

Keywords: diabetes, immune cells, inflammation, macrophages
Abstract
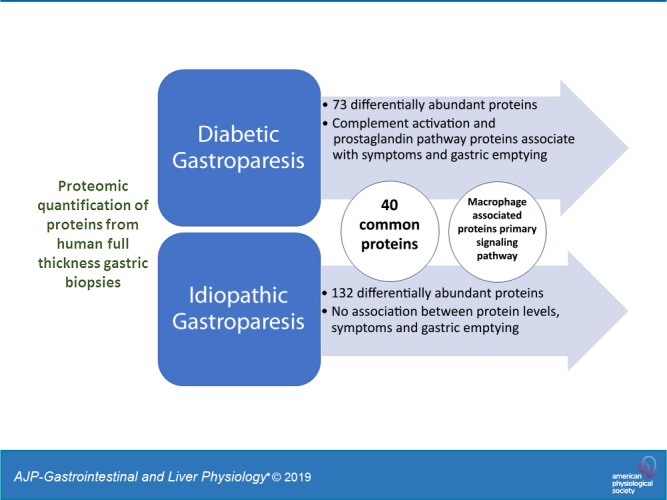
Macrophage-based immune dysregulation plays a critical role in development of delayed gastric emptying in diabetic mice. Loss of anti-inflammatory macrophages and increased expression of genes associated with pro-inflammatory macrophages has been reported in full-thickness gastric biopsies from gastroparesis patients. We aimed to determine broader protein expression (proteomics) and protein-based signaling pathways in gastric biopsies of diabetic (DG) and idiopathic gastroparesis (IG) patients. Additionally, we determined correlations between protein expressions, gastric emptying, and symptoms. Full-thickness gastric antrum biopsies were obtained from nine DG patients, seven IG patients, and five nondiabetic controls. Aptamer-based SomaLogic tissue scan that quantitatively identifies 1,305 human proteins was used. Protein fold changes were computed, and differential expressions were calculated using Limma. Ingenuity pathway analysis and correlations were carried out. Multiple-testing corrected P < 0.05 was considered statistically significant. Seventy-three proteins were differentially expressed in DG, 132 proteins were differentially expressed in IG, and 40 proteins were common to DG and IG. In both DG and IG, “Role of Macrophages, Fibroblasts and Endothelial Cells” was the most statistically significant altered pathway [DG false discovery rate (FDR) = 7.9 × 10−9; IG FDR = 6.3 × 10−12]. In DG, properdin expression correlated with GCSI bloating (r = −0.99, FDR = 0.02) and expressions of prostaglandin G/H synthase 2, protein kinase C-ζ type, and complement C2 correlated with 4 h gastric retention (r = −0.97, FDR = 0.03 for all). No correlations were found between proteins and symptoms or gastric emptying in IG. Protein expression changes suggest a central role of macrophage-driven immune dysregulation in gastroparesis, specifically, complement activation in diabetic gastroparesis.
NEW & NOTEWORTHY This study uses SOMAscan, a novel proteomics assay for determination of altered proteins and associated molecular pathways in human gastroparesis. Seventy-three proteins were changed in diabetic gastroparesis, 132 in idiopathic gastroparesis compared with controls. Forty proteins were common in both. Macrophage-based immune dysregulation pathway was most significantly affected in both diabetic and idiopathic gastroparesis. Proteins involved in the complement and prostaglandin synthesis pathway were associated with symptoms and gastric emptying delay in diabetic gastroparesis.
INTRODUCTION
Gastroparesis is defined by a delay in gastric emptying in association with upper gastrointestinal symptoms of nausea, vomiting, fullness, bloating, and abdominal pain. The etiologies can be diabetes, postsurgical, or idiopathic. The loss or injury of interstitial cells of Cajal (ICC), the “pacemaker” cells involved in generating electrical slow waves that drive rhythmic contraction of smooth muscle cells (SMCs), is described as the primary cellular abnormality in animal models of gastroparesis (12, 35). Nearly 50% of patients with diabetic and idiopathic gastroparesis have significant loss of ICC in the gastric body (21); however, almost all have a significant loss of ICC in the gastric antrum (20). Additionally, ultrastructural damage to ICC was seen in all gastroparesis patients studied (13). Finally, loss of enteric neural fibers and smooth muscle fibrosis was reported in a subset of patients with gastroparesis (21).
Recently, macrophage-mediated immune dysregulation has emerged as an important molecular abnormality in gastroparesis. In the nonobese diabetic (NOD) mouse model, the proportion of CD206+ macrophages expressing heme oxygenase-1 (HO1), considered traditionally as “M2” macrophages (alternatively activated or anti-inflammatory), increased with the development of diabetes and markedly decreased with the onset of delayed gastric emptying. The development of delayed gastric emptying was also associated with an increase in inducible nitric oxide synthase (iNOS) expression, a marker for “M1” phenotype of macrophages (classically activated or pro-inflammatory) (6). Additionally, HO1 expressed in CD206+ macrophages is negatively associated with delayed gastric emptying in diabetic mice, and polymorphisms in the HO1 gene (HMOX1) are associated with clinical symptoms in patients with gastroparesis (17).
In a report by Cipriani et al. (9), CSF1op/op mice, which lack macrophages due to a mutation in macrophage colony stimulating factor (CSF1), did not develop delayed gastric emptying despite severe diabetes, whereas their wild-type counterparts did. Interestingly, intraperitoneal injection of CSF1 to CSF1op/op mice restored the muscularis macrophages, and these mice acquired delayed gastric emptying upon induction of diabetes (8). These reports clearly suggest the crucial roles played by macrophages on the onset of gastroparesis in diabetic models.
Previously, we have shown in full-thickness gastric biopsies of body (3) and antrum (20) obtained from diabetic and idiopathic gastroparesis patients a significant reduction in the total number of CD206+ macrophages, and this reduction correlated significantly with the loss of ICC (20). The loss of ICC has also been shown to correlate with the severity of gastric emptying in patients with diabetic gastroparesis (19). It is still unclear how the loss of macrophages influences the loss of ICC; however, paracrine signaling through inflammatory mediators like TNFα or IL6 may play a role (8, 11).
More recently, we have shown by deep transcriptomic profiling that genes associated with M1 macrophage phenotype had an increased expression in gastric muscularis of patients with idiopathic gastroparesis (22). Further characterization of molecular changes in human gastroparesis is needed, especially in idiopathic gastroparesis, which lacks animal models. Thus, we aimed to determine the differentially abundant proteins in gastric full-thickness biopsies from diabetic and idiopathic gastroparesis patients using a proteomics approach. Second, we aim to determine proteomics-based signaling pathways in diabetic and idiopathic gastroparesis and determine associations between expressed proteins, gastric emptying delay, and symptom severity of gastroparesis.
METHODS
Tissue samples.
Full-thickness gastric antrum biopsies from nine diabetic gastroparesis and seven idiopathic gastroparesis patients were obtained during implantation of a gastric electrical stimulator at a clinical site of the National Institute of Diabetes and Digestive and Kidney Diseases Gastroparesis Clinical Research Consortium (GpCRC). Gastroparesis was defined by a 4-h gastric emptying scintigraphy test (%gastric retention >60% at 2 h and/or >10% at 4 h) and absence of gastric outlet obstruction on upper endoscopy. Control antrum tissue was obtained from five subjects undergoing bariatric surgery at Mayo Clinic. Tissue was obtained in a standardized location in gastric antrum from all subjects. Informed consent was obtained from all gastroparesis patients. Control tissue was obtained in an anonymous fashion as surgical waste. The study was approved by Institutional Review Boards at Temple University and the Mayo Clinic.
The full-thickness gastric biopsy tissue was procured in the operating room and snap-frozen no later than 30 min from procurement. Frozen gastroparesis samples were shipped overnight in dry ice from Temple University to the Mayo Clinic. All samples were stored at −70°C until ready for use. Single thawing was done at the time of protein extraction. Tissue was cryopulverized and protein extracted using T-Per tissue protein extraction agent. Halt protease inhibitor cocktail (Thermo Fisher Scientific) was used during protein extraction. Protein concentrations were calculated using Pierce bicinchoninic acid (BCA) microplate assay (Thermo Fisher Scientific), followed by quantification using Epoch plate reader. The Aptamer-based SOMAscan Assay Human Cell & Tissue kit (CLTH_4.2_20161012_1.5k) that quantitatively identifies 1,305 human proteins was used. A detailed review of nucleic acid aptamer-based proteomic strategy is provided elsewhere (52). Briefly, 120 µL of the lysate at 20 µg/mL was mixed with 2.0 µL of Z-block reagent and 1.2 µL of HALT protease, and then 100 uL of the mixture was input into the assays. The kitted controls and blank samples were treated the same way as the actual lysate samples for the assay QC and data normalization. After all assay steps were completed, the SOMAmers bound to and representative of specific proteins in the lysate samples were hybridized onto Agilent DNA microarray slides for the SOMAscan 1.3k assay. After washing and staining was completed, the slides were scanned with an Agilent SureScan microarray scanner at a 5-µm resolution to detect Cy3 fluorescence. Gridding and analysis of images was performed using Agilent Feature Extraction version 10.7.3.1 using the SOMAscan protocol.
Data normalization and differential expression.
SOMAscan raw data was processed following the manufacturer’s recommendations. In brief, hybridization normalization was performed using control probes to remove intra-array hybridization variation. Calibrator control samples were run in replicates on all arrays along with clinical samples, and the median values of calibrators were utilized to perform intensity scaling between plates. Normalized and scaled data were subjected to differential expression using Limma software (41). In brief, the intensity of each probe (i.e., protein) was compared between a pair of experimental groups using a linear model configured to compute moderated t-statistics via empirical Bayes moderation of standard errors. Protein fold changes (on log scale) were computed and differential expression P values corrected for multiple testing using the Benjamini-Hochberg [false discovery rate (FDR)] method. Proteins with a corrected P value of ≤0.05 were considered as significantly differentially expressed between groups. All computations were performed in R statistical programming environment (version 3.4.1).
A post hoc power calculation was done using Hierarchical Clustering Explorer (version 3.5). To accomplish this, we estimated the pooled variance observed for each probe. The minimum detectable effect size (i.e., fold change) was set to 1.25. Using a two-sample t-test as test statistic and setting the type I error to 0.05, we estimated that a minimum of five samples were required (per group) to detect the desirable effect size with a power of 80%. The pooled variance of 0.9% of the probes (i.e., 13 out 1,305 probes) was too high to detect the effect size of 1.25 with a minimum of five samples. However, the minimum detectable effect size for these probes when five samples per group were used was 1.35.
Pathway and gene set enrichment analysis.
Significantly differentially expressed proteins between a pair of experimental groups were uploaded to the Ingenuity Pathway Analysis (IPA) software (version 01-07), and pathways with a Benjamini-Hochberg (FDR) corrected P value of ≤0.05 were considered for interpretation.
Correlation with gastric emptying and symptoms.
Normalized and log-transformed protein probe intensities were correlated with percent gastric retention at 4 h, GCSI subscores, and overall score using Spearman rank correlation. P values of the correlations were estimated using “Algorithm AS 89” and corrected using Benjamini-Hochberg method. All computations were performed in an R statistical programming environment (version 3.4.1).
RESULTS
Demographic characteristics of study subjects.
Table 1 represents the demographic characteristics of study participants. The median age and the sex distribution were similar between patients with diabetic gastroparesis, idiopathic gastroparesis, and controls. The gastroparesis cardinal symptom index (GCSI) subscores (nausea, bloating and fullness) and the overall GCSI score were similar between diabetic and idiopathic gastroparesis patients.
Table 1.
Demographic characteristics of patients with diabetic gastroparesis, idiopathic, gastroparesis and controls
| Diabetic Gastroparesis | Idiopathic Gastroparesis | Controls | |
|---|---|---|---|
| Median age (range) | 38 (22–57) yr | 40 (26–69) yr | 43 (25–65) yr |
| Women/men | 7/2 | 5/2 | 3/2 |
| Biopsy location | Antrum | Antrum | Antrum |
| Site collected | Temple University | Temple University | Mayo Clinic |
| %Gastric retention at 4 h (means ± SD) | 37.5 ± 15.4 | 21.0 ± 17.8 | |
| GCSI (means ± SD) | |||
| Nausea | 3.3 ± 0.7 | 3.3 ± 0.6 | |
| Bloating | 4.0 ± 0.6 | 3.4 ± 1.2 | |
| Fullness | 4.0 ± 0.9 | 3.5 ± 1.7 | |
| Overall | 3.8 ± 0.5 | 3.7 ± 0.9 |
GCSI, gastroparesis cardinal symptom index.
Differentially expressed proteins.
Compared with controls, 73 proteins were differentially expressed in diabetic gastroparetics (Fig. 1A), and 132 proteins were differentially expressed in idiopathic gastroparetics (Fig. 1B). A volcano plot of differentially abundant proteins is shown (Fig. 2). Forty proteins were common to both diabetic and idiopathic gastroparesis. Of those, 15 proteins were overexpressed in both diabetic and idiopathic gastroparesis, whereas 25 were underexpressed in both. A Venn diagram with the number and names of uniquely expressing and overlapping proteins in diabetic and idiopathic gastroparesis is shown in Fig. 3. A complete list of all protein expressions in patients with diabetic and idiopathic gastroparesis is available in GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130672). Based on statistical significance, the top 10 differentially expressed proteins in diabetic and idiopathic gastroparesis, as compared with the controls, are listed in Table 2. Of the top 10 proteins, 9 were underexpressed in diabetic gastroparesis patients when compared with controls, whereas 1 was overexpressed. However, for the top 10 most different proteins in idiopathic gastroparetics, 4 showed decreased expression, whereas the rest showed an increased expression when compared with the controls. There were four proteins in the two lists, i.e., trefoil factor 2 (TFF2), fatty acid-binding protein 3 (FABP3), glycerol-3-phosphate dehydrogenase dehydrogenase 1 (GPD1), and granulocyte colony-stimulating factor 3 (CSF3), that were common between diabetic and idiopathic gastroparesis patients, and all had decreased expression in both groups with a similar fold change pattern.
Fig. 1.


Heat map showing differentially expressed proteins (false discovery rate < 0.05). A: abundance of 73 proteins was changed in diabetic gastroparesis [n = 9 diabetic gastroparesis, 5 controls (Ctrl)]. B: abundance of 132 proteins was changed in idiopathic gastroparesis (n = 7 idiopathic gastroparesis, 5 controls). Ctrl, control; DG, diabetic gastroparesis.
Fig. 2.

A: volcano plot showing expressed proteins. B: diabetic gastroparesis vs. controls. Idiopathic gastroparesis vs. controls. Proteins in red indicate false discovery rate (FDR) < 0.01.
Fig. 3.

Venn diagram showing unique and common differentially abundant proteins in diabetic and idiopathic gastroparesis. Thirty-three proteins were uniquely changed (17 overexpressed and 16 underexpressed) in diabetic gastroparesis as compared with controls. Ninety-two proteins were uniquely changed in idiopathic gastroparesis (66 overexpressed and 26 underexpressed). Forty proteins were common to both diabetic and idiopathic gastroparesis (15 overexpressed and 25 underexpressed in both). Protein names and log fold changes shown using colored codes.
Table 2.
Most significantly affected proteins in diabetic and idiopathic gastroparesis
| Proteins | Fold Change | Adjusted P Value |
|---|---|---|
| Diabetic gastroparesis vs. controls (n = 9 and 5) | ||
| Trefoil factor 2 | −11.3 | 1.1 × 10−10 |
| FABP3, heart | −2.8 | 1.9 × 10−5 |
| GPD1 | −12.2 | 7.8 × 10−5 |
| PRSS1 | −2.3 | 1.8 × 10−4 |
| IL-1R2 | −1.5 | 1.7 × 10−3 |
| CSF3 | −1.3 | 2.4 × 10−3 |
| CA3 | −8.3 | 2.8 × 10−3 |
| AHSG | −1.2 | 6.1 × 10−3 |
| CA1 | −4.7 | 6.7 × 10−3 |
| CAMK2B | 1.7 | 7.2 × 10−3 |
| Idiopathic gastroparesis vs. controls (n = 7 and 5) | ||
| TFF2 | −11.3 | 2.7 × 10−10 |
| SLIRK5 | 2.0 | 2.6 × 10−4 |
| PGD | 1.5 | 3.3 × 10−4 |
| FABP3, heart | −2.4 | 3.7 × 10−4 |
| GPD1 | −9.2 | 4.4 × 10−4 |
| NRG1 | 1.2 | 4.4 × 10−4 |
| FGF12 | 1.3 | 5.7 × 10−4 |
| CSF3 | −1.4 | 6.4 × 10−4 |
| AIP | 1.5 | 6.9 × 10−4 |
| FGF1 | 1.9 | 6.9 × 10−4 |
AHSG, α-2-HS-glycoprotein; AIP, AH receptor-interacting protein; CA1, carbonic anhydrase 1; CA3, carbonic anhydrase 3; CAMK2B, calcium/calmodulin-dependent protein kinase type II subunit-β; CSF3, granulocyte colony-stimulating factor; FABP3, fatty acid-binding protein; FGF1 and -12, fibroblast growth factor 1 and 12, respectively; GPD1, glycerol-3-phosphate dehydrogenase [NAD(+)], cytoplasmic; IL-1R2, interleukin-1 receptor type 2; NRG1, neuregulin-1; PGD, 6-phosphogluconate dehydrogenase, decarboxylating; PRSS1, trypsin-1; SLIRK5, SLIT and NTRK-like protein 5; TFF2, trefoil factor 2.
Pathway analysis.
Significantly different proteins in the two groups were subjected to IPA to determine the canonical biological pathways associated. The top most differentially expressed pathways in diabetic and idiopathic gastroparesis are shown in Table 3. In both diabetic and idiopathic gastroparesis, IPA identified the proteins belonging to the “role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis” as the most statistically significant among the pathways tested (diabetic gastroparesis FDR = 7.9 × 10−9; idiopathic gastroparesis FDR = 6.3 × 10−12). Proteins of this pathway that are unique to diabetic gastroparesis are found to be matrix metalloproteinase-3 (MMP3), secreted frizzled-related protein 1 (SFRP1), and frizzled related protein (FRZB), whereas proteins that are unique to idiopathic gastroparesis include colony-stimulating factor 2 (CSF2), proto-oncogene tyrosine-protein kinase Src (SRC), Akt serine/threonine-protein kinase 2 (AKT2), MAP kinase-activated protein kinase 2 (MAPKAPK2), interleukin 1 receptor type I (IL1R1), X-linked interleukin-1 receptor accessory protein-like 2 (IL1RAPL2), serine protease 2 (PRSS2), protein disulfide-isomerase A3 (PDIA3), and macrophage migration inhibitory factor (MIF). The proteins that were observed to be common in both diabetic and idiopathic gastroparetics are calcium/calmodulin-dependent protein kinase type II subunit-β (CAMK2B), serine protease 1 (PRSS1), interleukin 17 receptor A (IL17RA), calcium/calmodulin-dependent protein kinase type II subunit-δ (CAMK2D), interleukin 1 receptor type 2 (IL1R2), calcium/calmodulin-dependent protein kinase IIα (CAMK2A), glycoprotein 130 (IL16ST), and interleukin 17 receptor C (IL17RC). The next two most significant pathways in diabetic gastroparesis include “ERK5 signaling” and “Myc mediated apoptosis signaling,” whereas in idiopathic gastroparesis “role of cytokines in mediating communication between immune cells” and “cross-talk between dendritic cells and natural killer cells” are identified as the next two most significant pathways.
Table 3.
Summary of ingenuity pathway analysis in diabetic and idiopathic gastroparesis
| Ingenuity Canonical Pathways | Adjusted P Value | Molecules |
|---|---|---|
| Diabetic castroparesis vs. controls | ||
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | 7.94E-09 | CAMK2B, PRSS1, IL17RA, MMP3, SFRP1, CAMK2D, IL1R2, FRZB, CAMK2A, IL6ST, and IL17RC |
| ERK5 signaling | 8.91E-05 | YWHAE, YWHAZ, IL6ST, and BAD |
| Myc-mediated apoptosis signaling | 0.000115 | FASLG, YWHAE, YWHAZ, and BAD |
| Role of osteoblasts, osteoclasts, and chondrocytes in rheumatoid arthritis | 0.00012 | BGLAP, MMP3, SFRP1, IL1R2, FRZB and BAD |
| Cross-talk between dendritic cells and natural killer cells | 0.000214 | CAMK2B, FASLG, CAMK2D and CAMK2A |
| Acute-phase response signaling | 0.000288 | APCS, C3, HMOX2, AHSG and IL6ST |
| IGF-1 signaling | 0.000501 | IGFBP2, YWHAE, YWHAZ and BAD |
| iCOS-iCOSL signaling in T helper cells | 0.000741 | CAMK2B, CAMK2D, CAMK2A and BAD |
| Role of IL-17A in psoriasis | 0.000813 | IL17RA and IL17RC |
| PI3K signaling in B lymphocytes | 0.001072 | CAMK2B, C3, CAMK2D and CAMK2A |
| Idiopathic gastroparesis vs. controls | ||
| Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis | 6.31E-12 | CAMK2B, CSF2, SRC, CAMK2D, IL1R2, AKT2, MAPKAPK2, IL6ST, CAMK2A, PRSS1, IL17RA, IL1R1, IL1RAPL2, PRSS2, PDIA3, MIF, and IL17RC |
| Role of cytokines in mediating communication between immune cells | 7.41E-07 | IL2, CSF2, IL24, IL20, CSF3, and IFNB1 |
| Crosstalk between dendritic cells and natural killer cells | 9.33E-07 | CAMK2B, IL2, CSF2, CD209, CAMK2D, IFNB1, and CAMK2A |
| PPARα/RXRα activation | 1.55E-06 | TGFB3, AIP, IL1R1, IL1RAPL2, TGFB2, INSR, GPD1, PDIA3, and IL1R2 |
| Aryl hydrocarbon receptor signaling | 1.95E-06 | TGFB3, GSTA3, SRC, AIP, TFF1, TGFB2, MDM2, and GSTP1 |
| p38 MAPK signaling | 6.61E-06 | TGFB3, MAPKAPK3, IL1R1, IL1RAPL2, TGFB2, IL1R2, and MAPKAPK2 |
| LPS/IL-1-mediated inhibition of RXR function A | 6.92E-06 | CHST2, GSTA3, FABP3, IL1R1, IL1RAPL2, HS6ST1, IL1R2, GSTP1, and CHST15 |
| Apelin cardiac fibroblast signaling pathway | 7.59E-06 | TGFB3, ANGPT2, TGFB2, and AKT2 |
| Role of osteoblasts, osteoclasts, and chondrocytes in rheumatoid arthritis | 9.77E-06 | BGLAP, SRC, CSF2, IL1R1, ADAMTS5, GSN, IL1RAPL2, IL1R2, and AKT2 |
| Xenobiotic metabolism signaling | 1.02E-05 | CAMK2B, CHST2, GSTA3, AIP, CAMK2D, HS6ST1, GSTP1, ESD, CAMK2A, and CHST15 |
AIP, AH-interacting protein; ADAMTS5, a disintegrin and metalloproteinase with thrombospondin-like motifs; BGLAP, bone γ-carboxyglutamate protein; CAMK2A, -B, and -D; CHST, calcium/calmodulin-dependent protein kinase type II subunit 2α, β, and -δ, respectively; CSF1, -2, and 3, colony stimulating factor 1, 2, and 3; FRZB, and frizzled related protein; GSN, gelsolin; GSTA, glutathione-S-transferase-α; GTSP, glutathione-S-transferase P; ILR1, interleukin 1 receptor type I; IL1RAPL2, X-linked interleukin-1 receptor accessory protein-like 2; INSR, insulin receptor; MAPKAPK3, mitogen-activated protein kinase-activated protein kinase 2; PI3K, phosphatidylinositol 3-kinase; PRSS1, serine protease 1; RXR, retinoid X receptor; TFF1, trefoil factor 2; TGFB3, transforming growth factor-β.
Correlations with gastric emptying and symptoms.
The expression of protein properdin correlated negatively with GCSI-bloating (Spearman r = −0.99; FDR = 0.02) in patients with diabetic gastroparesis (Fig. 4A). No statistically significant correlations (after correction for multiple comparisons) were found between protein expressions and other subscores (nausea, fullness) or the overall GCSI score. Meanwhile, the expressions of proteins prostaglandin G/H synthase 2, protein kinase Cζ type (PKCζ), and complement C2 were also found to be negatively correlated with 4 h of gastric retention in patients with diabetic gastroparesis (Spearman, r = −0.97; FDR = 0.03 for all; Fig. 4, B–D). No statistically significant correlations were found between protein expressions and gastric retention or symptom scores in patients with idiopathic gastroparesis.
Fig. 4.

Correlations between protein expression (normalized and log transformed), symptoms, and gastric emptying in diabetic gastroparesis. A: properdin (CFP) abundance negatively correlated with gastroparesis cardinal symptom index (GCSI) bloating. No correlations were seen between other subscores (nausea, fullness) or overall GCSI. B–D: negative correlation between 4 h of gastric retention and expressions of prostaglandin G/H synthase 2 (PTGS2), complement 2 (C2), and protein kinase Cζ (PRKCZ). No correlations were seen between protein abundance, symptoms, and gastric retention in idiopathic gastroparesis. A complete list of all protein expressions in patients with diabetic and idiopathic gastroparesis is available in GEO database (GSE130672; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130672; Token: grwpcgikvfmvzgl). AU, arbitrary units. FDR, false discovery rate.
DISCUSSION
This study provides novel proteomics-based molecular characterization of full-thickness gastric tissue from patients with diabetic and idiopathic gastroparesis. Of the 1,305 proteins that can be assessed with this assay, 73 were altered in patients with diabetic gastroparesis and 132 in idiopathic gastroparesis, suggesting a fundamental organic and molecular basis for the pathophysiology of gastroparesis. Idiopathic gastroparesis patients had twice as many proteins altered. Additionally, more than half of the proteins changed in diabetic gastroparesis patients were also changed in patients with idiopathic gastroparesis. This suggests potential overlap of altered molecular pathways in diabetic and idiopathic gastroparesis. However, nonoverlapping protein alterations suggest the likely existence of unique molecular mechanisms, especially in patients with idiopathic gastroparesis. This study validates findings from mouse studies and transcriptomics in humans, suggesting macrophages to be a critical cellular component in the pathophysiology of gastroparesis. The correlation analysis suggests a strong association between lower expression of properdin, a positive regulator of complement with increasing severity of bloating. Finally, lower expressions of prostaglandin G/H synthase 2, protein kinase Cζ type, and complement C2 correlated with worse gastric emptying. Thus, impaired complement activation may play a role in mediating symptoms and gastric emptying delay in diabetic gastroparesis.
Previous studies in animal models and humans with gastroparesis have elucidated the crucial role of the innate immune system in the pathogenesis of gastroparesis (3, 8, 9, 20). Recently, we have reported the deep transcriptomic profiles of stomach tissues of diabetic and idiopathic gastroparesis patients, which highlight innate immune dysregulation as an important feature of gastroparesis (22). Of the commonly expressed proteins in diabetic and idiopathic gastroparesis, the most downregulated protein turned out to be TFF2, a spasmolytic polypeptide belonging to the protease-resistant protein family known as the trefoil factor family (10), well known to be secreted by goblet cells along with mucus to play an important role in mucosal homeostasis (27, 49). This surprised us, because we had used the mucosa-stripped muscularis layer of the gastric antrum for proteomic profiling. Interestingly, TFF2 is expressed not only by gastric epithelial cells but also by macrophages (23, 29). Resident peritoneal macrophages isolated from TFF2−/− mice display a highly activated phenotype and gene expression similar to wild-type macrophages incubated with IL-1β (29), suggesting a pro-inflammatory (M1) phenotype in the absence of TFF2. Intestinal lamina propria macrophages from TFF2−/− mice produce higher amounts of IL-12 (30), which is a pro-inflammatory cytokine often associated with M1 macrophages (38, 53), than their wild-type counterparts. TFF2 has also been shown to inhibit iNOS and nitric oxide (NO) in monocytes in response to bacterial endotoxins (18). Other studies have also shown a role for TFF2 in macrophage recruitment and protection against chemical (24) or infectious (44) injury in the intestinal tract. Lower expression of TFF2 in our samples may reflect a phenotypical switch toward M1 macrophage spectrum that was also seen in our transcriptomic analysis (22). Furthermore, administration of exogenous TFF2 to rat models of colitis (36) and gastric/duodenal ulceration (37) has been found to accelerate tissue repair. Hence, it would be worth considering examining the effects of exogenous TFF2 administration in mouse models of diabetic gastroparesis.
Diabetic and idiopathic gastroparesis patients also displayed 9- to 12-fold lower expression of GPD1, an enzyme involved in regulating the rate-limiting steps of carbohydrate and lipid metabolism. Downregulation of GPD1 has been shown to be influenced by IL-1β (16), a pro-inflammatory cytokine often secreted by M1 macrophages. However, the expression of IL-1β was not changed in our gastroparesis samples compared with the controls. Other downregulated proteins that were common between diabetic and idiopathic gastroparesis patients are associated with fatty acid metabolism (FABP3) and granulocyte proliferation and survival (CSF3). FABP3 or H-FABP has been shown to be expressed in a broad range of skeletal and smooth muscles as well as lipid and steroid synthesizing cells (55). In a recent study, CSF-3 decreased proinflammatory (M1)/anti-inflammatory (M2) macrophage ratio in bone marrow grafts from healthy donors (51). A lower CSF-3 may result in an increase in that ratio, suggesting a skew towards a proinflammatory macrophage state. CSF3 expression was increased upon treatment of human umbilical mesenchymal stem cells with neuronal-conditioned medium along with other markers for neuronal differentiation, pointing to a possible role in neural regeneration (7).
The overall GCSI score is determined by individual scores for each of the subtypes: nausea, bloating, and fullness (39). The etiology and region of bloating in gastroparesis is unclear; however, small intestinal bacterial overgrowth or dysbiosis in upper gastrointestinal tract can cause bloating (14). Here, we report the expression of properdin, a positive regulator of complement activation negatively correlating with bloating. Properdin has been known to be protective in peritonitis induced by sublethal cecal ligation and perforation (47). Gastric microbiome has not been studied in gastroparesis. It is possible that lower properdin promotes upper gastrointestinal microbial dysbiosis, which may induce symptoms of bloating. Alternatively, this may reflect a response to microbial dysbiosis in the upper gastrointestinal tract. In fact, tissue deposition of properdin and bacterial antigen-antibody complexes has been reported as a part of systemic immune complex disease and has responded to immunosuppressive therapy (15).
The mechanism of action for properdin depends on the complement system (45), a part of the innate immune system that involves the activation of several different components in bringing up pathogen clearance by phagocytosis (40). Activation of complement system leads to the infiltration of leukocytes in to the damaged tissue (2). We found the complement C2 expression correlating negatively with 4 h of gastric retention in diabetic gastroparetics. Complement C2 is the serum glycoprotein involved in the classical pathway that is cleaved by activated C1, with the resultant C2a combining with C4b to generate C3, a central component of complement system (28). The generated C3 may then lead to inflammatory (C3a, C5a), opsonization (C3b, C4b) or terminal lysis pathways (C5, C6, C7, C8, and C9) (32). A recent study demonstrated the role of complement 3a in enteric neural crest cell migration, providing initial observations for a possible role of complement activation in motility disorders like gastroparesis (5). Complement C3 was also decreased in our diabetic gastroparesis patients (fold change = −1.7, FDR = 0.03). Additionally, complement C1q was shown to be involved in polarizing macrophages to acquire M2 phenotype in an IL-33-dependent manner (2). Because we have seen the reduction of M2 macrophage content in patients with diabetic and idiopathic gastroparesis (3, 20), it would be beneficial to delineate the involvement of different pathways of complement system in macrophage polarization in the context of gastroparesis. The third protein showing a negative correlation with 4 h of gastric retention in patients with diabetic gastroparesis was the PKCζ, a serine/threonine kinase known to play a pivotal role in intestinal barrier function (1). However, the role of PKCζ in gastrointestinal motility has not been studied.
Gastric emptying is a highly complex process involving coordinated interactions of a variety of cell types and associated mediators. Two proteins that are connected to direct effects on the cell types that contribute to normal gastric contractility patterns, PRSS1 (trypsin-1) and prostaglandin G/H synthase 2 (COX-2), were identified. Trypsin-1 showed 2.3-fold lower expression in diabetic gastroparesis patients compared with controls. The role of proteases in the epithelium as well as endothelium is well established, but one study also found expression of protease-activated receptor RNA in smooth muscle cells, ICC, and platelet-derived growth factor receptor-α-positive cells in colonic tunica muscularis. Functional responses in colonic muscles to trypsin were biphasic and associated with activation of excitatory Ca2+-activated Cl− (anoctamin-1, TMEM16A) conductances in ICC and, separately, inhibitory Ca2+-activated K+ (SK3) conductances in platelet-derived growth factor receptor-α-positive cells (48). Therefore, lower expression of trypsin may result in impaired excitability of ICC or gastric smooth muscle cells. The enzyme prostaglandin G/H synthase 2 (COX-2) synthesizes prostaglandins from arachidonic acid in various organs, including the gastrointestinal tract (33), where the prostaglandins have well-documented effects on gastrointestinal contractility. In the stomach, PGE2 tonically suppresses the spontaneous electrical and mechanical activities of canine antral circular muscle and the responsiveness of the muscle to excitatory stimuli (43). Basal PGE was also found to limit resting and Ach-induced motility of the rat ileum (42). PGE2 has been shown to be involved in gastric dysrhythmias (26, 46), which have been associated with gastroparesis (34). A recent study of Lepob mice, a model of type 2 diabetes, showed intact ICC and enteric nerves but upregulation of various components of prostaglandin signaling. Inhibition of prostaglandin-endoperoxide synthase-2 increased slow-wave amplitudes and frequency of the antral contractions (4). We found a significant negative correlation between the amount of prostaglandin G/H synthase 2 enzyme and 4 h of gastric retention in diabetic gastroparetics but not in idiopathic gastroparesis patients. It is difficult to reconcile this with the known anti-motility effects of prostaglandins described above, but COX-2 inhibition is known to suppress the M2 phenotype in macrophages, and therefore, it may contribute to the pathogenesis of gastroparesis (31). Additionally, macrophage COX-2 was found to be protective against diabetic nephropathy, similar to higher expressions associating with lower gastric retention in our samples (50).
The SOMAscan proteomic platform allows the most extensive profiling of human tissue proteins in a reproducible manner (25). Significant advantages of SOMAmers over traditional aptamers include significantly enhanced binding force between target proteins and corresponding aptamers due to chemical modification. SOMAscan can quantify proteins over a dynamic range with high sensitivity and accuracy (52). Our attempt to identify differentially expressing proteins showed both common and unique proteins in patients with diabetic and idiopathic gastroparesis. Complementing our work that looked at transcriptomic expression profile (22), the proteomics-based IPA analysis to determine the possible involvement of differentially expressing proteins in various canonical pathways found “the role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis” as the most highly expressed pathway in both diabetic and idiopathic gastroparesis patients. In addition, most of the expressed proteins belonging to this pathway are common between diabetic and idiopathic gastroparetics (CAMK2B, PRSS1, CAMK2A, CAMK2D, IL17RC, IL16ST). The lower expression of calcium/calmodulin-dependent protein kinases (CAMKs) in both diabetic and idiopathic gastroparesis types is interesting and may result in alteration in calcium signaling that can affect both smooth muscle contractility and also other cell types, like macrophages and their response to injurious states (54).
The study must be seen in light of its limitations. Changes in protein expression profile reflect associations with gastroparesis and may be in response to the physiological and clinical changes in gastroparesis rather than playing a role in its pathophysiology. Second, although the SOMAscan 1.3k array provides an ability to simultaneously detect and quantify 1,305 human proteins, it still represents only 5% of the total human proteome. Thus, several relevant proteins could have been missed. The pathway analysis uses curated databases that are enriched for canonical cancer and immune biological pathways and thus could bias the interpretation of the observed changes. Furthermore, the array most often covers key proteins, but not all proteins, present in a given pathway, which dampens the pathway analysis. Finally, the sample size is small. However, we still saw a robust signal for the number of altered proteins that were significant after multiple correction testing and that make biological sense in terms of our current understanding of the cytopathology of gastroparesis. These should be seen as hypothesis generating for future experimental and clinical research to verify the role of the putative pathways identified in the pathogenesis of gastroparesis.
Conclusions.
In conclusion, this report using untargeted proteomics provides an in-depth determination of cellular and molecular targets in gastric muscle layers of patients with diabetic and idiopathic gastroparesis. In addition to validating the changes seen in mouse studies of diabetic gastroparesis as well as expression changes seen on human gastric RNA sequencing, this study provides novel findings, including possible involvement of the complement system in pathophysiology of gastroparesis. The pattern of identified targets is unique but coherent with activation of an injurious innate immune cell profile and damage to smooth muscle contractility, a pattern that is consistent with predicted causes for gastroparesis. In combination, these form a strong basis for investigation of immune-based targeting in gastroparesis. Looking at the large number of proteins altered in gastroparesis, this study confirms a strong molecular basis for this disorder. Further work is needed to validate and localize the proteins that were found to be differentially expressed. Additionally, their role in generation of symptoms and gastric dysfunction will need to be determined. This study also gets us one step closer toward determining the utility of proteomics in gastric and other motility disorders of the gut that lack biomarkers. With advancements in endoscopic approaches toward obtaining muscular tissue from gastrointestinal tract, protein-based markers may be further investigated for their potential as disease biomarkers. Finally, the detailed expression profiles presented should be used by us and other investigators in the field to develop hypotheses and targeted studies assessing the role these proteins play in gastroparesis pathophysiology.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants U01-DK-112194, U01-DK-073983, U01-DK-112193, U01-DK-073975, U01-DK-074035, U01-DK-074007, U01-DK-073974, and U01-DK-074008. Additionally, M. Grover is supported by NIDDK K23 DK 103911 and R03 DK 120745.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.G. and NIDDK GpCRC conceived and designed research; C.E.B., T.J.M., T.A.K., M.L.K., and H.P.P. performed experiments; M.G., S.D., and NIDDK GpCRC analyzed data; M.G., S.D., L.L.C., K.P.Y., P.J.P., I.S., R.W.M., K.L.K., T.L.A., B.K., R.J.S., S.J.G., T.J.M., T.A.K., M.L.K., J.T., F.A.H., H.P.P., G.F., and NIDDK GpCRC interpreted results of experiments; M.G. and SD prepared figures; M.G. and NIDDK GpCRC drafted manuscript; M.G., S.D., C.E.B., L.L.C., K.P.Y., P.J.P., I.S., R.W.M., K.L.K., T.L.A., B.K., R.J.S., S.J.G., T.J.M., T.A.K., M.L.K., J.T., F.A.H., H.P.P., G.F., and NIDDK GpCRC edited and revised manuscript; M.G., S.D., C.E.B., L.L.C., K.P.Y., P.J.P., I.S., R.W.M., K.L.K., T.L.A., B.K., R.J.S., S.J.G., T.J.M., T.A.K., M.L.K., J.T., F.A.H., H.P.P., G.F., and NIDDK GpCRC approved final version of manuscript.
ACKNOWLEDGMENTS
The members of the Gastroparesis Clinical Research Consortium are as follows: Clinical centers: Dr. Robert Shulman (principal investigator), Dr. Bruno Chumpitazi, (co-investigator), Cynthia Bouette, Heather Charron, and Nelufa Islam (Baylor College of Medicine, Houston, TX); BCM pediatric gastroparesis centers: Dr. Kent Williams (National Children’s Hospital, Columbus, OH); Dr. Samuel Nurko (Boston Children’s Hospital, Harvard Medical School, Boston, MA); Dr. James Varni (Texas A & M, College Station, TX); Dr. Pankaj Pasricha (principal investigator), Dr. Robert Barat (co-investigator), Dr. Jiande Chen, Guillermo Barahona Hernandez, Robert Burns, and Megan McKnight (Johns Hopkins University, Baltimore, MD); Dr. Branden Kuo (principal investigator), Dr. April Mendez, Dr. Kyle Staller, and Dr. Andrea Thurler (Massachusetts General Hospital, Boston, MA); Dr. Henry Parkman (principal investigator), Dr. Alan Mauer, Dr. Zubair Malik, Dr. Perry Orthey, and Dr. Amiya Palit (Temple University, Philadelphia, PA); Dr. Richard W. McCallum (principal investigator), Dr. Irene Sarosiek (co-investigator), Dr. Sean Connery, Marisol Ramirez, Natalia Vega, and Solmaz Sigaroodi (Texas Tech University Health Sciences Center, El Paso, TX); Dr. Thomas Abell (principal investigator), Dr. Abigail Stocker (co-investigator), Bridget Cannon, Lindsay Nowotny, and Tachisha Walls (University of Louisville, Louisville, KY); Dr. Kenneth Koch (principal investigator), Lynn Baxter, Anya Brown, Paula Stuart, and Amirah Abdullah (Wake Forest University, Winston-Salem, NC); Legacy Clinical Centers: Dr. William Snape (principal investigator), Nata DeVole, Dr. Karen Earle, Dr. Kjersti Kirkeby, Candice Lee, Dr. Mimi Lin, Doug Troyer, and Anna von Bakonyi [California Pacific Medical Center, San Francisco, CA (2008–2017)]; Dr. Jorge Calles-Escandon (principal investigator) [MetroHealth Medical Center, Cleveland, OH (2013–2016)]; Dr. Linda Nguyen (principal investigator), Dr. Emerald Adler, Chiara Orlando [Stanford University, Stanford, CA (2008–2017)]; Dr. William Hasler (principal investigator), Dr. William Herman, Dr. Andrew Kraftson, Dr. Amy E. Rothberg, and Sophanara Wootten [University of Michigan, Ann Arbor, MI (2006–2017)]; resource centers: Dr. James Tonascia (principal investigator), Pat Belt, and Dr. Michele Donithan (2006–2017), Dr. Linda Lee, Laura Miriel, Dr. Emily Sharkey, Dr. Alice Sternberg, Dr. Mark Van Natta, Dr. Laura Wilson, Dr. Goro Yamada, and Dr. Katherine Yates, [Scientific Data Research Center, Johns Hopkins University (Bloomberg School of Public Health), Baltimore, MD]; Dr. Gianrico Farrugia (principal investigator), Dr. Madhusudan Grover, and Cheryl Bernard (Pathology Resource Center, Mayo Clinic College of Medicine, Rochester, MN); Sponsor: Dr. Jose Serrano (program official), Dr. Frank Hamilton (project scientist), Dr. Sherry Hall, Dr. Stephen James, and Rebecca Torrance (National Institute of Diabetes, Digestive and Kidney Diseases, Bethesda, MD).
REFERENCES
- 1.Banan A, Fields JZ, Zhang Y, Keshavarzian A. Key role of PKC and Ca2+ in EGF protection of microtubules and intestinal barrier against oxidants. Am J Physiol Gastrointest Liver Physiol 280: G828–G843, 2001. doi: 10.1152/ajpgi.2001.280.5.G828. [DOI] [PubMed] [Google Scholar]
- 2.Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol 188: 5682–5693, 2012. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernard CE, Gibbons SJ, Mann IS, Froschauer L, Parkman HP, Harbison S, Abell TL, Snape WJ, Hasler WL, McCallum RW, Sarosiek I, Nguyen LA, Koch KL, Tonascia J, Hamilton FA, Kendrick ML, Shen KR, Pasricha PJ, Farrugia G; NIDDK Gastroparesis Clinical Research Consortium (GpCRC) . Association of low numbers of CD206-positive cells with loss of ICC in the gastric body of patients with diabetic gastroparesis. Neurogastroenterol Motil 26: 1275–1284, 2014. doi: 10.1111/nmo.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blair PJ, Hwang SJ, Shonnard MC, Peri LE, Bayguinov Y, Sanders KM, Ward SM. The role of prostaglandins in disrupted gastric motor activity associated with type 2 diabetes. Diabetes 68: 637–647, 2019. doi: 10.2337/db18-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broders-Bondon F, Paul-Gilloteaux P, Gazquez E, Heysch J, Piel M, Mayor R, Lambris JD, Dufour S. Control of the collective migration of enteric neural crest cells by the Complement anaphylatoxin C3a and N-cadherin. Dev Biol 414: 85–99, 2016. doi: 10.1016/j.ydbio.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi KM, Kashyap PC, Dutta N, Stoltz GJ, Ordog T, Shea Donohue T, Bauer AJ, Linden DR, Szurszewski JH, Gibbons SJ, Farrugia G. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 138: 2399–2409.e1, 2010. doi: 10.1053/j.gastro.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou SC, Ko TL, Fu YY, Wang HW, Fu YS. Identification of genetic networks during mesenchymal stem cell transformation into neurons. Chin J Physiol 51: 230–246, 2008. [PubMed] [Google Scholar]
- 8.Cipriani G, Gibbons SJ, Miller KE, Yang DS, Terhaar ML, Eisenman ST, Ördög T, Linden DR, Gajdos GB, Szurszewski JH, Farrugia G. Change in populations of macrophages promotes development of delayed gastric emptying in mice. Gastroenterology 154: 2122–2136.e12, 2018. doi: 10.1053/j.gastro.2018.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cipriani G, Gibbons SJ, Verhulst PJ, Choi KM, Eisenman ST, Hein SS, Ordog T, Linden DR, Szurszewski JH, Farrugia G. Diabetic Csf1op/op mice lacking macrophages are protected against the development of delayed gastric emptying. Cell Mol Gastroenterol Hepatol 2: 40–47, 2016. doi: 10.1016/j.jcmgh.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhar DK, Wang TC, Tabara H, Tonomoto Y, Maruyama R, Tachibana M, Kubota H, Nagasue N. Expression of trefoil factor family members correlates with patient prognosis and neoangiogenesis. Clin Cancer Res 11: 6472–6478, 2005. doi: 10.1158/1078-0432.CCR-05-0671. [DOI] [PubMed] [Google Scholar]
- 11.Eisenman ST, Gibbons SJ, Verhulst PJ, Cipriani G, Saur D, Farrugia G. Tumor necrosis factor alpha derived from classically activated “M1” macrophages reduces interstitial cell of Cajal numbers. Neurogastroenterol Motil 29: e12984, 2017. doi: 10.1111/nmo.12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrugia G. Interstitial cells of Cajal in health and disease. Neurogastroenterol Motil 20, Suppl 1: 54–63, 2008. doi: 10.1111/j.1365-2982.2008.01109.x. [DOI] [PubMed] [Google Scholar]
- 13.Faussone-Pellegrini MS, Grover M, Pasricha PJ, Bernard CE, Lurken MS, Smyrk TC, Parkman HP, Abell TL, Snape WJ, Hasler WL, Unalp-Arida A, Nguyen L, Koch KL, Calles J, Lee L, Tonascia J, Hamilton FA, Farrugia G; NIDDK Gastroparesis Clinical Research Consortium (GpCRC) . Ultrastructural differences between diabetic and idiopathic gastroparesis. J Cell Mol Med 16: 1573–1581, 2012. doi: 10.1111/j.1582-4934.2011.01451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foley A, Burgell R, Barrett JS, Gibson PR. Management strategies for abdominal bloating and distension. Gastroenterol Hepatol (NY) 10: 561–571, 2014. [PMC free article] [PubMed] [Google Scholar]
- 15.Gamble CN, Kimchi A, Depner TA, Christensen D. Immune complex glomerulonephritis and dermal vasculitis following intestinal bypass for morbid obesity. Am J Clin Pathol 77: 347–352, 1982. doi: 10.1093/ajcp/77.3.347. [DOI] [PubMed] [Google Scholar]
- 16.Gao D, Madi M, Ding C, Fok M, Steele T, Ford C, Hunter L, Bing C. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Endocrinol Metab 307: E289–E304, 2014. doi: 10.1152/ajpendo.00430.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbons SJ, Grover M, Choi KM, Wadhwa A, Zubair A, Wilson LA, Wu Y, Abell TL, Hasler WL, Koch KL, McCallum RW, Nguyen LA, Parkman HP, Sarosiek I, Snape WJ, Tonascia J, Hamilton FA, Pasricha PJ, Farrugia G. Repeat polymorphisms in the Homo sapiens heme oxygenase-1 gene in diabetic and idiopathic gastroparesis. PLoS One 12: e0187772, 2017. doi: 10.1371/journal.pone.0187772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giraud AS, Pereira PM, Thim L, Parker LM, Judd LM. TFF-2 inhibits iNOS/NO in monocytes, and nitrated protein in healing colon after colitis. Peptides 25: 803–809, 2004. doi: 10.1016/j.peptides.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 19.Grover M, Bernard CE, Pasricha PJ, Lurken MS, Faussone-Pellegrini MS, Smyrk TC, Parkman HP, Abell TL, Snape WJ, Hasler WL, McCallum RW, Nguyen L, Koch KL, Calles J, Lee L, Tonascia J, Ünalp-Arida A, Hamilton FA, Farrugia G; NIDDK Gastroparesis Clinical Research Consortium (GpCRC) . Clinical-histological associations in gastroparesis: results from the Gastroparesis Clinical Research Consortium. Neurogastroenterol Motil 24: 531–539, 2012. doi: 10.1111/j.1365-2982.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grover M, Bernard CE, Pasricha PJ, Parkman HP, Gibbons SJ, Tonascia J, Koch KL, McCallum RW, Sarosiek I, Hasler WL, Nguyen LAB, Abell TL, Snape WJ, Kendrick ML, Kellogg TA, McKenzie TJ, Hamilton FA, Farrugia G; NIDDK Gastroparesis Clinical Research Consortium (GpCRC) . Diabetic and idiopathic gastroparesis is associated with loss of CD206-positive macrophages in the gastric antrum. Neurogastroenterol Motil 29: e13018, 2017. doi: 10.1111/nmo.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grover M, Farrugia G, Lurken MS, Bernard CE, Faussone-Pellegrini MS, Smyrk TC, Parkman HP, Abell TL, Snape WJ, Hasler WL, Ünalp-Arida A, Nguyen L, Koch KL, Calles J, Lee L, Tonascia J, Hamilton FA, Pasricha PJ; NIDDK Gastroparesis Clinical Research Consortium . Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology 140: 1575–1585.e8, 2011. doi: 10.1053/j.gastro.2011.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grover M, Gibbons SJ, Nair AA, Bernard CE, Zubair AS, Eisenman ST, Wilson LA, Miriel L, Pasricha PJ, Parkman HP, Sarosiek I, McCallum RW, Koch KL, Abell TL, Snape WJ, Kuo B, Shulman RJ, McKenzie TJ, Kellogg TA, Kendrick ML, Tonascia J, Hamilton FA, Farrugia G; NIDDK Gastroparesis Clinical Research Consortium (GpCRC) . Transcriptomic signatures reveal immune dysregulation in human diabetic and idiopathic gastroparesis. BMC Med Genomics 11: 62, 2018. doi: 10.1186/s12920-018-0379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung LY, Sen D, Oniskey TK, Katzen J, Cohen NA, Vaughan AE, Nieves W, Urisman A, Beers MF, Krummel MF, Herbert DR. Macrophages promote epithelial proliferation following infectious and non-infectious lung injury through a Trefoil factor 2-dependent mechanism. Mucosal Immunol 12: 64–76, 2019. doi: 10.1038/s41385-018-0096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judd LM, Chalinor HV, Walduck A, Pavlic DI, Däbritz J, Dubeykovskaya Z, Wang TC, Menheniott TR, Giraud AS. TFF2 deficiency exacerbates weight loss and alters immune cell and cytokine profiles in DSS colitis, and this cannot be rescued by wild-type bone marrow. Am J Physiol Gastrointest Liver Physiol 308: G12–G24, 2015. doi: 10.1152/ajpgi.00172.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim CH, Tworoger SS, Stampfer MJ, Dillon ST, Gu X, Sawyer SJ, Chan AT, Libermann TA, Eliassen AH. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci Rep 8: 8382, 2018. doi: 10.1038/s41598-018-26640-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim CH, Zinsmeister AR, Malagelada JR. Mechanisms of canine gastric dysrhythmia. Gastroenterology 92: 993–999, 1987. doi: 10.1016/0016-5085(87)90975-9. [DOI] [PubMed] [Google Scholar]
- 27.Kinoshita K, Taupin DR, Itoh H, Podolsky DK. Distinct pathways of cell migration and antiapoptotic response to epithelial injury: structure-function analysis of human intestinal trefoil factor. Mol Cell Biol 20: 4680–4690, 2000. doi: 10.1128/MCB.20.13.4680-4690.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnan V, Xu Y, Macon K, Volanakis JE, Narayana SV. The structure of C2b, a fragment of complement component C2 produced during C3 convertase formation. Acta Crystallogr D Biol Crystallogr 65: 266–274, 2009. doi: 10.1107/S0907444909000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurt-Jones EA, Cao L, Sandor F, Rogers AB, Whary MT, Nambiar PR, Cerny A, Bowen G, Yan J, Takaishi S, Chi AL, Reed G, Houghton J, Fox JG, Wang TC. Trefoil family factor 2 is expressed in murine gastric and immune cells and controls both gastrointestinal inflammation and systemic immune responses. Infect Immun 75: 471–480, 2007. doi: 10.1128/IAI.02039-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBerry C, Egan CE, Rani R, Yang Y, Wu D, Boespflug N, Boon L, Butcher B, Mirpuri J, Hogan SP, Denkers EY, Aliberti J, Herbert DR. Trefoil factor 2 negatively regulates type 1 immunity against Toxoplasma gondii. J Immunol 189: 3078–3084, 2012. doi: 10.4049/jimmunol.1103374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Na YR, Yoon YN, Son DI, Seok SH. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One 8: e63451, 2013. doi: 10.1371/journal.pone.0063451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nauser CL, Farrar CA, Sacks SH. Complement recognition pathways in renal transplantation. J Am Soc Nephrol 28: 2571–2578, 2017. doi: 10.1681/ASN.2017010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Banion MK. Cyclooxygenase-2: molecular biology, pharmacology, and neurobiology. Crit Rev Neurobiol 13: 45–82, 1999. doi: 10.1615/CritRevNeurobiol.v13.i1.30. [DOI] [PubMed] [Google Scholar]
- 34.O’Grady G, Abell TL. Gastric arrhythmias in gastroparesis: low- and high-resolution mapping of gastric electrical activity. Gastroenterol Clin North Am 44: 169–184, 2015. doi: 10.1016/j.gtc.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ordög T, Takayama I, Cheung WK, Ward SM, Sanders KM. Remodeling of networks of interstitial cells of Cajal in a murine model of diabetic gastroparesis. Diabetes 49: 1731–1739, 2000. doi: 10.2337/diabetes.49.10.1731. [DOI] [PubMed] [Google Scholar]
- 36.Poulsen SS, Kissow H, Hare K, Hartmann B, Thim L. Luminal and parenteral TFF2 and TFF3 dimer and monomer in two models of experimental colitis in the rat. Regul Pept 126: 163–171, 2005. doi: 10.1016/j.regpep.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Poulsen SS, Thulesen J, Christensen L, Nexo E, Thim L. Metabolism of oral trefoil factor 2 (TFF2) and the effect of oral and parenteral TFF2 on gastric and duodenal ulcer healing in the rat. Gut 45: 516–522, 1999. doi: 10.1136/gut.45.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raggi F, Pelassa S, Pierobon D, Penco F, Gattorno M, Novelli F, Eva A, Varesio L, Giovarelli M, Bosco MC. Regulation of human macrophage M1–M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol 8: 1097, 2017. doi: 10.3389/fimmu.2017.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Revicki DA, Rentz AM, Dubois D, Kahrilas P, Stanghellini V, Talley NJ, Tack J. Gastroparesis Cardinal Symptom Index (GCSI): development and validation of a patient reported assessment of severity of gastroparesis symptoms. Qual Life Res 13: 833–844, 2004. doi: 10.1023/B:QURE.0000021689.86296.e4. [DOI] [PubMed] [Google Scholar]
- 40.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11: 785–797, 2010. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanders KM, Ross G. Effects of endogenous prostaglandin E on intestinal motility. Am J Physiol Endocrinol Metab 234: E204–E208, 1978. doi: 10.1152/ajpendo.1978.234.2.E204. [DOI] [PubMed] [Google Scholar]
- 43.Sanders KM, Szurszewski JH. Does endogenous prostaglandin affect gastric antral motility? Am J Physiol Gastrointest Liver Physiol 241: G191–G195, 1981. doi: 10.1152/ajpgi.1981.241.2.G191. [DOI] [PubMed] [Google Scholar]
- 44.Shah AA, Mihalj M, Ratkay I, Lubka-Pathak M, Balogh P, Klingel K, Bohn E, Blin N, Baus-Loncar M. Increased susceptibility to Yersinia enterocolitica Infection of Tff2 deficient mice. Cell Physiol Biochem 30: 853–862, 2012. doi: 10.1159/000341463. [DOI] [PubMed] [Google Scholar]
- 45.Somani R, Richardson VR, Standeven KF, Grant PJ, Carter AM. Elevated properdin and enhanced complement activation in first-degree relatives of South Asian subjects with type 2 diabetes. Diabetes Care 35: 894–899, 2012. doi: 10.2337/dc11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stein J, Zeuzem S, Uphoff K, Laube H. Effects of prostaglandins and indomethacin on gastric emptying in the rat. Prostaglandins 47: 31–40, 1994. doi: 10.1016/0090-6980(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 47.Stover CM, Luckett JC, Echtenacher B, Dupont A, Figgitt SE, Brown J, Männel DN, Schwaeble WJ. Properdin plays a protective role in polymicrobial septic peritonitis. J Immunol 180: 3313–3318, 2008. doi: 10.4049/jimmunol.180.5.3313. [DOI] [PubMed] [Google Scholar]
- 48.Sung TS, Kim HU, Kim JH, Lu H, Sanders KM, Koh SD. Protease-activated receptors modulate excitability of murine colonic smooth muscles by differential effects on interstitial cells. J Physiol 593: 1169–1181, 2015. doi: 10.1113/jphysiol.2014.285148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran CP, Cook GA, Yeomans ND, Thim L, Giraud AS. Trefoil peptide TFF2 (spasmolytic polypeptide) potently accelerates healing and reduces inflammation in a rat model of colitis. Gut 44: 636–642, 1999. doi: 10.1136/gut.44.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Yao B, Wang Y, Fan X, Wang S, Niu A, Yang H, Fogo A, Zhang MZ, Harris RC. Macrophage cyclooxygenase-2 protects against development of diabetic nephropathy. Diabetes 66: 494–504, 2017. doi: 10.2337/db16-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen Q, Kong Y, Zhao HY, Zhang YY, Han TT, Wang Y, Xu LP, Zhang XH, Huang XJ. G-CSF-induced macrophage polarization and mobilization may prevent acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 54: 1419–1433, 2019. doi: 10.1038/s41409-019-0449-9. [DOI] [PubMed] [Google Scholar]
- 52.Xiong H, Yan J, Cai S, He Q, Peng D, Liu Z, Liu Y. Cancer protein biomarker discovery based on nucleic acid aptamers. Int J Biol Macromol 132: 190–202, 2019. doi: 10.1016/j.ijbiomac.2019.03.165. [DOI] [PubMed] [Google Scholar]
- 53.Yu XL, Wu BT, Ma TT, Lin Y, Cheng F, Xiong HY, Xie CL, Liu CY, Wang Q, Li ZW. Overexpression of IL-12 reverses the phenotype and function of M2 macrophages to M1 macrophages. Int J Clin Exp Pathol 9: 8963–8972, 2016. [Google Scholar]
- 54.Zhang X, Guo L, Collage RD, Stripay JL, Tsung A, Lee JS, Rosengart MR. Calcium/calmodulin-dependent protein kinase (CaMK) Ialpha mediates the macrophage inflammatory response to sepsis. J Leukoc Biol 90: 249–261, 2011. doi: 10.1189/jlb.0510286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zschiesche W, Kleine AH, Spitzer E, Veerkamp JH, Glatz JF. Histochemical localization of heart-type fatty-acid binding protein in human and murine tissues. Histochem Cell Biol 103: 147–156, 1995. doi: 10.1007/BF01454012. [DOI] [PubMed] [Google Scholar]