.
Table 2.
Sus scrofa forward and reverse primer sequences used for RT-PCR with corresponding product sizes (bp)
| Primer Sequence 5′–3′ |
||||
|---|---|---|---|---|
| Sus Scrofa Gene | Protein | Forward Primer | Reverse Primer | Product Size, bp |
| Il1r1 | IL1R1 | TTACACAGAGACTACCGGTTGC | CAGTTTTATCACCCGGGAGACA | 263 |
| Il1r2 | IL1R2 | AAATGATTCCACGACCATCCCA | TCCCAAAACCAGGATGATGAGG | 874 |
| Tnfrsf1a | TNFRSF1A | GAAACAGGACACCATCTGCAAC | AAGCTAAGCCAACGAAGAGGAA | 218 |
| Tgfbr1 | TGFBR1 | GAGACAGGCCATTTGTATGTGC | TAAATCTCTGCCTCACGGAACC | 520 |
| Tgfbr2 | TGFBR2 | GGAAGCTGATGGAGTTCAGTGA | TCTCTGTCTTCCAGGAGGCATA | 252 |
| Tgfbr3 | TGFBR3 | ATATTCACCACAAGCCTGTCGT | ATACTCCTTTCGGGCCCAATTT | 224 |
| Il6r | IL6R | AAGATTCTGCGTCGGTACCATT | TGTTGCTGACATCATAAGGGCT | 312 |
| Il10ra | IL10RA | GGATGAAGTGACTCTGACGGTT | GCTTCACCCTGACACACAATTC | 257 |
| Il4r | IL4R | AGAGCTACCTGTACTCGGAACT | CCCTTTGTCTTCCTCCTCTTCC | 695 |
| Tlr1 | TLR1 | TGTCCCACAACAAGTTGGAGAA | TTGTGGGAAACTGAACACCTCA | 650 |
| Tlr2 | TLR2 | TCCCAAATCTGCGAATCCTGAA | ATGCAACCTCCGGACTGTTAAT | 512 |
| Tlr3 | TLR3 | CGATGACCTCCCGGCAAATATA | GAGATTTTCCAGTTGGAGCTGC | 382 |
| Tlr4 | TLR4 | TTCTCTCCTGCCTGAGATCTGA | ACTCCAGGTAGGTATTCCTGCT | 555 |
| Tlr5 | TLR5 | TCCTGTGGTCTCTCTGATGCTA | GGGTTCATACACTTCCCCCAAT | 284 |
| Tlr6 | TLR6 | AGACAATCTTGTGCCATCCCAT | GGCCCTTGAGTGAGTTCCAATA | 409 |
| Tlr7 | TLR7 | GACACTAAAGACCCAGCAGTGA | CTGAAGGGGCTTCTCAAGGAAT | 297 |
| Tlr8 | TLR8 | CTGAGGCAGAACAGGATTTCCT | TTCATCACCCAGTCTGTGACAG | 542 |
| Tlr9 | TLR9 | GCCTACGAACTCTCAACCTCAA | GGAAGTTCTCACTCAGGTCCAG | 696 |
| Tlr10 | TLR10 | TCAGGTGCTTGCCCAGAAATAT | TCTTGCCAGGATCAGAGTTTCC | 717 |
| Cyp2c42 | CYP2C42 | TCCTGTCTGCTTCTCCTTTCAC | GGGAGCACAGTCCAGGATAAAA | 489 |
| Cyp2e1 | CYP2E1 | TCGAGATTTCACTGACACCCTG | GTTAAAGTGCTGCAAGATGGCA | 592 |
| Alox5 | 5-LOX | TGACAGTGGATGAAGAACTGGG | TGTTTTTGCCGTGTTTCCAGTT | 259 |
| Alox12b | 12R-LOX | ACACCATCCAGATCAACAGCAT | CCAGAGGACCAATAGGACGATG | 602 |
| Alox15 | 12/15-LOX | CAGGAGGATGAACTCTTTGGCT | TCGAATATACCTCCGTCCGAGA | 578 |
| Alox15b | 15-LOX2 | GCGAAATGCTGAGTTCTCCATC | CAGGGTGGAATAGTTCAGCTGT | 283 |
| Cox1 | COX1 | TCACCCGCAATACTATGAGCTC | TGTGTGATAGGGAGGAGGACAT | 510 |
| Cox2 | COX2 | CCCTTTCCAACTAGGCTTCCAA | TAGTCGTCCTGGGATAGCATCT | 529 |
| Gapdh | GAPDH | TTTTAACTCTGGCAAAGTGGAC | CATTGTCGTACCAGGAAATGAG | 884 |
Three different primer sets were generated and tested for each gene. Primers included in this table were utilized for Fig. 1, A–C. GAPDH was used as a positive control.
Quantitative PCR.
Choroid plexus cells were grown on 0.4-µm polycarbonate filtered inserts (Millipore Sigma) until confluent (10–12 days). Twenty-four hours prior to use, experimental cells were treated with specific cytokines and allowed to incubate overnight. Cells were washed with cold 1× PBS twice, and total RNA was isolated using the Monarch Total RNA Miniprep Kit (New England Biolabs) according to the manufacturer’s instructions for cultured cells. Purified RNA concentration was measured using an ND2000 NanoDrop (Fisher Scientific, Waltham, MA). Approximately 100 ng of total RNA was reverse transcribed into cDNA using the Monarch LunaScript RT SuperMix Kit (New England Biolabs). cDNA was diluted 1:10 with nuclease-free water (New England Biolabs). Quantitative (q)PCR was carried out on a LightCycler 480 Instrument II real-time PCR system (Roche LifeScience, Penzberg, Germany), using LightCycler 480 SYBR Green I Master Mix (no. 04707516001; Roche LifeScience). qPCR cycle conditions were 95°C for 5 min, followed by 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s. Data are presented as relative expression fold change using the 2−ΔΔCT method (28) relative to the calibrator housekeeping genes GAPDH and Rps18. Data are shown as fold change of TRPV4 in treated cells relative to the normalized controls.
Electrophysiology.
Electrophysiological measurements were conducted by excising and mounting confluent (10–12 days) PCP-R cells grown on filter inserts in Ussing chambers that were attached to a DVC-1000 voltage/current clamp (World Precision Instruments, Sarasota, FL) using voltage and current electrodes. To each side of the chamber, 10 mL of 37°C serum-free media was added. The chambers were water-jacketed to allow the cells and media to be kept at a consistent physiological temperature (37°C). Furthermore, the media was continuously oxygenated and circulated by bubbling 5% CO2-95% O2 directly into the media. Following clamping of the spontaneous transepithelial potential difference to zero for an equilibration period of at least 30 min, experimental compounds were added to the apical and/or basolateral membranes by means of the serum-free media, and the cultures were monitored for changes in short-circuit current (SCC) and transepithelial resistance (TER). SCC is a measure of net transepithelial ion flux; a positive deflection indicates either cation absorption or anion secretion, and a negative deflection indicates the reverse. TER is a measure of changes in epithelial barrier function and was measured every 200 s through a 2 mV pulse. The instantaneous change in SCC from the pulse was used to calculate TERs by means of Ohm’s Law. These resistance calculations were converted to conductance by taking the inverse of the resistances. Conductance is a measure of transepithelial permeability. These parameters provided real-time information about transepithelial ion flux in response to modulators of ion channels or intracellular signaling molecules. In each experiment, the cells were preincubated on both basolateral and apical sides with the experimental compounds for either 10 or 20 min or 24 h prior to addition of TRPV4 agonist GSK1016790A, as indicated on the figures. In all experiments, control and effector-treated samples were grown and analyzed in parallel cultures.
Statistics.
Statistics were performed using two-tailed Student’s t-test in Sigma Plot 13. P < 0.05 was considered significant. Student’s t-test was used to compare experimental groups with the control group as well as the experimental groups with each other, as indicated by the symbols defined in the figure captions.
RESULTS
To determine the ability of the PCP-R cell line to respond to and/or produce various cytokines and inflammatory mediators, RT-PCR was performed to identify specific genes of interest. First, we focused on the receptors for the various cytokines tested in the cell line, of which only the mRNA for the second isoform of the IL-1 receptor (Il1r2) and the receptor for IL-10 (Il10ra) were absent (Fig. 1A). Toll-like receptors (TLRs) are the major membrane-bound proteins necessary for the detection of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). These receptors can also play a role in the activation of pathways responsible for cytokine production (1, 34). Within the pig genome, 10 different Tlr genes have been described (Tlr1–Tlr10) (6). Somewhat surprisingly, all of these receptors were found to be present in the PCP-R cell line (Fig. 1B).
Fig. 1.

RT-PCR in porcine choroid plexus-Riems (PCP-R) cells. A: mRNA for the various cytokine receptors. All were present except for the second isoform of the IL-1 receptor (Il1r2) and the IL-10 receptor (Il10ra). B: mRNA for porcine Toll-like receptors (TLR) 1–10 is present in the PCP-R cell line. C: RT-PCR results for various enzymes capable of arachidonic acid (AA) metabolism. Cytochrome P (CYP)450 epoxygenases, Cyp2c42 and Cyp2e1, are both absent. mRNA for lipoxygenase (Alox)15 and Alox15b are present whereas Alox5 and Alox12 are absent. Cyclooxygenase (Cox)1 and 2 are both present. Gapdh was utilized as positive controls for each gel. Band product sizes can be found in Table 2. Ladder, 100bp; T, template. Lanes denoted (+) or (−) “T” signify addition or no addition of template control. D: diagram of AA metabolism via the three different pathways, pathway inhibitors, and subsequent metabolites.
Arachidonic acid (AA) is a substrate for the production of various inflammatory mediators, some of which have been reported to stimulate TRPV4 (5, 32, 47). Therefore, we investigated which of the enzymes necessary to metabolize AA were present in the cells (Fig. 1C). Figure 1D provides a simplified summary of AA metabolism. Cytochrome P450 epoxygenases (CYP) are responsible for the production of EETs from AA. EETs have been reported as being activators of TRPV4 (47). While there are many known isoforms of Cyp genes, only two of these have currently been identified in the pig: Cyp2c42 and Cyp2e1. Neither of these isoforms were present in the porcine CP cells (Fig. 1C). Another pathway of AA metabolism utilizes lipoxygenases to produce hydroperoxides. In pigs, there are currently four lipoxygenase (Alox) gene sequences available: Alox5, Alox12b, Alox15, and Alox15b. Of these, mRNAs from Alox15 and Alox15b were present in the PCP-R cells. Finally, prostaglandins are formed when AA is metabolized by cyclooxygenases (COX) 1 or 2. Both Cox1 and Cox2 mRNA were found in the cell line (Fig. 1C).
Utilizing electrophysiology to measure changes in transepithelial ion flux and epithelial barrier permeability, several pro- and anti-inflammatory cytokines and mediators that are hypothesized to be important in inflammation-related neurological diseases were tested to assess how they would affect the basal characteristics of the PCP-R cells and subsequently their effect on the activation of TRPV4 via its agonist GSK1016790A. In each figure, controls were conducted by adding a vehicle treatment either 10 min or 24 h prior to the addition of the TRPV4 agonist (GSK1016790A). Vehicle treatments consisted of only the solvent by which each cytokine was solubilized and administered. Concentrations for each cytokine were determined by either by the manufacturer’s instruction or by the specific IC50. All cytokines were initially added 10 min prior to adding the agonist. No effects were seen with any of the tested cytokines with this short preincubation (data not shown). In immune cells, cytokine stimulation has been reported to activate innate immune genes after 24 h postinfection (43). Therefore, incubations were lengthened to 24 h. Interleukin (IL)-1β and tumor necrosis factor (TNF)-α are two of the most prominent proinflammatory cytokines seen in neurodegenerative diseases (2, 7, 15, 41, 48). After 24 h of preincubation of the PCP-R cells with either cytokine, both effectors had significant effects on TRPV4-mediated ion flux, measured as short-circuit current (SCC), albeit on different phases of the ion transport response. IL-1β caused an attenuation of the agonist response measured as SCC in the early (0–15 min) time frame while TNF-α caused an expedited increase in SCC toward baseline in the late response (15–30 min) to the agonist. Interestingly, these two proinflammatory cytokines had opposite effects on transepithelial permeability, measured as conductance. TNF-α preincubated cells showed ion transport and conductances that were statistically the same as controls after a 24 h incubation. IL-1β caused a significant attenuation in the TRPV4 agonist-mediated change in conductance, which paralleled the effect on transepithelial ion flux measured as SCC (Figs. 2 and 3).
Fig. 2.
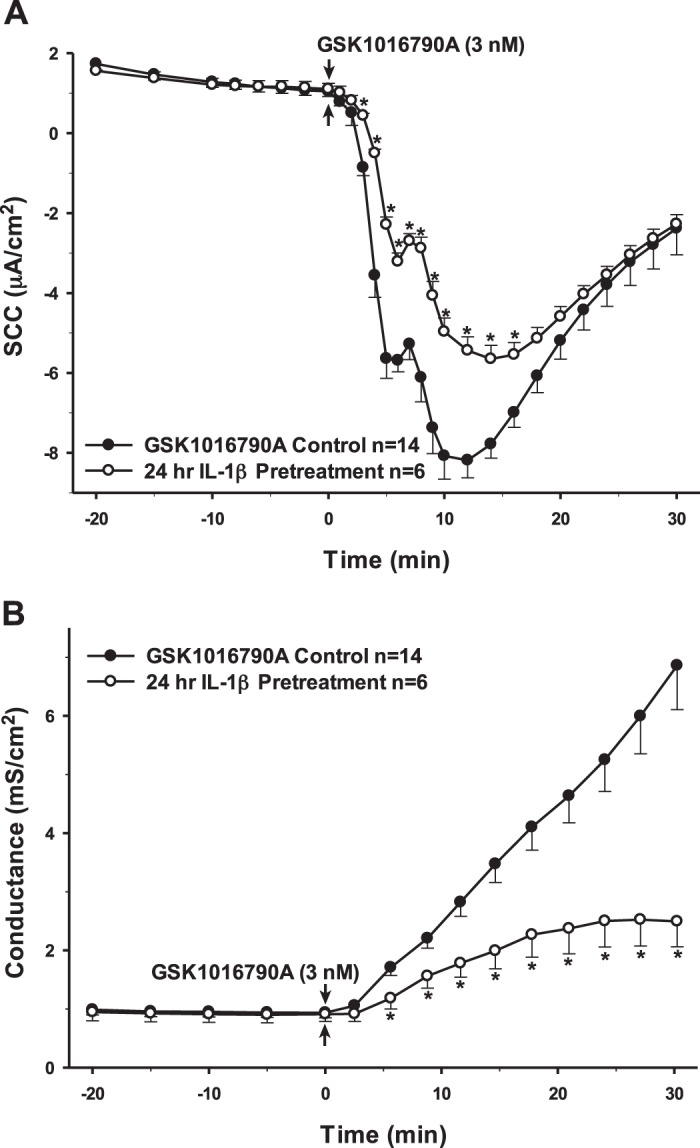
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with proinflammatory cytokine IL-1β for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. IL-1β (10 ng/mL in PBS) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with IL-1β is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control.
Fig. 3.
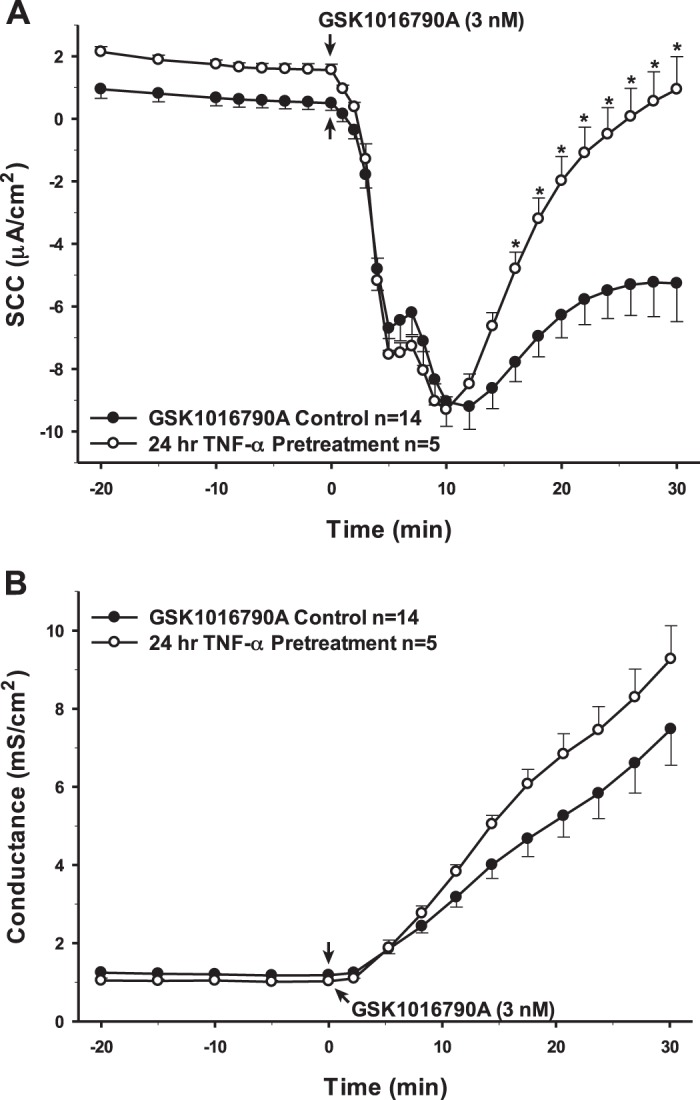
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with proinflammatory cytokine TNF-α for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. TNF-α (0.15 ng/mL in deionized water) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with TNF-α is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control.
Interestingly, 24 h preincubation with TGF-β1, which has been reported to have both pro- and anti-inflammatory effects, showed changes in SCC and conductance that were remarkably similar to IL-1β (Fig. 4). However, the same treatment with IL-6, another dual pro- and anti-inflammatory cytokine, yielded no significant change in the response to the TRPV4 agonist (Fig. 5). Finally, the cells were preincubated with the cytokines IL-10 and IL-4. Neither of these anti-inflammatory cytokines had any long-term effects on either transepithelial ion flux or conductance changes in response to agonist stimulation (Figs. 6 and 7). The receptor for IL-6 is present; however, there is no apparent receptor for IL-10 (Fig. 1A).
Fig. 4.
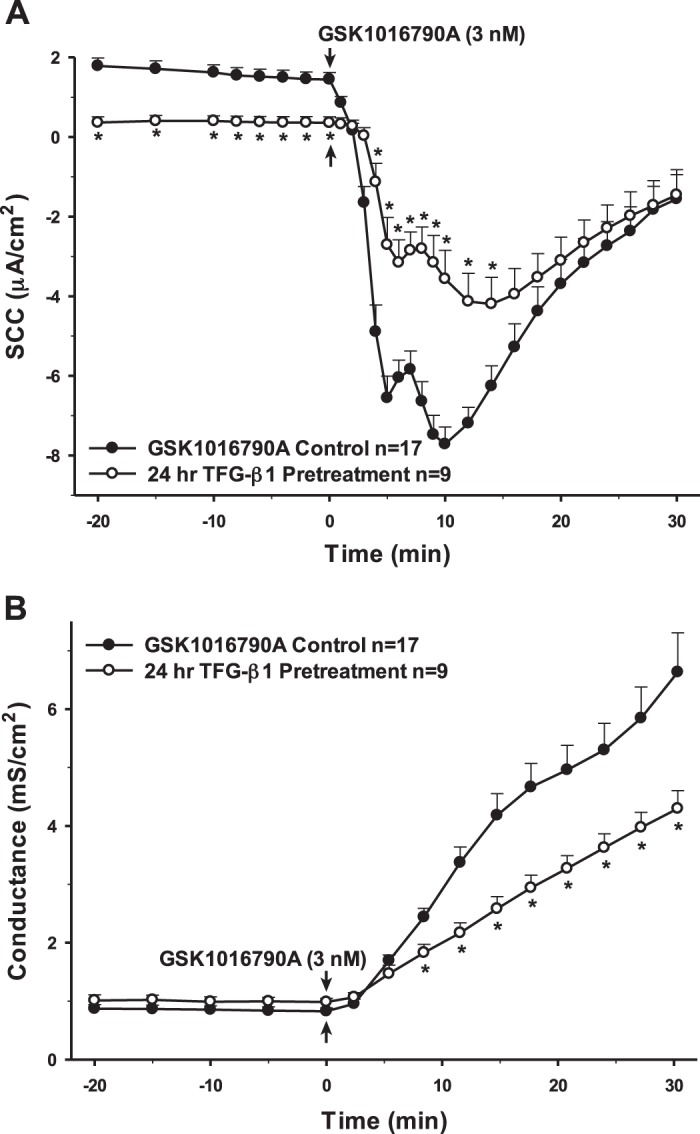
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with pro- and anti-inflammatory cytokine TGF-β1 for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. TGF-β1 (2 ng/mL in 4 mM HCl) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with TGF-β1 is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control.
Fig. 5.
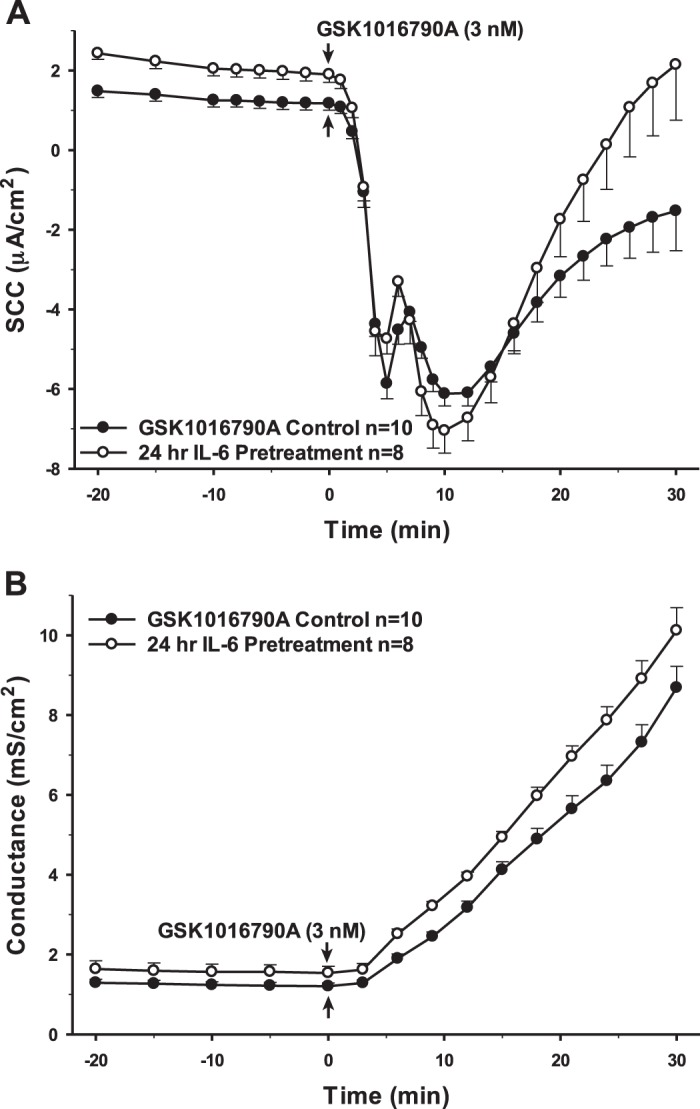
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with anti-inflammatory cytokine IL-6 for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. IL-6 (10 ng/mL in 4 mM HCl) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with IL-6 is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current.
Fig. 6.
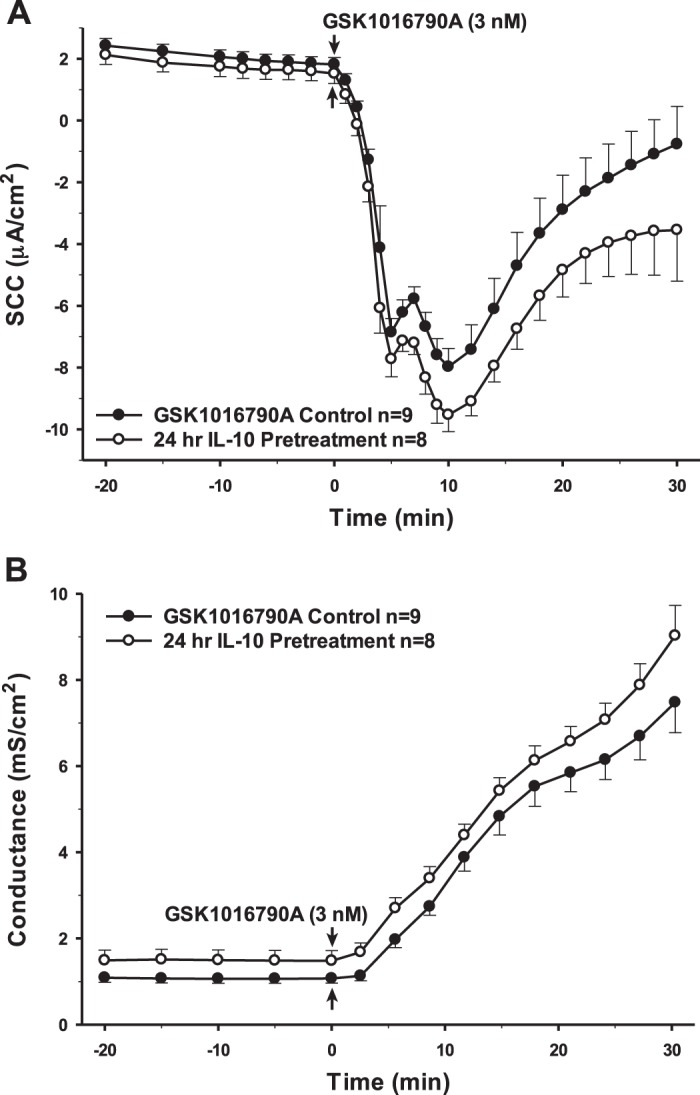
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with anti-inflammatory cytokine IL-10 for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. IL-10 (5 ng/mL in PBS) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with IL-10 is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current.
Fig. 7.
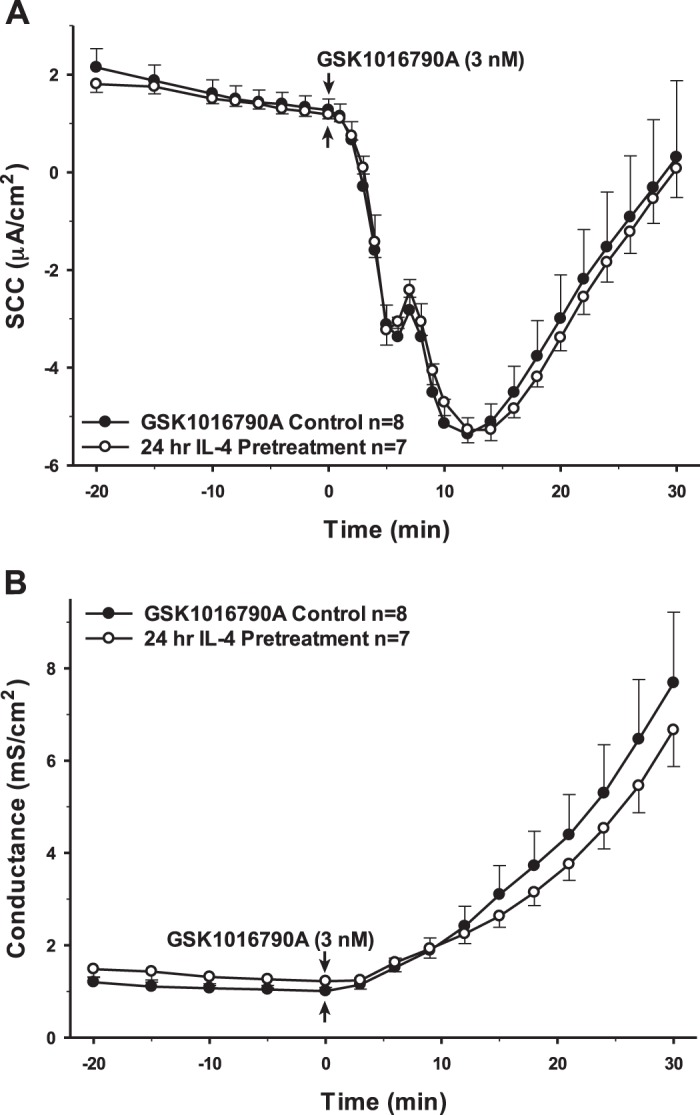
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with pro- and anti-inflammatory cytokine IL-4 for 24 h before transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. IL-4 (20 ng/mL in PBS) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with IL-4 is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current.
NF-κB is a transcription factor often associated with proinflammatory signaling pathways. Proinflammatory cytokines, mainly IL-1β and TNF-α, are considered activators of NF- κB, thereby mediating downstream effects on the activation of inflammation and further production of cytokines (23). As such, the PCP-R cells were preincubated with a selective inhibitor for NF-κB, pyrrolidine dithiocarbamate (PDTC) (52). Inhibiting NF-κB 10 min prior to adding the TRPV4 agonist caused a potentiation of the intermediate (5–10 min) phase of TRPV4-mediated ion flux. There was also a corresponding increase in permeability compared with the control that is entirely consistent with the time course of the SCC response. However, this change did not persist; permeability returned to control levels 30 min after agonist addition (Fig. 8). As NF-κB is a transcription factor, it might take longer for its true effects to become evident. Therefore, we also inhibited the proinflammatory mediator 24 h prior to adding the TRPV4 agonist (Fig. 9). In this instance, a very different result occurred. TRPV4 agonist-stimulated transepithelial ion flux was significantly reduced compared with the control. Furthermore, the 24-h incubation resulted in a substantial increase in basal conductance prior to agonist addition. In contrast to the control, the cellular permeability did not increase substantially after agonist addition in the PDTC-pretreated cells.
Fig. 8.
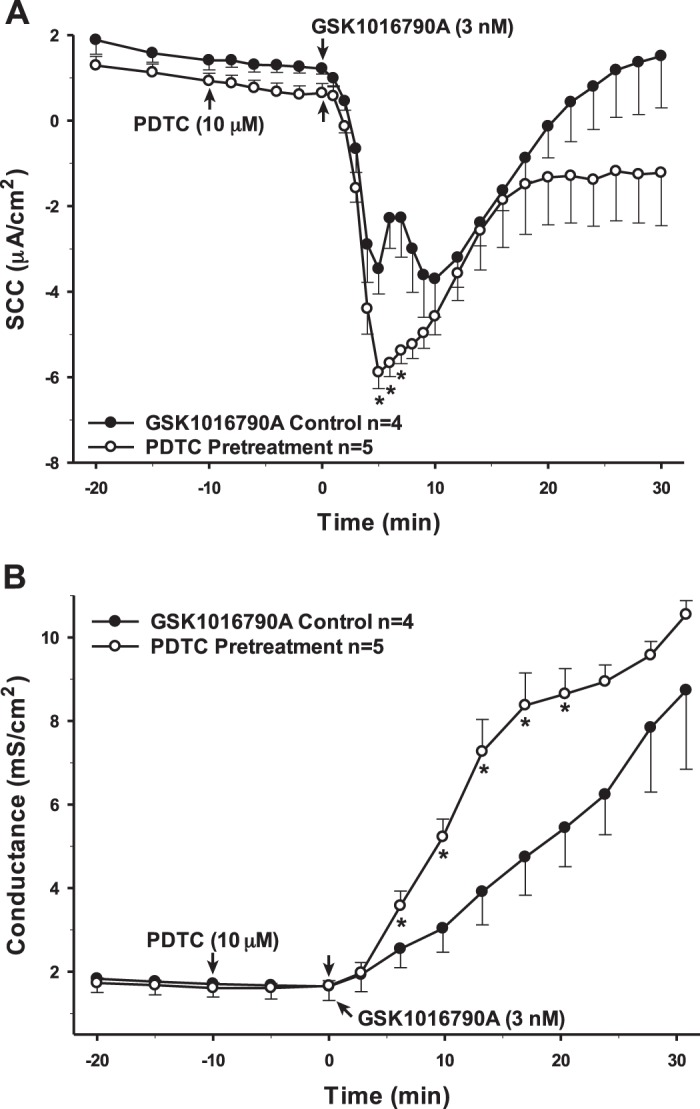
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) for 10 min prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. PDTC (10 µM in DMSO) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −10 min. TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 10 min with PDTC is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control.
Fig. 9.
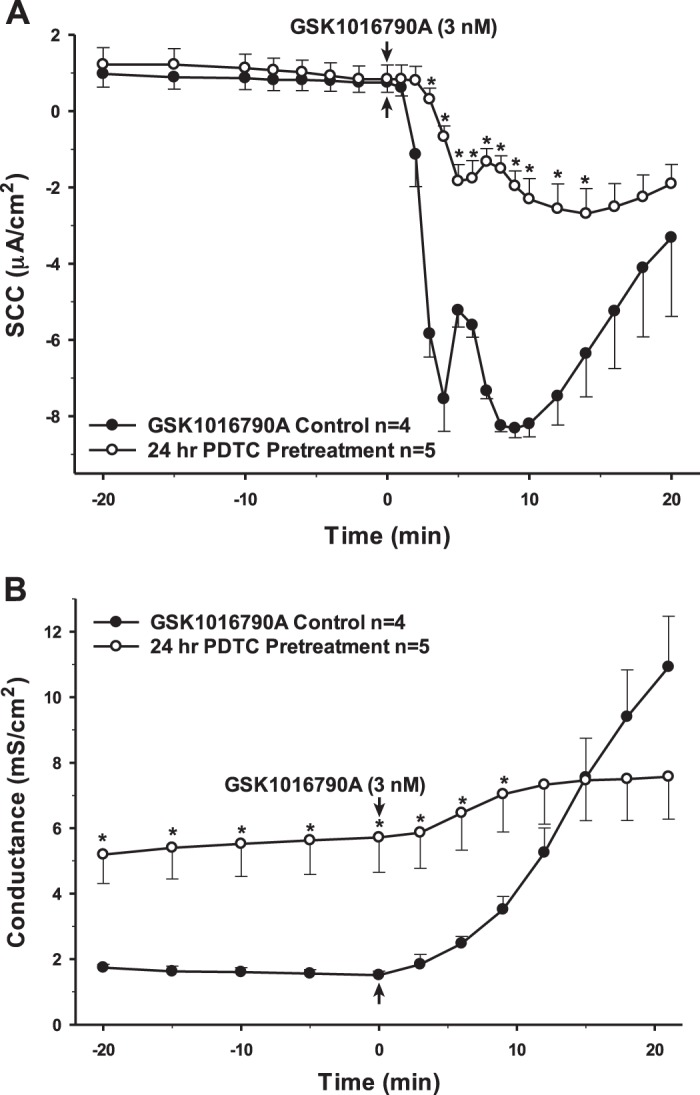
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC) for 24 h prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. PDTC (10 µM in DMSO) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −24 h (not shown). TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 24 h with PDTC is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control.
To determine whether the observed changes in TRPV4 activity may be due to changes in transporter abundance, the quantitative effect of specific cytokines on the mRNA expression of TRPV4 in PCP-R cells was examined (Fig. 10). The cells were treated with specific cytokines for 24 h and qPCR performed. There were no significant changes in TRPV4 expression levels when incubated for 24 h with the proinflammatory cytokines TNF-α and IL-1β. Similarly, cells treated with the anti-inflammatory cytokines IL-4 and IL-10 showed no statistically significant perturbations in TRPV4 expression at the mRNA level. Finally, neither TGF-β1 nor IL-6, shown to be both pro- and anti-inflammatory, had marked effects on TRPV4 expression.
Fig. 10.
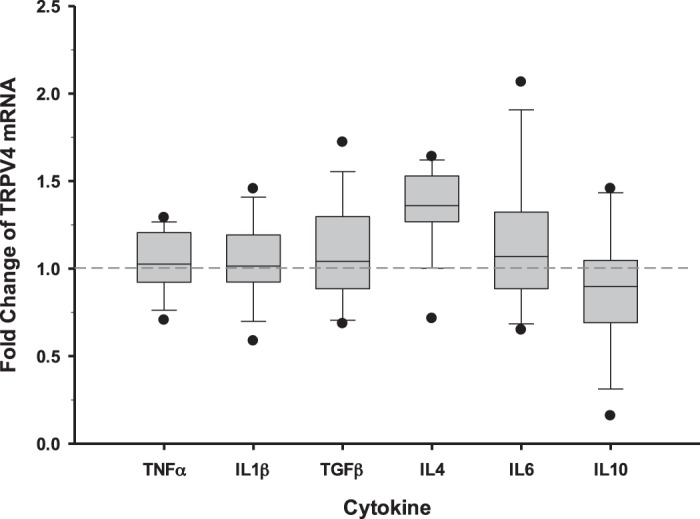
Relative fold change of transient receptor potential vanilloid 4 (TRPV4) mRNA in porcine choroid plexus-Riems (PCP-R) cells 24 h after incubation with specific cytokines known to be involved in inflammatory responses. TNF-α (0.15 ng/mL), IL-1β (10 ng/mL), TGF-β1 (2 ng/mL), IL-4 (10 ng/mL), IL-6 (20 ng/mL), and IL-10 (5 ng/mL) were added both apically and basolaterally in individual wells (n = 5 for each cytokine). Fold change of TRPV4 in treated wells is presented relative to the normalized controls (n = 6). GAPDH and RPS18 were used as housekeeping genes to determine the 2−ΔΔCT fold change in TRPV4. The minimum and maximum fold-change values are shown as individual points on the graph. Circles signify the maximum and minimum values. Lines within the boxes signify mean values and bars signify ±SE for number of experiments indicated.
AA and its metabolite, 5,6-EET, are often cited as being activators of TRPV4 (5, 7, 31, 32, 47). Specifically, AA has been reported to be able to activate TRPV4 at concentrations between 3 and 10 µM (5, 31, 47). However, when AA was added to the PCP-R cells at 10 µM, we did not see an increase in TRPV4-mediated ion flux or permeability changes (Fig. 11). Therefore, we performed a dose response with AA in the PCP-R cells in relation to TRPV4 activation via its agonist GSK1016790A. With increasing concentrations of AA, the change in TRPV4-mediated ion flux decreased significantly (Fig. 12). At the highest concentration tested (100 µM), we saw a complete inhibition of both TRPV4-mediated ion flux and change in permeability (Fig. 11).
Fig. 11.
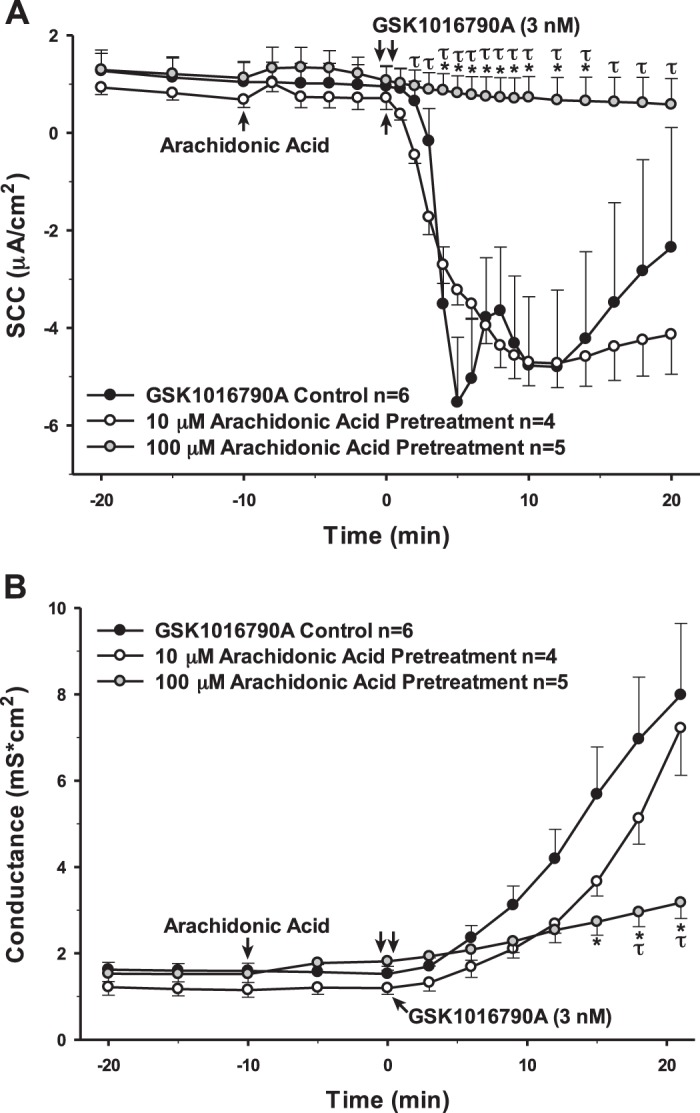
Effect of a range of concentrations of arachidonic acid (AA) on transient receptor potential vanilloid 4 (TRPV4)-mediated transepithelial ion flux and cellular permeability. AA solubilized in 100% ethanol was added to the porcine choroid plexus-Riems (PCP-R) media on both basolateral and apical sides of the membrane at time (t) = −10 min. TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at time (t) = 0 min. AA pretreatment at 10 µM is signified by white circles. AA pretreatment at 100 µM is signified by gray circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control; τP < 0.05 against 10 µM AA pretreatment.
Fig. 12.
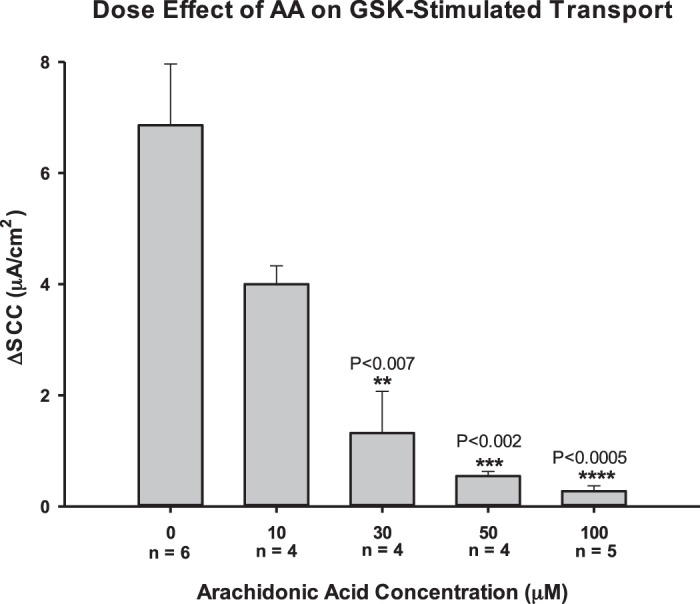
Dose effect of arachidonic acid (AA) on the change in transient receptor potential vanilloid 4 (TRPV4)-mediated transepithelial ion flux in response to TRPV4 agonist GSK1016790A. Porcine choroid plexus-Riems (PCP-R) cells were pretreated with arachidonic acid (AA) solubilized in 100% ethanol at various concentrations 10 min prior to TRPV4 agonist addition. AA was added to the PCP-R media on both basolateral and apical sides of the membrane. The change in transepithelial ion flux was measured 5 min after TRPV4 agonist addition (ΔSCC). Error bars signify the SE for number of experiments indicated. ΔSCC, change in short-circuit current. **, ***, and ****Significant against 0 µM AA control with P values given.
To further elucidate the role of the AA pathway in the function of TRPV4, we utilized various inhibitors for the enzymes involved in AA metabolism to determine which of the pathways is involved in TRPV4 activity. Indomethacin is a nonsteroidal anti-inflammatory drug (NSAID) that is able to prevent the metabolism of AA into the proinflammatory prostaglandins by nonselectively inhibiting both cyclooxygenases (22, 25, 32, 33, 51). However, preincubation with indomethacin did not change the TRPV4-mediated ion movement or permeability responses (data not shown). Eicosatetraynoic acid (ETYA) is an inhibitor of all cyclooxygenases and lipoxygenases, which produce prostaglandins and leukotrienes, respectively. When added to the cells 20 min prior to the TRPV4 agonist, this inhibitor caused a significant attenuation to both the SCC and conductance changes. Finally, we preincubated the cells with SKF-525A, a nonspecific inhibitor of cytochrome P450 responsible for the production of EETs. This inhibitor caused a complete inhibition of both transepithelial ion flux and changes in cellular permeability caused by TRPV4 activity (Fig. 13).
Fig. 13.
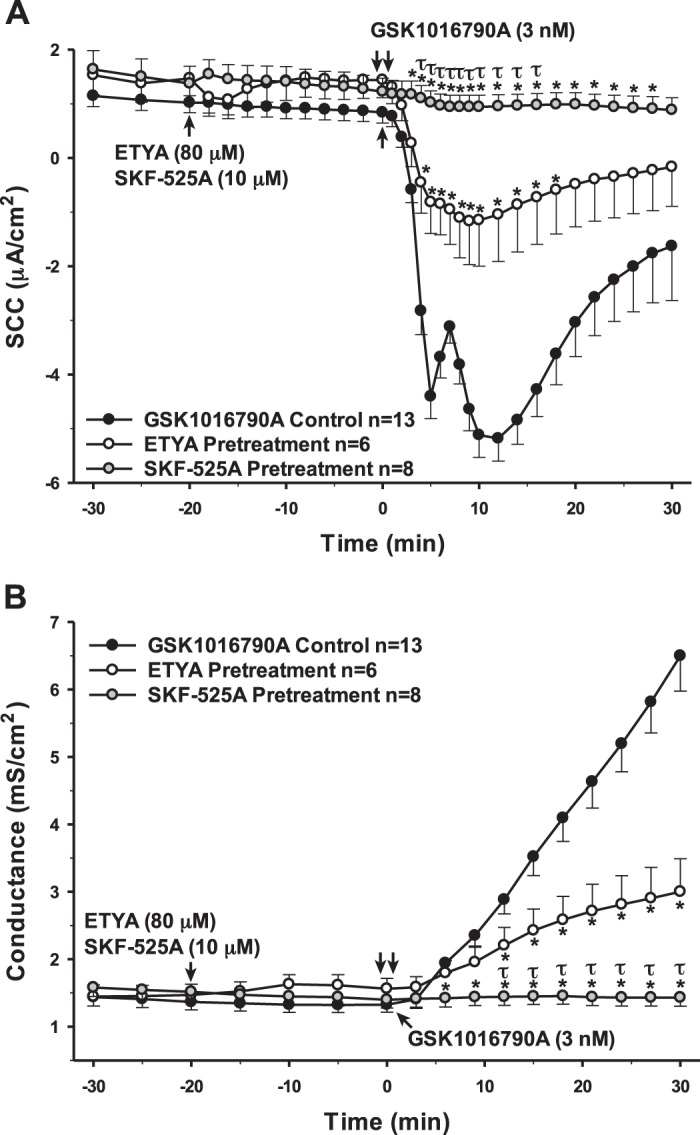
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with arachidonic acid (AA) metabolism inhibitors 20-min prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect of inhibitor preincubation on TRPV4-stimulated transepithelial ion flux and conductance was measured. All inhibitors were solubilized in 100% ethanol. Cyclooxygenase and lipoxygenase inhibitor eicosatetraynoic acid (ETYA) was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −20 min. Cytochrome P450 inhibitor SKF-525A was added to the PCP-R media on both basolateral and apical sides of the membrane at t = −20 min. TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. ETYA pretreatment is signified by white circles. SKF-525A pretreatment is signified by gray circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current. *P < 0.05 against control; τP < 0.05 against ETYA pretreatment.
As previously mentioned, the anti-inflammatory AA metabolite EET is considered largely to be the most probable metabolite involved in TRPV4 activation (5, 7, 32, 47). However, when the PCP-R cells were incubated with the isoform 5,6-EET for 10 min prior to adding the agonist, we did not see significant evidence of a change in TRPV4-mediated activity. While the 10- to 30-min phase after agonist addition appeared to be inhibited with both SCC and conductance measurements, this difference was not statistically different (Fig. 14). The conductance change is similar to the change seen with higher doses of AA.
Fig. 14.
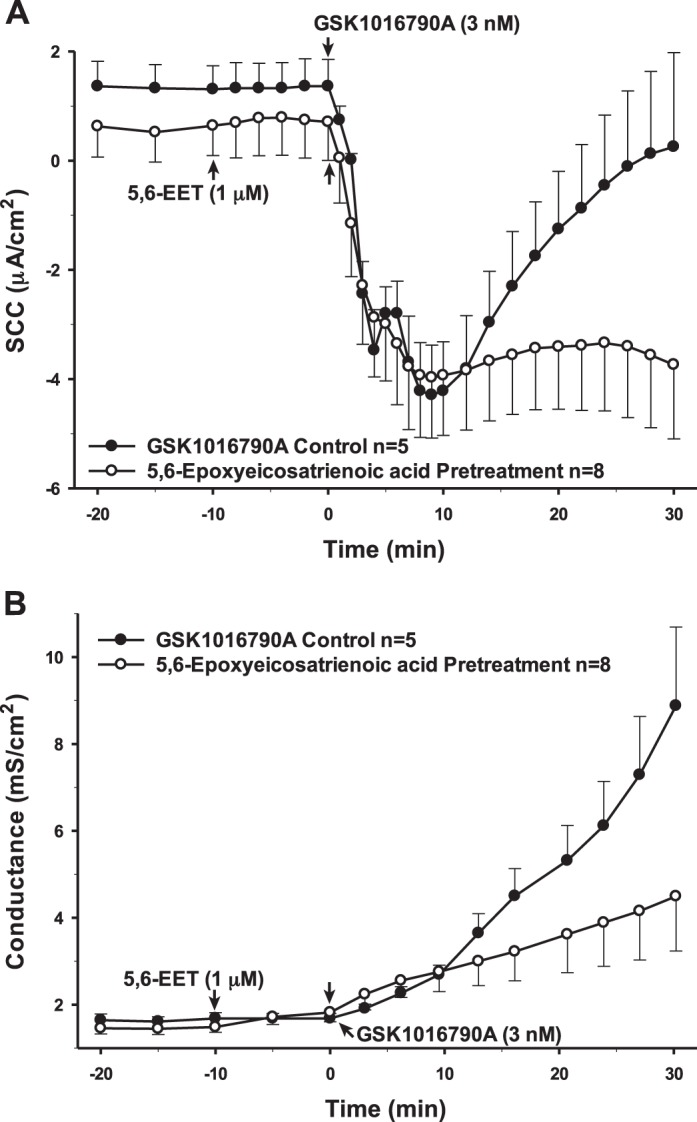
Pretreatment of porcine choroid plexus-Riems (PCP-R) cells with arachidonic acid metabolite 5,6-epoxyeicosatrienoic acid (5,6-EET) at 10 min prior to transient receptor potential vanilloid 4 (TRPV4) agonist addition. Effect on transepithelial ion flux and conductance was measured. 5,6-EET solubilized in 100% ethanol was added to the PCP-R media on both basolateral and apical sides of the membrane at time (t) = −10 min. TRPV4 agonist GSK1016790A was added to basolateral side of the membrane only at t = 0 min. Incubation for 10 min is signified by white circles. GSK1016790A control is signified by black circles. Circles signify mean values and bars signify ±SE for number of experiments indicated. SCC, short-circuit current.
DISCUSSION
TLRs are found on microglia, astrocytes, and other immune cells and are well known for playing a key role in the initiation of innate immunity in response to PAMPs and DAMPs. The TLRs not only play a major role in a cell’s ability to respond to inflammatory reactions but also enable the cells to contribute to the inflammation. When the TLRs detect PAMPs or DAMPs, this can cause a cascade of events leading to activation of transcription factors NF-κB and AP-1 and subsequent cytokine production (6, 34). In particular, TLR4 is best known for producing key pro- and anti-inflammatory cytokines, such as TNF-α, IL-6, and IL-10, and is most often associated with inflammation (6, 34). It is also of interest that expression levels of TLRs have been found to change during aging (34). Specifically, proinflammatory cytokine production increases while secretion of anti-inflammatory cytokines decreases. TLRs have been associated with age-related inflammatory neurodegenerative diseases, such as ischemic stroke, Alzheimer’s disease, and multiple sclerosis (34).
The PCP-R cell line contains mRNA for the receptors of most cytokines tested, except for IL-10, and all 10 of the known porcine TLRs. This indicates that CP cells have the potential to respond to cytokines and inflammatory signals as well as produce several different cytokines, which would be secreted into the CSF. While we did not explore cytokine production in these studies, it is likely that autocrine or paracrine cytokines will have effects on CSF production by the CP cells.
In studies using lung and endothelial tissue, TRPV4 antagonists have been found to reduce pulmonary edema and protect against sepsis, respectively. In both studies, TRPV4 inhibition was also found to decrease inflammation and cytokine production (3, 9). This would suggest that TRPV4 acts in a proinflammatory manner. To confirm this in the brain, we looked at various types of cytokines, including proinflammatory, anti-inflammatory, and paradoxical pro- and anti-inflammatory cytokines. Indeed, long-term (24 h) incubation with the proinflammatory cytokines had significant effects on TRPV4 activity. However, the responses seen with the two prominent proinflammatory cytokines were not the same. Furthermore, the paradoxical cytokines TGF-β1 and IL-6 had varying effects, with TGF-β1 causing strikingly similar results as IL-1β while IL-6 had no significant effect.
The exact nature of the electrophysiological response to TRPV4 agonists is undoubtedly complex. We have previously shown that a component of the response is due to potassium secretion via the intermediate conductance (IK) channel (37). However, the complex nature of the SCC response, as well as the differential effects of cytokines on this response, strongly suggests a multitude of intersecting ion fluxes. SCC is a measure of net transepithelial ion movements. Although the current returns to “baseline” ∼30 min after treatment with a TRPV4 agonist, the sustained high conductance suggests a continued transepithelial ion flux with anions and cations moving in opposite directions, resulting in an electroneutral short circuit current or net zero electrogenic flux. The cytokines that have an effect on net ion fluxes appear to be doing so by altering different components of the complex response.
While it cannot be assumed that all proinflammatory pathways cause an attenuation or inhibition of the response, it is most likely, based on our observations, that TGF-β1 is acting in a proinflammatory manner while IL-6 is acting in an anti-inflammatory manner in the CP cells. Together, these data imply that proinflammatory cytokines diminish TRPV4 function. While upstream activators TNF-α and IL-1β required 24 h to mediate a measurable effect on the PCP-R cells, the finding that short-term inhibition of the proinflammatory transcription factor NF-κB caused a potentiation of TRPV4-mediated ion flux and permeability changes cannot be easily explained. This may indicate a preexisting tone of NF-κB activation in the PCP-R cells. The inhibition of this baseline activity may cause an increase in barrier permeability, as ions are more easily able to move across the membrane. After 24 h of inhibition, the basal conductance is substantially higher than the control, which further suggests underlying NF-κB activity. However, the attenuated response upon TRPV4 stimulation suggests that NF-κB activation, and possibly its downstream inflammatory effectors, can be considered contributors to TRPV4-mediated ion flux and permeability changes.
Effects seen after long-term cytokine incubations raised the question of whether the changes in TRPV4-mediated activity were due to an alteration of the transcription of TRPV4. However, there were no statistical differences in the quantitative PCR of TRPV4 in response to 24-h cytokine incubations, indicating that the cytokines are having an effect on the downstream targets of the ion channel.
Several publications have reported that AA and its metabolites are able to cause a substantial elevation of [Ca2+]i by activating TRPV4 with concentrations as low as 3 µM AA (5, 31, 47). Specifically, these studies also cite EETs as the predominant activator of the cation channel (5, 7, 32, 47). However, it has also been reported that endogenous levels of AA can differ depending on the tissue type, with physiological concentrations varying from 2 to 16 µM. In pancreatic islet cells, the concentration was found to be as high as 75 µM (31). Based on previous studies, we hypothesized that TRPV4 activity would be potentiated after preincubation with either physiological concentrations of AA or its metabolites. Surprisingly, concentrations of AA comparable with previous publications (10 µM) failed to potentiate TRPV4-mediated activity. Rather, AA (up to 100 µM) caused an inhibition of the TRPV4-mediated response to transepithelial ion flux and cell permeability in a dose-dependent manner. AA inhibition of TRPV4-mediated ion flux in the PCP-R cells suggests a cell-type specific effect on TRPV4. Taken together with our cytokine data, in which selective proinflammatory cytokines downregulate TRPV4-mediated activity, this would imply that inflammation acts to decrease TRPV4 function.
There is controversy concerning which isoform of EET is responsible for activating TRPV4 (12). It is possible that different isoforms of EET affect TRPV4 in a cell-specific manner. While 5,6-EET is most often considered to be the activating isoform in other cell types, this does not appear to be the case when it is added exogenously in CP cells. It is possible that the 5,6-EET is only effective when produced inside the cells. However, it is unknown whether the PCP-R cell line is able to produce EETs. The production of EETs occurs when AA is metabolized by the enzymes cytochrome P450 epoxygenase, of which there are multiple isoforms (40). Currently, only two of these enzymes have been sequenced in pigs, Cyp2c42 and Cyp2e1, neither of which were present in the cells. However, there are several other isoforms still untested, including those most associated with EET production, Cyp2c8, Cyp2c9, Cyp2c19, and Cyp2j2 (40). Therefore, these data cannot lead to the conclusion that there are no cytochrome P450 epoxygenases present. Furthermore, the dramatic inhibitory effects of the SKF-525A CYP450 inhibitor imply that endogenous EET production may activate TRPV4 in these cells. It is possible that a different EET isoform other than 5,6-EET is affecting TRPV4 in the CP or that the production occurs within a membrane-limited compartment that is closely linked to the channel.
We determined the potential effects of the other AA metabolites in the CP cells. Both isoforms of cyclooxygenase were present in the PCP-R cells, and therefore, the cells are able to produce prostaglandins, prostacyclin, and thromboxane A2 (36). However, inhibition of both COX1 and COX2 with the NSAID indomethacin did not alter the effects of TRPV4 activation in the cells. Thus, these metabolites likely do not play a role in TRPV4-mediated pathways.
The lipoxygenase (LOX) pathway contains seven known gene isomers: Aloxe3, -5, -5ap, -12, -12b, -15, and -15b (30). Of the enzymes, ALOX12B, -E3, and -15B are found in the skin and other epithelial cells, and ALOX5, -12, and -15 are located primarily in immune cells. Many of the isomers have been reported in different forms of cancers, but all of them can be involved in inflammation (30). Of the four sequenced porcine isomers, only ALOX15 and ALOX15B were found in our cell line. Interestingly, ALOX15 is considered one of the isoforms that can be partially controlled by cytokines. Anti-inflammatory cytokines such as IL-4 and IL-13 have the potential to increase expression of this gene, which can then decrease production of proinflammatory cytokines, such as IL-12 (30). The isoform has also been associated with maintaining dermal integrity through anti-inflammatory suppression of inflammation in the skin (19). On the other hand, ALOX15 is also capable of increasing proinflammatory matrix metalloproteinase (MMP) expression and contributing to arthritic disease progression (49, 53). Once again, this leads to the question of whether TRPV4 is part of a pro- or anti-inflammatory process. Less is known about the ALOX15B isoform, but it has been implicated in decreasing epithelial barrier permeability (30). Thus, increases in this isoform could contribute to increases in transepithelial ion flux and conductance in the CP tissue.
As previously stated, inhibiting cyclooxygenases alone does not affect the cellular response to TRPV4 agonists. However, when both cyclooxygenases and lipoxygenases are inhibited, there is a partial inhibition of the TRPV4-mediated response. As shown above, inhibiting the CYP450s and subsequent EET production results in complete inhibition of the TRPV4-mediated response. Thus, the interaction between the AA metabolites and TRPV4 is complicated and may involve both positive and negative effectors produced in a stimulus-dependent manner. Future experiments will investigate how these metabolites interact with the channel.
In summary, TRPV4-mediated transepithelial ion flux and increases in conductance are negatively affected by proinflammatory cytokines and mediators. We also found that, in contrast to previously reported studies, arachidonic acid does not increase TRPV4-related activity. Rather, when added to choroid plexus cells, arachidonic acid diminished TRPV4-mediated transepithelial ion flux and changes in permeability. Finally, we confirmed that in the choroid plexus, inhibition of EET production causes a complete inhibition of TRPV4-mediated transepithelial ion flux and cellular permeability changes. Therefore, in the choroid plexus, TRPV4 regulation by inflammatory mediators is complex, consistent with the role of this channel as a hub protein that can integrate multiple extracellular signals and subsequently mediate a multiphasic change in transepithelial ion transport.
GRANTS
This research was supported by Indiana Clinical and Translational Sciences Institute Predoctoral Award UL1TR001108 (S. Simpson) and the Department of Defense Investigator-Initiated Research Award W81XWH-16-PRMRP-IIRA (B. Blazer-Yost).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S. and B.B.-Y. conceived and designed research; S.S. and D.P. performed experiments; S.S. and D.P. analyzed data; S.S., D.P., and B.B.-Y. interpreted results of experiments; S.S. and D.P. prepared figures; S.S. drafted manuscript; S.S., D.P., C.S., H.S., and B.B.-Y. edited and revised manuscript; S.S., D.P., C.S., H.S., and B.B.-Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nicolas Berbari and Patrick Antonellis for input in optimizing and troubleshooting the qPCR assay methods.
REFERENCES
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 4: 499–511, 2004. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Azizi G, Navabi SS, Al-Shukaili A, Seyedzadeh MH, Yazdani R, Mirshafiey A. The role of inflammatory mediators in the pathogenesis of Alzheimer’s disease. Sultan Qaboos Univ Med J 15: e305–e316, 2015. doi: 10.18295/squmj.2015.15.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76: 77–98, 2005. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Cao S, Anishkin A, Zinkevich NS, Nishijima Y, Korishettar A, Wang Z, Fang J, Wilcox DA, Zhang DX. Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. J Biol Chem 293: 5307–5322, 2018. doi: 10.1074/jbc.M117.811075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clop A, Huisman A, van As P, Sharaf A, Derdak S, Sanchez A. Identification of genetic variation in the swine toll-like receptors and development of a porcine TLR genotyping array. Genet Sel Evol 48: 28, 2016. doi: 10.1186/s12711-016-0206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrigan F, Mander KA, Leonard AV, Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation 13: 264, 2016. doi: 10.1186/s12974-016-0738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J Neuroimmunol 101: 211–221, 1999. doi: 10.1016/S0165-5728(99)00148-4. [DOI] [PubMed] [Google Scholar]
- 9.Dalsgaard T, Sonkusare SK, Teuscher C, Poynter ME, Nelson MT. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Sci Rep 6: 33841, 2016. doi: 10.1038/srep33841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93: 1847–1892, 2013. doi: 10.1152/physrev.00004.2013. [DOI] [PubMed] [Google Scholar]
- 11.Erickson K, Baron IS, Fantie BD. Neuropsychological functioning in early hydrocephalus: review from a developmental perspective. Child Neuropsychol 7: 199–229, 2001. doi: 10.1076/chin.7.4.199.8737. [DOI] [PubMed] [Google Scholar]
- 12.Filosa JA, Yao X, Rath G. TRPV4 and the regulation of vascular tone. J Cardiovasc Pharmacol 61: 113–119, 2013. doi: 10.1097/FJC.0b013e318279ba42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golomb J, Wisoff J, Miller DC, Boksay I, Kluger A, Weiner H, Salton J, Graves W. Alzheimer’s disease comorbidity in normal pressure hydrocephalus: prevalence and shunt response. J Neurol Neurosurg Psychiatry 68: 778–781, 2000. doi: 10.1136/jnnp.68.6.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han G, Li F, Singh TP, Wolf P, Wang XJ. The pro-inflammatory role of TGFβ1: a paradox? Int J Biol Sci 8: 228–235, 2012. doi: 10.7150/ijbs.8.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrero MT, Estrada C, Maatouk L, Vyas S. Inflammation in Parkinson’s disease: role of glucocorticoids. Front Neuroanat 9: 32, 2015. doi: 10.3389/fnana.2015.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jankovic J, Newmark M, Peter P. Parkinsonism and acquired hydrocephalus. Mov Disord 1: 59–64, 1986. doi: 10.1002/mds.870010108. [DOI] [PubMed] [Google Scholar]
- 17.Kant S, Stopa EG, Johanson CE, Baird A, Silverberg GD. Choroid plexus genes for CSF production and brain homeostasis are altered in Alzheimer’s disease. Fluids Barriers CNS 15: 34, 2018. doi: 10.1186/s12987-018-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, Furey CG, Zhou X, Mansuri MS, Montejo J, Vera A, DiLuna ML, Delpire E, Alper SL, Gunel M, Gerzanich V, Medzhitov R, Simard JM, Kahle KT. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med 23: 997–1003, 2017. doi: 10.1038/nm.4361. [DOI] [PubMed] [Google Scholar]
- 19.Kim SN, Akindehin S, Kwon HJ, Son YH, Saha A, Jung YS, Seong JK, Lim KM, Sung JH, Maddipati KR, Lee YH. Anti-inflammatory role of 15-lipoxygenase contributes to the maintenance of skin integrity in mice. Sci Rep 8: 8856, 2018. doi: 10.1038/s41598-018-27221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krauss JK, Regel JP, Droste DW, Orszagh M, Borremans JJ, Vach W. Movement disorders in adult hydrocephalus. Mov Disord 12: 53–60, 1997. doi: 10.1002/mds.870120110. [DOI] [PubMed] [Google Scholar]
- 21.Kushel’ YV, Belova YD, Tekoev AR. [Intramedullary spinal cord tumors and hydrocephalus: an analysis of the results of surgical treatment in 541 patients]. Vopr Neirokhir 81: 56–60, 2017. doi: 10.17116/neiro201781456-60. [DOI] [PubMed] [Google Scholar]
- 22.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 83: 483–492, 1995. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651, 2009. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee P, Monaco EA III, Friedlander RM. Blocking TGF-β activity and associated inflammation may halt hydrocephalus. Neurosurgery 73: N13–N14, 2013. doi: 10.1227/01.neu.0000438332.72566.f5. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272: 3406–3410, 1997. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 26.Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535, 2000. doi: 10.1016/S0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin CT, Riva-Cambrin JK. Management of posterior fossa tumors and hydrocephalus in children: a review. Childs Nerv Syst 31: 1781–1789, 2015. doi: 10.1007/s00381-015-2781-8. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Lu KT, Huang TC, Tsai YH, Yang YL. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J Neurochem 140: 718–727, 2017. doi: 10.1111/jnc.13920. [DOI] [PubMed] [Google Scholar]
- 30.Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol 6: 297–310, 2015. doi: 10.1016/j.redox.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meves H. Arachidonic acid and ion channels: an update. Br J Pharmacol 155: 4–16, 2008. doi: 10.1038/bjp.2008.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205, 2004. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 33.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285: 1276–1279, 1999. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Brain Res Rev 59: 278–292, 2009. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ott BR, Jones RN, Daiello LA, de la Monte SM, Stopa EG, Johanson CE, Denby C, Grammas P. Blood-cerebrospinal fluid barrier gradients in mild cognitive impairment and Alzheimer’s disease: relationship to inflammatory cytokines and chemokines. Front Aging Neurosci 10: 245, 2018. doi: 10.3389/fnagi.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palumbo S. Pathogenesis and progression of multiple sclerosis: the role of arachidonic acid–mediated neuroinflammation. In: Multiple Sclerosis: Perspectives in Treatment and Pathogenesis, edited by Zagon IS and McLaughlin PJ. Brisbane, Australia: Codon Publications, 2017, chapt. 7. doi: 10.15586/codon.multiplesclerosis.2017.ch7. [DOI] [PubMed] [Google Scholar]
- 37.Preston D, Simpson S, Halm D, Hochstetler A, Schwerk C, Schroten H, Blazer-Yost BL. Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am J Physiol Cell Physiol 315: C357–C366, 2018. doi: 10.1152/ajpcell.00312.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813: 878–888, 2011. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 39.Schroten M, Hanisch FG, Quednau N, Stump C, Riebe R, Lenk M, Wolburg H, Tenenbaum T, Schwerk C. A novel porcine in vitro model of the blood-cerebrospinal fluid barrier with strong barrier function. PLoS One 7: e39835, 2012. doi: 10.1371/journal.pone.0039835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shahabi P, Siest G, Meyer UA, Visvikis-Siest S. Human cytochrome P450 epoxygenases: variability in expression and role in inflammation-related disorders. Pharmacol Ther 144: 134–161, 2014. doi: 10.1016/j.pharmthera.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Sosvorova L, Mohapl M, Vcelak J, Hill M, Vitku J, Hampl R. The impact of selected cytokines in the follow-up of normal pressure hydrocephalus. Physiol Res 64, Suppl 2: S283–S290, 2015. [DOI] [PubMed] [Google Scholar]
- 42.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2: 695–702, 2000. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 43.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76: 16–32, 2012. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toft-Bertelsen TL, Križaj D, MacAulay N. When size matters: transient receptor potential vanilloid 4 channel as a volume-sensor rather than an osmo-sensor. J Physiol 595: 3287–3302, 2017. doi: 10.1113/JP274135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tully HM, Dobyns WB. Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur J Med Genet 57: 359–368, 2014. doi: 10.1016/j.ejmg.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vadivelu S, Rekate HL, Esernio-Jenssen D, Mittler MA, Schneider SJ. Hydrocephalus associated with childhood nonaccidental head trauma. Neurosurg Focus 41: E8, 2016. doi: 10.3171/2016.8.FOCUS16266. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424: 434–438, 2003. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 48.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol 4: 18, 2013. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu MY, Lin TH, Chiu YC, Liou HC, Yang RS, Fu WM. Involvement of 15-lipoxygenase in the inflammatory arthritis. J Cell Biochem 113: 2279–2289, 2012. doi: 10.1002/jcb.24098. [DOI] [PubMed] [Google Scholar]
- 50.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 35: 419–432, 2002. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto T, Nozaki-Taguchi N. Analysis of the effects of cyclooxygenase (COX)-1 and COX-2 in spinal nociceptive transmission using indomethacin, a non-selective COX inhibitor, and NS-398, a COX-2 selective inhibitor. Brain Res 739: 104–110, 1996. doi: 10.1016/S0006-8993(96)00817-7. [DOI] [PubMed] [Google Scholar]
- 52.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Sitaraman SV, Merlin D. Ste20-related proline/alanine-rich kinase (SPAK) regulated transcriptionally by hyperosmolarity is involved in intestinal barrier function. PLoS One 4: e5049, 2009. doi: 10.1371/journal.pone.0005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao L, Cuff CA, Moss E, Wille U, Cyrus T, Klein EA, Praticò D, Rader DJ, Hunter CA, Puré E, Funk CD. Selective interleukin-12 synthesis defect in 12/15-lipoxygenase-deficient macrophages associated with reduced atherosclerosis in a mouse model of familial hypercholesterolemia. J Biol Chem 277: 35350–35356, 2002. doi: 10.1074/jbc.M205738200. [DOI] [PubMed] [Google Scholar]