

Abstract
Changes in reactive oxygen species and extracellular matrix seem to participate in pulmonary hypertension development. Because we recently reported evidence for chronic hypoxia decreasing expression of cartilage oligomeric matrix protein (COMP) and evidence for this controlling loss of pulmonary arterial smooth muscle bone morphogenetic protein receptor-2 (BMPR2) and contractile phenotype proteins, we examined if changes in superoxide metabolism could be an important factor in a bovine pulmonary artery (BPA), organoid cultured under hypoxia for 48 h model. Hypoxia (3% O2) caused a depletion of COMP in BPA, but not in bovine coronary arteries. Knockdown of COMP by small-interfering RNA (siRNA) increased BPA levels of mitochondrial and extra-mitochondrial superoxide detected by MitoSOX and dihydroethidium (DHE) HPLC products. COMP siRNA-treated BPA showed reduced levels of SOD2 and SOD3 and increased levels of NADPH oxidases NOX2 and NOX4. Hypoxia increased BPA levels of MitoSOX-detected superoxide and caused changes in NOX2 and SOD2 expression similar to COMP siRNA, and exogenous COMP (0.5 μM) prevented the effects of hypoxia. In the presence of COMP, BMPR2 siRNA-treated BPA showed increases in superoxide detected by MitoSOX and depletion of SOD2. Superoxide scavengers (0.5 μM TEMPO or mitoTEMPO) maintained the expression of contractile phenotype proteins calponin and SM22α decreased by 48 h hypoxia (1% O2). Adenoviral delivery of BMPR2 to rat pulmonary artery smooth muscle cells prevented the depletion of calponin and SM22α by COMP siRNA. Thus, COMP regulation of BMPR2 appears to have an important role in controlling hypoxia-elicited changes in BPA superoxide and its potential regulation of contractile phenotype proteins.
Keywords: extracellular matrix, NADPH oxidases, pulmonary hypertension, smooth muscle phenotype, superoxide dismutase
INTRODUCTION
The extracellular matrix (ECM) is thought to play an important role in the development of pulmonary hypertension (PH; see Ref. 23). Evidence has been emerging suggesting that cartilage oligomeric matrix protein (COMP) is found in the ECM of arteries and that it appears to influence vascular smooth muscle remodeling (9, 25, 26). Our very recent work has detected evidence that arteries from a rat model of hypoxia-induced pulmonary hypertension and cultured pulmonary artery smooth muscle cells exposed to hypoxia show evidence for hypoxia depleting COMP and the loss of COMP promoting a phenotypic shift from a contractile to a synthetic migratory/proliferative phenotype associated with a shift in key contractile marker proteins (26).
It is well established that changes in reactive oxygen species (ROS) are associated with the actions of hypoxia on pulmonary arteries, vascular remodeling, and pulmonary hypertension development (4, 7, 14, 16, 18). For example, vascular tissue from various pulmonary hypertension models have been associated with increased ROS generation by NOX2 (14), NOX4 (16), and mitochondria and with depletion of mitochondrial SOD2 (4) and extracellular SOD3 (18) scavengers of superoxide. However, there is minimal evidence for COMP having an influence on ROS in vascular regulation. There is recent evidence for COMP interacting with vascular integrins (25) and for COMP maintaining expression of bone morphogenetic protein-2 and bone morphogenetic protein receptor-2 (BMPR2; see Refs. 9 and 26). In contrast, there is evidence for both integrins and BMPR2 being important in the modulation of vascular superoxide generation and pulmonary hypertension development (13, 15, 23, 24). Thus, the focus of this study is to examine relationships between hypoxia modulating COMP and COMP potentially influencing remodeling protein changes through alterations in BMPR2 and aspects of ROS metabolism in isolated bovine pulmonary arteries.
MATERIALS AND METHODS
Materials.
All salts used for making physiological solutions were analyzed reagent grade from Baker Chemical. All gases were purchased from Airgas (Bronx, NY). Recombinant human COMP was obtained from Abcam (ab174082). Specific antibodies were purchased from the companies indicated: anti-COMP (ab74524, 1:2,000; Abcam), anti-SOD1 (ab13498, 1:1,000; Abcam), anti-SOD2 (ab13533, 1:10,000; Abcam), anti-SOD3 (07–704, 1:10,000; Upstate Millipore), anti-NOX2 (sc-130543, 1:1,000; Santa Cruz), anti-NOX4 (sc-55142, 1:200; Santa Cruz), anti-SM22α (ab14106, 1:10,000; Abcam), anti-calponin [C2687, 1:5,000 (see Fig. 5) (Sigma) or ab175572, 1:500 (see Fig. 6) (Abcam)], anti-BMPR2 (sc-5682, 1:200; Santa Cruz), VDAC (PA1–954A, 3:1,000; Invitrogen), anti-GAPDH (MAB374, 1:1,000; Millipore Sigma), and anti-β-actin (A5441, 1:10,000; Sigma Chemical). The ability of these antibodies to detect the proteins studied in the bovine species for the molecular weights indicated have been verified by the supplier and/or by multiple reports in the scientific literature, including data included in our previous studies (1–3, 20). In addition, the COMP and BMPR2 antibodies are also verified by small-interfering RNA (siRNA) data and by conformation of previous observations (26) reporting their depletion in pulmonary arteries by hypoxia that is included in this study. Superoxide scavengers and detection probes were purchased from the companies indicated: mitoTEMPO (ALX-430–150-M005; Enzo), TEMPO (214000; Sigma-Aldrich), MitoSOX (M36008; Life Technologies) and dihydroethidium (DHE, D11347; Life Technologies), SOD from bovine blood (S7571; Sigma), and gp91ds-tat (NH2-RKKRRQRRRCSTRIRRQL-COOH; Tufts University Peptide Synthesis Core Facility).
Fig. 5.

Effects of scavengers of mitochondrial (mitoTEMPO) or extra-mitochondrial (TEMPO) superoxide on the hypoxia-elicited decreases in the levels of SM22α and calponin. Representative Western blot bands and quantitative analysis of cartilage oligomeric matrix protein (COMP) level under 1% and 3% O2 (*P < 0.05, one-way ANOVA with Newman-Keuls correction, n = 5) (A) and calponin (n = 8) and SM22α (n = 9) under 1% O2 (*P < 0.05, **P < 0.01, one-way ANOVA with Newman Keuls correction) (B). CTL, normoxia; H48, hypoxia for 48 h; T, 0.5 μM TEMPO under normoxia; H48 + T, 1% O2 for 48 h in the presence of 0.5 μM TEMPO; MT, 0.5 μM mitoTEMPO under normoxia; H48 + MT, 1% O2 for 48 h in the presence of 0.5 μM mitoTEMPO.
Fig. 6.

Effects of bone morphogenetic protein receptor-2 (BMPR2) on the expression of contractile phenotype proteins associated with the actions of cartilage oligomeric matrix protein (COMP) under normoxia. Representative Western blot bands and quantitative analysis of BMPR2 in bovine pulmonary artery (BPA) cultured under 3% O2 for 48 h (*P < 0.05, **P < 0.01, two-way ANOVA with Sidak correction, n = 6) (A); BMPR2 and calponin in BPA treated with scramble siRNA, BMPR2 siRNA, or BMPR2 siRNA plus COMP (*P < 0.05, one-way ANOVA with Newman Keuls correction, n = 6) (B); and the levels of BMPR2, SM22α, and calponin in cultured pulmonary artery smooth muscle cells (PASMCs) derived from male rats treated with adenovirus-vector (Ad-V or V) or adenovirus-BMPR2 (Ad-B or B) without or with scramble siRNA (S) or COMP siRNA (Ci) [*P < 0.05, **P < 0.01, ***P < 0.001, BMPR2: (n = 3) one-way ANOVA with Newman Keuls correction and SM22α (n = 5) or calponin (n = 5): two-way ANOVA with Sidak correction] (C). CTL, normoxia; H48, hypoxia for 48 h; H48 + C, 1% O2 for 48 h in the presence of 0.5 μM COMP; Bi, BMPR2 siRNA; Bi + C, COMP was added to the culture media for 24 h after BMPR2 siRNA treatment for 24 h; Ad-V, treatment with 1.6 × 10−5 plaque-forming units (PFU) of adenovirus-vector for 48 h; Ad-B, treatment with 1.2 × 10−5 PFU of adenovirus-BMPR2 for 48 h; S + Ad-V/Ad-B, treatment with scramble siRNA for 24 h and then adding Ad-V/Ad-B for another 48 h; Ci + Ad-V/Ad-B, treatment with COMP siRNA for 24 h and then adding Ad-V/Ad-B for another 48 h).
Animal handling and exposure to chronic hypoxia.
Male C57BL/6J mice (8–10 wk) were purchased from Jackson Laboratories (Bar Harbor, ME). All animal protocols were approved by the Institutional Animal Care and Use Committee at New York Medical College. Mice were either exposed to normoxic (21% O2) or normobaric (10% O2) hypoxic conditions for 21 days in a hypoxic in vivo cabinet (Coy Laboratories, Grass Lake, MI) setup for measuring O2 and recording time-dependent changes (3, 21). After 21 days, lungs were isolated from each mouse after they were anesthetized with 50 mg/kg pentobarbital sodium. Pulmonary arteries were carefully isolated from each mouse in ice-cold buffer and then frozen in liquid nitrogen. Frozen lungs of mice were pulverized and then homogenized in lysis buffer containing protease and phosphatase inhibitors, as previously described (22). Bradford method was used for protein quantification assay, and samples were prepared for gel electrophoresis.
Tissue preparations.
Bovine lungs and hearts were obtained from a slaughterhouse in ice-cold phosphate buffer saline and prepared as previously described (10, 11, 17). Although the gender of these animals is not known, the animals are thought to be predominately male. Left anterior descending and left circumflex bovine coronary arteries (BCA) and the second or third main branches of bovine pulmonary arteries (BPA) were used in experiments. BCA and BPA were cleaned of their connective tissue and then cut into rings of 2–3 mm in diameter and width. The endothelium was removed by rubbing the lumen. Organoid-cultured BCA were used in experiments of Western blot protein analysis. BPA rings were used in experiments for superoxide measurements and Western blot protein analysis. As indicated in results, BPA rings were organoid cultured in the absence and presence of hypoxia (3 or 1% O2) in a COY chamber (Coy Laboratory Products), in the absence or presence of agents including 0.5 μM COMP, 1 μM SOD, 1 μM catalase, 50 μM gp91ds-tat, 0.5 μM mitoTEMPO, and 0.5 μM TEMPO with Dulbecco's modified Eagle medium containing 10% fetal bovine serum and 1% antibiotics (penicillin, streptomycin, and amphotericin B) for 48 h at 37°C with 5% CO2. Studies initially used 3% O2 based on our previous study in cultured rat pulmonary artery cells (26). In some experiments, 1% O2 was then used for hypoxia to obtain more robust responses.
Detection of changes in mitochondrial and extra-mitochondrial superoxide.
HPLC measurement of the superoxide-specific hydroxylated products of MitoSOX and DHE were employed for quantifying changes in mitochondrial matrix and extra-mitochondrial matrix superoxide, using previously described methods (3). At the beginning of the experiment, a standard curve was generated by injecting increasing concentrations of mito-2-hydroxyethidium or 2-hydroxyethidium in the column. After 48 h of organoid culture, BPA rings were incubated with either 5 μM MitoSOX or DHE for 1 h in the dark to measure mitochondrial and extra-mitochondrial superoxide, respectively. They were washed several times with Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4) and then flash-frozen with liquid nitrogen. Tissues were first weighed and then pulverized in the presence of liquid nitrogen, dissolved in a solution of 100% acetonitrile (HPLC grade). These samples were incubated at −20°C for 1 h. After 1 h samples were centrifuged, and the supernatant was used for HPLC analysis of the superoxide-specific hydroxylated product of MitoSOX (mito-2-hydroxyethidium) or of DHE (2-hydroxyethidium) (27) using an HPLC system with a Jasco FP-1520 fluorescence detector and a Beckman ultrasphere reverse-phase column (C18, 5 μm, 250 × 4.6 mm; see Ref. 3).
Western blot analysis.
Frozen BPA or BCA were pulverized and then homogenized in lysis buffer containing protease and phosphatase inhibitors, as previously described (17). Bradford method was used to assay protein quantification, and samples were prepared for gel electrophoresis. Proteins were separated using a 10% SDS-polyacrylamide gel under reducing and denaturing conditions. Gels were transferred to polyvinylidene difluoride membranes, and the membranes were blocked with Tris-buffered saline with Tween 20 + 5% milk for 1 h. After this the membranes were exposed to primary and secondary antibodies as per the manufacturer’s protocol. Protein bands were visualized with an enhanced chemiluminescence kit (Pierce, Rockford, IL) on X-OMAT autoradiography paper (Kodak, Rochester, NY) in a dark room. Protein levels were measured using densitometry analysis with the UN-SCAN-IT gel software by Silk Scientific (Orem, UT). Molecular mass (kDa) of different proteins are as follows: COMP, 72; SOD1, 18; SOD2, 25; SOD3, 45; NOX2, 61; NOX4, 70; calponin, 35; SM22α, 22, BMPR2, 115; GAPDH, 38; and β-actin, 42. For detection of changes in the expression of mitochondrial proteins (SOD2), the mitochondrial protein VDAC (32 kDa) was used as a loading control. For detection of changes in the expression of COMP, SOD1, NOX2, NOX4, calponin, SM22α, and BMPR2, β-actin was used as a loading control. GAPDH was used as a loading control for SOD3. The expression of these loading controls was not altered under the conditions examined in results.
Transfection of siRNA.
siRNA was purchased from GenePharma (Shanghai). The sequences employed for the siRNA of COMP were sense, 5′-AGAAACUUGAGCUGUGUUGAUGCC-3′, and anti-sense, 5′- GGCUAUCAAGACAGCUCAAGUUUCU-3′. The sequences employed for the siRNA of BMPR2 were sense, 5′-GCUUGUGAUGGAGUAUUAUTT-3′, and anti-sense, 5′-AUAAUACUCCAUCACAAGCTT-3′. A scramble stealth RNAi duplex served as a negative control. The siRNA sequences used for BPA were based on previously designed sequences for knockdown of COMP in mice (26) because the siRNA for the equivalent bovine COMP mRNA sequence was the same as the mouse COMP sequence. In vitro siRNA transfection (50 pM) of organoid-cultured BPA was performed using RNAi MAX (Invitrogen). The transfection procedures followed the manufacturers' instructions, and data in results confirm that the siRNA sequences from mice successfully knock down the levels of COMP and the siRNA sequences from bovine successfully knock down the levels of BMPR2 in BPA.
Recombinant adenovirus construction and infection.
The adenovirus for BMPR2 (Ad-BMPR2; NM_080407) was constructed and amplified by GENECHEM. An adenovirus (CMV-MCS-3FLAG-SV40-EGFP, Ad-vector) was used as a negative control. Pulmonary artery smooth muscle cells originating from male rats (26) cultured at ~80% confluency were infected with adenovirus for 48 h or transfected with siRNA for 24 h and then adding Ad-Vector/Ad-BMPRR2 for another 48 h.
Statistical analysis.
Statistical analysis was performed with GraphPad Prism 5 software. All values are expressed as means ± SE, with the exception of data reported in a box-and-whisker plot format. Statistical analyses between two groups were performed with Student’s t test or paired t test, and a two-way ANOVA or one-way ANOVA was used for comparison between multiple groups. Paired t tests or 2-way ANOVA were used to analyze data from vascular segments from the same animal that were run together in the same experimental analysis, and this helped compensate for variability in some of the measurements, which showed consistent patterns of changes. A value of P < 0.05 was used to establish statistical significance.
RESULTS
Hypoxia decreased the level of COMP in the lungs of mice and BPA.
To investigate the role of COMP in hypoxia-induced pulmonary hypertension, we first examined the level of COMP in the lungs of a chronic hypoxia-induced PH mouse model to confirm observations previously described in rats exposed to chronic hypoxia (26). Because COMP in lungs is mainly expressed in the medial layer of pulmonary arteries (26), changes detected in the expression levels of COMP in lungs should reflect changes in this vascular segment. As shown in Fig. 1A, the protein level of COMP significantly decreased (P < 0.01) in the chronic hypoxia mouse PH model compared with mice exposed to normoxia. Organoid-cultured bovine arteries were then used to examine the effects of hypoxia (3% O2) on the level of COMP in BPA (Fig. 1B) and circumflex (Fig. 1C) and left anterior descending (Fig. 1D) BCA. The data show that the level of COMP decreased in BPA (P < 0.05, Fig. 1B) and not in the BCA segments studied (Fig. 1, C and D). These results provide evidence that COMP is normally expressed in BPA and that it is lowered by hypoxia under both in vitro and in vivo conditions.
Fig. 1.

Effects of hypoxia on the level of cartilage oligomeric matrix protein (COMP) in the lungs of mice and organoid-cultured bovine arteries. Representative Western blot bands and quantitative analysis of COMP protein in the lungs from 3 wk chronic hypoxia (10% O2)-exposed male mice with pulmonary hypertension (PH) and control normoxic (21% O2) mice (**P < 0.01, PH mice versus control mice, Student’s t test, n = 5) (A); and in bovine pulmonary artery (BPA) organoid cultured under 3% or 21% O2 for 48 h (*P < 0 0.05, 3% versus 21% O2, paired t test, n = 5 (B); and in left anterior descending (paired t test, n = 5) (C); and left circumflex coronary arteries (n = 7) organoid cultured under 3% vs. 21% O2 (paired t test, n = 7) (D).
COMP attenuated the increased level of MitoSOX induced by hypoxia in BPA.
We tried to identify aspects of the relationship between COMP and the level of superoxide in smooth muscle using endothelium-denuded BPA. The picomole per milligram tissue levels of superoxide-derived specific hydroxylated products of MitoSOX and DHE (27) were measured by HPLC (3). The data showed that the level of superoxide detected by MitoSOX was elevated in BPA after organoid culture under hypoxia (3% O2) for 48 h (P < 0.05), and this was prevented by the presence of exogenous COMP (P < 0.05, Fig. 2A). Whereas the hypoxia group showed evidence of variability in the levels of mitochondrial superoxide detected by MitoSOX, the level increased in pulmonary arteries from 9 of 10 animals studied compared with controls from the same vascular segment studied. There was no increase in the detection of superoxide by DHE after culture of BPA under hypoxia for 48 h. Knockdown of COMP with siRNA (Fig. 2B) significantly increased the level of superoxide detected by MitoSOX and DHE (P < 0.01, Fig. 2C).
Fig. 2.

Effect of hypoxia and cartilage oligomeric matrix protein (COMP) on the levels of mitochondrial matrix and extra-mitochondrial matrix superoxide detected by MitoSOX and dihydroethidium (DHE), respectively. A: level of superoxide-specific metabolites of MitoSOX and DHE detected by HPLC in bovine pulmonary artery (BPA) cultured under 21 or 3% O2 for 48 h in the absence or presence of COMP. **P < 0.01 (two-way ANOVA with Sidak correction, n = 10; A). B: COMP protein level deceased by COMP siRNA (*P < 0.05, paired t test, n = 5; B). C: level of MitoSOX (n = 14) and DHE (n = 8) superoxide detected by HPLC in BPA treated with COMP siRNA or scramble siRNA (*P < 0.05, **P < 0.01 for COMP siRNA versus scramble siRNA, paired t test).
Effects of hypoxia and COMP on the expression of SOD family members.
To clarify the mechanism of increases in superoxide detection by MitoSOX and DHE levels caused by COMP-siRNA, we measured the expression levels of the forms of SOD known to be present in vascular tissue. Organoid culture of BPA under 3% O2 for 48 h did not alter the level of SOD1 (Fig. 3A), but it significantly decreased the expression of SOD2 (P < 0.05, Fig. 3B). Supplement of exogenous COMP reversed the decreased level of SOD2 (P < 0.05, Fig. 3B). Moreover, treatment of BPAs with COMP siRNA decreased both SOD2 and SOD3 (P < 0.05, Fig. 3D), and it was confirmed that depletion of COMP did not influence expression of the mitochondrial marker VDAC normalized to β-actin (data not shown).
Fig. 3.

Effects of hypoxia and cartilage oligomeric matrix protein (COMP) on the expression of the different forms of SOD present in bovine pulmonary artery (BPA). The level of individual forms of SOD measured by Western blotting in BPA cultured under 21% (CTL) or 3% O2 for 48 h (H48) in the absence and presence of COMP (C, 0.5 μM) is shown. A–C: representative Western blot bands and quantitative analysis of Cu,Zn-SOD1 (A) (n = 8), mitochondrial SOD2 (B) (n = 6), and extracellular SOD3 (C) (n = 6). *P < 0.05, two-way ANOVA with Sidak correction. D: representative Western blot bands and quantitative analysis showing the effect of knockdown of COMP with siRNA decreasing protein levels of SOD2 (n = 9) and SOD3 (n = 7). *P < 0.05, COMP-siRNA vs. scramble siRNA, paired t test. (H48, 3% O2 for 48 h; H48 + C, 3% O2 for 48 h and treated with 0.5 μM COMP at the same time; COMPi, siRNA-COMP.) B and C were edited between the 2nd and 3rd channels that are shown separated by a line to remove two additional channel bands from the original imaging capture of the same membrane.
Effects of hypoxia and COMP on the expression of NOX2 and NOX4.
We also measured the levels of NOX enzymes previously reported (NOX2 and NOX4; see Ref. 11) to be expressed in BPA to clarify the mechanisms causing changes in the detection of superoxide-derived products of MitoSOX and DHE increased by COMP-siRNA. Hypoxia (3% O2) for 48 h significantly increased NOX2 (P < 0.05, Fig. 4A). The increase was reversed by exogenous COMP. There was no significant increase of NOX4 after culture of BPA under hypoxia for 48 h. Under aerobic conditions, exogenous COMP altered the expression of NOX2 (P < 0.001) but not NOX4 (Fig. 4, A and B). Compared with scramble siRNA, COMP-siRNA increased both NOX2 (P < 0.001) and NOX4 (P < 0.01, Fig. 4C).
Fig. 4.

Effects of hypoxia and cartilage oligomeric matrix protein (COMP) on the expression of NADPH oxidases NOX2 and NOX4 detected by Western blotting. Bovine pulmonary artery (BPA) were cultured under 21% (CTL) or 3% O2 (H48) for 48 h in the absence and presence of COMP (0.5 μM). A and B: representative Western blot bands and quantitative analysis of changes in NOX2 (A) and NOX4 (B). *P < 0.05, two-way ANOVA with Sidak correction, n = 7. C: effects of siRNA knockdown of COMP on detection of NOX2 (n = 8) and NOX4 (n = 6) protein levels. Representative Western blot bands and quantitative analysis of NOX2 and NOX4. **P < 0.01, ***P < 0.001, COMP siRNA vs. scramble siRNA, paired t test. (H48 + C, 3% O2 for 48 h and treated with 0.5 μM COMP at the same time; COMPi, COMP siRNA.)
Treatments decreasing superoxide in mitochondrial or extra-mitochondrial regions of BPA attenuated changes in SM22α and calponin levels elicited by hypoxia.
The above data showed that hypoxia decreased the expression of COMP, and the deficiency of COMP led to the increased level of superoxide. Our previous study reported that the deficiency of COMP in pulmonary artery smooth muscle cells led to the decrease of smooth muscle contractile phenotype proteins (26). Here, we tried to find relationships between superoxide in mitochondrial or extra-mitochondrial regions of BPA and changes in smooth muscle contractile phenotype protein expression by employing the superoxide scavengers mitoTEMPO (0.5 μM) or TEMPO (0.5 μM), respectively (8). As shown in Fig. 5A, 1% O2 significantly enhanced the decrease in COMP compared with the decrease seen with 3% O2. Thus, organoid culture of BPA under hypoxia (1% O2) was used to examine the effects of a more robust decrease in COMP, and this was associated with significant decrease (Fig. 5B) in SM22α and calponin of the levels of these proteins observed under the control conditions of 21% O2. Under this more severe hypoxic condition, the presence of the superoxide scavenger mitoTEMPO or TEMPO increased expression of calponin and SM22α under hypoxia to levels that did not differ from those seen in the absence of hypoxia (Fig. 5B).
Effects of BMPR2 on the expression of contractile phenotype proteins associated with the actions of COMP under normoxia.
Our previous study reported that the deficiency of COMP in pulmonary artery smooth muscle cells led to the decrease of BMPR2, and overexpression of COMP reversed the reduced level of BMPR2 induced by hypoxia (26). As shown in Fig. 6A, 3% O2 significantly reduced BMPR2 protein levels in BPA, which could be restored by exogenous COMP. Here, we tried to find relationships between BMPR2 and the expression changes of calponin and SM22α, contractile phenotype proteins controlled by COMP (26), by employing the BMPR2 siRNA to deplete BMPR2 (Fig. 6B) or adenovirus-BMPR2 (Fig. 6C). Knockdown of BMPR2 by siRNA under aerobic conditions seemed to reduce the protein level of calponin (Fig. 6B). Exogenous COMP did not restore the decreased calponin induced by BMPR2 siRNA (Fig. 6B). Overexpression of BMPR2 by adenovirus under aerobic conditions reversed the levels of calponin and SM22α reduced by COMP siRNA in rat pulmonary artery smooth muscle cells (Fig. 6C).
Influence of BMPR2 in maintaining decreased superoxide levels.
We further tried to find relationships between BMPR2 and the changes in smooth muscle superoxide. Knockdown of BMPR2 by siRNA increased mitochondrial matrix and extra-mitochondrial matrix superoxide (P < 0.05 or 0.01) and exogenous COMP did not prevent detection of the increased superoxide observed in BPA treated with siRNA to deplete BMPR2 (Fig. 7A). Knockdown of BMPR2 by siRNA decreased SOD2 protein level, which could not be improved by exogenous COMP (P < 0.05, Fig. 7B). The depletion of BMPR2 did not alter the expression of SOD1, SOD3, NOX2, or VDAC (Fig. 7B). In contrast, COMP decreased the level of NOX2 under normoxia in arteries treated with scramble siRNA compared with the scramble siRNA group not treated with COMP (by 51.6%, P < 0.05, n = 5, data not shown) in a manner similar to measurements shown in Fig. 3A made in the absence of scramble siRNA. Thus, siRNA depletion of BMPR2 appears to prevent detection of COMP decreasing NOX2 expression. As shown in Fig. 7C, 3% O2 significantly reduced BMPR2 protein levels in BPA, which could be restored by exogenous SOD and gp91 ds-tat but not by catalase.
Fig. 7.

Effects of bone morphogenetic protein receptor-2 (BMPR2) on the detection of superoxide and levels of SOD and NADPH oxidase NOX2. A: levels of MitoSOX (n = 10) and dihydroethidium (DHE, n = 7)-detected superoxide by HPLC analysis in bovine pulmonary artery (BPA) treated with scramble siRNA, BMPR2 siRNA, or BMPR2 siRNA plus cartilage oligomeric matrix protein (COMP). *P < 0.05, **P < 0.01, analysis by one-way ANOVA with Newman-Keuls correction. B: representative Western blot bands and quantitative analysis of SOD1, SOD2, SOD3, and NOX2 in BPA treated with scramble siRNA, BMPR2 siRNA, or BMPR2 siRNA plus COMP. *P < 0.05, analysis of one-way ANOVA with Newman-Keuls correction, n = 6. C: representative Western bands and quantitative analysis of BMPR2 in BPA cultured under 3% O2 for 48 h treated with or without catalase, SOD, and gp91 ds-tat. *P < 0.05, analysis of one-way ANOVA with Newman-Keuls correction, n = 8. Bi, BMPR2 siRNA; Bi + C, COMP added to the culture media for 24 h after BMPR2 siRNA treatment for 24 h; CTL, normoxia; H48, hypoxia for 48 h; H-cat, 3% O2 for 48 h in the presence of 1 μM catalase; H-SOD, 3% O2 for 48 h in the presence of 1 μM SOD; H-gp91, 3% O2 for 48 h in the presence of 50 μM gp91 ds-tat. Note that the box-and-whisker format of the SOD data in B shows the median, upper, and lower quartile box and extreme data points.
DISCUSSION
Data in the present study showed that COMP was decreased in lungs from mice exposed to 3 wk of hypoxia and bovine pulmonary artery organoid cultured under hypoxia for 48 h, which extend our previous observations made in rat preparations documenting that hypoxia lowers the expression of COMP in pulmonary arterial smooth muscle cells. A key set of new observations reported in this study support COMP normally functioning to maintain low levels of superoxide by a combination of suppressing the expression of NOX2 and by maintaining mitochondrial SOD2 expression. Data in the study also support COMP potentially functioning to attenuate aspects of pulmonary arterial smooth muscle dedifferentiation by processes functioning to attenuate increases in vascular mitochondrial and extra-mitochondrial sources of superoxide, and a potential role for BMPR2 functioning to maintain the expression of SOD2 in controlling these processes. Potential relationships between data reported in this study and hypothesized mechanisms considered in this discussion are included in Fig. 8.
Fig. 8.
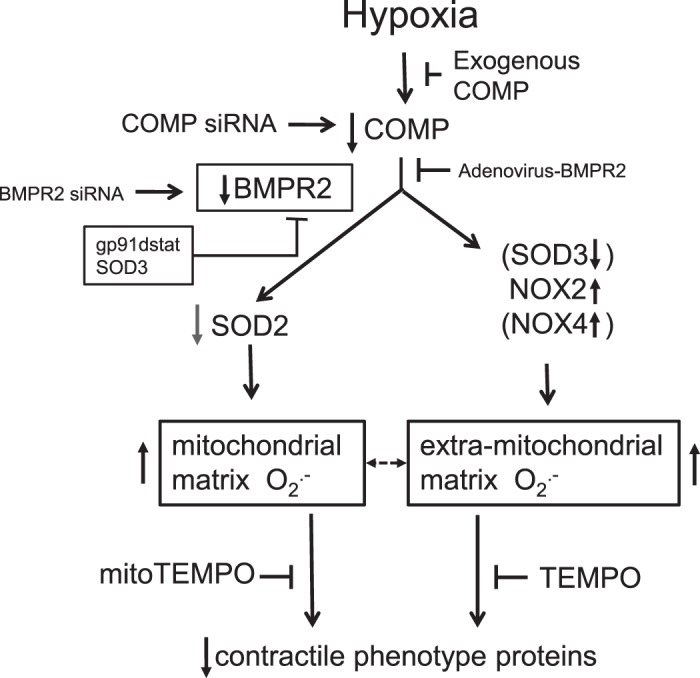
Model showing some of the hypothesized actions resulting from a depletion of cartilage oligomeric matrix protein (COMP) from bovine pulmonary smooth muscle resulting from organoid culture under hypoxia for 48 h that are potentially contributing to the observed increases in mitochondrial and extra-mitochondrial matrix in superoxide that seem to participate in a depletion of contractile phenotype proteins based on the actions of siRNA depletion of COMP and attenuation of the actions of hypoxia by treatment with COMP. COMP depletion promotes decreases in SOD2 and increases in NADPH oxidase NOX2, associated with increases in mitochondrial superoxide (detected by MitoSOX) and extra-mitochondrial superoxide [detected by dihydroethidium (DHE)]. The actions of superoxide scavengers TEMPO and mitoTEMPO on inhibiting the effects of hypoxia suggest roles for increases in the different sources of superoxide contribute to depleting contractile phenotype protein levels. Depletion of bone morphogenetic protein receptor-2 (BMPR2) by siRNA promoting decreased levels of calponin and adenovirus-BMPR2 reversal of reduced calponin and SM22α caused by COMP siRNA indicate COMP regulation of BMPR2 expression is potentially a key process maintaining the contractile phenotype. Moreover, BMPR2 siRNA increases superoxide (as supported by detection of MitoSOX and DHE) and promotes contractile protein depletion in bovine pulmonary artery, which cannot be prevented by exogenous COMP. Thus, protective effects of COMP on the contractile phenotype of pulmonary artery smooth muscle cells (PASMCs) appear to be via maintenance of BMPR2 signaling. SOD3 and NOX4 are shown in parentheses because changes in the expression of these proteins in pulmonary arteries observed with siRNA depletion of COMP or by more prolonged in vivo exposure of animals to hypoxia reported in the literature were not observed in the 48-h organoid culture hypoxia model used in the present study.
The data in the present study in isolated BPA organoid cultured under hypoxia for 48 h, our recently published observations in cultured rat pulmonary artery smooth muscle cells, and in vivo chronic hypoxia studies in rats (26) and mice (Fig. 1A) all provide evidence for hypoxia decreasing the expression of COMP in the pulmonary vasculature (Fig. 1B). Interestingly, hypoxia did not cause a detectable decrease in the expression of COMP in isolated circumflex (Fig. 1C) and left anterior descending (Fig. 1D) BCA. Although the reasons for differences in the effects of hypoxia on the depletion of COMP in pulmonary versus coronary arterial smooth muscle are not known, there is substantial evidence for major differences in the effects of hypoxia in these vascular segments. For example, acute hypoxia causes contraction of BPA and relaxation of BCA in a manner associated with mechanisms linked to differences in changes in intracellular calcium, cytosolic NADPH redox, levels of reactive oxygen species, and the function of signaling by protein kinase G (10, 11, 17). Thus, differences in one or more of these regulatory processes could be controlling the detected changes in COMP expression.
The consequences of COMP depletion by siRNA on detecting increases of superoxide in mitochondrial and extra-mitochondrial matrix regions of BPA, and effects on the expression of NADPH oxidases and the various forms of SOD, all suggest that COMP has a major influence on reactive oxygen species and redox signaling associated with changes in these species. For example, increased expression of NOX2 is likely to increase superoxide and hydrogen peroxide in cytosolic and extracellular regions. The detected decreases in SOD2 should favor detection of increased mitochondrial superoxide, which could be associated with a somewhat diminished conversion to hydrogen peroxide. These changes could potentially influence signaling mechanisms controlling aspects of pulmonary arterial smooth muscle phenotype by processes such as decreasing the protective effects (6) of regulation by protein kinase G and enhancing pulmonary hypertension-promoting signaling mechanisms such as Rho kinase (5, 19).
Consideration of the consequences of changes in aspects of superoxide metabolism across all of the experiments in this study may provide new insight into regulatory processes linking COMP and hypoxia to controlling phenotype-associated changes in the expression of contractile proteins. Hypoxia caused prominent increases in mitochondrial matrix superoxide and decreases in SOD2 that were prevented by the presence of COMP. Treatment with COMP seemed to deplete NOX2 both in the absence and presence of hypoxia. Hypoxia also appeared to increase the activation of NOX2 and extracellular superoxide based on the inhibitory actions of gp91 ds-tat and addition of SOD to the extracellular incubation media on the depletion of BMPR2 by hypoxia, and this source of extracellular superoxide might not be detected by the HPLC method used with DHE. Regulatory interactions between mitochondrial and NOX-generated oxidant species (8) could also be a factor with differences in the magnitudes of detected changes in subcellular sources of superoxide. Organoid culture with siRNA for COMP in the absence of hypoxia caused increases in NOX2 and NOX4 and decreases in SOD2 and SOD3 resembling the pattern of the effects of organoid culture of BPA with hypoxia for 48 h. In contrast, the increases in extra-mitochondrial superoxide with COMP siRNA treatment seemed more prominent than increases in mitochondrial matrix superoxide, which differs from the pattern seen with hypoxia. Thus, hypoxia appears to have effects that go beyond depleting COMP in influencing the levels of superoxide and its potential impact on phenotype-shifting processes. Because superoxide scavenger TEMPO and mitoTEMPO had significant effects in reversing the depletion of SM22α and calponin caused by hypoxia, increased superoxide and/or decreased peroxide derived from superoxide may be an important factor in controlling signaling systems regulating the depletion of these proteins.
The BMPR2 receptor has been suggested to be a component of the signaling effects of COMP (9, 26). In addition, human genetic mutations in BMPR2 and depletion of BMPR2 in animal models have been associated with increased superoxide and/or oxidant stress in multiple systems (12). Interestingly, the pattern of changes in superoxide with COMP depletion resembled some of the patterns seen with BMPR2 depletion with siRNA, with increases in extra-mitochondrial superoxide being greater in magnitude than mitochondrial matrix increases. Data in Fig. 7 showing that COMP did not significantly inhibit the increases in either source of superoxide observed in BMPR2 siRNA-treated BPA suggest that maintaining BMPR2 receptor signaling may be potentially a system through which COMP functions to lower superoxide in BPA. Although siRNA for both COMP and BMPR2 depleted mitochondrial SOD2, only COMP appeared to have effects on the expression of NOX2. However, the lowering of NOX2 expression by COMP appeared to depend on the expression of BMPR2. Thus, there may be differences in processes regulated by COMP and BMPR2 that need to be further clarified.
The observed decrease in COMP resulting from organoid culture of BPA under hypoxia was associated with detecting many of the changes observed with siRNA depletion of COMP. For example, both treatments promoted the detection of increased mitochondrial superoxide, increased NOX2, and decreased SOD2. Moreover, the presence of added COMP to the organoid culture of BPA under hypoxia essentially prevented detection of these effects of hypoxia, suggesting that COMP depletion by hypoxia was contributing to causing the increases in superoxide through processes such as increasing NOX2 and decreasing SOD2. However, hypoxia also appears to influence other processes that shift changes in the magnitude of superoxide to favor the mitochondrial increases, and perhaps the extracellular generation of superoxide by NOX2. Because the effects of hypoxia are associated with SOD2 depletion, and decreased extra-mitochondrial peroxide is thought to be a driving factor in pulmonary arterial smooth muscle phenotype shifting (4), COMP could also be functioning to attenuate the loss of contractile proteins through increasing extra-mitochondrial hydrogen peroxide. Previous studies in the mouse chronic hypoxia model of pulmonary hypertension and other models of this disease process have documented evidence for increases in mitochondrial superoxide, and NOX2 and NOX4 expression, and decreases in mitochondrial SOD2 and extracellular SOD3 expression (4, 14, 16, 18), suggesting decreases in COMP expression in vivo could have pulmonary hypertension development-associated roles in changing the function of these systems. Thus, therapeutic approaches to increase COMP in the pulmonary vasculature could potentially be beneficial in treating aspects of the progression of pulmonary hypertension. Whereas COMP shows patterns of effects suggesting it could function to promote mechanisms that are protective against pulmonary hypertension, further studies are needed to determine its role in this disease process and in forms of pulmonary hypertension that are promoted by factors other than hypoxia.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-115124 and HL-12979 and by grants from the Natural Science Foundation of the People’s Republic of China (31400989) and the Natural Science (C2015069) and Postdoctoral Science (LBHQ15087) Foundation of Heilongjiang Province.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.Y. and M.S.W. conceived and designed research; H.Y., N.A., B.H., M.R.K., and B.Z. performed experiments; H.Y., N.A., B.H., B.Z., D.S., and M.S.W. analyzed data; H.Y., N.A., D.S., and M.S.W. interpreted results of experiments; H.Y. and D.S. prepared figures; H.Y. and M.S.W. drafted manuscript; H.Y., N.A., D.S., and M.S.W. edited and revised manuscript; H.Y., N.A., B.H., M.R.K., B.Z., D.S., and M.S.W. approved final version of manuscript.
REFERENCES
- 1.Ahmad M, Kelly MR, Zhao X, Kandhi S, Wolin MS. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol 298: H1879–H1888, 2010. doi: 10.1152/ajpheart.01228.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad M, Zhao X, Kelly MR, Kandhi S, Perez O, Abraham NG, Wolin MS. Heme oxygenase-1 induction modulates hypoxic pulmonary vasoconstriction through upregulation of ecSOD. Am J Physiol Heart Circ Physiol 297: H1453–H1461, 2009. doi: 10.1152/ajpheart.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alhawaj R, Patel D, Kelly MR, Sun D, Wolin MS. Heme biosynthesis modulation via δ-aminolevulinic acid administration attenuates chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L719–L728, 2015. doi: 10.1152/ajplung.00155.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thébaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010. doi: 10.1152/ajplung.00276.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012. doi: 10.1152/ajplung.00002.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009. doi: 10.1152/ajplung.90568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Y, Wang Y, Wang L, Liu B, Tian Q, Liu CJ, Zhang T, Xu Q, Zhu Y, Ake O, Qi Y, Tang C, Kong W, Wang X. Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ Res 108: 917–928, 2011. doi: 10.1161/CIRCRESAHA.110.234328. [DOI] [PubMed] [Google Scholar]
- 10.Gao Q, Wolin MS. Effects of hypoxia on relationships between cytosolic and mitochondrial NAD(P)H redox and superoxide generation in coronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 295: H978–H989, 2008. doi: 10.1152/ajpheart.00316.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 12.Lane KL, Talati M, Austin E, Hemnes AR, Johnson JA, Fessel JP, Blackwell T, Mernaugh RL, Robinson L, Fike C, Roberts LJ II, West J. Oxidative injury is a common consequence of BMPR2 mutations. Pulm Circ 1: 72–83, 2011. doi: 10.4103/2045-8932.78107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KM, Tsai KY, Wang N, Ingber DE. Extracellular matrix and pulmonary hypertension: control of vascular smooth muscle cell contractility. Am J Physiol Heart Circ Physiol 274: H76–H82, 1998. doi: 10.1152/ajpheart.1998.274.1.H76. [DOI] [PubMed] [Google Scholar]
- 14.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 15.Madden JA, Christman NJ. Integrin signaling, free radicals, and tyrosine kinase mediate flow constriction in isolated cerebral arteries. Am J Physiol Heart Circ Physiol 277: H2264–H2271, 1999. doi: 10.1152/ajpheart.1999.277.6.H2264. [DOI] [PubMed] [Google Scholar]
- 16.Mittal M, Roth M, König P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hänze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101: 258–267, 2007. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 17.Neo BH, Kandhi S, Wolin MS. Roles for redox mechanisms controlling protein kinase G in pulmonary and coronary artery responses to hypoxia. Am J Physiol Heart Circ Physiol 301: H2295–H2304, 2011. doi: 10.1152/ajpheart.00624.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008. doi: 10.1152/ajplung.90293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 20.Patel D, Alhawaj R, Kelly MR, Accarino JJ, Lakhkar A, Gupte SA, Sun D, Wolin MS. Potential role of mitochondrial superoxide decreasing ferrochelatase and heme in coronary artery soluble guanylate cyclase depletion by angiotensin II. Am J Physiol Heart Circ Physiol 310: H1439–H1447, 2016. doi: 10.1152/ajpheart.00859.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel D, Alhawaj R, Wolin MS. Exposure of mice to chronic hypoxia attenuates pulmonary arterial contractile responses to acute hypoxia by increases in extracellular hydrogen peroxide. Am J Physiol Regul Integr Comp Physiol 307: R426–R433, 2014. doi: 10.1152/ajpregu.00257.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel D, Kandhi S, Kelly M, Neo BH, Wolin MS. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G 1α. Am J Physiol Lung Cell Mol Physiol 306: L383–L391, 2014. doi: 10.1152/ajplung.00301.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, Appleby S, Shanahan CM, Bloch KD, Pepke-Zaba J, Upton P, Morrell NW. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med 192: 859–872, 2015. doi: 10.1164/rccm.201408-1509OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu CJ, Zhu Y, Gao Y, Xu Q, Kong W, Wang X. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res 106: 514–525, 2010. doi: 10.1161/CIRCRESAHA.109.202762. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Jia Q, Feng X, Chen H, Wang L, Ni X, Kong W. Hypoxia decrease expression of cartilage oligomeric matrix protein to promote phenotype switching of pulmonary arterial smooth muscle cells. Int J Biochem Cell Biol 91, Pt A: 37–44, 2017. doi: 10.1016/j.biocel.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 27.Zielonka J, Hardy M, Kalyanaraman B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: ramifications in superoxide measurements. Free Radic Biol Med 46: 329–338, 2009. doi: 10.1016/j.freeradbiomed.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]