

Abstract
Hyperoxia exposure in premature infants increases the risk of subsequent lung diseases, such as asthma and bronchopulmonary dysplasia. Fibroblasts help maintain bronchial and alveolar integrity. Thus, understanding mechanisms by which hyperoxia influences fibroblasts is critical. Cellular senescence is increasingly recognized as important to the pathophysiology of multiple diseases. We hypothesized that clinically relevant moderate hyperoxia (<50% O2) induces senescence in developing fibroblasts. Using primary human fetal lung fibroblasts, we investigated effects of 40% O2 on senescence, endoplasmic reticulum (ER) stress, and autophagy pathways. Fibroblasts were exposed to 21% or 40% O2 for 7 days with etoposide as a positive control to induce senescence, evaluated by morphological changes, β-galactosidase activity, and DNA damage markers. Senescence-associated secretory phenotype (SASP) profile of inflammatory and profibrotic markers was further assessed. Hyperoxia decreased proliferation but increased cell size. SA-β-gal activity and DNA damage response, cell cycle arrest in G2/M phase, and marked upregulation of phosphorylated p53 and p21 were noted. Reduced autophagy was noted with hyperoxia. mRNA expression of proinflammatory and profibrotic factors (TNF-α, IL-1, IL-8, MMP3) was elevated by hyperoxia or etoposide. Hyperoxia increased several SASP factors (PAI-1, IL1-α, IL1-β, IL-6, LAP, TNF-α). The secretome of senescent fibroblasts promoted extracellular matrix formation by naïve fibroblasts. Overall, we demonstrate that moderate hyperoxia enhances senescence in primary human fetal lung fibroblasts with reduced autophagy but not enhanced ER stress. The resulting SASP is profibrotic and may contribute to abnormal repair in the lung following hyperoxia.
Keywords: autophagy, endoplasmic reticulum stress, lung development, oxygen, senescence
INTRODUCTION
In recent years, advances in neonatal care have significantly reduced the mortality of preterm infants (51), but conditions such as wheezing, asthma, and bronchopulmonary dysplasia (BPD) remain major concerns for morbidity associated with preterm birth (37, 45). The use of antenatal corticosteroids, early administration of exogenous surfactant, and gentle ventilation strategies have led to a decreased reliance on high-concentration oxygen and increased use of moderate hyperoxia (40–60%) to treat neonatal hypoxemia (8). The pathology associated with oxygen-induced parenchymal lung disease has gradually changed to the “new BPD,” characterized by arrested or disordered lung development, enlarged distal airspaces, alveolar simplification, and vascular dysmorphogenesis, with lesser changes in the bronchial airways (16). However, such changes are typically associated with high oxygen exposures, while moderate hyperoxia continues to contribute to bronchial airway changes per se (54). Indeed, moderate hyperoxia largely influences the bronchial airways (54). Clinically, even with moderate hyperoxia, wheezing and asthma continue to remain a concern (7). Thus, in the context of current clinical practice, the mechanisms by which moderate hyperoxia affects the perinatal lung become important.
Lung fibroblasts play a crucial role in the response to lung injury and in maintaining the integrity of bronchial and alveolar structures during lung development (26). A number of studies have already identified fibroblasts as a target of hyperoxia, although most studies have tended to use much higher O2 concentrations, and typically, these studies have used animal models of hyperoxia-induced BPD (2, 28, 40). Fibroblasts exposed to hyperoxia show a reduced proliferation rate (15), suggesting that O2 may both injure the lung and impair healing. However, the mechanisms of hyperoxia-induced fibroblast growth inhibition in the neonatal period are unclear.
Cellular stressors promote formation of senescent lung fibroblasts that further secrete factors, including proinflammatory cytokines, chemokines, and extracellular matrix (ECM) proteases (collectively termed senescence-associated secretory phenotype, or SASP), which enhance chronic lung diseases, such as COPD and pulmonary fibrosis (32). Cells can be induced to undergo premature senescence by various types of cellular stress, including oxidative stress, oncogene activation, endoplasmic reticulum (ER) stress, and DNA damage (21). The SASP generated by senescent fibroblasts stimulates a fibrotic phenotype, leading to prolonged inflammation and progressive fibrosis (44).
Previous studies have demonstrated induction of cellular senescence with hypoalveolarization using high concentrations of O2 in both in vitro and in vivo models (19, 23). Using a similar model of murine hyperoxia to induce BPD, Teng et. al. (50) demonstrated elevated markers of ER stress. Interestingly, decreases in cellular autophagic function have been demonstrated to induce cellular senescence (10). Finally, ER stress, a known inducer of senescence can both inhibit and stimulate cellular autophagy (38). While these findings all point to cellular senescence in the context of hyperoxia, lack of consistency in experimental paradigms, including the level of O2 and duration of exposure, make it difficult to determine the relevance of senescence in the context of clinically utilized moderate hyperoxia (<60%). In this study, we hypothesized that even prolonged moderate hyperoxia induces premature senescence in developing airways in the context of promoting lung inflammation and fibrosis. Using canalicular stage human fetal lung fibroblasts, we explored cellular senescence, the SASP portfolio, production of extracellular matrix (ECM), and activation of ER stress and autophagy mechanisms.
MATERIALS AND METHODS
Reagents.
Antibodies used for Western blotting, In-Cell Western, and immunofluorescence were purchased from Abcam (Cambridge, MA): p-γH2A.X (ab11174, 1:200), p21 (ab109520, 1:1000), beclin 1(ab62557, 1:1000), collagen I (ab34710, 1:100), collagen III (ab7778, 1:100), collagen IV (ab6586, 1:100), fibronectin (ab2413, 1:100); α-SMA (ab21027, 1:1000); Cell Signaling Technology (Danvers, MA): GAPDH (2118, 1:1000), p53 (9282S, 1:1000), p-p53 (ser15) (9284S, 1:1000), LC3B (2775, 1:1000), Bip (2775, 1:1000), PERK (5683T, 1:1000), p-PERK (3179S, 1:1000), cleaved Caspase3 (9664, 1:1000), p-Rb (3179S, 1:1000), CDK2 (3179S, 1:1000), CDK4 (3179S, 1:1000), CD45 (13917T, 1:100), CD73 (13160S, 1:100), CD105 (14606S, 1:100); Millipore (Burlington, MA): Ki-67 (AB9260, 1:100); NOVUS Biologicals (Centennial, CO): CD44 (NBP1–31488, 1:200); Santa Cruz Biotechnology (San Jose, CA): Bax (sc-6236, 1:1000), Bcl2 (sc-783, 1:1000); and Sigma Aldrich (St. Louis, MO): vimentin (V5255, 1:1000), and etoposide (E1383).
Cell isolation and culture.
Primary human fetal lung fibroblasts were isolated from canalicular-stage (18 to 22 wk gestation) lung tissue of human abortuses (Stemcell Express; exempt per Mayo Institutional Review Board) (14, 33, 42). Fetal lung tissue was washed with PBS, and airways, which were cut into 2–3-mm pieces, were placed in Dulbecco’s minimum essential medium (DMEM) with 10% fetal bovine serum (FBS; HyClone) with 1% human AB serum (ABS), and 1 mg/mL of collagenase IV was added for 1 h at 37°C. The media were replaced with DMEM 10% FBS, 5% ABS and cells were plated in T25 flasks for 24 h prior and the media then replaced with DMEM 10% FBS, 1% ABS. Cells were maintained at 37°C in 5% CO2. In this study, primary fetal lung fibroblasts were used between passages 2 and 8, thus avoiding confounding effects of replicative aging. Cells were seeded at 1 × 104 cells/mL density.
Since the developing lung contains multiple progenitor cells that are also isolated using the explant approach, we used immunofluorescence staining to determine surface marker expression in the isolated cells (Supplemental Fig. S1; Supplemental Material is available at https://doi.org/10.6084/m9.figshare.8796818). Cells were found to be negative for CD45 (hematopoietic source), but positive for CD44, CD73, and CD105. These positive markers happen to overlap for mesenchymal stem cells, as well as fibroblasts (6); however, the concurrent presence of vimentin and α-smooth muscle actin as well as the generation of ECM and lack of colony formation suggest a fibroblast phenotype.
Fibroblasts were maintained at 37°C in 5% CO2 in DMEM containing 10% FBS and 1% penicillin/streptomycin. Cells were exposed to moderate hyperoxia (40% O2) or normoxia (21% O2) for 7 days. Oxygen levels were monitored using a MiniOx 3000 oxygen monitor (MSA Medical Products, Pittsburgh, PA). As a positive control, cells were treated with 10 μM etoposide for 48 h to induce senescence and were collected on day 7 together with cells in hyperoxia and normoxia groups (33). Etoposide is a potent inducer of cellular senescence acting via DNA-damage pathways (22). Since senescence is induced by a low dose of etoposide, whereas apoptosis is triggered at higher doses (30), 10 μM etoposide was used as a positive control for senescence throughout this study.
Conditioned media.
Fetal fibroblasts (1 × 105 cells) were cultured on 100 mm dishes (10 ml medium/dish). The supernatant was collected after hyperoxia, normoxia, or etoposide exposure on day 7 and centrifuged at 4,000 g at 4°C for 30 min and filtered through a 0.2-μm membrane; its concentration was normalized to the number of cells on day 7. Before adding to naïve cells, conditioned media were diluted with normal media to reflect equivalent source cell concentration.
Cell morphology and proliferation.
Cell morphology was assessed under light microscopy at ×20 and ×40 magnifications with cell and nuclear size determined using automated delineation from thresholded images in Nikon NIS Elements.
The CellTiter 96 AQueous One Solution cell proliferation assay (MTS) (G358B; Promega, Madison, WI) was performed for cell proliferation. Cells were seeded into 96-well plates at a density of 1 × 103 cells/well. After adherence overnight, cells were treated with 21% O2, 40% O2 or for 1, 3, 5, and 7 days, respectively. At the end point, 0.5 mg/mL tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] was incubated at 37°C for 2 h, and a Flexstation 3 microplate reader (Molecular Devices, Sunnyvale, CA) was used to record absorbance at 490 nm. Cell proliferation was determined using a standard curve.
Senescence-associated β-galactosidase staining.
Senescence-associated β-galactosidase (SA-β-gal) staining, a marker of cellular senescence (29), was performed using senescence detection kit (9860S; Cell Signaling), according to the manufacturer’s instructions. Briefly, fetal fibroblasts were fixed in 2% formaldehyde containing 0.2% glutaraldehyde for 15 min and then incubated with 1 mg/ml X-gal (pH 6.0) overnight at 37°C. Cell nuclei were stained with DAPI. As a percentage of positive cells, blue-stained cells were counted under a microscope at ×40 magnification, and 20 random high-power fields (HPF) were analyzed per experiment. Images were captured using fluorescence microscopy using a Cytation 5 (Biotek) in the same field for both the SA-β-gal staining and DAPI staining.
Immunostaining.
Fetal lung fibroblasts in eight-well chamber slides (2 × 103 cells per well) were exposed to normoxia or hyperoxia for 7 days, or 10 μM etoposide for 48 h and were fixed with 4% paraformaldehyde, permeabilized, immunostained with p-γH2A.X [ab11174, 1:200; Abcam)] and Ki 67 (AB9260, 1:200; Millipore) using standard procedures (nuclei counterstained with DAPI) and imaged using fluorescence microscopy (13). Positive cells were determined in each sample from 10 randomly selected fields of view at ×40 magnification blindly, using a visual threshold by ImageJ software, and are expressed as percentages of total cell number. All assays were done in triplicate, and at least 200 cells were counted.
Cell cycle analysis.
Fetal lung fibroblasts in the normoxia, hyperoxia, and etoposide groups were subjected to cell cycle analysis by flow cytometry. After treatment, the cells were washed with 1× PBS, trypsinized with 0.25% Trypsin-EDTA, centrifuged at 800 rpm for 5 min. The cell pellet was washed once with 1× PBS and resuspended in ice-cold 70% ethanol. The cells were stored at 4°C until samples from all time points were collected (not more than 4 wk) (60). One million cells from each sample were processed for flow cytometry. Cells were washed and resuspended in 1× PBS, incubated with RNase A (Promega) at 0.5 μg/mL concentration and propidium iodide (ab139418; Abcam) 50 μg/mL for 2 h at 37°C. Propidium iodide-labeled cells were detected with flow cytometry, and results were analyzed using ModFit LT 5.0 (BD; Topsham, ME).
Western blot assay.
Immunoblotting was performed as previously described (13). Briefly, cells were harvested in lysis buffer supplemented with a protease inhibitor tablet (Roche, Indianapolis, IN). Standard SDS-PAGE (4–12%) with nitrocellulose membrane transfer techniques were used. Blots were probed with appropriate primary antibodies p21 (Abcam; ab109520), Beclin 1 (Abcam; ab62557), GAPDH (Cell Signal; 2118), p53 (Cell Signal; 9282S), p-p53 (ser15) (Cell Signal; 9284S), LC3B (Cell Signal; 2775), Bip (Cell Signal; 2775), and PERK (Cell Signal; 5683T) and p-PERK (Cell Signal; 3179S). Following IRDye secondary antibodies (LI-COR) incubation for 1 h at room temperature, membranes were imaged on an ODYSSEY Infrared Imaging System (LI-COR), and quantitative densitometry (quantification and intensity) was performed by Image Studio software (Li-Cor). Protein blots were normalized to GAPDH. Results were reported as fold change from control.
Quantitative real-time PCR.
After treatment, total RNA was isolated using RNeasy mini kit (Qiagen, Valencia, CA). Complementary DNA (cDNA) was prepared using the Transcriptor reverse transcription kit (Roche, Indianapolis, IN) and was used as a template for RT-PCR optimized for the Roche LC480 Light Cycler, with GAPDH as an internal control. The ΔΔCt method, as above, was used to determine changes in expression of mRNA of interest. Unexposed control was used as the calibrator for quantification (1). Primer assays were obtained from Qiagen for IL1-α (QT00001127), IL-1β (QT00021385), IL-6 (QT00083720), CDKN1a/p21 (QT00062090), CDKN2a/p16 (QT00089964), TP53 (QT00060235), MMP3 (QT00060235), TGFβ (QT00060235). Forward and reverse primers for GAPDH (forward: AAGGTGAAGGTCGGAGTCAACGGATT and reverse: CCATGGAATTTGCCATGGGAGGAATC), IL-8 (forward: CCTGATTTCTGCAGCTCTGTGTGA and reverse: AATTTCTGTGTTGGCGCAGTGTGG) and TNF-α (forward: ATGAGCACTGAAAGCATGATCCGG and reverse: ATCACTCCAAAGTGCAGCCAGAA) were obtained from Integrated DNA Technologies (Coralville, IA).
Monodansylcadaverine staining.
Fetal fibroblasts (2 × 104 cells/well) were exposed to normoxia or hyperoxia for 7 days, or 10 μM etoposide for 48 h. Monodansylcadaverine (MDC) staining was performed using autophagy detection kit (ab133075; Abcam), according to the manufacturer’s instructions. Tamoxifen, a known inducer of autophagy, was included as a positive control. Briefly, cells were washed with cell-based assay buffer and incubated at 37°C in 0.05 mM MDC for 10 min. Following incubation, cells were washed with cell-based assay buffer again at 37°C. All staining procedures were performed in the dark. Autophagic vacuoles stained by MDC were detected with a UV filter designed to detect DAPI. Fluorescence intensity analysis is determined by ImageJ software in each sample from 10 randomly selected fields of view at ×40 magnification blindly.
Enzyme-linked immunoassay.
Media from cells exposed for 7 days to 21%, 40% O2, or etoposide were collected and concentrated for analysis by Procartaplex 7-plex ELISA (Thermo Fisher Scientific) to assess the SASP profile (IL1-α, IL1-β, IL-6, IL-8, TNF-α, LAP, and PAI-1) with standard procedures to determine concentrations. A Magpix multiplex plate reader (Luminex Corporation, Madison, WI) was used to detect optical density, and standard curves were generated to determine concentrations in media.
Modified In-Cell Western for ECM.
Fetal lung fibroblasts plated at fixed density were treated with 1% FBS DMEM media (control), or conditioned media from cells exposed to normoxia or hyperoxia for 7 days, or treated with 10 μM etoposide for 48 h. After a 48-h treatment with media, the cells were lysed, and deposition of fibronectin, collagen I, collagen III, and collagen IV was determined using semiquantitative immunofluorescence expressed as a fold change from control (52). Protein concentrations were normalized to cell count, as determined by performing MTS proliferation assay on each plate before cell lysis.
Statistical analysis.
All experiments involved at least five separate primary lung fibroblasts samples obtained from individual fetus (n presents number of fetuses). Where appropriate, protocols were repeated three times to ensure reproducibility (technical replicates). Results are expressed as means ± SE. Statistical significance was determined by Students t-test or ANOVA with Holm-Sidak multiple comparison post hoc test. (GraphPad Software, San Diego, CA). Statistical significance was considered when P value was <0.05.
RESULTS
Hyperoxia decreases fibroblast proliferation.
Primary human fetal lung fibroblasts were allowed to proliferate for 7 days with exposure to normoxia (21% O2), normoxia with 10 μM etoposide for 48 h, or hyperoxia (40% O2). MTS assays indicated that both moderate hyperoxia and etoposide led to a loss of cell-proliferative capacity (Fig. 1, A and B). After exposure to hyperoxia for 7 days, fibroblasts demonstrated decreased cell density with altered morphology similar to that with etoposide treatment, consisting of large flattened cells that differed substantially from the expected spindle-shaped cells in the normoxia group (Fig. 1A). Consistent with the morphological and cell density data, hyperoxia and etoposide-treated fibroblasts exhibited >40% reduction in Ki-67-positive cells (Fig. 1, C and D).
Fig. 1.
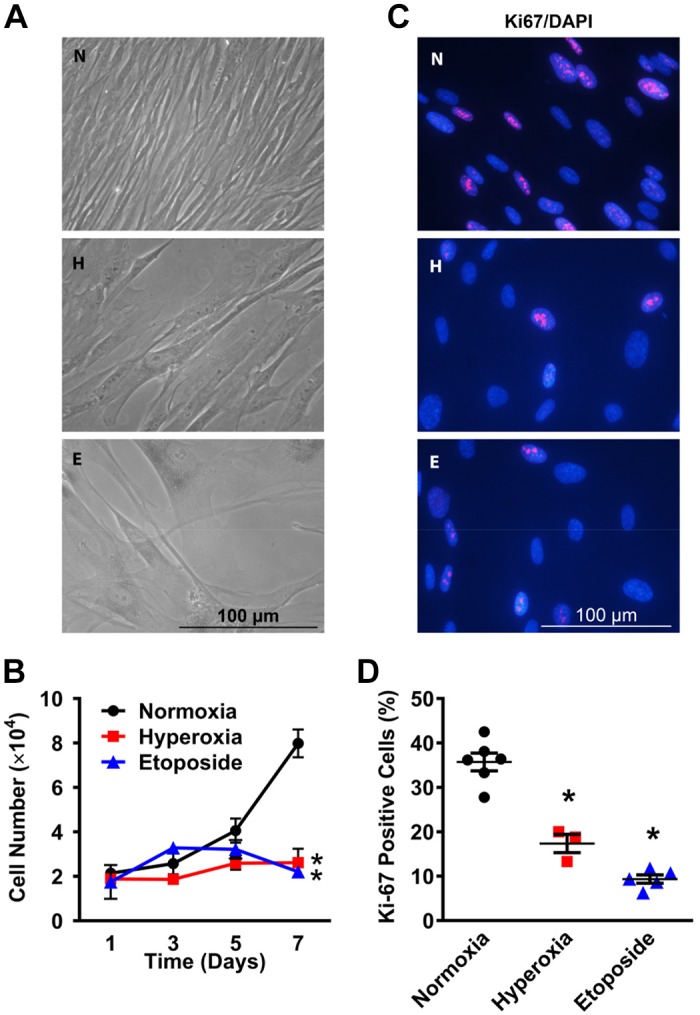
Moderate hyperoxia alters cellular proliferation and morphology of primary human fetal lung fibroblasts. A: fibroblasts were exposed to normoxia (21% O2; N) or hyperoxia (40% O2; H) for 7 days or treated with 10 μM etoposide (E) for 48 h (inducer of senescence). Phase-contrast microscopy (×400 magnification) shows enlarged and flattened cellular morphology following hyperoxia or etoposide, suggesting senescence. B: fetal lung fibroblasts were exposed to normoxia or hyperoxia for 1, 3, 5, and 7 days. MTS assay was performed to assess cellular proliferative capacity. C: after 7 days of exposure, cells were immunostained for nuclear Ki-67 (×400 magnification). D: cells positive for Ki-67 were counted in 10 random fields and are expressed as percentages of total cell number. Hyperoxia reduced fibroblast proliferation to levels comparable to that with etoposide. Values are means ± SE from n = 5 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
Hyperoxia induces premature senescence and DNA damage in fibroblasts.
Since hyperoxia altered fetal fibroblast cellular morphology and reduced proliferative capacity, we tested the possibility that induction of senescence was involved. Normoxia, hyperoxia, and etoposide-treated cells were stained for SA-β-gal, an accepted marker of senescence. Increased SA-β-gal staining was visible within both hyperoxia and etoposide-treated cells (Fig. 2A). Indeed, more than 40% of the cell population in the hyperoxia group displayed SA-β-gal positivity with a flattened and enlarged morphology, findings similar to that seen with the etoposide group (Fig. 2A). These data helped correlate the altered morphology to enhanced senescence in the same cells.
Fig. 2.

Moderate hyperoxia induces premature senescence in human fetal lung fibroblasts. A: fibroblasts were exposed to normoxia (21%; N) or hyperoxia (40%; H) for 7 days or treated with 10 μM etoposide (E) for 48 h. Cellular senescence was estimated based on expression of senescence-associated β-galactosidase (SA-β-Gal) and cell morphology. Representative pictures of SA-β-Gal–stained cells were examined under brightfield and fluorescence microscopy. Flattened and enlarged cells colored with blue-green were considered senescent (×400 magnification). Quantification of SA-β-Gal–positive cells was based on automated microscopic analysis. B: after exposure, as above, cells were fixed and immunolabeled with an antibody against p-γ-H2A.X, a marker of DNA double-strand breaks, and counterstained with DAPI (×400 magnification). Graph shows the ratio of p-γ-H2A.X–positive cells to total. Values are means ± SE from n = 5 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
DNA damage response (DDR) is a major pathway leading to induction of cellular senescence (49). Therefore, we explored induction of DDR within the context of hyperoxia. Cells were fixed and immunolabeled to detect phospho-γH2A.X foci, an established marker of double-stranded DNA breaks and a hallmark of DDR (41). Phospho-γH2A.X foci-positive cells were observed in a much larger fraction of fetal fibroblasts exposed to hyperoxia or etoposide (~50%) compared with normoxia (less than 10%) (Fig. 2B).
Hyperoxia, cell cycle, and senescence-associated cell checkpoint proteins.
A characteristic of senescent cells is an irreversible arrest of the cell cycle (36). Flow cytometry analysis of cell cycle progression using propidium iodide showed that hyperoxia and etoposide exposure resulted in increased accumulation of cells in the G2/M phase, compared with the normoxia group (Fig. 3, A and B). The S phase did not significantly differ between groups. In addition, with a smaller percentage of cells in the G1 phase, it appears as if the cells are not completing the M phase and reentering the G1 phase in the hyperoxia and etoposide groups compared with usual cycling with normoxia (Fig. 3, A and B), again highlighting hyperoxia-induced arrest primarily in the G2/M phase. Consistent with the DNA damage-induced cell cycle arrest properties of senescent cells, mRNA expression of p21 was noted to be elevated, while p16 and p53 levels remained statistically unchanged (Fig. 3E). However, Western blot analysis indicated that hyperoxia and etoposide treatments of fetal fibroblasts resulted in overexpression of cell cycle regulatory proteins phosphorylated p53 (p-p53), total p53, and total p21 (Fig. 3, C and D). p21 has previously been demonstrated to cause arrest both in the G1 and the G2/M phase (31). Overall, these data suggest that prolonged moderate hyperoxia exposure activates a senescent program characterized by arrest of the cell cycle in G2/M phase, with increased gene and protein expression of p53 and p21.
Fig. 3.

Moderate hyperoxia activates a cellular senescence program in human fetal lung fibroblasts. A: flow cytometry analyses of DNA content of fetal fibroblasts exposed to normoxia, hyperoxia for 7 days, or to etoposide for 48 h. B: histograms showing the percentages of cells at G1, S, and G2/M phases. C: representative Western blot analyses using antibodies against phosphorylated p53, p53, or p21. GAPDH immunoblots were used as loading controls. D: graphs represent relative expression of p-p53, p53, or p21 to GAPDH expression. E: total RNA was isolated from cells and mRNAs for p53, p21, and p16 were quantified by quantitative PCR. Values are expressed as means ± SE from n = 5–7 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
Hyperoxia suppresses autophagy but does not induce ER stress.
Alterations in autophagy and ER stress have been reported in models of cellular senescence previously (3, 17). Accordingly, we explored alterations in these pathways in the response of fetal fibroblasts to hyperoxia.
MDC is a specific in vivo marker for autophagic vacuoles (58). Fetal fibroblasts were treated as described earlier and stained with MDC to observe autophagic vacuoles. In the hyperoxia and etoposide groups, only certain cells stained positive for MDC and the fluorescence intensity of MDC staining in the cells, representing autophagy, was markedly decreased compared with normoxia and tamoxifen-treated positive control groups (Fig. 4A). Further, Western blot analysis of autophagic protein LC3B showed reduced expression of LC3B-II in the hyperoxia group compared with the normoxia group (P < 0.05). The protein expression of beclin-1, another marker for autophagy, tended towards a decrease, but was not statistically significant (Fig. 4B).
Fig. 4.

Moderate hyperoxia suppresses autophagy but not endoplasmic reticulum (ER) stress in primary human fetal lung fibroblasts. A: monodansylcadaverine (MDC) staining of primary fetal lung fibroblasts exposed to normoxia, hyperoxia for 7 days, or to etoposide for 48 h. Tamoxifen was used as a positive control for induction of autophagy (×400 magnification). Graph shows fluorescence measurements of autophagic level (MDC fluorescence). B: representative Western blot using antibodies against autophagy markers LC3B and beclin-1. Graphs represent relative expression of LC3B-II or beclin-1 to GAPDH expression. C: representative Western blot using antibodies against ER stress markers IRE1α, Bip, p-PERK, PERK, and GAPDH. Graphs represent relative expression of IRE1α, Bip, p-PERK, or PERK to GAPDH expression. Values are means ± SE from n = 5–7 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
Several studies have identified the regulatory role of ER stress in autophagy and in senescent cells (11, 48). Altered autophagy may be a downstream effect of ER stress. To test whether moderate hyperoxia and etoposide had a deleterious effect on the endoplasmic network, we performed Western blot analysis to compare expression of several proteins implicated in the ER stress response and found changes in levels of binding immunoglobulin protein (BiP) and IRE1α under some conditions. IRE1α is a proximal sensor of the ER stress and contributes to monitoring the quality of the proteins synthesized in the ER, while BiP acts as a chaperone in the ER stress pathway. There was a decrease in the levels of IRE1α in cells exposed to hyperoxia and etoposide compared with normoxia (Fig. 4C). However, no statistical changes were noted in p-PERK or PERK. Overall, hyperoxia did not appear to be an inducer of ER stress in fetal fibroblasts, but did appear to downregulate autophagy. This is consistent with the overall picture of a senescent cell, which is resistant to apoptosis and remains metabolically active, secreting important paracrine and autocrine factors.
Hyperoxia-induced senescent fetal lung fibroblasts have a secretory phenotype.
It is well known that senescent cells demonstrate a senescence-associated secretory phenotype (SASP) (59). To assess the secretory phenotype of hyperoxia-induced senescent fetal lung fibroblasts, cells were exposed to normoxia or hyperoxia for 7 days or 10 μM etoposide for 48 h. Conditioned medium (CM) was collected, concentrated, and processed by customized ELISA to assess the SASP profile. Compared with cells exposed to normoxia, the protein concentrations of IL-1α, IL-1β, IL-6, TNF-α, LAP, and PAI-1 in the hyperoxia- and etoposide- treated groups were significantly increased (Fig. 5A). Transcript levels of the SASPs in fetal fibroblasts were measured as well, revealing significantly higher levels of IL-1β, IL-8, and MMP3 in hyperoxia group compared with normoxia controls (Fig. 5B). These results showed that the secretome of hyperoxia-induced senescent fetal lung fibroblasts is proinflammatory and profibrotic and may promote remodeling.
Fig. 5.

Hyperoxia-induced senescent fetal lung fibroblasts have a secretory phenotype. A: concentrated medium of fetal fibroblasts exposed to normoxia (21% O2) or hyperoxia (40% O2) for 7 days or to etoposide was assessed by customized ELISA for protein levels of IL-1α, IL-1β, IL-6, IL-8, TNF-α, LAP, and PAI-1. B: total RNA was isolated from primary human fetal fibroblasts from these groups, and mRNA for IL-1α, IL-1β, IL-8, TGFβ, and MMP3 was quantified by quantitative PCR. Hyperoxia increased both mRNA and protein for a variety of proinflammatory and profibrotic mediators. Values are means ± SE from n = 5–7 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
Hyperoxia induces a pro-fibrotic SASP.
Extracellular matrix (ECM) is a tissue-specific macromolecular structure that plays important roles in shaping cell behavior and influencing lung development and growth. Changes to ECM quality in the lung likely promote fibroblast behavior in the early and late wound repair response (56). To assess the impact of moderate hyperoxia on ECM, modified In-Cell Westerns were performed to analyze deposition of key ECM proteins: collagen I, III, IV, and fibronectin. Hyperoxia significantly increased deposition of collagen IV, while collagen I, collagen III, and fibronectin deposition was not affected. Meanwhile, treatment with etoposide for 48 h significantly increased deposition of all the four ECM proteins (Fig. 6). Thus, hyperoxia effects on fibroblasts may be ECM protein-specific.
Fig. 6.

Effect of moderate hyperoxia on extracellular matrix (ECM) deposition. A modified Li-Cor In-Cell Western technique (semiquantitative immunofluorescence) was used to show deposition of collagen I, III, IV, and fibronectin by fibroblasts exposed to normoxia (21% O2) or hyperoxia (40% O2) for 7 days or treated with etoposide. Hyperoxia increased collagen IV while etoposide increased other ECM proteins as well. Values are means ± SE from n = 7 or 8 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
To examine potential profibrotic paracrine effects of the SASP, normal primary human fetal lung fibroblasts (i.e., naïve cells) were treated for 48 h with CM from fetal fibroblasts exposed to normoxia (Con-CM) or hyperoxia (SASP-CM), or treated with etoposide (Etop-CM). Proliferative capacity was detected using MTS assay. Naïve cells treated with SASP-CM demonstrated significantly increased proliferation compared with cells treated with Con-CM (Fig. 7A). Further, naïve cells treated with SASP-CM showed significantly increased collagen III deposition compared with cells treated with Con-CM (Fig. 7B). Meanwhile, naïve cells treated with Etop-CM showed significantly increased collagen I, collagen III, and collagen IV deposition compared with cells treated with Con-CM. Thus, our results demonstrate that the secretome of senescent fibroblasts is profibrotic and could induce a profibrotic phenotype in neighboring tissue as well.
Fig. 7.

Profibrotic effects of the senescence-associated secretory phenotype (SASP) on naïve human fetal lung fibroblasts. A: naïve fibroblasts were treated for 48 h with conditioned media (concentrated supernatant) from fibroblasts exposed to normoxia (Con-CM), hyperoxia (SASP-CM), or etoposide-conditioned media (Etop-CM). B: MTS assay was used to assess proliferative changes of naïve cells exposed to CM. C: In-Cell Western was used to detect naïve cell deposition of extracellular matrix (ECM). CM from hyperoxia-exposed fibroblasts enhanced ECM production by naïve cells, showing a profibrotic SASP portfolio. Values are means ± SE from n = 5–7 samples. *P < 0.05 compared with normoxia group (one-way ANOVA).
DISCUSSION
Advances in neonatal care have been associated with a marked increase in survival of very preterm infants (gestational age <28 wk), who are already predisposed to significant morbidities, including pulmonary diseases, such as asthma and BPD. Here, physiologically and clinically necessary administration of oxygen with or without mechanical ventilation is a known contributor to perinatal lung disease in prematurity. Even with improved clinical care involving moderate rather than severe hyperoxia, wheezing and asthma remain a major problem (37, 45), highlighting the importance of bronchial changes. Using fibroblasts from the canalicular phase, representing the phase of pulmonary development in very preterm neonates, we explored molecular mechanisms responsible for this pathology. Using clinically relevant levels of hyperoxia (40% O2), we demonstrated induction of premature senescence in primary fetal fibroblasts. Exposure to hyperoxia for 7 days resulted in flattened and wide cells, with reduced proliferative capacity. Other evidence of senescence included increased SA-β-gal and decreased Ki67 staining. In addition, overexpression of p53 and p21, along with demonstration of cell cycle arrest at the G2/M phase, further support the induction of senescence. Interestingly, the secretome of senescent fibroblasts is profibrotic and proinflammatory and activates surrounding tissue in a paracrine fashion to enhance these effects, potentially contributing to the pathogenesis of oxygen-induced airway changes in premature infants. From the mechanistic perspective, our model demonstrated elevated levels of p-γH2a.X, indicating induction of DNA damage; however, ER stress is unaltered, although there is a reduction in autophagy.
Our study demonstrated that both hyperoxia- and etoposide-treated primary fetal lung fibroblasts produced a cell cycle arrest in G2/M phase. This result is inconsistent with the concept that cellular senescence is due to an irreversible arrest in the G1 phase of the cell cycle (46, 47). However, several studies have reported that senescence-inducing stimuli can activate p21-dependent signaling, resulting in permanent cell cycle arrest in G2/M phase by inhibiting mitotic Cdk complexes and pRb phosphorylation (53), an observation confirmed in our studies where we find inhibition of p-Rb and CDK2 expression (Supplemental Fig. S2). Several recent publications have validated the idea that the senescence program can be launched after G2 arrest (12, 35). Thus, we speculate that cells in our model slip from long-term G2 phase without mitosis directly into the G0/G1 phase without dividing.
The interaction between senescence and autophagy in pulmonary fibrotic disease has been extensively reported (21). Autophagy is considered to be one of the most important defense responses against aging and cellular senescence (39). Insufficient autophagy is involved in accelerated cell senescence in adult chronic obstructive pulmonary disease, suggesting a novel protective role for autophagy in smoke-induced senescence (9). In contrast, it was also reported that activation of autophagy results in an increase in cellular senescence (35). In our study, we found autophagy was defective in primary human fetal lung fibroblasts that were exposed to hyperoxia or treated with etoposide. We found reduced expression of LC3B-II and decreased MDC staining that point to a role for impaired autophagy. However, there were no changes in beclin-1 and LC3B-I. With respect to autophagy, the importance of LC3B-I is debatable, while LC3B-II is a direct indicator of autophagic vacuoles (as confirmed by MDC staining). While beclin-1 did not increase, this is not a direct indicator of autophagy per se. Thus, in spite of differences in changes of different typical markers of autophagy, we conclude that autophagy was, in fact, reduced with hyperoxia. Such a decrease in autophagic mechanisms may allow for continued metabolic activity and secretion of SASP in the context of increased senescence.
While the proportion of cells in the S phase was slightly decreased by hyperoxia and etoposide, there was no statistically significant difference compared with normoxia. We reanalyzed the portion of apoptotic cells in these three groups as sub-G1 using ModFit LT 5.0 (Supplemental Fig. S2). The proportion of cells in the sub-G1 phase of the hyperoxia group and etoposide group was higher than that of the normoxia group, but the proportion was relatively small and there is no significant difference because of the variation, which indicates that apoptosis was a mechanism, but may be not the major pathway for hyperoxia and etoposide-induced pathological changes.
Previous studies have reported a profibrotic role for ER stress in fibroblasts, and hyperoxia-induced hypoalveolarization has also been described (50). We hypothesized that hyperoxia-induced cellular stress activated the unfolded protein response in the ER. This mechanism is initiated by BiP, which in turn activates three pathways (IRE1α, PERK/eIF2, and AFT), which ultimately contribute to decreasing the burden on the ER (57). Evaluation of ER stress in our study showed a decrease in protein expression of IRE1α, without changes in BiP, p-PERK, and PERK, suggesting that the unfolded protein response is likely not sufficiently activated in our model. This difference from other studies may be due to the lower level of O2 being used in our study. Prior investigations have reported that ER stress may induce senescence (18), and while our model cannot corroborate such a finding, it is possible that the unfolded protein response is activated at an earlier time point during hyperoxic exposure (since we took readouts only at 7 days), as noted in other studies (4, 24, 25, 50). After induction of senescence, fibroblasts may be optimally adapted to their current environment, leading to the continued production of intra and extracellularly destined proteins. Regardless of these issues, our studies show that at a time point when cellular senescence is clearly present, the extent of ongoing ER stress is not high.
An important pathological feature of senescent cells is the SASP, and while the SASP of lung fibroblasts in COPD and IPF has previously been described (20, 44), its characterization was limited to the gene expression of a few inflammatory markers. In this study, both hyperoxia- exposed and etoposide-treated cells exhibited increased production of cytokines typically associated with the SASP, as well as a markedly proinflammatory gene expression profile. The levels of IL-1, IL-8, TNFα, LAP, and PAI-1 in hyperoxia-exposed CM were found to be higher than normoxia-exposed CM. PAI-1 activity has been reported to be a specific SASP factor elevated up to 50-fold in models of senescence among endothelial cells and lung and skin fibroblasts (5, 55). Latent-associated peptide (LAP) is located in the anterior region between the NH2-terminal peptide and the COOH-terminal peptide of TGF-β1 precursor peptide, and it has been used as a surrogate for TGF-β1; elevation suggests increased activation of fibrotic activity of fibroblasts in both an autocrine and paracrine fashion. Interestingly, although hyperoxia induced changes in LAP, indirectly suggesting extracellular TGFβ activity, we did not observe a change in cellular TGFβ expression per se, although this discordance does not necessarily imply lack of altered TGFβ activity, depending on the extent of TGFβ secreted versus activated by cleavage. Indeed, the SASP profile does include proteases that could enhance TGFβ activity regardless of changes in levels secreted.
In our study, the secretome of senescent fibroblasts stimulated a fibrotic phenotype in naïve healthy cells, which is characterized by ECM deposition and cell proliferation. The secretion of these senescence-associated factors has the potential to alter the local pulmonary microenvironment, leading to a phenotype similar to oxygen-induced injury noted during the neonatal life of premature infants. Collagen and elastin networks produced by mesenchymal cells are deposited and extensively remodeled, and are closely related to the developmental maturation of alveolar epithelial structures (27). The inflammatory mediator IL-1 is a key component of the inflammasome, and IL-8 is chemotactic to various immune cells, both of which may lead to systemic and local inflammation and tissue damage. Taken together, these senescent lung fibroblasts with increased expression of proinflammatory and profibrotic factors would amplify aberrant wound repair processes that follow remodeled ECM, exacerbating the fibrotic response, which may play a role in abnormal repair of lung injury and development.
While our studies point to novel mechanisms by which hyperoxia influences the developing airway, we recognize limitations in our approach. Our studies were entirely in vitro, and all specimens were obtained from abortuses at 18–22 wk. On the one hand, this window represents a period of rapid bronchial airway growth and also an age proximate to survival, and thus, canalicular stage fibroblasts represent lung development in very preterm neonates. Thus, our results are most appropriate for understanding hyperoxia’s effects in the context of bronchial disease. On the other hand, most premature infants with high-risk BPD are born in the saccular stage of lung development, and thus, the relevance of fibroblasts from an earlier stage to BPD per se may be less clear. However, it is very difficult to obtain primary fibroblasts from the saccular stage, and this study, which encapsulates the canalicular period, allows us to understand the overall biology of oxygen-related insults. Here, alterations in vimentin and α-smooth muscle actin following hyperoxia are associated with BPD, which we do observe with 40% O2 exposure in our fibroblast model (Supplemental Fig. S3). Regardless of these observations, it is important to note that given an entirely in vitro approach, further studies are needed to correlate fibroblast changes to structural changes in vivo in the context of both bronchial thickening, as well as alveolar and vascular changes of BPD. Indeed, even in the context of in vitro work, our studies were conducted using standard two-dimensional cell culture techniques based on plastic surfaces. However, these models do not reflect all the complexity of native tissues regarding molecular composition and structure. Therefore, a multitude of three-dimensional (3D) cell culture systems, organoids, or in vivo work would represent next steps for further investigation.
In our studies, we used 21% O2 as normoxia, given this is a standard cell culture condition. However, fibroblasts are adapted to lower O2 concentration (34), and therefore, 21% O2 could be seen as relatively hyperoxic. Indeed, such O2 levels are also above that normally encountered in vivo in the fetus (43). In an effort to address this concern, we cultured fetal fibroblasts in 21% O2 vs. 5% O2 for 7 days and compared proliferation and senescence to find there was no difference in fibroblast proliferation at day 7, but if anything, senescent cell proportion was less in the 5% O2 group (Supplemental Fig. S4). This phenomenon meets our expectations that fetal fibroblasts are optimized for growth at normal oxygen levels for a fetus (5% O2) and that even transitioning to normoxia (21% O2), which represents a clinically relevant level induces senescence. Further exposure to iatrogenic supplemental oxygenation, such as 40% O2, worsens the senescence phenotype of the tissue.
In conclusion, our study demonstrates that prolonged moderate hyperoxia induces premature senescence in primary human fetal lung fibroblast, with evidence of altered morphology, decreased cellular proliferation, DNA damage, and cell cycle arrest at G2/M phase with the overexpression of p53 and p21. Mechanistically, the unfolded protein response may not be triggered, but lower levels of autophagy were noted. Finally, the secretome of senescent fibroblasts remodels ECM and stimulates a fibrotic phenotype in naive healthy cells, which may play a role in abnormal repair of lung injury and development, contributing to the pathogenesis of BPD.
GRANTS
This work was supported by the National Natural Science Foundation of China (81501292; K. You), Mayo Clinic Department of Obstetrics and Gynecology (P. Parikh), the Mayo Clinic Center for Biomedical Discovery (R. D. Britt and C. M. Pabelick), Mayo Clinic National Center for Advancing Translational Sciences (Grant UL1TR002377; R. D. Britt and C. M. Pabelick), and NIH/National Heart, Lung, and Blood Institute Grants R00 HL131682 (R. D. Britt), R01 HL056470 (Y. S. Prakash), and R01 HL138402 (C. M. Pabelick).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.Y., P.P., K.K., R.D.B., and Y.S.P. conceived and designed research; K.Y., P.P., K.K., S.W., L.J.M., B.Y., A.M.R., B.R., and J.J.T. performed experiments; K.Y., P.P., K.K., L.J.M., A.M.R., B.R., J.J.T., and Y.S.P. analyzed data; K.Y., P.P., S.W., R.D.B., C.M.P., and Y.S.P. interpreted results of experiments; K.Y., L.J.M., B.R., J.J.T., and Y.S.P. prepared figures; K.Y., P.P., and Y.S.P. drafted manuscript; K.Y., P.P., K.K., S.W., A.M.R., B.R., R.D.B., C.M.P., and Y.S.P. edited and revised manuscript; K.Y., P.P., K.K., S.W., L.J.M., B.Y., A.M.R., B.R., J.J.T., R.D.B., C.M.P., and Y.S.P. approved final version of manuscript.
REFERENCES
- 1.Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J Cell Physiol 232: 1053–1068, 2017. doi: 10.1002/jcp.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balaji S, Dong X, Li H, Zhang Y, Steen E, Lingappan K. Sex-specific differences in primary neonatal murine lung fibroblasts exposed to hyperoxia in vitro: implications for bronchopulmonary dysplasia. Physiol Genomics 50: 940–946, 2018. doi: 10.1152/physiolgenomics.00075.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blazanin N, Son J, Craig-Lucas AB, John CL, Breech KJ, Podolsky MA, Glick AB. ER stress and distinct outputs of the IRE1α RNase control proliferation and senescence in response to oncogenic Ras. Proc Natl Acad Sci USA 114: 9900–9905, 2017. doi: 10.1073/pnas.1701757114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choo-Wing R, Syed MA, Harijith A, Bowen B, Pryhuber G, Janér C, Andersson S, Homer RJ, Bhandari V. Hyperoxia and interferon-γ-induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress-dependent pathway. Am J Respir Cell Mol Biol 48: 749–757, 2013. doi: 10.1165/rcmb.2012-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comi P, Chiaramonte R, Maier JA. Senescence-dependent regulation of type 1 plasminogen activator inhibitor in human vascular endothelial cells. Exp Cell Res 219: 304–308, 1995. doi: 10.1006/excr.1995.1232. [DOI] [PubMed] [Google Scholar]
- 6.Denu RA, Nemcek S, Bloom DD, Goodrich AD, Kim J, Mosher DF, Hematti P. Fibroblasts and mesenchymal stromal/stem cells are phenotypically indistinguishable. Acta Haematol 136: 85–97, 2016. doi: 10.1159/000445096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Fiore JM, Martin RJ, Li H, Morris N, Carlo WA, Finer N, Walsh M, Jobe AH, Caplan MS, Polin RA, Laptook AR, Oh W, Hensman AM, Gingras D, Barnett S, Lillie S, Francis K, Andrews D, Angela K, Fanaroff AA, Newman NS, Siner BS, Zadell A, Schibler K, Donovan EF, Bridges K, Alexander B, Grisby C, Mersmann MW, Mincey HL, Hessling J, Goldberg RN, Cotten CM, Wallace DK, Freedman SF, Auten KJ, Fisher KA, Foy KA, Stoll BJ, Piazza AJ, Buchter S, Carlton DP, Hutchinson AK, Hale EC, Higgins RD, Archer SW, Poindexter BB, Lemons JA, Hamer F, Herron DE, Miller LC, Wilson LD, Berberich MA, Blaisdell CJ, Gail DB, Kiley JP, Gantz MG, Das A, Crawford MM, Hastings BK, Irene AR, O’Donnell Auman J, Huitema CP, Pickett JW II, Wallace D, Zaterka-Baxter KM, Van Meurs KP, Stevenson DK, Ball MB, Proud MS, Frantz ID III, Fiascone JM, Furey A, MacKinnon BL, Nylen E, Ambalavanan N, Collins MV, Cosby SS, Phillips VA, Rasmussen MR, Wozniak PR, Rich W, Arnell K, Bridge R, Demetrio C, Bell EF, Widness JA, Klein JM, Johnson KJ, Duara S, Everett-Thomas R, Watterberg KL, Ohls RK, Rohr J, Lacy CB, Phelps DL, Laroia N, Markowitz GD, Reubens LJ, Burnell E, Sánchez PJ, Rosenfeld CR, Salhab WA, Allen J, Grau L, Guzman A, Hensley G, Lepps MH, Martin M, Miller NA, Solis A, Vasil DM, Wilder K, Kennedy KA, Tyson JE, Morris BH, Harris BF, Lis AE, Martin S, McDavid GE, Tate PL, Wright SL, Yoder BA, Faix RG, Burnett J, Jensen JJ, Osborne KA, Spencer C, Weaver-Lewis K, O’Shea TM, Peters NJ, Shankaran S, Sood BG, Bara R, Billian E, Johnson M, Ehrenkranz RA, Narendran V, Bhandari V, Jacobs HC, Cervone P, Gettner P, Konstantino M, Poulsen JA, Taft J; SUPPORT Study Group of the Eunice Kennedy Shriver National Institute of Child Health, and Human Development Neonatal Research Network . Patterns of oxygenation, mortality, and growth status in the surfactant positive pressure and oxygen trial cohort. J Pediatr 186: 49–56.e1, 2017. doi: 10.1016/j.jpeds.2017.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dumpa V, Bhandari V. Surfactant, steroids and non-invasive ventilation in the prevention of BPD. Semin Perinatol 42: 444–452, 2018. doi: 10.1053/j.semperi.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, Kawaishi M, Hirano J, Odaka M, Morikawa T, Nishimura S, Nakayama K, Kuwano K. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. OncoImmunology 1: 630–641, 2012. doi: 10.4161/onci.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42, 2016. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK, O’Brien M, Mau T, Yung R. Toll-like receptor 4 (TLR4)-deficient mice are protected from adipose tissue inflammation in aging. Aging (Albany NY) 9: 1971–1982, 2017. doi: 10.18632/aging.101288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gire V, Dulic V. Senescence from G2 arrest, revisited. Cell Cycle 14: 297–304, 2015. doi: 10.1080/15384101.2014.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartman W, Helan M, Smelter D, Sathish V, Thompson M, Pabelick CM, Johnson B, Prakash YS. Role of hypoxia-induced brain-derived neurotrophic factor in human pulmonary artery smooth muscle. PLoS One 10: e0129489, 2015. doi: 10.1371/journal.pone.0129489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartman WR, Smelter DF, Sathish V, Karass M, Kim S, Aravamudan B, Thompson MA, Amrani Y, Pandya HC, Martin RJ, Prakash YS, Pabelick CM. Oxygen dose responsiveness of human fetal airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 303: L711–L719, 2012. doi: 10.1152/ajplung.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussain N, Wu F, Christian C, Kresch MJ. Hyperoxia inhibits fetal rat lung fibroblast proliferation and expression of procollagens. Am J Physiol Lung Cell Mol Physiol 273: L726–L732, 1997. doi: 10.1152/ajplung.1997.273.4.L726. [DOI] [PubMed] [Google Scholar]
- 16.Kalikkot Thekkeveedu R, Guaman MC, Shivanna B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir Med 132: 170–177, 2017. doi: 10.1016/j.rmed.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang C, Elledge SJ. How autophagy both activates and inhibits cellular senescence. Autophagy 12: 898–899, 2016. doi: 10.1080/15548627.2015.1121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HS, Kim Y, Lim MJ, Park YG, Park SI, Sohn J. The p38-activated ER stress-ATF6α axis mediates cellular senescence. FASEB J 33: 2422–2434, 2019. doi: 10.1096/fj.201800836R. [DOI] [PubMed] [Google Scholar]
- 19.Klimova TA, Bell EL, Shroff EH, Weinberg FD, Snyder CM, Dimri GP, Schumacker PT, Budinger GR, Chandel NS. Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. FASEB J 23: 783–794, 2009. doi: 10.1096/fj.08-114256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar M, Seeger W, Voswinckel R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 51: 323–333, 2014. doi: 10.1165/rcmb.2013-0382PS. [DOI] [PubMed] [Google Scholar]
- 21.Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S, Kobayashi K, Nakayama K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig 54: 397–406, 2016. doi: 10.1016/j.resinv.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2: 924–935, 2010. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Londhe VA, Sundar IK, Lopez B, Maisonet TM, Yu Y, Aghai ZH, Rahman I. Hyperoxia impairs alveolar formation and induces senescence through decreased histone deacetylase activity and up-regulation of p21 in neonatal mouse lung. Pediatr Res 69: 371–377, 2011. doi: 10.1203/PDR.0b013e318211c917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozon TI, Eastman AJ, Matute-Bello G, Chen P, Hallstrand TS, Altemeier WA. PKR-dependent CHOP induction limits hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 300: L422–L429, 2011. doi: 10.1152/ajplung.00166.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu HY, Zhang J, Wang QX, Tang W, Zhang LJ. Activation of the endoplasmic reticulum stress pathway involving CHOP in the lungs of rats with hyperoxia-induced bronchopulmonary dysplasia. Mol Med Rep 12: 4494–4500, 2015. doi: 10.3892/mmr.2015.3979. [DOI] [PubMed] [Google Scholar]
- 26.McGowan S. Understanding the developmental pathways pulmonary fibroblasts may follow during alveolar regeneration. Cell Tissue Res 367: 707–719, 2017. doi: 10.1007/s00441-016-2542-3. [DOI] [PubMed] [Google Scholar]
- 27.Mižíková I, Ruiz-Camp J, Steenbock H, Madurga A, Vadász I, Herold S, Mayer K, Seeger W, Brinckmann J, Morty RE. Collagen and elastin cross-linking is altered during aberrant late lung development associated with hyperoxia. Am J Physiol Lung Cell Mol Physiol 308: L1145–L1158, 2015. doi: 10.1152/ajplung.00039.2015. [DOI] [PubMed] [Google Scholar]
- 28.Morty RE. Recent advances in the pathogenesis of BPD. Semin Perinatol 42: 404–412, 2018. doi: 10.1053/j.semperi.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496, 2014. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 30.Nagano T, Nakano M, Nakashima A, Onishi K, Yamao S, Enari M, Kikkawa U, Kamada S. Identification of cellular senescence-specific genes by comparative transcriptomics. Sci Rep 6: 31758, 2016. doi: 10.1038/srep31758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niculescu AB III, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 18: 629–643, 1998. doi: 10.1128/MCB.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol 35: 681–688, 2006. doi: 10.1165/rcmb.2006-0169OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parikh P, Britt RD Jr, Manlove LJ, Wicher SA, Roesler A, Ravix J, Teske J, Thompson MA, Sieck GC, Kirkland JL, LeBrasseur N, Tschumperlin DJ, Pabelick CM, Prakash YS. Hyperoxia-induced cellular senescence in fetal airway smooth muscle cells. Am J Respir Cell Mol Biol 61: 51–60, 2019. doi: 10.1165/rcmb.2018-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patrick A, Seluanov M, Hwang C, Tam J, Khan T, Morgenstern A, Wiener L, Vazquez JM, Zafar H, Wen R, Muratkalyeva M, Doerig K, Zagorulya M, Cole L, Catalano S, Lobo Ladd AA, Coppi AA, Coşkun Y, Tian X, Ablaeva J, Nevo E, Gladyshev VN, Zhang ZD, Vijg J, Seluanov A, Gorbunova V. Sensitivity of primary fibroblasts in culture to atmospheric oxygen does not correlate with species lifespan. Aging (Albany NY) 8: 841–847, 2016. doi: 10.18632/aging.100958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Neut M, Haar L, Rao V, Santha S, Lansu K, Rana B, Jones WK, Gentile S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget 7: 21991–22004, 2016. doi: 10.18632/oncotarget.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polewska J, Skwarska A, Augustin E, Konopa J. DNA-damaging imidazoacridinone C-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J Pharmacol Exp Ther 346: 393–405, 2013. doi: 10.1124/jpet.113.203851. [DOI] [PubMed] [Google Scholar]
- 37.Priante E, Moschino L, Mardegan V, Manzoni P, Salvadori S, Baraldi E. Respiratory outcome after preterm birth: a long and difficult journey. Am J Perinatol 33: 1040–1042, 2016. doi: 10.1055/s-0036-1586172. [DOI] [PubMed] [Google Scholar]
- 38.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy 11: 1956–1977, 2015. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435, 2010. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 40.Rehan VK, Torday JS. The lung alveolar lipofibroblast: an evolutionary strategy against neonatal hyperoxic lung injury. Antioxid Redox Signal 21: 1893–1904, 2014. doi: 10.1089/ars.2013.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riballo E, Kühne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Löbrich M. A pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to gamma-H2AX foci. Mol Cell 16: 715–724, 2004. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 42.Roesler AM, Wicher SA, Ravix J, Britt RD Jr, Manlove L, Teske JJ, Cummings K, Thompson MA, Farver C, MacFarlane P, Pabelick CM, Prakash YS. Calcium sensing receptor in developing human airway smooth muscle. J Cell Physiol 234: 14,187–14,197, 2019. doi: 10.1002/jcp.28115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandbichler AM, Jansen B, Peer BA, Paulitsch M, Pelster B, Egg M. Metabolic plasticity enables circadian adaptation to acute hypoxia in zebrafish cells. Cell Physiol Biochem 46: 1159–1174, 2018. doi: 10.1159/000489058. [DOI] [PubMed] [Google Scholar]
- 44.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skromme K, Leversen KT, Eide GE, Markestad T, Halvorsen T. Respiratory illness contributed significantly to morbidity in children born extremely premature or with extremely low birthweights in 1999–2000. Acta Paediatr 104: 1189–1198, 2015. doi: 10.1111/apa.13165. [DOI] [PubMed] [Google Scholar]
- 46.Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science 273: 63–67, 1996. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- 47.Stein GH, Dulić V. Origins of G1 arrest in senescent human fibroblasts. BioEssays 17: 537–543, 1995. doi: 10.1002/bies.950170610. [DOI] [PubMed] [Google Scholar]
- 48.Su Y, Wang P, Shen H, Sun Z, Xu C, Li G, Tong T, Chen J. The protein kinase D1-mediated classical protein secretory pathway regulates the Ras oncogene-induced senescence response. J Cell Sci 131: jcs207217, 2018. doi: 10.1242/jcs.207217. [DOI] [PubMed] [Google Scholar]
- 49.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62: 1876–1883, 2002. [PubMed] [Google Scholar]
- 50.Teng RJ, Jing X, Michalkiewicz T, Afolayan AJ, Wu TJ, Konduri GG. Attenuation of endoplasmic reticulum stress by caffeine ameliorates hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol 312: L586–L598, 2017. doi: 10.1152/ajplung.00405.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Victora JD, Silveira MF, Tonial CT, Victora CG, Barros FC, Horta BL, Santos ISD, Bassani DG, Garcia PCR, Scheeren M, Fiori HH; Pelotas Cohorts Study Group . Prevalence, mortality and risk factors associated with very low birth weight preterm infants: an analysis of 33 years. J Pediatr (Rio J). In press. doi: 10.1016/j.jped.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vogel ER, VanOosten SK, Holman MA, Hohbein DD, Thompson MA, Vassallo R, Pandya HC, Prakash YS, Pabelick CM. Cigarette smoke enhances proliferation and extracellular matrix deposition by human fetal airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L978–L986, 2014. doi: 10.1152/ajplung.00111.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, Schreiber M, Wagner EF, Nishina H, Penninger JM. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat Cell Biol 6: 215–226, 2004. doi: 10.1038/ncb1098. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Jafri A, Martin RJ, Nnanabu J, Farver C, Prakash YS, MacFarlane PM. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am J Physiol Lung Cell Mol Physiol 307: L295–L301, 2014. doi: 10.1152/ajplung.00208.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West MD, Shay JW, Wright WE, Linskens MH. Altered expression of plasminogen activator and plasminogen activator inhibitor during cellular senescence. Exp Gerontol 31: 175–193, 1996. doi: 10.1016/0531-5565(95)02013-6. [DOI] [PubMed] [Google Scholar]
- 56.White ES. Lung extracellular matrix and fibroblast function. Ann Am Thorac Soc 12, Suppl 1: S30–S33, 2015. doi: 10.1513/AnnalsATS.201406-240MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Z, Wang H, Fang S, Xu C. Roles of endoplasmic reticulum stress and autophagy on H2O2−induced oxidative stress injury in HepG2 cells. Mol Med Rep 18: 4163–4174, 2018. doi: 10.3892/mmr.2018.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xue E, Zhang Y, Song B, Xiao J, Shi Z. Effect of autophagy induced by dexamethasone on senescence in chondrocytes. Mol Med Rep 14: 3037–3044, 2016. doi: 10.3892/mmr.2016.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu S, Wang X, Geng P, Tang X, Xiang L, Lu X, Li J, Ruan Z, Chen J, Xie G, Wang Z, Ou J, Peng Y, Luo X, Zhang X, Dong Y, Pang X, Miao H, Chen H, Liang H. Melatonin regulates PARP1 to control the senescence-associated secretory phenotype (SASP) in human fetal lung fibroblast cells. J Pineal Res 63: e12405, 2017. doi: 10.1111/jpi.12405. [DOI] [PubMed] [Google Scholar]
- 60.Yuzwa SA, Borrett MJ, Innes BT, Voronova A, Ketela T, Kaplan DR, Bader GD, Miller FD. Developmental emergence of adult neural stem cells as revealed by single-cell transcriptional profiling. Cell Rep 21: 3970–3986, 2017. doi: 10.1016/j.celrep.2017.12.017. [DOI] [PubMed] [Google Scholar]