

Abstract
Pulmonary arterial hypertension (PAH) is a morbid disease characterized by progressive right ventricle (RV) failure due to elevated pulmonary artery pressures (PAP). In PAH, histologically complex vaso-occlusive lesions in the pulmonary vasculature contribute to elevated PAP. However, the mechanisms underlying dysfunction of the microvascular endothelial cells (MVECs) that comprise a significant portion of these lesions are not well understood. We recently showed that MVECs isolated from the Sugen/hypoxia (SuHx) rat experimental model of PAH (SuHx-MVECs) exhibit increases in migration/proliferation, mitochondrial reactive oxygen species (ROS; mtROS) production, intracellular calcium levels ([Ca2+]i), and mitochondrial fragmentation. Furthermore, quenching mtROS with the targeted antioxidant MitoQ attenuated basal [Ca2+]i, migration and proliferation; however, whether increased mtROS-induced [Ca2+]i entry affected mitochondrial morphology was not clear. In this study, we sought to better understand the relationship between increased ROS, [Ca2+]i, and mitochondrial morphology in SuHx-MVECs. We measured changes in mitochondrial morphology at baseline and following inhibition of mtROS, with the targeted antioxidant MitoQ, or transient receptor potential vanilloid-4 (TRPV4) channels, which we previously showed were responsible for mtROS-induced increases in [Ca2+]i in SuHx-MVECs. Quenching mtROS or inhibiting TRPV4 attenuated fragmentation in SuHx-MVECs. Conversely, inducing mtROS production in MVECs from normoxic rats (N-MVECs) increased fragmentation. Ca2+ entry induced by the TRPV4 agonist GSK1017920A was significantly increased in SuHx-MVECs and was attenuated with MitoQ treatment, indicating that mtROS contributes to increased TRPV4 activity in SuHx-MVECs. Basal and maximal respiration were depressed in SuHx-MVECs, and inhibiting mtROS, but not TRPV4, improved respiration in these cells. Collectively, our data show that, in SuHx-MVECs, mtROS production promotes TRPV4-mediated increases in [Ca2+]i, mitochondrial fission, and decreased mitochondrial respiration. These results suggest an important role for mtROS in driving MVEC dysfunction in PAH.
Keywords: calcium, endothelium, mitochondria, PAH
INTRODUCTION
In pulmonary arterial hypertension (PAH), progressive increases in pulmonary artery pressure lead to right ventricular dysfunction, failure, and eventually death. Endothelial cell (EC) dysfunction is thought to contribute to vaso-occlusive lesion formation and increased pulmonary artery pressures in PAH. For instance, ECs in PAH patients exhibit increased proliferation and evidence of oxidant stress (12). The hyperproliferative ECs in PAH are thought to be of microvascular origin, referring to ECs that originate from the small-diameter vessels in the lung (46). Similar to what is observed in vivo, ECs isolated from human lungs with PAH also exhibit exuberant growth capacity, with increased migration and proliferation in vitro (18, 75–77). However, the mechanisms underlying the changes seen in PAH ECs, such as increased migration and proliferation in vivo and in vitro, are not fully understood.
To better understand the pathobiological mechanisms of EC dysfunction in PAH, we recently isolated microvascular ECs (MVECs) from the Sugen/hypoxia (SuHx) rat model of experimental PAH (53). In this model, rats are given a one-time injection of SU5416, a vascular endothelial growth factor receptor-2 (VEGFR2) inhibitor, and placed in hypoxia for 3 wk, followed by a return to normoxia. As shown by numerous laboratories (29, 39, 62), SuHx rats exhibit hemodynamic and histological changes, including increased right ventricular systolic pressure (RVSP) and the presence of vaso-occlusive lesions, similar to human PAH (12).
Using MVECs isolated from normoxic (N-MVEC) and SuHx (SuHx-MVEC) rats, we recently observed evidence of endothelial-to-mesenchymal transition (EndMT), a process associated with an oncogenic cell phenotype characterized by increased migration, proliferation, and metabolic changes such as glycolytic shift (19, 35, 57). EndMT, increased migration, and proliferation in ECs have been noted by other laboratories using various PAH models (57, 60, 73). To determine the mechanistic basis for these functional changes, we measured reactive oxygen species (ROS) and intracellular calcium concentration ([Ca2+]i) levels, signaling molecules that play key roles in promoting migration and proliferation in ECs (2, 20, 61, 65). We found that baseline ROS levels and [Ca2+]i were both increased in SuHx-MVECs (53) and that SuHx-MVEC ROS levels were normalized by quenching mitochondrial ROS (mtROS), while basal [Ca2+]i was attenuated by inhibiting either mtROS or the Ca2+ channel transient receptor potential vanilloid-4 (TRPV4) (53), suggesting a possible mechanistic link between mtROS production and TRPV4 activation in SuHx-MVECs. Furthermore, inhibiting either mtROS or TRPV4 similarly attenuated migration and proliferation in SuHx-MVECs. Collectively, these data suggested a link between mtROS and [Ca2+]i in facilitating SuHx-MVEC migration and proliferation, although the exact interactions between these two pathways remain incompletely understood.
As part of our study of mtROS in SuHx-MVECs, we recently examined mitochondrial structure and function and observed increased fragmentation (53). Mitochondrial dysfunction has been extensively studied in pulmonary artery smooth muscle cells (PASMCs) in PAH; for instance, a distinct role for mitochondrial dysfunction in promoting PASMC dysfunction has been previously established (5–7, 11, 37, 49, 64). Similar to PASMCs, mitochondrial dysfunction has been observed in ECs isolated from humans with PAH (77), but unlike in PASMCs, the mechanistic details underlying mitochondrial dysfunction in MVECs are still under investigation.
In PASMCs, one critical aspect of mitochondrial dysfunction is the dysregulation of mitochondrial fission/fusion dynamics (5, 44). Mitochondria are dynamic organelles that constantly undergo fission (i.e., fragmentation) and fusion. At baseline, fission and fusion are in equilibrium. However, a shift in the balance between fusion and fission can occur in situations of cellular stress (44). Interestingly, although increases in fission and mtROS production often occur together (32), the mechanistic links connecting mtROS production to changes in mitochondrial morphology are not fully known, particularly in ECs. Similarly, increased [Ca2+]i is associated with mitochondrial dysfunction in a variety of cell types (16, 28, 31, 43), but the specific effect of changes in cytosolic Ca2+ on mitochondrial morphology in vascular ECs has not been fully resolved.
In addition to increased levels of ROS and [Ca2+]i, changes in mitochondrial morphology can also be induced by shifts in mitochondrial bioenergetics. For instance, a shift toward fatty acid oxidation is associated with both increased fragmentation (48, 52) and fusion (33), depending on the cell type. Previously, we found that basal and maximal oxygen consumption rate (OCR) were decreased in SuHx-MVECs, while extracellular acidification rate (ECAR) was increased, suggestive of a glycolytic shift in the mitochondrial energetic profile (53) similar to that observed in cancer cells and other cell types with high migratory/proliferative capacity (70), including human PAH ECs (75). Taken together, our prior oxidative phosphorylation and mitochondrial fragmentation data reinforced the hypothesis that significant mitochondrial dysfunction was present in SuHx-MVECs. However, the role played by elevated [Ca2+]i, and the interaction between mtROS and [Ca2+]i, in regulating mitochondrial dynamics and energetics remain unknown. Thus we hypothesized that mtROS production activates TRPV4 and increases [Ca2+]I in SuHx-MVECs, promoting fragmentation. We reasoned that this feed-forward mechanism might explain increased basal levels of mitochondrial fragmentation, mtROS, and [Ca2+]i in SuHx-MVECs maintained in culture, even in the absence of exogenous injurious agonists. Thus, in this study, we measured the effect of changing mtROS or [Ca2+]i on mitochondrial structure (i.e., morphology) and function (i.e., respiration) in N- and SuHx-MVECs to determine the mechanistic links by which mtROS and [Ca2+]i may be contributing to mitochondrial dysfunction.
METHODS
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine.
Drugs and reagents.
SU5416 was obtained from Tocris. Baculoviral constructs for MitoRFP and roGFP were obtained from Life Technologies. Antimycin A (AA) was obtained from Abcam. MitoQ (MQ) was provided by Antipodean Pharmaceuticals. GSK2193874 (GSK2) was obtained from SelleckChem, and HC-067047 (HC) was obtained from EMD Millipore. Cyclosporine A (CSA) was obtained from Sigma. Drug treatment times were 1 h (GSK2, MQ, HC), 20 min (AA), or 30 min (CSA).
PAH animal model.
Induction of the SuHx model was performed by injecting SU5416 prepared in a carboxymethylcellulose (CMC)-containing diluent as described earlier (29). Rats (male, Wistar, 250–350 g, 4 mo old) were injected with 20 mg/kg of SU5416 subcutaneously, then exposed to 10% hypoxia for 3 wk before being returned to normoxia for an additional 2 wk. Control rats were injected with vehicle and maintained at room air for 5 wk. All animals were kept in the same room and thus were exposed to the same light-dark cycles and room temperatures. Animals were housed in standard rat cages at three rats/cage. No breeding was performed.
Hemodynamics.
Following induction of anesthesia (pentobarbital sodium ip, 43 mg/kg) and confirmation (via paw pinch) of sufficient sedation depth, RVSP measurements were made using a transdiapragmatic approach as previously described (29, 53). Animals were euthanized (via exsanguination) after hemodynamic measurement and before the start of MVEC isolation.
Isolation and culture of MVECs.
MVECs were isolated, grown to confluence, and phenotyped at each passage as described previously (53, 55). Briefly, after hemodynamic measurements were made, peripheral strips of rat lung were dissected and digested with collagenase (type 1A; 1 mg/mL) dissolved in a DMEM-based media containing 20% fetal bovine serum (FBS; Hyclone). Following digestion, cells were incubated with CD31-conjugated dynabeads (Invitrogen), magnetically selected, grown to confluence, and then reselected using Griffonia simplicifolia-conjugated beads (Vector). Following dual selection, cells were again phenotyped for smooth muscle (smooth muscle actin, myosin heavy chain) and EC (von Willebrand factor, Griffonia lectin) markers before being frozen at passage 3. All experiments were performed on cells at passages 3–4. All cells were grown in DMEM media containing antibiotics, endothelial growth factor supplement (Millipore), and 5% FBS as described previously (53).
Western blotting.
Confluent flasks of N- and SuHx-MVECs were trypsinized, pelleted, and lysed with lysis buffer (T-PER) containing a protease inhibitor cocktail tablet (Roche). Protein levels were quantified using a BCA kit (Pierce). Electrophoresis of equal amounts of protein was performed before transfer to polyvinylidene difluoride membranes, blocking (5% BSA in TBS-T), incubation with primary antibody (phospho-Drp1, total Drp1, and Mfn-2: 1:2,000, overnight 4°C, CST) and appropriate secondary antibody (goat anti-rabbit or anti-mouse: 1:10,000, 1 h, RT, KPL) and ECL (3 min, Amersham). Membranes were then stripped and reprobed for GAPDH (GAPDH-HRP: 1:10,000, Bio-Rad).
Antibody validation.
We have previously provided full-length gels and validation information for the phosphor- and total Drp1 antibodies (pSer616: 3455Ss, Cell Signaling; total Drp1: 8570, Cell Signaling) (53). The mitofusin-2 (Mfn-2) antibody used in this study (9482, Cell Signaling) has been previously validated using small interfering (si) RNA (38). We have also provided a representative full-length Mfn-2 immunoblot lane in Supplemental Fig. S1 (Supplemental Material is available at https://doi.org/10.5281/zenodo.3249000).
Mitochondrial morphology.
Mitochondrial morphology measurements were made on semiconfluent monolayers of cells seeded on glass coverslips for 24 h in DMEM media with 5% FBS. Cells were incubated with Mitotracker (100 nM) for 20 min before being imaged on an Olympus fluorescence microscope (×40 oil, frame rate: 5 frames/min). All images were obtained under identical image conditions (light intensity, binning, gain, and magnification). Image processing was performed using an automated algorithm as recently described (53). Briefly, images of individual cells stained for mitochondria were analyzed using an automated image-processing algorithm as described recently by Ouellet et al. (41). After background subtraction, binning and threshold were automatically determined before determination of mitochondrial network length using the Momito program. Output mitochondrial length distributions were collected and processed in R (45).
Oxygen consumption measurements.
N- and SuHx-MVECs (20,000 cells/well) were plated on a XF96 V3 PS cell culture microplate and incubated overnight in DMEM containing 5% serum. The cells were then washed and incubated with base media (Agilent) containing glutamine, pyruvate, and glucose as described previously (53). Cells and treatment solutions (2 µM oligomycin, 0.5 µM FCCP, and 0.5 µM rotenone/antimycin A) were loaded into a XFe96 Seahorse Flux Analyzer. Normalization of values to cell number was done using a CyQuant assay (Thermo). Data analysis was performed using Wave (Agilent) and GraphPad Prism.
Extracellular lactate measurement.
Extracellular lactate measurement was performed using a YSI STAT 2300 Lactate analyzer. Buffer and calibrant solutions were prepared per the manufacturers’ directions. A fresh lactate membrane was inserted and allowed to calibrate for 24 h. Stable resting lactate membrane currents were ensured before measurements. D35s dishes were plated with N- and SuHx-MVECs with and without MQ or HC treatment (24 h). At 24 h, the media supernatant was extracted and cells were trypsinized and counted (Scepter cell counter, Millipore). The supernatant samples were stored at −80°C. Samples were run in batches with untreated and treated N- and SuHx samples run sequentially. The results were normalized for cell count and to the normoxic control.
Targeted metabolomics.
N- and SuHx-MVECs were washed three times with ice-cold phosphate-buffered saline (PBS). Samples were lysed using ice-cold 80% HPLC-grade methanol (Fisher Scientific) diluted with 20% mass-spec (MS)-grade water. Samples were centrifuged (and cell pellets saved for total protein quantification), and the supernatant was stored overnight at −80°C, then evaporated using a speed vacuum followed by lyophilization. The dried metabolites were resuspended in 50% (vol/vol) acetonitrile diluted with MS-grade water. An Agilent 1260 HPLC and 6490 triple-quadrupole (QQQ) mass spectrometer were used to assess the metabolites associated with glycolysis, the TCA cycle, and energetics. Full details of the LC/MS parameters are provided in Supplemental Table S1. Table Agilent MassHunter and Agilent Qualitative and Quantitative Analysis Software packages were used to assess and quantify the metabolic profiles of the samples. To identify the relevant glycolytic, TCA cycle, and energetic compounds, pure standards were assessed under the same conditions as the samples to determine the optimal precursor/product ion transitions, collision energies, and ion polarity for each metabolite. Metabolite peaks were integrated for raw intensities. The total protein concentration of each sample was determined via a FilterMax F5 microplate reader and a bovine serum albumin (BSA) standard. This total protein concentration was then used to normalize the raw intensities determined for each of the samples.
Intracellular Ca2+ measurements.
Cells were seeded on glass coverslips (50–60% confluence; 24-h incubation in DMEM 5% FBS media) and loaded with fura 2-AM (5 µM; 1 h) before being placed in a temperature-controlled flow chamber as described previously (53). The perfusate used was a modified Krebs buffer containing (in mM) 118 NaCl, 4.7 KCl, 0.57 MgSO4, 1.18 KH2PO4, 25 NaHCO3, 2.5 CaCl2 and 10 glucose gassed with 16% O2 and 5% CO2. After establishment of a stable baseline, F380/F380 was recorded (Incytim2 software). [Ca2+]i was estimated from F340/F380 measured in calibration solutions with Ca2+ concentrations of 0–1,350 nM (Molecular Probes, Eugene, OR).
Intracellular ROS measurements.
MVECs were infected with a roGFP-GRX1 plasmid delivered in a baculovirus vector (Premo ROS sensor, Life Biotechnologies) at 80 MOI. At 48 h, cells were imaged, and the F400/F490 was calculated as described previously (53).
Data analysis/statistics.
All values are expressed as means ± SE. For roGFP measurements, data were collected from up to 30 cells and the values were averaged to obtain a single value for each experiment. All experiments were performed using cells isolated from at least five different rats. Cells from different animals were not pooled during isolation, so each n represents cells isolated exclusively from one animal. For SeaHorse experiments, technical replicates from four to six wells were averaged to yield one value per animal. Similarly, mitochondrial length distribution data from multiple microscopy images using cells isolated from the same animal were averaged to yield a single biological n. Data were compared using unpaired a Student’s t test, Mann–Whitney U/Kruskal–Wallis tests for nonparametric data, or two-way ANOVA (with post hoc Tukey’s test) to determine the effect of treatment across multiple groups.
The earthmover distance (EMD) metric was used to compare differences between the length/number distributions between N- and SuHx-MVEC mitochondria. This statistic can be used to evaluate changes in the distribution of nonparametric histogram data (40, 41). EMD was calculated using the Momito program written by Ouellet et al. (41). As described previously (41), the difference between two length distribution curves was considered significant if the EMD between the two groups was significantly higher than the intragroup differences in EMD within each individual group (compared using ANOVA, with Holm–Sidak post hoc). A P value <0.05 was accepted as statistically significant for all experiments.
RESULTS
Following induction of experimental PAH, SuHx rats exhibited significant increases in RVSP, RV/LV+S, and RV/body weight at the end of 5 wk (Fig. 1, A–D). N- and SuHx-MVECs obtained from these animals were used for all subsequent studies. Similar to previous results in confocal images of Mito-RFP-tagged MVECs (53), fragmentation was significantly increased in MitoTracker-labeled SuHx-MVEC mitochondria (Fig. 1, E and F). We previously showed that application of MQ acutely normalized total ROS levels in SuHx-MVECs (53). To determine whether mtROS contributes to mitochondrial fragmentation, we imaged N- and SuHx-MVECs following MQ treatment. As shown in Fig. 2A, MQ treatment had no effect on mitochondrial fragmentation in N-MVECs; however, MQ significantly attenuated fragmentation (evidenced by a downward shift of the mitochondrial length-distribution curve) in SuHx-MVECs (Fig. 2, B and C).
Fig. 1.

Hemodynamics and microvascular endothelial cell (MVEC) mitochondrial fragmentation in the Sugen/hypoxia (SuHx) model of pulmonary arterial hypertension (PAH). A: representative tracings of closed-chest right ventricular systolic pressure (RVSP) measurement in normoxic (N) and SuHx rats. Scatter plots showing means ± SE for RVSP (B), right ventricle/left ventricle+septal weight (RV/LV+S; C), and RV/body weight in N- and SuHx rats; D). *Significant difference from normoxic animals (t test). E: representative photomicrographs and reconstructed mitochondrial network images for N- and SuHx-MVECs. F: length distribution curves for N- and SuHx-MVEC mitochondria. *Significant difference from N-MVECs (ANOVA); n = 5/group.
Fig. 2.

Effect of mitochondrial reactive oxygen species (mtROS) quenching on mitochondrial fragmentation. A: Representative mitochondrial network images in N- and SuHx-MVECs in the absence and presence of MitoQ (MQ; 1µM – 1 h). B and C: length distribution curves (with SE at each distribution length) for mitochondria from N- and SuHx-MVECs with and without MQ treatment; n = 5 per/group (ANOVA). D and E: Western blot and densitometry showing pSer616Drp1 and total Drp1 protein levels in N- and SuHx-MVECs with and without MQ treatment, each n from a different animal. *Significant difference from normoxic control. **Significant difference from SuHx-MVECs (ANOVA).
We correlated these imaging findings with phosphorylation of dynamin-related protein-1 (Drp1), a GTPase that is critical for initiation of mitochondrial fission (14, 67). Similar to prior reports in other cell types (37, 68, 72), we previously found that fragmentation in SuHx-MVECs was accompanied by increased pSer616Drp1 levels (53). MQ treatment had no effect on Drp1 levels (phosphorylated or total) in N-MVECs but attenuated Drp1 phosphorylation in SuHx-MVECs (Fig. 2, D and E).
To further determine whether increased mtROS could promote fragmentation in SuHx-MVECs, we performed a “gain-of-function” experiment where N-MVECs were treated with AA, a drug that inhibits electron transport at complex III and induces mtROS production (10, 27, 50). Increasing ROS levels with AA (10 µM; 20 min) treatment in N-MVECs was confirmed using the ratiometric redox sensor roGFP (Fig. 3A). To determine whether AA-induced mtROS production caused fragmentation, we measured mitochondrial fragmentation in AA-treated N-MVECs. As shown in Fig. 3, B and C, mitochondrial fragmentation was significantly increased in AA-treated N-MVECs and was similar to values observed in untreated SuHx-MVECs. We previously observed that mtROS increases [Ca2+]i by activating the TRPV4 channel (53). To determine whether AA-induced mtROS generation promoted fission via TRPV4, we measured the effect of AA on mitochondrial fragmentation in N-MVECs treated with a TRPV4 inhibitor (HC) and observed attenuation of AA-induced mitochondrial fragmentation (HC+AA).
Fig. 3.
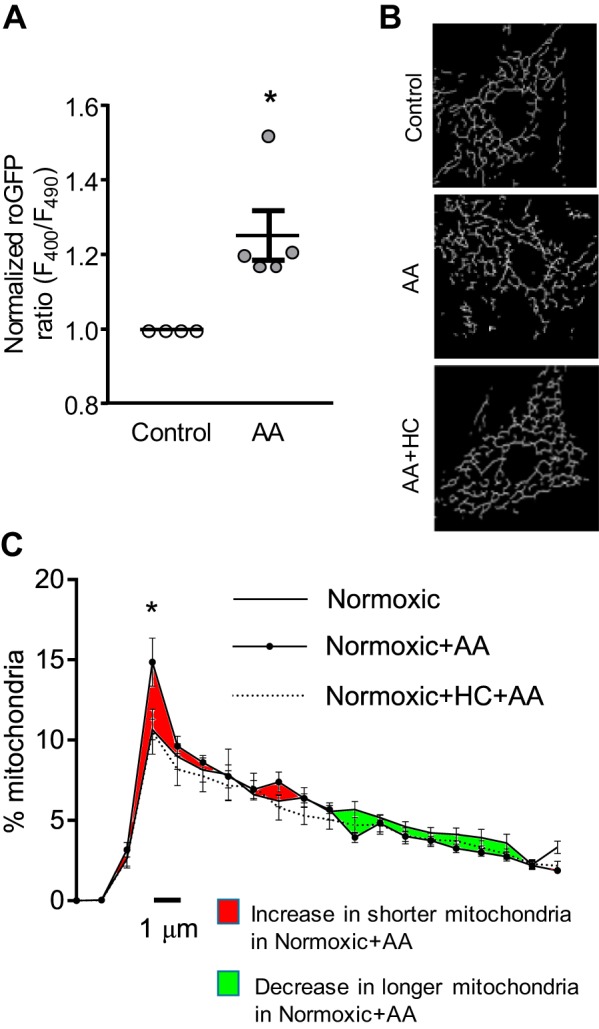
mtROS production in N-MVECs. A: scatter plot showing means ± SE for roGFP ratios in N-MVECs with and without treatment with antimycin A (AA; 10 µM). B: representative mitochondrial network images in untreated, AA-, and HC+AA-treated N-MVECs. C: length distribution curves for N-MVEC mitochondria in untreated, AA-, and HC+AA-treated N-MVECs. *Significant difference from normoxic control (ANOVA); n = 5/group.
Since impairment or loss of counterregulatory fusion mechanisms may be sufficient to increase fission, we examined whether loss of fusing capacity was contributing to increased SuHx-MVEC mitochondrial fragmentation. To initiate fusion, we used brief serum starvation (2-h incubation in DMEM without FBS), which has previously been shown to potently induce fusion (47). We observed significantly more elongated mitochondria in N-MVECs with serum starvation (Fig. 4, A and B). Serum starvation of SuHx-MVECs also significantly improved mitochondrial fragmentation (Fig. 4, Cand D). As shown in Fig. 4, E and F, we also measured levels of mitofusin-2 (Mfn-2), a critical regulator of mitochondrial fusion, and found no differences in Mfn-2 expression between N- and SuHx-MVECs. These data suggest that fusion mechanisms were intact and impairment of fusion was not contributing to mitochondrial fragmentation in SuHx-MVECs.
Fig. 4.

Induction of fusion in N- and SuHx-MVECs. Representative images (A and C) and length distribution curves (B and D) in N- and SuHx-MVECs in basal (5% FBS) serum and after incubation in serum-free media for 2 h.*Significant difference from untreated SuHx control; n = 10–15 images from 5 different animals. E and F: representative immunoblots and densitometry showing mitofusin-2 (Mfn-2) protein levels in N- and SuHx-MVECs. *Significant difference from N-MVEC control. **Significant difference from SuHx control (ANOVA); n = 5/group.
Our previous work showed that, in MVECs, both endogenous and exogenous ROS can activate TRPV4 to increase [Ca2+]i and that quenching mtROS with MQ normalized [Ca2+]i in SuHx-MVECs (53–55). Thus we hypothesized that mtROS may promote mitochondrial fragmentation by activating TRPV4. First, we measured ROS levels when TRPV4 was inhibited to determine whether increased [Ca2+]i itself can contribute to ROS formation in N- and SuHx-MVECs. As shown in Fig. 5A, treatment with GSK22193875 (GSK2; 30 nM) a specific inhibitor of TRPV4 previously shown to decrease [Ca2+]i in SuHx-MVECs (53), had no effect on ROS levels in SuHx-MVECs. These data are consistent with previous results obtained using HC-067047 (HC; 10 μM), a different TRPV4 inhibitor (53). Next, we measured fragmentation following treatment with both GSK2 and HC. As shown in Fig. 5, B–F, similar to MQ treatment, TRPV4 inhibition with either HC or GSK2 attenuated fragmentation in SuHx-MVECs.
Fig. 5.

Transient receptor potential vanilloid-4 (TRPV4) and mitochondrial fragmentation in SuHx-MVECs. A: scatter plot showing means ± SE for roGFP ratios in N- and SuHx-MVECs with and without treatment with GSK2193874 (GSK2; 30 nM); n = 5–6 (from different animals)/group. Representative network images (B) and length distribution curves (C–F) for N- and SuHx-MVEC mitochondria with and without treatment with 2 TRPV4 inhibitors: HC-067047 (HC; 10 µM) and GSK2 (30 nM). *Significant difference from SuHx-MVECs (ANOVA).
Since we hypothesized that mtROS-induced Ca2+ influx via TRPV4 was responsible for mitochondrial fragmentation, we questioned whether TRPV4 activation alone would be sufficient to induce fragmentation in N-MVECs. Our previous work showed that total TRPV4 protein levels were similar in N- and SuHx-MVECs (53), but the effect of direct TRPV4 activation on [Ca2+]i in N- and SuHx-MVECs was unknown. Thus we measured [Ca2+]i in N- and SuHx-MVECs following treatment with GSK1016790A (GSKA; 1.5 µM) a specific TRPV4 agonist. Surprisingly, [Ca2+]i was not changed in N-MVECs following GSKA exposure (Fig. 6A). In contrast, GSKA induced a large increase in [Ca2+]i in SuHx-MVECs (Fig. 6B), suggesting that although total TRPV4 protein expression was similar in N- and SuHx-MVECs, TRPV4 activation was enhanced in SuHx-MVECs. To confirm this finding and determine whether mtROS were involved, we measured GSKA-induced Ca2+ influx after MQ treatment. Similar to our prior report (53), treatment of SuHx-MVECs with MQ decreased basal [Ca2+]i (Fig. 6C). Interestingly, MQ treatment also attenuated the GSKA-induced increase in [Ca2+]i in SuHx-MVECs (Fig. 6, C and D), suggesting that mtROS, in addition to directly activating TRPV4, might also contribute to sensitization of TRPV4 to chemical agonists.
Fig. 6.

GSK1016790A (GSKA)-induced Ca2+ influx in SuHx-MVECs. A and B: representative traces showing intracellular Ca2+ concentration ([Ca2+]i) in N- and SuHx-MVECs with and without MQ treatment at baseline and following perfusion with TRPV4 agonist GSKA (1.5 µM). C: scatter plot showing means ± SE baseline and GSKA-induced changes in [Ca2+]i in N- and SuHx-MVECs in the absence and presence of MQ. D: scatter plot showing means ± SE change in [Ca2+]i (nM) in N-MVECs before and after GSKA (N vs. N+GSKA), untreated SuHx-MVECs before and after GSKA (S vs. S+GSKA), and MQ-treated SuHx-MVECs before and after GSKA (S+MQ vs. S+MQ+GSKA). *Significant difference from N-MVECs. **Significant difference from SuHx-MVECs (ANOVA).
Given our [Ca2+]i data, we next hypothesized that TRPV4 agonism might worsen fragmentation in SuHx-MVECs. GSKA treatment did not significantly alter mitochondrial fragmentation in N-MVECs, likely due to the lack of effect on GSK on [Ca2+]i in these cells. In SuHx-MVECs, GSKA treatment increased the percentage of shorter mitochondria, but this small shift in the length/number distribution was not statistically significant (Supplemental Fig. S2).
Last, since TRPV4 blockade and mtROS quenching similarly improved mitochondrial fragmentation, we questioned whether these changes in mitochondrial fragmentation were accompanied by improvement in mitochondrial respiration. Consistent with our previously reported data (53), basal OCR was decreased while extracellular acidification rate (ECAR) was increased in SuHx-MVECs (Fig. 8). Despite improving mitochondrial fragmentation and [Ca2+]i, 1-h treatment with MQ or HC did not reverse changes in SuHx-MVEC OCR and ECAR (data not shown). Hypothesizing that changes to mitochondrial respiration following ROS quenching might occur later than changes to mitochondrial fragmentation, we measured OCR and ECAR after 24-h MQ or HC treatment. We encountered significant batch variation in the raw values of OCR and ECAR across animals; thus we normalized values to untreated normoxic controls, but have presented the raw values in the traces shown in Fig. 7, A–D, and in Supplemental Table S2. As shown in Fig. 7, 24-h MQ treatment improved OCR and ECAR in SuHx-MVECs. Interestingly, HC treatment decreased ECAR, but not OCR, in SuHx-MVECs.
Fig. 8.

A: heat map showing fold-change differences in glycolysis and TCA metabolites in N- and SuHx-MVECs (n = cells isolated from 8 individual animals). B: table showing median fold-change (and IQR) for metabolite levels; n = 8 biological replicates. C: scatter plot showing fold-change in ATP levels in SuHx-MVECs. D: scatter plot showing fold-change in extracellular lactate at baseline and following treatment (24 h) with MitoQ (MQ) or HC-067047 (HC). *Significant difference from normoxic control (t test). **Significant difference from untreated SuHx-MVEC control (ANOVA).
Fig. 7.

Effect of mtROS and Ca2+ inhibition on mitochondrial respiration. Curves of mitochondrial oxygen consumption rate (OCR) in N- and SuHx-MVECs at baseline and following treatment with MQ (A and B) and HC (C and D). Scatter plots showing means ± SE for basal OCR (E) and extracellular acidification rate (ECAR; F) in N-MVECs normalized to untreated N-MVEC controls. *Significant difference from N-MVEC control. **Significant difference from SuHx-MVECs (ANOVA).
While the ECAR results, combined with the decreased basal/maximum OCR, were suggestive of glycolytic shift, we sought to confirm these observations using additional measurements of glycolysis at baseline and with drug treatment. Targeted metabolomic analysis of key glycolysis metabolites in N- and SuHx-MVECs revealed upregulation of glycolysis metabolites as well as increased ATP (Fig. 8, A–C). In addition to increased levels of intracellular lactate, as shown in Fig. 8D, extracellular lactate levels were increased at baseline in SuHx-MVECs, but were reduced with MQ and HC (24 h) treatment (Fig. 8D).
DISCUSSION
In this study, we show that MVECs isolated from a rodent model of PAH exhibit significant mitochondrial fragmentation that can be modulated by mtROS-induced Ca2+ influx via the TRPV4 channel. Furthermore, our data suggest that quenching mtROS attenuates both morphological and functional abnormalities in SuHx-MVECs. Mitochondrial dysfunction has been implicated in the pathobiology of many diseases, including PAH (49). For instance, decreasing fission by either genetic silencing of Drp1 or use of the Drp1 inhibitor P110 attenuated pulmonary artery smooth muscle cell proliferation (37) and RV dysfunction (64) in animal models of PAH, suggesting that restoring mitochondrial morphology to a more networked state is sufficient to ameliorate cellular dysfunction. Unlike PASMCs, less is known about the mechanisms regulating mitochondrial fission/fusion in MVECs during PAH. This point is relevant because MVECs are a component of plexiform lesions (30), a key feature of PAH (36), and we, along with several other groups, recently reported significant endothelial dysfunction, including EndMT, in multiple PAH models (35, 57, 60). Work in a variety of cell types, including MVECs, epithelial and cancer cells, suggests that the underpinnings of EndMT may be metabolic in origin, as induction of EndMT is typically accompanied by significant metabolic reprogramming (26, 73). Our current work also suggests an association between altered mitochondrial energetics and mitochondrial fragmentation in SuHx-MVECs, with mitochondrial structure being modified by inhibiting Ca2+ entry or quenching mtROS (53). While similar manipulations also normalize SuHx-MVEC migration and proliferation (53), it remains to be seen whether improvement in mitochondrial fragmentation alone is sufficient to reverse abnormal PAH EC cell function. Further experiments measuring migration and proliferation in SuHx-MVECs using fusion-inducing agents will be needed to distinguish whether the effects of inhibiting mtROS-induced Ca2+ entry on migration and proliferation are due to improvement of mitochondrial structure or whether mitochondrial fragmentation is a simply marker in cells with high [Ca2+]i.
A link between Ca2+ homeostasis and mitochondrial structure/function has long been appreciated. Mitochondria act as a local sink for rising Ca2+, but increasing [Ca2+]i also alters mitochondrial function and morphology (31, 58). We show that inhibition of TRPV4 in SuHx-MVECs attenuates baseline [Ca2+]i and fission to an extent similar to quenching mtROS. That TRPV4 activation is downstream of mtROS suggests that ROS-induced Ca2+ influx via TRPV4 contributes to increased fission in SuHx-MVECs. Since we observed similar attenuation of mitochondrial fragmentation using two different inhibitors of TRPV4 previously shown to be specific for this channel (63, 71), it is unlikely that the observed effect of TRPV4 inhibition on mitochondrial fragmentation is due to nonspecific blockade of other Ca2+ channels. However, a contribution to increased basal [Ca2+]i in SuHx-MVECs via increased ER release and/or decreased mitochondrial Ca2+ uptake mechanisms cannot be definitively excluded based on the current studies. With regard to the pharmacokinetics of TRPV4 inhibition, we previously showed that perfusing SuHx-MVECs with either HC or GSK2 decreased basal [Ca2+]i levels within a few minutes (53); thus, in our current mitochondrial morphology studies, we initially used a short incubation time (1 h) to study the effect of TRPV4 inhibition on mitochondrial fragmentation. TRPV4 has been previously implicated in various models of lung injury and in regulating hypoxic pulmonary vasoconstriction (4, 9, 24, 25) and recent evidence from the cerebral vasculature suggests that mtROS may activate TRPV4 to promote brain EC dysfunction after traumatic brain injury (59). However, a role for this channel in regulating mitochondrial fragmentation has, to our knowledge, not been previously described.
The mechanism by which increased [Ca2+]i induces mitochondrial fission is not fully understood; however, emerging evidence in neurons suggests that activation of plasma membrane channels leads to Ca2+-dependent phosphorylation of Drp1, which in turn promotes fission (13, 28). In this study, we quantified mitochondrial network architecture using a newer, unsupervised algorithm to better understand the relationship between [Ca2+]i and mitochondrial structure in SuHx-MVECs. To ensure image quality of mitochondrial networks, we obtained mitochondrial images of live, subconfluent MVEC monolayers imaged in a temperature-controlled, gassed chamber, to minimize cellular stresses that may occur with specimen processing and can impact mitochondrial morphology/dynamics. Additionally, we corroborated changes in mitochondrial fission with corresponding increase or decrease in levels of phosphorylated Drp1. Specifically, differences in mitochondrial fragmentation between N-MVECs, SuHx-MVECs, and MQ-treated SuHx-MVECs correlated with corresponding changes in phosphorylation of Drp1 at the Ser616 residue, which is critical for Drp1 recruitment to the mitochondria. In addition to Ser616, the 637 serine residue of Drp1 also plays a role in regulating Drp1 activity. The role of Ser637 phosphorylation in Drp1 recruitment to mitochondria in ECs in MVECs remains under investigation. We previously reported that phosphorylation of Ser637 was increased in SuHx-MVECs (53). This finding was in contrast to the role of Ser637 in regulating fission in HeLa cells, where dephosphorylation of Ser637 by calcineurin has been shown to be important for initiation of fission (13). Of note, the opposite (i.e., increased Ser637 phosphorylation associated with fission) has been shown in kidney ECs (68). To determine whether changes in Ser637 phosphorylation were contributing to increased fission in SuHx-MVECs, we treated SuHx-MVECs with cyclosporine A, a calcineurin inhibitor, reasoning that if dephosphorylation of Ser637 by calcineurin was contributing to increased fission, inhibiting this pathway would improve fragmentation in SuHx-MVECs. However, we did not observe any change in SuHx-MVEC fragmentation (Supplemental Fig. S3). These data, along with our previous data showing increased Ser637 phosphorylation of Drp1 in SuHx-MVECs at baseline (53), suggest that dephosphorylation of Ser637 does not appear to be playing a major role in promoting increased fission in SuHx-MVECs.
Other proteins involved in mitochondrial machinery, including the Rho-GTPase Miro1, which regulates mitochondrial movement on microtubules, are also regulated by [Ca2+]i (8). Furthermore, Drp1 is one part of a complex of proteins, such as mitochondrial fission factor (Mff) (21) and mitochondrial dynamics protein 49/51 (MiD49/51) (42), that are required for successful execution of fission. Further work is needed to determine which, if any, of the other components of the fission machinery are also abnormal in SuHx-MVECs.
Our serum starvation data suggest that induction of fusion via alternative pathways is able to partially overcome increased mitochondrial fragmentation in SuHx-MVECs. Furthermore, levels of the critical fusion regulator Mfn-2 do not appear to be significantly different in SuHx-MVECs. These data suggest that the primary deficit in mitochondrial structure in SuHx-MVECs is increased fission rather than inability to fuse. However, more work using alternative fusion-inducing stimuli and/or overexpression of proteins involved in fusion is needed to conclusively determine the role of fusion in SuHx-MVECs. Importantly, fission/fusion dynamics exist within the larger context of biogenesis (i.e., generation of new mitochondria) and mitophagy (i.e., disposal of severely dysfunction mitochondria). Thus, while fission is clearly increased and fusion does not appear to be impaired in SuHx-MVECs, more studies are needed to determine whether additional deficits in initiation of mitophagy and/or biogenesis are contributing to the continued presence of fragmented dysfunctional mitochondria in SuHx-MVECs.
Similar to increased [Ca2+]i, significant evidence points to the role of increased ROS in promoting mitochondrial fission (22, 32, 66, 78). Our data suggest a link between these two regulators of mitochondrial fragmentation; that is, the effects of ROS on mitochondrial fragmentation may be regulated via ROS-induced increases in [Ca2+]i via TRPV4. That inhibiting mtROS decreased, and increasing mtROS enhanced, fragmentation, while MQ treatment had no effect in N-MVECs, suggests that the effect of MQ on mitochondrial fragmentation is unlikely to be due to nontargeted effects of this drug. The dose of MQ used has previously been shown to effectively quench ROS production at complex III (10). Although we show complex III ROS generation (i.e., AA treatment) was sufficient to cause fragmentation, it is possible at mtROS generation at other complexes may also produce similar fragmentation and changes in [Ca2+]i. Furthermore, we used short time points (1 h) in our MQ treatment experiments since we previously observed that MQ decreases basal [Ca2+]i acutely in SuHx-MVECs. However, we did not perform time course experiments with MQ; thus it is possible that improvement in mitochondrial structure may occur even earlier than our studied time point.
Our data suggest that mtROS might increase the sensitivity of TRPV4 to GSKA, since agonist-induced Ca2+ influx was greater in untreated SuHx-MVECs, which have higher mtROS levels at baseline. Moreover, quenching mtROS attenuated the GSKA response in SuHx-MVECs. The lack of GSKA-induced Ca2+ influx in N-MVECs provides an explanation as to why mitochondrial fragmentation did not significantly change in GSKA-treated N-MVECs. Interestingly, although GSKA induced a significant Ca2+ influx in SuHx-MVECs, additional fragmentation was not observed, possibly because the mitochondrial network was already extensively fragmented at baseline, and further fission was not possible. Another possibility is that the transient GSKA-induced Ca2+ spike in MVECs (1) may not be long enough to induce fission. In neurons, for instance, a sustained [Ca2+]i elevation is needed for induction of fission (31). With regard to increased GSKA sensitivity in SuHx-MVECs at baseline, and MQ treatment attenuating this response, we hypothesize that this may be due to regulation of TRPV4 translocation. Recent evidence in macrophages suggests that TRPV4 translocates to the cell surface in response to injurious stimuli (51). Caveolin-1 (Cav-1) is a key protein that regulates membrane trafficking of proteins, including Ca2+ channels, into the cell membrane. Regulated in part by redox-sensitive tyrosine phosphorylation (15), Cav-1 was recently shown to compartmentalize (and immunoprecipitate) with TRPV4 (23); thus we hypothesize that the mechanism behind our observed findings may involve trafficking of TRPV4 to the membrane by ROS-induced activation of Cav-1. Of note, however, we (54) and others (69, 74) have previously shown that TRPV4 activity can also be modulated by phosphorylation. Thus another possible explanation for our GSKA-induced Ca2+ influx data is that mtROS may directly oxidize intermediary kinases that then regulate the sensitivity to TRPV4 to activating stimuli by phosphorylating this channel. Further experiments, such as membrane fractionation to look for changes in TRPV4 protein, phospho-tyrosine coimmunoprecipitation, or measurement of GSKA-induced Ca2+ influx following inhibition of Cav-1 and/or kinases known to phosphorylate TRPV4, are needed to further dissect the mechanisms by which ROS activate TRPV4 in SuHx-MVECs.
While both inhibition of mtROS and reduction of [Ca2+]i improved mitochondrial fragmentation, only MQ improved mitochondrial function (i.e., respiration). Even after ensuring cell viability and number before and after our respiration measurements, baseline respiration values for SuHx-MVECs were low, especially compared with other cell types. Interestingly, HC did not improve mitochondrial respiration in SuHx-MVECs. One possibility is that while Ca2+-dependent mechanisms regulate mitochondrial movement, in the presence of ongoing increased mtROS generation, improving fission by inhibiting Ca2+ entry may be insufficient to rescue the metabolic abnormalities in SuHx-MVECs and would simply lead to refragmentation at a later time point as ROS levels continue to rise. Another possibility is that mtROS may be affecting mitochondrial signaling pathways related to respiration independently of [Ca2+]i, in which case decreasing [Ca2+]i alone may be insufficient to reverse mtROS-induced decreases in oxidative phosphorylation, especially since we showed that TRPV4 inhibition decreases [Ca2+]i, but not mtROS, in SuHx-MVECs. Interestingly, despite not improving mitochondrial respiration, TRPV4 inhibition significantly improved ECAR, suggesting that TRPV4-mediated Ca2+ entry may also regulate glycolysis directly, independently of mitochondrial fission. While our ECAR data, taken together with our metabolomic and extracellular lactate data, implicate shifts in glycolysis, additional experiments aimed at specifically interrogating glycolytic flux in SuHx-MVECs are needed.
Together, our respiration and roGFP data suggest that mtROS generation is upstream of increased [Ca2+]i in SuHx-MVECs. However, whether mtROS production in SuHx-MVECs is triggered by underlying shifts in cell metabolism is unclear. For instance, evidence in cancer and immune cells suggests that, in the presence of glycolytic shift, increased dependence on glutamine as a source for TCA intermediates may fuel ROS production (34, 70). On the other hand, Diebold et al. (17) showed that, in lung ECs, mtROS production at complex III was sufficient to decrease basal respiration (similar to our current findings) and induce metabolic dysfunction. As mentioned earlier, restoration of mitochondrial fusion/fission dynamics toward a more fused state was sufficient to ameliorate cardiac dysfunction in PAH and other models of injury (37, 64). However, if the underlying metabolic dysfunction is not corrected, inhibiting pathways such as mtROS, [Ca2+]i, or even Drp1 may provide short-term restoration of cellular function, but mitochondrial dysfunction may return as the inhibitors of these downstream pathways are degraded or exported out of the cell. Thus further work is needed to more mechanistically understand the driving forces behind the ROS- and Ca2+-mediated dysfunction in mitochondrial dynamics and metabolism described herein.
In summary, our current study suggests an interplay among mtROS, [Ca2+]i, and mitochondrial fragmentation and function in MVECs in PAH (Fig. 9). Our data suggest that this process may be self-propagating, with increased production of mtROS inducing Ca2+ influx via TRPV4, which in turn promotes mitochondrial fission and mtROS production. On the other hand, while inhibition of TRPV4 improves mitochondrial fragmentation without any effects on respiration, inhibiting mtROS restores both mitochondrial fragmentation and respiration in SuHx-MVECs, suggesting that mtROS production may be fueled by additional, possibly metabolic, factors. Additional studies aimed at understanding the metabolic underpinnings of mtROS production in SuHx-MVECs will be informative in further elucidating the mechanisms of microvascular endothelial dysfunction in PAH. Nonetheless, given the beneficial effects of reducing mtROS in mitochondrial and cellular function, specifically targeting mtROS in MVECs may be an attractive candidate for treating PAH.
Fig. 9.
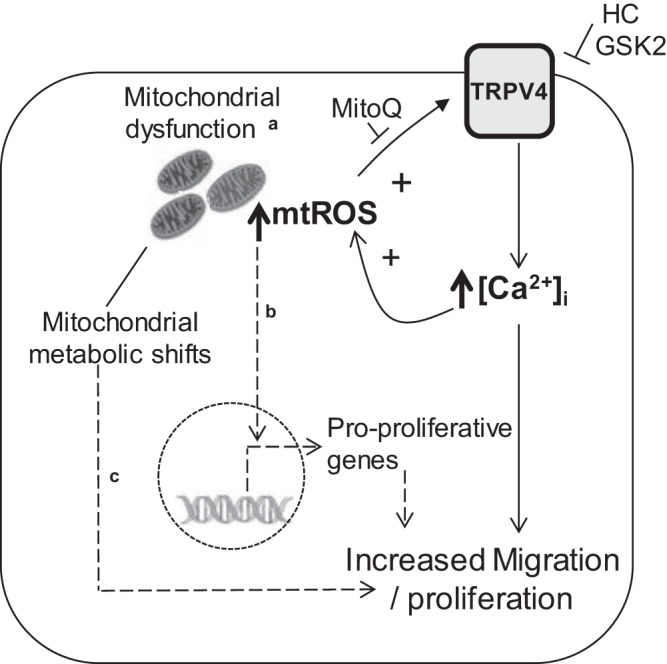
Schematic describing our proposed pathway of interaction between TRPV4 and mitochondrial fragmentation in SuHx-MVECs (solid lines) as well as other established pathways linking mitochondrial dysfunction to EC migration/proliferation (dashed lines). aIncreased ROS production, decreased basal/maximal respiration, increased fission, and evidence of glycolytic shift. bDirect effects of mtROS on transcription (3). cChanges in fuel utilization following glycolytic shift providing carbons (i.e., anaplerosis) for generation of metabolites essential for biosynthetic activities such as proliferation (79).
GRANTS
Support for this study was provided by National Heart, Lung and Blood Institute Grants F32HL124930 and K08HL132055 (K. Suresh), R01HL073859, R25HL084762, and R01HL126514 (L. A. Shimoda), F32HL124727 and K08HL133475 (J. C. Huetsch), and T32HL007534 (K. Suresh and J. C. Huetsch). Additional support includes a Gilead PAH Research Scholar Award and Parker B. Francis Research Opportunity Award (K. Suresh), as well as NIH S10OD016374 (S. Kuo, JHUSOM Microscope facility).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.S., Z.B., J.Z., A.L., M.D., and L.A.S. conceived and designed research; K.S., L.S., H.J., Z.B., J.Z., J.C.H., C.K., M.G.A., and B.J.K. performed experiments; K.S., L.S., Z.B., J.Z., J.C.H., C.K., M.G.A., and B.J.K. analyzed data; K.S., B.J.K., S.M.C., A.L., M.D., and L.A.S. interpreted results of experiments; K.S. prepared figures; K.S. drafted manuscript; K.S., J.C.H., M.G.A., S.M.C., M.D., and L.A.S. edited and revised manuscript; S.M.C., A.L., M.D., and L.A.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Jimmie Sylvester and David Pearse for thoughtful comments and guidance on experimental design and data analysis. We also thank Dr. Michael Caterina for providing expertise regarding TRPV4 and the Johns Hopkins School of Medicine (JHUSOM) Microscope Facility for assistance with fluorescence confocal imaging.
REFERENCES
- 1.Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, Thodeti CK. PKCα mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol 301: H757–H765, 2011. doi: 10.1152/ajpheart.00142.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol 3: 1011–1034, 2013. doi: 10.1002/cphy.c120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5: ra47, 2012. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res 99: 988–995, 2006. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369: 2236–2251, 2013. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 6.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 7.Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73: 1100–1112, 1993. doi: 10.1161/01.RES.73.6.1100. [DOI] [PubMed] [Google Scholar]
- 8.Bagur R, Hajnóczky G. Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Mol Cell 66: 780–788, 2017. doi: 10.1016/j.molcel.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: 1029–1036, 2007. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 12.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–769, 2004. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 13.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105: 15803–15808, 2008. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho B, Choi SY, Cho HM, Kim HJ, Sun W. Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system. Exp Neurobiol 22: 149–157, 2013. doi: 10.5607/en.2013.22.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coelho-Santos V, Socodato R, Portugal C, Leitão RA, Rito M, Barbosa M, Couraud PO, Romero IA, Weksler B, Minshall RD, Fontes-Ribeiro C, Summavielle T, Relvas JB, Silva AP. Methylphenidate-triggered ROS generation promotes caveolae-mediated transcytosis via Rac1 signaling and c-Src-dependent caveolin-1 phosphorylation in human brain endothelial cells. Cell Mol Life Sci 73: 4701–4716, 2016. doi: 10.1007/s00018-016-2301-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta 1797: 607–618, 2010. doi: 10.1016/j.bbabio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, Chandel NS. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab 1: 158–171, 2019. doi: 10.1038/s42255-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duong HT, Comhair SA, Aldred MA, Mavrakis L, Savasky BM, Erzurum SC, Asosingh K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm Circ 1: 475–486, 2011. doi: 10.4103/2045-8932.93547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7: 11853, 2016. [Erratum in Nat Commun 8: 14710, 2017.] doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, Paria B, Munaron L, Ambudkar IS. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31: 200–212, 2012. doi: 10.1038/onc.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402–2412, 2008. doi: 10.1091/mbc.e07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radic Biol Med 52: 348–356, 2012. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goedicke-Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, Bänfer S, Jacob R, Grgic I, Hoyer J. Evidence for functional and dynamic microcompartmentation of Cav-1/TRPV4/K(Ca) in caveolae of endothelial cells. Eur J Cell Biol 94: 391–400, 2015. doi: 10.1016/j.ejcb.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Goldenberg NM, Ravindran K, Kuebler WM. TRPV4: physiological role and therapeutic potential in respiratory diseases. Naunyn Schmiedebergs Arch Pharmacol 388: 421–436, 2015. doi: 10.1007/s00210-014-1058-1. [DOI] [PubMed] [Google Scholar]
- 25.Goldenberg NM, Wang L, Ranke H, Liedtke W, Tabuchi A, Kuebler WM. TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology 122: 1338–1348, 2015. doi: 10.1097/ALN.0000000000000647. [DOI] [PubMed] [Google Scholar]
- 26.Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol 7: 295, 2017. doi: 10.3389/fonc.2017.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase downregulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol Cell Biol 31: 3546–3556, 2011. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182: 573–585, 2008. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huetsch JC, Jiang H, Larrain C, Shimoda LA. The Na+/H+ exchanger contributes to increased smooth muscle proliferation and migration in a rat model of pulmonary arterial hypertension. Physiol Rep 4: e12729, 2016. doi: 10.14814/phy2.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humbert M, Montani D, Perros F, Dorfmüller P, Adnot S, Eddahibi S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol 49: 113–118, 2008. doi: 10.1016/j.vph.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Jeyaraju DV, Cisbani G, Pellegrini L. Calcium regulation of mitochondria motility and morphology. Biochim Biophys Acta 1787: 1363–1373, 2009. doi: 10.1016/j.bbabio.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: the yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel) 7: E13, 2018. doi: 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanna A, Dustin ML. Mitochondrial fusion fuels T cell memory. Cell Res 26: 969–970, 2016. doi: 10.1038/cr.2016.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, Dang CV. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15: 110–121, 2012. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mammoto T, Muyleart M, Konduri GG, Mammoto A. Twist1 in hypoxia-induced pulmonary hypertension through transforming growth factor-β-Smad signaling. Am J Respir Cell Mol Biol 58: 194–207, 2018. doi: 10.1165/rcmb.2016-0323OC. [DOI] [PubMed] [Google Scholar]
- 36.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res 68: 75–103, 2004. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM, Trempe JF, Fon EA. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7: e32866, 2018. doi: 10.7554/eLife.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 40.Orlova DY, Zimmerman N, Meehan S, Meehan C, Waters J, Ghosn EE, Filatenkov A, Kolyagin GA, Gernez Y, Tsuda S, Moore W, Moss RB, Herzenberg LA, Walther G. Earth Mover’s Distance (EMD): a true metric for comparing biomarker expression levels in cell populations. PLoS One 11: e0151859, 2016. doi: 10.1371/journal.pone.0151859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouellet M, Guillebaud G, Gervais V, Lupien St-Pierre D, Germain M. A novel algorithm identifies stress-induced alterations in mitochondrial connectivity and inner membrane structure from confocal images. PLOS Comput Biol 13: e1005612, 2017. doi: 10.1371/journal.pcbi.1005612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12: 565–573, 2011. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng T-I, Jou M-J. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201: 183–188, 2010. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 44.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol 304: R393–R406, 2013. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R Development Core Team R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2018. https://www.r-project.org. [Google Scholar]
- 46.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195, 2011. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 61: 2074–2083, 2012. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl) 93: 229–242, 2015. doi: 10.1007/s00109-015-1263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanders SP, Zweier JL, Kuppusamy P, Harrison SJ, Bassett DJ, Gabrielson EW, Sylvester JT. Hyperoxic sheep pulmonary microvascular endothelial cells generate free radicals via mitochondrial electron transport. J Clin Invest 91: 46–52, 1993. doi: 10.1172/JCI116198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, McDonald C, Hamilton TA, Olman MA. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J Immunol 196: 428–436, 2016. doi: 10.4049/jimmunol.1501688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem 285: 5748–5758, 2010. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suresh K, Servinsky L, Jiang H, Bigham Z, Yun X, Kliment C, Huetsch J, Damarla M, Shimoda LA. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 314: L893–L907, 2018. doi: 10.1152/ajplung.00430.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, Pearse DB, Shimoda LA. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309: L1467–L1477, 2015. doi: 10.1152/ajplung.00275.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suresh K, Servinsky L, Reyes J, Undem C, Zaldumbide J, Rentsendorj O, Modekurty S, Dodd-O JM, Scott A, Pearse DB, Shimoda LA. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 312: L143–L153, 2017. doi: 10.1152/ajplung.00361.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K, West J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 314: L118–L126, 2018. doi: 10.1152/ajplung.00296.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta 1763: 442–449, 2006. doi: 10.1016/j.bbamcr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 59.Szarka N, Pabbidi MR, Amrein K, Czeiter E, Berta G, Pohoczky K, Helyes Z, Ungvari Z, Koller A, Buki A, Toth P. Traumatic brain injury impairs myogenic constriction of cerebral arteries: role of mitochondria-derived H2O2 and TRPV4-dependent activation of BKCa channels. J Neurotrauma. In press. doi: 10.1089/neu.2017.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, Wang Z, Gupta A, Zhou T, Sun X, Dash S, Wang Z, Balistrieri A, Zheng Q, Cordery AG, Desai AA, Rischard F, Khalpey Z, Wang J, Black SM, Garcia JGN, Makino A, Yuan JX-J. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 314: L256–L275, 2018. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang H, Yamamura A, Yamamura H, Song S, Fraidenburg DR, Chen J, Gu Y, Pohl NM, Zhou T, Jiménez-Pérez L, Ayon RJ, Desai AA, Goltzman D, Rischard F, Khalpey Z, Black SM, Garcia JGN, Makino A, Yuan JXJ. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L846–L859, 2016. doi: 10.1152/ajplung.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 63.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 159ra148, 2012. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 64.Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, Chen KH, Hong Z, Sharp WW, Kutty S, Archer SL. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J Mol Med (Berl) 95: 381–393, 2017. doi: 10.1007/s00109-017-1522-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Touyz RM. Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal 7: 1302–1314, 2005. doi: 10.1089/ars.2005.7.1302. [DOI] [PubMed] [Google Scholar]
- 66.Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res 122: 58–73, 2018. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186: 805–816, 2009. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15: 186–200, 2012. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wegierski T, Lewandrowski U, Müller B, Sickmann A, Walz G. Tyrosine phosphorylation modulates the activity of TRPV4 in response to defined stimuli. J Biol Chem 284: 2923–2933, 2009. doi: 10.1074/jbc.M805357200. [DOI] [PubMed] [Google Scholar]
- 70.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107: 8788–8793, 2010. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, Liedtke W, Sham JS. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol 305: C704–C715, 2013. doi: 10.1152/ajpcell.00099.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18: 501–510, 2015. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira II, Brittain EL, Wolfgang MJ, Jurczak MJ, Fessel JP, Finkel T. A metabolic basis for endothelial-to-mesenchymal transition. Mol Cell 69: 689–698.e7, 2018. doi: 10.1016/j.molcel.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu H, Zhao H, Tian W, Yoshida K, Roullet JB, Cohen DM. Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase-dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J Biol Chem 278: 11520–11527, 2003. doi: 10.1074/jbc.M211061200. [DOI] [PubMed] [Google Scholar]
- 75.Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr Physiol 1: 357–372, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu W, Kaneko FT, Zheng S, Comhair SAA, Janocha AJ, Goggans T, Thunnissen FBJM, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 18: 1746–1748, 2004. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 77.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 104: 1342–1347, 2007. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658, 2006. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zecchin A, Kalucka J, Dubois C, Carmeliet P. How endothelial cells adapt their metabolism to form vessels in tumors. Front Immunol 8: 1750, 2017. doi: 10.3389/fimmu.2017.01750. [DOI] [PMC free article] [PubMed] [Google Scholar]