

Abstract
Cell death is a fundamental process in cardiac pathologies. Recent studies have revealed multiple forms of cell death, and several of them have been demonstrated to underlie adverse cardiac remodeling and heart failure. With the expansion in the area of myocardial cell death and increasing concerns over rigor and reproducibility, it is important and timely to set a guideline for the best practices of evaluating myocardial cell death. There are six major forms of regulated cell death observed in cardiac pathologies, namely apoptosis, necroptosis, mitochondrial-mediated necrosis, pyroptosis, ferroptosis, and autophagic cell death. In this article, we describe the best methods to identify, measure, and evaluate these modes of myocardial cell death. In addition, we discuss the limitations of currently practiced myocardial cell death mechanisms.
Listen to this article's corresponding podcast at https://ajpheart.podbean.com/e/guidelines-for-evaluating-myocardial-cell-death/.
Keywords: apoptosis, autophagic cell death, cardiovascular disease, ferroptosis, heart, mitochondrial-mediated necrosis, necroptosis, pyroptosis
INTRODUCTION
A cell succumbs to death either immediately after exposure to multiple stimuli extremely unfavorable to it, or it dies gradually in a regulated manner. In the pathological heart, myocardial cell death is controlled by several receptors and signaling. Based on intrinsic and extrinsic stimuli, myocardial cells adopt different mechanisms of cell death. A myocardial cell may have either an intact or disrupted plasma membrane during cell death. Death by apoptosis and autophagy maintains cell membrane integrity. On the other hand, pyroptosis and necroptosis lead to disruption of the cell membrane, resulting in the release of cytoplasmic contents, including inflammatory cytokines. Release of these cytokines from the dying cells provides stress signals that alert the neighboring cells and mobilize protective mechanisms in the organism. However, massive myocardial cell death is observed in ischemia-reperfusion injury, which is detrimental to the heart. Out of the multiple reported forms of cell death (73), the six major forms observed in the heart are apoptosis, necroptosis, mitochondrial-mediated necrosis, pyroptosis, ferroptosis, and autophagic cell death. We will describe the best practices to measure these six forms of myocardial cell death.
Programmed cell death is an essential process in the developmental stage and is required for the formation of different structures, such as separation of the digits in hands and legs. In the adult stage, cell death is an important process to prevent infection against viruses and microbes, because proliferation of viruses and microbes requires a host cell, and thus host cell death would prevent their replication (20). However, cell death in key organs such as the heart is detrimental, because myocardial cells are terminally differentiated, have limited capacity for division, and perform important functions. Death of myocardial cells such as cardiomyocytes compromises contractility of the heart muscle, which in turn instigates adverse remodeling that ultimately leads to cardiac dysfunction and subsequent heart failure. Thus, cell death that results in loss of myocardial cells is a critical step in cardiac pathology, and targeting cell death mechanisms is a promising therapeutic option to mitigate and/or revert cardiomyopathy.
The research area of cell demise has advanced rapidly in recent years and multiple forms of cell death mechanisms have been identified (73). However, there is a lack of standardization regarding the evaluation of myocardial cell death. In the heart, cell death is a critical step to initiate pathological remodeling. With the increased emphasis on the rigor and reproducibility and the importance of cell death in cardiac pathophysiology, it is necessary to set guidelines for the best practices to measure myocardial cell death. This guidelines article describes different methods to measure apoptosis, pyroptosis, necroptosis, mitochondrial-mediated necrosis, ferroptosis, and autophagic cell death in the heart, limitations of some of the methods, and recommendations for the best practices to assess cell death mechanisms in the heart.
APOPTOSIS
Apoptosis is the most studied form of cell death in the heart. Although low levels of myocyte apoptosis (80–250 myocytes/105 nuclei) are observed in the failing human heart, the persistent dropout of myocytes is a causal mechanism of heart failure (270). This programmed cell death mechanism plays a crucial role in cardiovascular embryogenesis and cardiac pathology (109, 115, 257). Apoptosis is characterized by cell shrinkage, chromatin condensation, and systematic DNA cleavage (56) and may equally afflict cardiomyocytes, interstitial cells, and endothelial cells. This slow apoptotic death of cardiomyocytes in adult life is of particular importance, as it may contribute to the pathogenesis of numerous myocardial diseases (such as ischemia, inflammation, infection, and immunologically mediated insults), where it may precede occurrence of necrosis/necroptosis. Two central pathways mediate apoptosis, an extrinsic pathway involving cell surface death receptors and an intrinsic pathway initiated by mitochondria and the endoplasmic reticulum (Fig. 1) (41, 109). BH3-only proteins of the BCL2-family are initiators of the intrinsic pathway of apoptosis that exert pro-apoptotic functions through Bak/Bak (80, 83, 157, 269). Due to the fact that cardiomyocytes have little potential for division, prevention of cell death is essential in the treatment of cardiovascular disease, such as myocardial infarction and heart failure (109). Both the expansion of our knowledge on the role of apoptosis in the complex cardiovascular environment and the evaluation of the efficacy on new therapeutic interventions in treating cardiovascular disease depend strongly on proper measurements and analyses of myocardial cell death (257). Because myocardial apoptosis and necrosis may constitute a spectrum, attempts to retard any one of the processes may reduce the extent of cell death. Other forms of cell death, such as autophagy and necroptosis, may cross-talk within the spectrum and further justify the necessity of intervention.
Fig. 1.

Apoptosis pathway. There are 2 major pathways that lead to apoptosis: extrinsic and intrinsic. The extrinsic pathway is triggered when ligands bind to the death receptors (such as tumor necrosis factor receptor I and Fas) on the surface of the cell. The binding induces conformational changes of the receptors that, with the help of the adaptor protein Fas-associated protein with death domain (FADD), activate pro-caspase-8 to caspase-8. The activated caspase-8 then activates pro-caspases-3,-6, and -7, the activation of which eventually leads to apoptosis. The intrinsic pathway is mitochondria dependent and happens in response to insults such as DNA damage, oxidative stress, and high calcium concentration. Activation of pro-apoptotic proteins (such as BAX) neutralizes the effects of the antiapoptotic proteins of the Bcl-2 family and leads to the release of apoptogenic factor cytochrome c from the mitochondria. Cytochrome c binds to APAF1 (apoptosis protease activating factor-1), and together they bind to and activate pro-caspase-9. The complex of cytochrome c, APAF1, and activated caspase-9 forms apoptosome, which also activates pro-caspases-3, -6, and -7, and leads to apoptosis. The cross-talk between the extrinsic and intrinsic pathways is through BH3-interacting domain (BID). When BID is cleaved by activated caspase-8, it is activated to the truncated form of BID (tBID) and translocates to mitochondria, promoting cytochrome c release. See Refs. 91 and 210.
Biochemical Tests
Several common biochemical methods can be used to detect apoptosis (Table 1). Terminal transferase dUTP nick-end labeling (TUNEL) staining and DNA laddering assay can detect the presence of DNA fragmentation (108). Annexin V staining can label phosphatidylserine residues that become exposed to outer cell surface during apoptosis (108). Caspase isoform activation is detected by showing caspase cleavage at specific sites that generate processed (active) caspase fragments (108).
Table 1.
Evaluation of myocardial apoptosis
| Method | Detection Target | Apoptotic Cell Presentation | Appropriate Studies | References |
|---|---|---|---|---|
| Biochemical | ||||
| TUNEL | DNA fragmentation | TUNEL positive | In vitro | 236 |
| DNA laddering | DNA fragmentation | Appearance of nucleosomal DNA ladder in agarose | In vitro | 92 |
| Light and electron microscopy | Morphological changes | Rounded-up phenotype | In vitro | 1, 246 |
| Flow cytometry/confocal imaging using annexin V | Exteriorization of phospholipids (PE/PS) | Annexin V positive | In vitro | 184 |
| Activity assay/Western blot/immunohistochemistry for target proteins | Protease activation/mitochondrial dysfunction | Caspase-3/7 activity assay: increased fluorescence, Western blotting, and immunohistochemistry; detection of activated proteins involved in the signaling pathway, e.g., caspase-3/6/7/8/9 | In vitro | 196, 217 |
| Bioimaging | ||||
| Imaging using annexin V probes labeled with 99mTc | Exteriorization of phospholipids (PE/PS) | Annexin V positive | In vivo | 113, 156 |
| Cardiovascular magnetic resonance (CMR) imaging | Cytokinetics | Myocardial hypokinesis | In vivo | 8, 172, 237 |
CMR, cardiovascular magnetic resonance; PE, phosphatidylethanolamine; PS, polyunsaturated ω-6 fatty acids TUNEL, terminal transferase dUTP nick end labeling.
TUNEL staining utilizes the activity of the terminal deoxynucleotidyl transferase enzyme to label the 3′ ends of DNA strand breaks, which are then identified in individual nuclei by microscopy (68). However, the specificity of TUNEL assay has been challenged, as TUNEL positive cells could be apoptotic, necrotic, or normal cells undergoing DNA repair (195). The same issue is shared by the DNA laddering assay (37). Therefore, it is recommended that when detecting the presence of apoptosis in the heart, one should perform TUNEL assays using triple staining- for the nuclei, TUNEL to detect DNA fragmentation in the apoptotic nuclei, and cardiomyocyte-specific markers (271) in the suspected ischemic, infarcted, and nonischemic areas. To further confirm the presence of apoptosis, it is also recommended that one or more additional indices of apoptosis [such as caspase activity, poly ADP ribose polymerase (PARP) cleavage, apoptosis inducing factor and cytochrome c translocation from the mitochondria] be measured (109).
Caspases are a group of cysteine proteases that play a crucial role in initiating and executing apoptosis. Initiator caspases, such as caspase-8 and -9, act upstream to initiate and regulate apoptosis, whereas the effector caspases, such as caspase-3, -6, and -7, act downstream of both the extrinsic and intrinsic pathways to carry out the final biochemical changes in apoptosis (109). The activity of specific caspases can be measured by Western blotting, immunohistochemical techniques, or caspase activity assays using specific fluoremetric substrate or antibodies against the cleaved forms of their target proteins, such as PARP (140, 219, 276).
Phospholipids, such as phosphatidylserine (PS) and phosphatidylethanolamine (PE), which normally reside on the inner leaflet, are exteriorized to the outer layer of the plasma membrane during apoptosis (257). Annexin V is a positively charged protein that binds to negatively charged PS on the cell surface of apoptotic cells (257). When annexin V is fluorescently conjugated, the translocation of phospholipids can be detected by flow cytometry (256). When used together with the membrane-impermeant dyes, apoptotic cells, displayed with only annexin V staining, can be distinguished from necrotic cells that display annexin V concurrently with vital dye uptake, because the externalization of PS is an early event of apoptosis that occurs with intact plasma membrane (68). Similar to annexin V, antibiotic duramycin is able to selectively target PE and can also be used for the detection of apoptosis (169, 229, 291). Therefore, proper controls and gating strategies need to be applied in this detection method. However, it should be noted that PS exposure before membrane compromise is also observed in oncotic cells (137). The identification of morphological changes using histological stains or fluorescent dyes and visualization under light or electron microscopy is very useful in the characterization of cell death (68).
Imaging In Vivo
Visualization of apoptosis through noninvasive imaging could be useful for understanding a disease, monitoring its course, prognostication, defining targeted therapy, and measuring the efficacy of therapeutic intervention (229). A number of targets, including cell surface markers, membrane permeability, intracellular apoptotic machinery activation (e.g., caspase), and mitochondrial imaging, can be investigated for noninvasive imaging of apoptotic cell death (229). Two kinds of noninvasive strategies have been employed for imaging of apoptosis: imaging of apoptosis-specific exteriorized targets and targeting of intracellular processes specific for apoptosis (Table 1).
Based on the affinity between annexin V and PS, or duramycin and PE, annexin V and duramycin tracers radiolabeled with 99mTc can be used for imaging apoptotic cells in many cardiovascular disorders (229). The main disadvantage of the annexin V probe is the significant off-target radiation, leading to the slow adoption of annexin V into clinical practice (229). Radiolabeled duramycin may obviate this shortcoming of the radionuclide imaging. Magnetic resonance imaging has also been employed for annexin V targeting through late gadolinium enhancement (LGE) for T1 relaxation enhancement or iron oxide nanoparticles for T2 relaxation enhancement (229). Currently, nanoparticles are being developed to image apoptosis without radiation and enable better topographic localization by using an apoptosis-sensing ligand linked with an imaging molecule (229). One of the strengths of this approach is the ability to localize apoptosis, determine its extent, and correlate it with morphology and functional consequences (229). However, because nanoparticles can be taken up by macrophages, such techniques may be limited to cardiac disease without significant inflammation or macrophage infiltration (229).
Clinical apoptosis imaging in acute myocardial infarction or transplant rejection has mainly employed 99mTc annexin V with single photon emission computed tomography (SPECT) (229). Although the imaging of nonexteriorized targets has not been attempted in cardiovascular diseases, data from the cancer imaging field suggest the possibility of developing radiolabeled substrates specific for executioner caspases; mitochondrial membrane potential imaging also seems feasible (229).
Recommendations to Evaluate Apoptosis
Taking together different characteristics that cardiac apoptosis possesses, we recommend that investigators should combine different biochemical methods in vitro and immunohistochemical methods (such as TUNEL or activated caspase-3 staining), or ultrastructural examination (such as immunogold staining for cytochrome c leakage from mitochondria in cardiomyocytes) in tissue specimens (Table 1). In addition, noninvasive imaging methods could offer great tools for in vivo detection of cardiac apoptosis and monitoring disease progression and therapeutic efficacy. Cleavage of caspase-8 should be measured for death receptor-mediated apoptosis, whereas cytochrome c release and caspase-9 cleavage should be measured to determine mitochondria-mediated apoptosis (Fig. 1).
In cell culture, apoptosis may often transition to necrosis over time with plasma membrane breakdown because there is no phagocytosis in cell culture. Thus, we recommend that apoptosis should be determined at early time points in cell culture or blocked by pan-caspase inhibitor such as zVAD-FMK.
NECROPTOSIS
The paradigm that cells with a ruptured plasma membrane die due to an accidental, chaotic, and unregulated cell death was discredited in 2005 with the recognition that tumor necrosis factor-α (TNFα), a cytokine known to trigger the canonical extrinsic apoptosis pathway, can cause cell loss manifesting with the necrotic morphotype (50). Regulated forms of necrosis include necroptosis, mitochondrial-mediated necrosis, pyroptosis, and ferroptosis. Necroptosis has been found to underlie pathomechanisms of inflammation (177, 208), malignancies (36, 102), and microbial and viral infections (102, 122, 197), as well as ischemic injury of the heart (3, 4, 159, 243, 286), brain (213, 279), retina (116), and kidneys (151). This form of necrotic cell death has also been identified in various types of human heart failure independently on the etiology (243) and has been shown to underlie, at least in part, some phenotypes of this cardiac damage (3, 4). In addition, several animal models of heart failure, such as drug/chemical-induced cardiomyopathy (286, 287) and after left anterior descending artery (LAD) ligation (77, 159, 194), have highlighted a role for necroptosis in adverse cardiac remodeling and worsening heart function. Although the precise mechanisms of necroptosis induction and execution under conditions of myocardial damage are still not fully known, inflammation and oxidative stress, both mediators of cardiac disease, have been associated with a pronecroptotic environment (159, 194). Very recently, it has been reported that NLRP3-associated inflammasome may play a role in both noninfarcted and infarcted areas of post-myocardial infarction, whereas a canonical necroptosis signaling has been detected in the latter one only (149). Thus, necroptosis exhibits not only an adaptive function upon failing of cellular response to stress, but also, it is involved in developmental safeguard programs and the innate immune response.
Necroptosis can be induced by several triggering molecules, mainly through the stimulation of death receptors [TNFR, FasR, TNF-related apoptosis-inducing ligand receptor (TRAIL-R)] by the TNF family cytokines, and Toll-like receptors (TLRs) (49, 50, 72, 181, 261). Likewise, type I interferons and certain pathogens as well genotoxic (67) and oxidative stresses (44, 59, 118) promote this type of regulated necrosis. Depending on the triggering stimulus, pathways that execute necroptotic cell loss are likely to be different, and the current understanding of its mechanisms is based largely on experiments with TNFR1 signaling, which follows the RIP1-RIP3-MLKL signaling cascade terminating with plasma membrane disruption and cell lysis. However, the death receptor and RIP1 requirement can be bypassed, and the core components of the necroptotic pathway are RIP3 and MLKL.
TNF-induced stimulation of TNFR1 leads to receptor trimerization and the recruitment of cytosolic adaptor proteins to form a membrane-bound complex known as complex I (175, 223). It is composed of TNFR-associated death domain (TRADD), TNFR-associated factors 2 (TRAF2) and 5 (TRAF5), receptor-interacting protein kinase 1 (RIP1), and cellular inhibitor of apoptosis 1/2 (cIAP1/2). Loss of cIAP1/2 or increased activity of its counterregulatory enzymes, such as cylindromatosis (CYLD) (181), causes deubiquitination of RIP1 and internalization of membrane-bound complex I, where it turns into a cell death-promoting complex by recruiting Fas-associated death domain (FADD) and caspase-8 (175, 223, 260). This complex, named complex II, determines cell mortality through either apoptosis (complex IIa) or necroptosis (complex IIb) (Fig. 2). In addition, the ripoptosome, a complex similar to complex II, is formed by genotoxic stress (loss of cIAP) or FasR stimulation and contains RIP1, FADD, cellular FLICE-like inhibitory protein (cFLIP) isoforms, and caspase-10 and caspase-8, can also act as a death-inducing complex (67, 193).
Fig. 2.

Necroptosis pathway. TNF receptors induce formation of complex I. If receptor-interacting protein kinase (RIP)1 remains ubiquitinated, the cell survives. Complex I forms complex II by deubiquitination of RIP1, and it is mediated by loss of cellular inhibitor of apoptosis 1/2 (cIAP1/2). Caspase 8 activation in complex II inhibits RIP3 and promotes apoptosis, whereas caspase inactivation promotes necroptosis by phosphorylating RIP1, which in turn phosphorylates RIP3. Phosphorylated RIP3 causes mixed-lineage kinase domain-like (MLKL) phosphorylation that triggers MLKL oligomerization and subsequently translocation to the cell membrane, where it makes pore and release damage-associated molecular patterns (DAMPs).
What particular cell death mode is promoted is controlled by the activity/presence of caspase-8. In fact, caspase-8 cleaves the kinase domain of RIP kinases, thereby making them inactive and inhibiting necroptosis signaling, but at the same time, it can promote apoptosis by the activation of apoptotic downstream signals (47, 66). In contrast, the insufficiently active caspase-8 or its genetic and pharmacological inhibition (106, 193) prevents RIP1 cleavage, thereby promoting its association with RIP3 to form an amyloid-like protein complex known as the necrosome (259). In addition, cFLIP isoforms, which are predominantly expressed as either long (cFLIPL) or short (cFLIPS) isoforms, serve as an intracellular regulator of caspase-8 activity. The former isoform blocks caspase-8 to a degree that is sufficient to cleave and thus inactivate RIP1 but is insufficient to promote the activation of procaspase-3 to caspase-3 (55, 67, 221, 239). On the other hand, cFLIPS completely blocks the proteolytic activity of caspase-8, therefore preventing RIP1 proteolysis and leaving it active for necroptosis signaling (67, 111) (Fig. 2).
The active RIP3 favors necroptosis by phosphorylation of other RIP1 molecules at Ser227, which in turn phosphorylates the pseudokinase mixed-lineage kinase domain-like (MLKL) at Thr357 and/or Ser358 (at Ser358 in mouse MLKL) (34, 240, 290). Phosphorylation of only one residue of MLKL is sufficient to cause a conformational change, which triggers the oligomerization of MLKL molecules and its translocation to membrane compartments (Fig. 2). Phosphorylated MLKL assembling into oligomers and their subsequent translocation and insertion into the plasma membrane have been suggested to be the key drivers of necroptosis. However, precise MLKL stoichiometry and mechanism of action are still only partially understood. Several partial events of MLKL being associated with necroptosis have been studied; nevertheless, it cannot be ruled out that they can occur concurrently. This includes the following: 1) MLKL forms homo-oligomers (tetramers and/or octamers) or hetero-oligomers with RIP3 (96, 243, 263, 268); 2) MLKL oligomerization and translocation to the plasma membrane occur due to a specific and nonredundant role of the heat shock protein 90-kDa α-family class A member-1 (99); 3) MLKL oligomerization induces phosphatidyl serine exposure, resulting in the phosphatidyl serine-exposing plasma membrane bubbles, which attract cell surface proteases of the a disintegrin and metalloprotease (ADAM) family inducing shedding of the diverse cell surface proteins, including receptors, adhesion molecules, growth factors, and cytokines (28); 4) MLKL oligomers bind to the plasma membrane due to electrostatic interactions between their conserved positively charged regions and negative headgroups of membrane phosphatidylinositol lipids and cardiolipin (214, 240, 263) [phosphatidylinositol transfer protein-α (PITPα) seems to facilitate this interaction (102)]; 5) MLKL oligomers in the plasma membrane directly form nonspecific pores (58) or modify sodium (27, 33), magnesium, and calcium influx through transient receptor potential cation channel, subfamily M member 7 (27); 6) upon the translocation to the plasma membrane, MLKL responds to RIP3-mediated phosphorylation by the activation of an oxidative burst produced by NADPH oxidase 1 (NOX1) (118, 120); 7) MLKL translocate to the nucleus (243) and nuclear p-MLKL associates wth p-RIP3 to form lower-order oligomers that further exported from the nucleus to contribute to the formation of cytoplasmic p-RIP3-p-MLKL higher order oligomers (268); and 8) MLKL induces NLRP3 inflammasome formation and caspase-1 activation, resulting in IL-1β cleavage before cell lysis, thereby suggesting that inflammation associated with necroptosis can occur independently of the release of proinflammatory cellular contents due to plasma membrane rupture (38).
As indicated above, necroptosis execution is consistently associated with plasma membrane mechanisms; nevertheless, mitochondria participation in this process has also been reported. RIP3-mediated phosphorylation of phosphatase phosphoglycerate mutase (PGAM-5) inducing the dephosphorylation of dynamin-related protein 1 (DRP1) with resultant mitochondrial fission and mitochondrial fragmentation has been suggested to mediate necroptosis (266). Two independent laboratories have indicated that this RIP3-PGAM5-Drp1 axis can promote necroptosis in response to ischemia-reperfusion (94, 228). On the other hand, Pgam5−/− mouse embryonic fibroblasts have been reported to respond normally to multiple inducers of necroptosis, thereby suggesting a dispensable role of PGAM5 in necroptosis signaling, at least in some cell types (182). The significance of mitochondria in necroptosis remains controversial because nearly complete depletion of the mitochondria did not influence necroptotic cell loss (247).
From the aforementioned discussion, it is evident that TNFR-induced necroptosis requires both RIP1 and RIP3 kinase activities. However, the formation of a pronecroptotic protein complex, which is independent of RIP1 and occurs through the association of RIP3 with other RIP homotypic interaction motif-containing proteins, such as DAI (DNA-dependent activator of IFN regulatory factor), ICP6 (infected-cell protein 6), and TRIF (TIR-domain-containing adapter-inducing interferon-β), due to M45/vIRA mutant cytomegalovirus (MCMV) (255), human herpes simplex virus type 1 infection (HV-1) (264), and stimulation of TLR (89), has also been reported. Thus, RIP3, unlike RIP1, seems to be a central signal-transducing element in necroptosis signaling. It receives pronecroptotic upstream signals and transduces the signal by activating MLKL to execute cell death.
Although the TNFR1-RIP1 link can result in cell death, it is necessary to mention that this signaling can also lead to cell survival under conditions when RIP1 ubiquitination at Lys377 is catalyzed by ubiquitin ligases cIAP1/2 (181, 260). In this scenario, RIP1 serves as a scaffold protein for the recruitment of transforming growth receptor-β-activated kinase 1 (TAK1) and nuclear factor-κB essential modulator (NEMO), which are known to induce survival genes, including nuclear factor-κB (NF-κB) (61, 144, 192). Of note is that unlike its cell death promoting function, this scaffolding function of RIP1 is independent of the kinase activity.
Identification of Necroptosis
An integrated approach involving the assessment of morphological, biochemical, and molecular features of necroptosis is essential for its identification and discrimination from the other necrosis-like cell death modes described in this review, such as the late phase of apoptosis and/or passive necrosis, autophagy, pyroptosis, ferroptosis, and mitochondrial-mediated necrosis (39, 57, 73). This indicates that the identification of specific necroptosis biomarkers should be combined with the traditional monitoring of these cell death modes and live/dead cell exclusion analysis. It is also worthy to note that a real-time morphological analysis could be considered because cells can readily time-dependently shift from apoptosis to necroptosis (and vice versa), or secondary necrosis can occur in later phases of apoptosis. In this section, the identification of specific necroptosis biomarkers is primarily discussed, whereas other methods, which provide additional supportive information, are listed briefly, as they have been nicely reviewed previously (43, 129, 258) and are also referred to in other sections of this review.
Identification of Specific Necroptosis Markers
A general way to assess necroptosis signaling is to measure mRNA expression and/or protein levels of RIP3, MLKL, and, alternatively, RIP1 (78, 151, 159, 242, 245, 286). However, the increased levels of these proteins, and mainly RIP1 and RIP3, do not necessarily indicate necroptosis. In support, as aforementioned, these proteins possess a variety of pleiotropic effects. For instance, RIP1 serves as an essential platform for induction of NF-κB activation and the production of proinflammatory cytokines (TNF, IL-1) as well as proteins producing some prosurvival effects (antioxidant enzyme manganese superoxide dismutase and the anti-apoptotic protein BCL2) (31, 61, 79, 93, 192). RIP3 is necessary for NLRP3 inflammasome activation with resultant production of IL-1β and IL-8 (110). In addition, both RIP1 and RIP3 are also associated with the intrinsic apoptosis pathway (162, 284). Total levels of MLKL do not provide clear evidence of active necroptosis because assembly and function of the NLRP3 inflammasome have also been shown to be regulated by MLKL (110), although another study has indicated independent action of NLRP3 in the inflammatory response (136). Based on these effects of RIP kinases and MLKL, additional measurements, such as their phosphorylation state, as well as necrosome formation and MLKL localization within the plasma membrane, are essential for the robust identification of necroptosis (Table 2).
Table 2.
Evaluation of necroptosis
| Methods to Detect the Particular Necroptotic Signaling | Key Notes | References | |
|---|---|---|---|
| Specific molecular events strongly suggesting necroptosis being activated and/or executed | |||
| Activation of canonical RIP1-RIP3-MLKL axis through phosporylation | 1) Detection: immunostaining; 2) inhibitors of RIP1 (necrostatins, GSK2982772), RIP3 (GSK`872, GSK`843), and MLKL inhibitors (necrosulfonamide); 3) knockdown approaches | Phosphorylated RIP1 and RIP3 suggest necroptosis activation; however, phosphorylation of either residue of MLKL robustly identifies necroptotic environment | 42, 49–51, 64, 102, 105, 114, 146, 159, 165, 191, 203, 242, 244, 263 |
| MLKL phosphorylation and translocation to the plasma membrane | 1) Detection: immunostaining, subcellular fractionation; 2) inhibitors: MLKL inhibitors (necrosulfonamide) | Evidence of MLKL in the membrane fraction indicates its prior activation by the phosphorylation and execution of necroptosis | 27, 33, 59, 240, 242, 243, 263, 268 |
| MLKL oligomerization in the nucleus and plasma membrane | Immunofluorescence, nonreducing SDS-PAGE, native page gels | Phosphorylation of MLKL is a prerequisite for homo/hetero-oligomerization and translocation to the membrane/nucleus | 96, 242, 263 |
| Necrosome formation: RIP1-RIP3 interaction, RIP3-MLKL interaction | Immunofluorescence, coimmunoprecipitation | Necrosome identification indicates necroptosis activation; however, end-stage effector of the necroptosis (p-MLKL) should be also provided | 34, 139, 194, 268, 286 |
| Other more nonspecific characteristics of necroptosis | Methods to detect nonspecific features of necroptotic cell death | ||
| Loss of cellular integrity, plasma membrane rupture | LDH, HMGB1 release, staining with impermeant dyes, annexin V/PI staining | ||
| Altered cell viability | XTT, MTT assay, etc. | ||
HMGB1, high-mobility group box 1 protein; LDH, lactate dehydrogenase; MLKL, mixed-lineage kinase domain-like; p-MLKL, phosphorylated mixed-lineage kinase domain-like; RIP1 and -3, receptor-interacting protein kinase 1 and -3, respectively; XTT, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Specific antibodies recognizing posttranslational modification via phosphorylation of particular residues have been developed, and thus phosphorylation of hRIP3 at Ser227 (Thr231/Ser232 in mRIP3) and hMLKL at Thr357 and Ser358 (at Ser358 in mMLKL) can be assessed by immunoblotting (102, 243, 245). Unlike in humans and mice, there is no commercial antibody recognizing necroptosis residues of rat MLKL, and therefore, Phos-tag Western blotting can be eligible for this purpose. It is important to note that while phosphorylation of MLKL is a strong indicator of necroptotic cell death, it can be questionable in some conditions (59, 243, 268). Necrosulfonamide, an MLKL inhibitor, was shown to affect neither phosphorylation nor oligomerization of MLKL, but it was capable of preventing MLKL membrane translocation and, consequently, membrane rupture. However, necrosulfonamide only appears to inhibit human MLKL but not mouse MLKL (240, 263). Given this fact, other approaches for necroptosis identification involving the detection of necrosome formation, and MLKL oligomers in nuclear and membrane fraction, might be more conclusive.
Necrosome formation can be detected by immunofluorescence or by coimmunoprecipitation as RIP1-RIP3 interaction (34, 139, 268, 286), and RIP3-MLKL interaction is also a powerful option (194, 268). The higher-molecular-weight bands running around 160–300 kDa representing MLKL tetramers and octamers can be used to identify MLKL oligomerization on a nonreducing SDS-PAGE or native PAGE gel (96, 242, 263). As the so-far accepted terminal event of the necroptosis pathway, membrane translocation of MLKL can be identified by cellular fractionation with subsequent Western blotting or by immunohistochemical/immunocytochemical staining (Table 2) (27, 33, 242, 263).
Additional Supportive Methods For Necroptosis Monitoring
As a form of necrosis, the main morphological hallmarks of necroptosis, monitored by a transmission electron microscope, time-lapse video microscopy with epifluorescence, or a phase-contrast inverted microscope, include an increase in cell size due to swelling, prominent nuclear swelling, swelling of other cytoplasmic organelles, and loss of mitochondrial and endoplasmic reticulum mass followed by rapid loss of plasma membrane integrity (19). Notably, optimizing the light exposure and fluorescent probe should be an important consideration to minimize technique-induced cytotoxicity. Likewise, depending on the necroptosis triggering stimuli and a particular cell line and thereby the kinetics of its cell cycle, the time points at which real-time morphology analysis is intended to be performed, should be individually optimized.
The conventional biochemical cytotoxicity assays for necroptosis identification are based on the evaluation of the leakage of intracellular molecules through the ruptured plasma membrane, such as lactate dehydrogenase (LDH), damage-associated molecular patterns (DAMPs) such as high-mobility group box 1 protein (HMGB1), IL-1α, and IL-33, and mitochondrial DNA (19). However, the presence of these molecules in in vivo experiments can also indicate a late phase of apoptosis/secondary necrosis or suggest macrophage activation. The loss of cellular integrity can be also identified by impermeant dyes [trypan blue, propidium iodide (PI), and 4′,6-diamidino2-phenylindole (DAPI)] that differentially enter the cytosol and/or are bound to DNA in dying cells, although they do not cross through the plasma membrane of the healthy cells. Similar to late apoptotic/secondary necrotic, necroptotic cells are annexin V and PI positive. To distinguish late apoptotic/secondary necrosis from necroptosis, image-based analysis can identify some specific morphological features. For the necroptotic cells, a diffuse PI staining and an intense fluorescence signal are characteristic, whereas late apoptotic/secondary necrotic cells, which are shrunken in size, with condensed chromatin, exhibit spots of PI staining and annexin V-positive blebs.
Another potential approach to discriminate the early stage of necroptosis from primary/secondary necrosis could be the assessment of mitochondrial membrane potential with fluorescent probes (JC-1, Rhodamine 123) that incorporate into mitochondria and fluoresce brightly, depending on the inner transmembrane potential (206). In cells with ruptured plasma membrane, which dissipates the potential, a decrease in fluorescence can be detected. This method is unlikely to be effective in distinguishing the later phase of necroptosis from necrosis, as both are known to exhibit the same loss of the plasma membrane integrity. In addition, as apoptotic cells can also exhibit a loss of mitochondrial membrane potential and thereby a decrease in fluorescent signal, additional analyses must be performed to avoid misinterpretation. However, in this regard, it should be noted that role of mitochondria in necroptosis is unclear, and although some authors have reported necroptosis execution due to mitochondrial fission associated with RIP3-PGAM5-Drp1 link (94, 228) and/or mitochondrial permeability transition (MPT) pore opening (150, 235) mediated by RIP3-CaMKII axis (287), others have provided contradictory data (186, 216). Thus, the above-mentioned fluorescent staining and other methods assessing dissipation of mitochondrial membrane potential, which are more indicative for mitochondrial-mediated necrosis, can be used as a supportive method to investigate mitochondrial involvement in necroptotic signaling.
Importantly, inhibitor and knockdown approaches, which allow rapid dissection of divergent cell death pathways, should be considered to test the involvement of necroptosis. Several pharmacological inhibitors of RIP1, RIP3, and MLKL have been synthetized. RIP1 inhibitors include a group of small-molecule chemicals named necrostatins, of which necrostatin-1 and its more selective analog necrostatin-1s have been extensively used in preclinical studies (49, 50, 117, 139, 152, 242, 244). They inhibit the death domain of RIP1, whereas other necrostatins target other steps in the necroptosis pathway through different mechanisms (49–51). In contrast to these molecules, another potent RIP1 inhibitor, GSK2982772, which binds in an allosteric pocket of the RIP1 kinase domain, possesses an exquisite kinase specificity and is a clinical candidate for treatment of inflammatory diseases, psoriasis, rheumatoid arthritis, and ulcerative colitis (86). RIP3 kinase inhibitors such as GSK`872, GSK`843, HS-1371, dabrafenib, and ponatinib (42, 64, 105, 146, 203) and necrosulfonamide, an MLKL inhibitor, which, however, blocks only human MLKL by targeting Cys86 (240, 263), have also been appropriated for studying conditions exhibiting necroptosis. Nevertheless, some limitations such as protein selectivity and species specificity must be considered when selecting them for studies. Thus, additional knockdown strategies can be conducted to confirm data from pharmacological experiments. Nowadays, MLKL-deficient (273) and RIP3-deficient animals (159) are available. In contrast, as indicated above, RIP1 deficiency may not be a good choice for necroptosis study. RIP1−/− mice have been found to display neonatal lethality (114), whereas mice with kinase-dead knock-in mutation of RIP1 are viable (191), supporting the evidence that this kinase has important pleotropic effects and that RIP1 with the dead kinase domain is a prosurvival factor affecting the downstream molecules (31, 79, 93).
Recommendations to Evaluate Necroptosis
Although the development of several accurate techniques for cell death detection has advanced, the precise determination of the extent of cell death due to necroptosis remains challenging. In addition, experimental conditions and material undergoing analysis (in vivo, in situ, in vitro, and ex vivo experiments), as well as the fact that some cells rapidly change phenotype, should be carefully considered when choosing particular methods of analysis. Regardless, the identification of necroptotic cell death is based on a set of analyses, with a cardinal focus on the monitoring of RIP3-MLKL necroptotic signaling-induced plasma membrane disruption (Table 2). Detailed analysis of necroptosis-associated relocalization of these proteins within individual cellular compartments as well as the formation of intracellular necroptotic complexes is recommended alongside indices of activation of the key necroptotic molecules (Table 2). Likewise, it is highly recommended that analyses of cell/tissue viability be provided in addition to conventional, nonspecific methods assessing cell membrane rupture. Importantly, pharmacological approaches and/or genetic engineering tools, such as ablation or transgenesis of particular necroptotic proteins, are needed to elucidate the exact role of this mode of cell death in certain cardiac pathologies. The specific molecular events strongly suggesting necroptosis, and their detection methods are described in Table 2.
MITOCHONDRIAL-MEDIATED NECROSIS
Just as mitochondria are key mediators of the intrinsic apoptotic pathway, they can also play an essential role in the execution of necrotic cell death, especially in cardiac myocytes. The archetypal mitochondrial-dependent necrotic pathway is that driven by the MPT pore. The MPT pore is a channel that spans the inner mitochondrial membrane (IMM) (25, 133, 211). Although not sufficient in size to enable the passage of proteins across the IMM, when open, the MPT pore allows the free movement of molecules <1.5 kD in size. Since this includes protons, pore opening will dissipate the mitochondrial membrane potential (ΔΨm) and, therefore, inhibit a variety of mitochondrial functions, most critical of which is ATP synthesis (25, 133, 211). In addition, water can enter the mitochondrial matrix down its osmotic gradient, causing the mitochondria to swell. This will disrupt cristae structure, further impairing oxidative phosphorylation (25, 133, 211). If mitochondral swelling is too excessive, the outer mitochondrial membrane can also rupture. Combined, these sequelae of pore opening ultimately cause the necrotic demise of the cell (Fig. 3).
Fig. 3.

Mitochondrial permeability transition (MPT)-mediated necrosis. Because of calcium overload and/or extremely high oxidative stress, the mitochondrial membrane becomes permeable and results in dissipation of mitochondrial membrane potential, leading to cessation of ATP synthesis. Increased permeability also causes influx of cytoplasmic water molecules, which causes matrix swelling that ultimately results in mitochondrial membrane rupture and release of apoptotic factors. In the presence of ATP, it leads to apoptosis. However, depletion of ATP causes MPT necrosis.
The primary inducers of MPT are Ca2+ and oxidants. In both cases, these stimulants act on the matrix side of the MPT pore (Fig. 3) (25, 85, 133, 211). Ca2+ needs to be taken up across the IMM through the mitochondrial Ca2+ uniporter (MCU) until a concentration of Ca2+ that is sufficient to induce MPT pore opening is reached. Oxidants react with vicinal thiols exposed to the matrix (85). However, which protein(s) constitutes the specific Ca2+ and/or redox sensor has yet to be established (see below). The induction of pore opening by these agents is further facilitated by any reductions in adenine nucleotides, which suppress MPT pore opening (85). Reactive oxygen species and/or Ca2+ are frequently elevated in multiple cardiac pathologies, and MPT has been reported to contribute to the cardiac myocyte necrosis underlying ischemia-reperfusion injury (9, 13, 35, 82, 188), mitochondrial, dystrophic, and myocarditic cardiomyopathies (166, 176, 183), and anthracycline cardiotoxicity (14, 165, 190).
The model of the proteins that make up the MPT pore is in constant flux with the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), and phosphate carrier (PiC), and the ATP synthase itself being proposed as the channel-forming protein (25, 133, 211). However, genetic studies have questioned the identity of these proteins as the components of the MPT channel (14, 84, 87, 88, 125). As such, the only pore component that has stood up to genetic testing is the matrix isomerase cyclophilin-D (CypD) (13, 16, 188, 222). However, it should be remembered that although CypD is a critical positive regulator of the MPT pore, it is not the channel-forming protein itself, and its ablation can have several MPT-independent effects on mitochondrial metabolism and function (62, 211). We will describe the most commonly used techniques for measuring MPT in the following sections, but we would point out that additional methods can be employed (2, 76, 294).
Identification of MPT and MPT-Dependent Necrosis In Vitro
Several methods have been developed and established as indices of MPT pore opening in both isolated mitochondria and intact cells (Table 3). The oldest method is the spectrophotometric measurement of Ca2+ or oxidant-induced swelling in mitochondria (13, 14, 35). Here, mitochondria are isolated from cultured myocytes or excised hearts, typically by differential centrifugation in sucrose-based medium. Energized mitochondria are then challenged with a large bolus of Ca2+ in the micromolar range and swelling measured over time as a decrease in absorbance at 520–540 nM. The advantages of this system are its simplicity and reproducibility. However, it requires a relatively large number of mitochondria, which can be problematic when using cultured myocytes or mouse hearts such that often only a single concentration of Ca2+ can be tested when ideally a dose curve should be constructed.
Table 3.
Evaluation of mitochondrial permeability transition-dependent necrosis
| Characteristics of MPT | Differences Between MPT-Dependent and Other Forms Of Cell Death | Methods to Detect Characteristics | References |
|---|---|---|---|
| Loss of ΔΨm | Opening of the MPT pore will dissipate the ΔΨm; however, other interventions can also induce loss of ΔΨm and therefore appropriate controls must be used. | 1) Detection: assessment of using potentiometric fluorophores such as TMRE or JC-1 both in cultured myocytes and epicardially in isolate perfused hearts; 2) inhibitors: cyclosporine-A, bongkrekic acid, CypD depletion, positive controls: Ca2+, H2O2, ionomycin | 9, 13, 35, 48, 82, 165, 166, 168, 176, 180, 183, 188, 190 |
| Increased IMM permeability | Rapid increase in IMM permeability ≤1.5 kD, leading to swelling, separate from the increase in OMM permeability induced during intrinsic apoptosis. | 1) Detection: measurement of swelling and CRC in isolated cardiac mitochondria and CRC and calcein CoCl2 in intact myocytes. Use of 2-DOG uptake or loss of mitochondrial NAD+ in vivo; 2) inhibitors: cyclosporine-A, bongkrekic acid, CypD depletion, positive controls: Ca2+, H2O2, ionomycin | 9, 13, 35, 54, 81, 82, 165, 166, 176, 183, 188, 190, 207 |
| Inhibition of mitochondrial ATP synthesis | By dissipating the ΔΨm, opening of the MPT pore inhibits ATP synthesis, leading to a depletion in adenine nucleotides. | 1) Detection: measurement of mitochondrial ATP levels in vitro and in vivo using commercially available kits, measurement of ATP synthesis rates in isolated mitochondria; 2) inhibitors: cyclosporine-A, bongkrekic acid, CypD depletion, positive controls: Ca2+, H2O2, ionomycin | 48, 53, 84, 227 |
CRC, Ca2+ retention capacity; CypD, cyclophilin-D; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; MPT, mitochondrial permeability transition; TMRE, tetramethylrhodamine ethyl ester.
An alternative method for measuring MPT is the Ca2+ retention capacity (CRC) assay (13, 87, 88, 188). This is an improvement on the swelling assay, as it can be done in intact (albeit permeabilized) cells, and the amount of starting material is considerably less, thereby making it more amenable to high throughput screens. In this assay, energized isolated mitochondria or permeabilized myocytes are incubated with the cell-impermeable Ca2+-sensitive dye Calcium Green-5N, which measures extramitochondrial Ca2+. Small boluses of Ca2+ are then added to the mix, which results in a spike in fluorescence as the ion binds the dye. The fluorescence then decays as the Ca2+ is taken up into the mitochondria via the MCU. This is repeated until the threshold required to open the MPT pore is reached. Opening of the pore allows the accumulated matrix Ca2+ to rapidly escape, resulting in a quick, large-amplitude increase in fluorescence. Because only small amounts of Ca2+ are added each time, a more precise calculation of the MPT pore’s Ca2+ sensitivity is obtained compared to that using the swelling assay. Moreover, depending on the fidelity of the detection system, it can also provide additional information regarding mitochondrial Ca2+ uptake kinetics.
Because both mitochondrial volume and CRC can be altered independently of direct changes in MPT, a role for the MPT pore should be verified using pharmacological inhibitors of the pore such as cyclosporine-A or bongkrekic acid, or RNAi-mediated depletion of CypD (Table 3). Pharmacological and/or genetic inhibition of MPT will attenuate swelling and greatly increase the amount of Ca2+ required to induce MPT in the CRC assay. However, it needs to be noted that the swelling and CRC assays measure only the sensitivity of the pore to Ca2+ and are typically done under “basal” conditions. Thus, although they can still provide useful information, they do not provide insight as to how MPT is occurring in response to a variety of noxious stimuli in intact cardiac myocytes.
In this regard, two commonly used methods for the assessment of MPT in intact myocytes are the measurement of ΔΨm and the calcein/Co2+ assay. ΔΨm can be measured using potentiometric dyes such as JC-1 or the rhodamine-based fluorophores such as tetramethylrhodamine ethyl ester (TMRE) (48). These dyes accumulate and fluoresce in the mitochondrial matrix due to the high negative charge. Opening of the MPT pore dissipates the ΔΨm, which reduces mitochondrial fluorescence. Typically, loading of the cell is done at the end of the stimulus incubation period, and microscopy, fluorimetry, or FACS can be used to measure differences between control and treated cells. Moreover, this assay is also amenable to measurement of ΔΨm over time once an intervention has been added. The major caveat is that although MPT pore opening will always reduce ΔΨm, ΔΨm can be reduced independently of MPT. Therefore, pharmacological and/or genetic inhibition of MPT, as described above, is required to ascribe any changes in ΔΨm to modulation of the MPT pore. H2O2 (oxidative stress) and ionomycin (Ca2+ overload), which induce MPT-dependent necrosis (13, 188), are also useful as positive controls.
To circumvent MPT-independent loss of mitochondrial membrane potential, Petronilli et al. (207) developed the calcein/CoCl2 assay. In this method, cells are loaded with calcein-AM dye, which distributes between the cytosol and mitochondria. However, when co-loaded with CoCl2, which cannot penetrate the mitochondria, the cytosolic signal is quenched so that only the mitochondria fluoresce. Opening of the MPT pore allows the equilibration of the mitochondrial calcein with the cytosolic CoCl2 such that mitochondrial fluorescence is then reduced. Therefore, a reduction in calcein intensity is an index of pore opening. Like the ΔΨm-based assay, this can be analyzed by microscopy, fluorimetry, or FACS in real time. Again, established MPT inhibitors and activators should ideally be used as negative and positive controls, respectively. Unfortunately, the ΔΨm and calcein methods cannot be done simultaneously as TMRE quenches the calcein signal (207), and there is wavelength overlap between calcein and JC-1.
Although the assays described above can provide insight as to whether a compound/intervention sensitizes the MPT pore or induces its opening, they do not tell whether such changes in MPT are the causative driver of necrotic death in this context. There are no specific measures of MPT-driven necrosis. As described for the other pathways, general necrosis end point measurements such as vital dye exclusion or LDH and HMGB1 release are typically used in conjunction with genetic or pharmacological MPT inhibition.
Identification of MPT and MPT-Dependent Necrosis In Vivo
Although multiple indices have been established for assessing MPT in isolated mitochondria and cultured myocytes, reliable assays of MPT in vivo are lacking. Typically, mitochondria are isolated from control and affected hearts, and then MPT is assessed by swelling or CRC assays. But this does not indicate what is happening in situ. Epicardial fluorimetry of TMRE or JC-1 has been utilized in isolated perfused hearts as a real-time measure of MPT (168, 180) with the caveat of measuring surface mitochondria only in a limited area. Griffiths and Halestrap (81) utilized an isotope approach whereby rats were injected with 3H-2-deoxyglucose (DOG) and then subjected to cardiac ischemia/reperfusion injury. DOG is taken up into the myocyte but cannot enter mitochondria under normal circumstances. However, opening of MPT pore allows the uptake of the DOG into mitochondria. The authors isolated cardiac mitochondria following injury and found that mitochondrial 3H levels were increased upon reperfusion, but not during ischemia alone. However, the fact that DOG is an inhibitor of glycolysis and results in a tritiated animal greatly hampers the use of this model. Di Lisa et al. (54) took a reverse approach and measured the reduction in mitochondrial NAD+ due to its release through the MPT pore and subsequent hydrolysis in the cytosol after ischemia-reperfusion. While avoiding radioactive isotopes, cytosolic NAD+ can change independently of the release of mitochondrial pool. Moreover, both assays are really only amenable to interventions that induce massive MPT-driven necrosis, and pathologies where a more subtle loss of myocytes due to MPT occurs may not be detectable.
Thus, as with the cells, determination of MPT-dependent necrosis in vivo is reliant on manipulation of the MPT pore. As such chemical inhibitors of CypD such as cyclosporine-A, NIM-811, and sanglifehrin-A can be (and have been) used. Nevertheless, such drugs inhibit other cyclophilins, and cyclosporine-A inhibits calcineurin, which plays its own role in cardiac myocyte death and pathology (204). Consequently, any long-term use of these compounds as pore inhibitors can be problematic. Although the adenine nucleotide translocase (ANT) inhibitor bongkrekic acid inhibits the pore independently of CypD, its expense precludes its use in vivo, and long-term inhibition of ANT would be toxic. The still uncertain composition of the MPT pore also makes it hard to genetically inhibit in vivo. The primary approach has been to use mice lacking CypD, and useful data has been obtained with these animals. However, constitutive loss of CypD can also alter mitochondrial biology in ways that are likely MPT independent (62, 211), and care should be taken in data interpretation when using these mice. Fortunately, mice with a floxed CypD allele are available (134), and acute deletion of cardiac myocyte CypD using an inducible Cre may prove more informative as compensatory or non-pore-related changes may be circumvented, or at least reduced.
Recommendations for Evaluating Mitochondrial Permeability Transition-Dependent Necrosis
We recommend that at least two indices of MPT be measured in response to a drug or intervention (such as the swelling, CRC, ΔΨm, and/or calcein/CoCl2 techniques described above), with or without MPT inhibition, in in vitro experiments. Measurement of ATP levels can also be used as a surrogate given that MPT inhibits ATP synthesis (53, 227). Similarly, pharmacological and/or genetic inhibition of the pore can provide insight for establishing MPT as a causative agent in increased cell necrosis. Given the intricacies and limitations of measuring MPT in vivo, we also recommend that two indices of MPT such as swelling or CRC be assessed ex vivo. As with in vitro studies, the role of MPT in myocyte injury and pathology would need to be substantiated by re-assessing the phenotype in the presence of MPT inhibition, either pharmacologically or using CypD-deficient mice. The characteristic features of MPT and detection methods of MPT-mediated necrosis are described in Table 3.
PYROPTOSIS
Pyroptosis is a lytic type of regulated cell death that is triggered by the formation of plasma membrane pores by members of the gasdermin (GSDM) family of proteins as a consequence of activation of inflammatory caspases, mainly caspase-1, but also murine caspase-11 and its human homologs caspase-4 and caspase-5 (70, 73, 230). Caspase-1 is activated by an intracellular multiprotein complex called the inflammasome that is formed in response to pathogen-associated molecular patterns (PAMPs) derived during infection and DAMPs derived during cellular stress and damage. Although several types of the inflammasome have been reported, the NLRP3 inflammasome is the best characterized and involves sterile inflammation in the pathophysiology of cardiovascular diseases, including myocardial infarction (248, 249, 251). The NLRP3 inflammasome is composed of NLRP3 [Nod-like receptor (NLR) family pyrin domain containing 3], the adaptor protein ASC (apoptosis-speck like protein containing a caspase recruiting domain, also known as PYCARD), and the cysteine protease caspase-1, and its formation induces caspase-1 activation (Fig. 4). Recent studies identified a pyroptosis executioner GSDMD that is a substrate of both caspase-1 and caspase-11-4/5 (127, 230). These caspases proteolytically cleave GSDMD into an NH2-terminal domain (GSDMD-N) and COOH-terminal domain (GSDMD-C). Cleaved GSDMD-N binds to phosphoinositides in the plasma membrane (or cardiolipin of a bacterial membrane) and forms oligomeric death-inducing pores with an inner diameter of 10–20 nm. Activated caspase-1 can also process the pro-forms of inflammatory cytokines IL-1β and IL-18 into their mature forms (Fig. 4). Although IL-1β and IL-18 lack a secretion signal, the GSDMD-forming pore allows passage of these cytokines (∼5 nm) through the cell membrane, which probably explains the caspase-1-dependent secretory mechanism of IL-1β and IL-18.
Fig. 4.

The canonical NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome pathway of pyroptosis. A number of substances generated during ischemia-reperfusion (termed signal 2) trigger the assembly of the NLRP3 inflammasome, which results in activation of the cysteine-aspartic protease caspase-1. Simultaneous activation of Toll-like receptors (signal 1) increases the expression of pro-interleukins and NLRP3 via NF-κB. Caspase-1 activates pro-interleukins and releases the NH2-terminal domain of gasdermin D, which makes large pores in the plasma membrane. Those pores allow the proinflammatory interleukins to be secreted, and the loss of membrane integrity also kills the cell (pyroptosis). Caspase 1 is also reported to attack many other proteins in the cell, including some vital enzymes like GAPDH, which could also contribute to its toxicity.
In contrast to caspase-1, caspase-11-4/5 directly binds cytosolic lipopolysaccharide (LPS) via its CARD (caspase activation and recruitment domain) domain, leading to the cleavage of GSDMD and subsequent pyroptosis. Although caspase-11-4/5 cannot process IL-1β and IL-18 directly, they induce GSDMD-mediated potassium efflux, which is sufficient to cause NLRP3 inflammasome activation and subsequent release of IL-1β and IL-18; this process is known as the noncanonical inflammasome pathway. Furthermore, recent studies indicated that pyroptosis can also be driven by caspase-3-induced cleavage of GSDME/DFNA5 (gasdermin E is also known as deafness associated tumor suppressor) (265). Because the pore-forming ability of GSDMD is considered to be conserved throughout GSDM family proteins (65), it is possible that other GSDM proteins are involved in the process of pyroptosis. The role of GSDMD, GSDME, and the noncanonical inflammasome pathway in myocardial ischemia-reperfusion injury is still poorly characterized.
Pyroptosis is characterized by loss of plasma membrane integrity, leading to cell swelling and rupture of the cell membrane, nuclear condensation, and release of inflammatory cytokines and intracellular DAMPs (Fig. 4). Some of these features are also observed in necrosis, apoptosis, and necroptosis. We herein propose several experimental models that are likely to involve pyroptosis and could be used to collect biochemical and pharmacological evidence that could either support or refute the presence and contributing role of pyroptosis on cardiac injury, with an emphasis on using multiple techniques (with assessment of expression and function) and models (both ex vivo and in vivo).
Canonical Mechanism of Caspase 1 Activation Within the Inflammasome
The inflammasome is a macromolecular structure that acts as a sensor for extracellular and intracellular injury, and as a mechanism for the initiation and amplification of the inflammatory response (90, 215). The inflammasome is formed by the aggregation of three main molecular entities. NLR or absent in melanoma 2-like receptors (ALR) constitute intracellular sensors of damage or danger signals that trigger the formation of the inflammasome by recruiting the adaptor protein ASC. The ASC scaffold allows for recruitment of caspase-1 and leads to oligomerization of pro-caspase-1 molecules and autocleavage into active caspase-1. The most widely studied sensor for the inflammasome is the NLRP3, also known as NALP3 or cryopyrin. A large number of unrelated stimuli activates NLRP3, including bacterial toxins, K+ efflux, and lysosomal failure (90).
An Alternative Mechanism of Caspase-1 Activation Independent of NLRP3 Inflammasome
The components of the NLRP3 inflammasome are not constitutively expressed in the heart. Formation of the NLRP3 inflammasome requires priming (e.g., during ischemia), followed by triggering by DAMPs released during ischemia (251–253). However, studies on LDH release with and without the use of a caspase-1/4 inhibitor, VRT-043198 (an active metabolite of VX-765), highlight a role for caspase-1 and -4 in membrane failure and cell death (Table 4). A benefit is seen with the introduction of a caspase-1/4 inhibitor given at the onset of reperfusion and assessment of infarct size measured after 2-h reperfusion (10). A smaller infarct size is seen after 24 h of reperfusion with a NLRP3 inhibitor, but the same benefit is not seen with an NLRP3 inhibitor given at reperfusion and infarct size is assessed only 3 h later (251). Therefore, the data suggest an NLRP3-independent yet rapid caspase-1/4 dependent membrane failure in this context.
Table 4.
Evaluation of pyroptosis
| Characteristics of Pyroptosis | Differences Between Pyroptosis and Other Forms of Cell Death | Methods to Detect Characteristic Features | References |
|---|---|---|---|
| Human caspase-1/4/5 (or murine caspase-1/11)-mediated cell death by membrane failure that allows intracellular contents to be lost; pyroptosis is often associated with DNA fragmentation. | Pyroptosis is believed to result from caspase activation of GSDMD, which then forms large pores in the plasma membrane; proof of ischemia-triggered pyroptosis is complicated by the fact that loss of energetics alone during ischemia can cause membrane failure by necrosis, and inflammatory components may contribute to that energy deficit. | Cell death should be associated with caspase-1 activation and GSDMD cleavage; current evidence is that caspase-1 or inflammasome inhibition reduces ischemia-induced cell death; however, proof of a pyroptosis mechanism would also require a demonstration that the cell death was GSDMD dependent. Models to detect loss of membrane integrity include: • Isolated cells: trypan blue or propidium iodide exclusion assay. • Cells and heart tissue: release of cytosolic enzymes (CK-MB, troponin I or T, or LDH) • Whole heart: infarct size by pathology or by enzyme release |
10, 112, 163, 167 |
GSDMD, gasdermin D; LDH, lactate dehydrogenase.
One possible explanation is that calpain activation upon reperfusion is releasing pro- caspase-1 sequestered in the actin cytoskeleton of the heart cells (288). Activation of calpain in macrophages by elevated Ca2+ digests a protein called flightless-1, which is a caspase-1 (and caspase-11)-binding protein and anchor to the cytoskeleton. Caspase-1 can be self-activated, but flightless-1 and other inhibitors function as caspase-1 pseudosubstrates preventing autocleavage. Calpain activation reduces flightless-1 levels, causing rapid self-activation of caspase-1 (145, 288). Whether calpain-dependent activation of caspase-1 also occurs in reperfused cardiac tissue is currently unknown.
Measuring Caspase-1 Activation in the Heart Tissue
Basal caspase-1 activity in the healthy heart is rather low; therefore, small changes in caspase-1 activation may be easily detected. Caspase-1 activation can be measured by detection of the appearance of the low-molecular weight active caspase-1 in a Western blot and expressed as its ratio to its heavier pro-caspase-1 precursor (22, 23). Caspase-1 activity can also be measured by the appearance of low-molecular weight fragments of endogenous caspase-1 substrates like IL-1, IL-18, GSDMD (Fig. 4), and others measured in the heart tissue or in the plasma (52), as well as by the cleavage of a fluorogenic substrate in which the heart tissue is incubated (22, 163, 164, 173). An intervention that reduces caspase-1 activity measured with one of the methods described and is associated with a reduction in infarct size may again be interpreted as evidence of possible pyroptosis-mediated cell death (Table 4). One limitation of this examination of biopsies in in vivo hearts is that it is not able to distinguish activation of caspase-1 in cardiomyocytes versus other resident or infiltrating cells.
Measuring Inflammasome Formation and Aggregation in the Heart Tissue
The components of the inflammasome are minimally expressed in the healthy heart, and the expression increases during insults like ischemia and infarction. The active inflammasome appears as a macromolecular structure or specks using fluorescence microscopy after staining for the scaffold protein ASC or for the mostly commonly involved effector of caspase-1, NLRP3 (18, 173, 254). Staining for one or more of these structures allows for the measurement of inflammasomal specks in the heart tissue, and using cell-specific costaining it allows for the determination of formation in cardiomyocytes versus other resident or nonresident cells. While the formation of the inflammasome specks can hypothetically occur without leading to activation of caspase-1, i.e., in presence of a powerful caspase-1 inhibitor, it is accepted that appearance of inflammasome specks can serve as a surrogate of caspase-1 activation and possible pyroptosis (173, 254). One of the limitations of this method is that it does not consider an inflammasome-independent mechanism of caspase-1 activation such as calpain activity, as discussed above. In rodent models of myocardial infarction, inflammasome specks have been detected in cardiomyocytes, cardiac fibroblasts, endothelial cells, and infiltrated leukocytes (173). Furthermore, activation of the NLRP3 inflammasome induces IL-1β release in cardiac fibroblasts and endothelial cells through pyroptosis, whereas in cardiomyocytes it only induces washout of cytosolic enzymes but not IL-1β (131, 132, 134). Therefore, pyroptosis may occur without secretion of IL-1β.
Supplementary Methods for Pyroptosis Monitoring
Triphenyl tetrazolium chloride (TTC) that stains viable myocardium red and nonviable myocardium unstained can be used to determine pyroptosis because it measures loss of dehydrogenase enzymes due to membrane failure. However, this method cannot distinguish pyroptosis from other forms of cell death where cell membrane integrity is compromised, such as necroptosis.
The release of the soluble cytosolic protein LDH is observed in pyroptosis. LDH release is a classical marker of necrosis, and therefore, it is not sufficient to identify, by itself, caspase-1 triggered pyroptosis. Exposing the isolated rat heart perfused with Krebs buffer to >20 min of global ischemia and 2–3 h of reperfusion will cause a large amount of the left ventricle to infarct (∼75% after 45 min of ischemia). Inhibiting caspase-1/4 with the highly selective inhibitor VRT-043198 will reduce that to ∼45% infarction, revealing a large component of caspase-1/4-dependent injury (10), which could reflect pyroptosis (Table 4). An advantage of the Krebs-perfused heart is that the venous effluent can be collected. The original demonstration of pyroptosis was the release of the soluble cytosolic protein LDH into the medium of cultured macrophages in which NLRP3 inflammasome formation had been stimulated (69). A reduction in LDH in the venous effluent with suppression of caspase-1/4 supports but still does not prove pyroptosis in this model, as caspase 1 is also known to digest important glycolytic enzymes that could also cause necrosis through loss of ATP required for cellular volume regulation (10, 226).
The time course of the membrane failure can be monitored in the isolated rat heart by measuring the rate of LDH washout during reperfusion with timed samples of effluent. By multiplying the volume collected for each sample by the LDH concentration and dividing by the sample duration, the rate of LDH release from the entire heart for each sample time can be calculated. This should then be normalized to the heart weight. Note that in hearts, which were reperfused with VRT-043198 containing buffer, the LDH begins to fall in the third minute of reperfusion, and the two curves continue to diverge until the end of the experiment (10). Thus, this model should be appropriate for studying mouse hearts that have been genetically manipulated to critically test the role of pyroptosis in this model.
Using inhibitors of caspase-1/4 or other components of the inflammasome followed by measurement of troponin T or I levels could also provide an indirect measure of pyroptosis.
Recommendations to Evaluate Pyroptosis
To date, the following parameters have been used to detect the presence of pyroptosis: 1) lytic cell death assessed by the release of cytoplasmic contents (e.g., LDH) and by the entry of cell-impermeable dyes [e.g., propidium iodide (PI)]; 2) activation of the inflammasome assessed by immunofluorescence (e.g., ASC speck) and activation of caspase-1 (and/or caspase-11–4/5) assessed by immunoblotting (e.g., processed fragments, p10/p33 of caspase-1) or assessment of enzymatic activity through cleavage of fluorogenic substrates; and 3) cleavage of GSDMD by immunoblotting (e.g., GSDMD-N). At present, however, a protocol to specifically differentiate pyroptosis from other forms of necrotic death is unavailable, especially considering a potential overlap between the different modes of necrosis. We recommend using multiple techniques described above to evaluate pyroptosis. The strongest evidence that pyroptosis is causing cell death would be a demonstration that the membrane failure is GSDMD dependent.
Additional Methods for Determining Necroptosis, Necrosis, and Pyroptosis
The measurement of viable and nonviable myocardium using TTC is the standard methodology to measure infarct size in laboratory animals (282). The infarct size is measured as the ratio of nonviable myocardium to the entire amount of myocardium that experienced ischemia (area of risk) or left ventricle (LV) by examining transverse sections of the heart from base to apex to evaluate infarct size (IS)/AR and IS/LV, respectively. TTC measures loss of dehydrogenase enzymes due to membrane failure and cannot distinguish between necrosis, necroptosis, pyroptosis, or any other modality of membrane failure.
Measuring LDH release from the isolated heart is a classical marker for necrosis and necroptosis in the damaged heart, for instance, in ischemia-reperfusion injury.
Detection of elevated troponin I or T levels is the mainstay of the diagnosis of acute myocardial infarction (AMI) in patients. As for other methods, plasma troponin levels provide a surrogate marker of infarction in in situ heart models of ischemia-reperfusion. The measurement of troponin levels at a given time point between 1 and 24 h after ischemia or, even better, measuring the area under the curve after three or more time points represents a good estimate of infarct size in the animal in vivo (163). Similarly to the other above-mentioned markers, increased levels of plasma troponin do not differentiate between necrosis, necroptosis, or pyroptosis.
FERROPTOSIS
Ferroptosis is a relatively new form of regulated cell death that was first described less than a decade ago (57). Since its discovery, ferroptosis has been shown to occur in several other disease models, including cancer (119), neurodegeneration (97), liver disease (289), kidney failure (5), and cardiovascular disease (63, 74, 148). The mechanisms underlying ferroptosis were discovered through small-molecule screens of anti-cancer drugs (57). Ferroptosis has characteristics that are distinct from other forms of regulated cell death: iron dependency, disturbances in redox balance mediated by glutathione (GSH) and glutathione peroxidase 4 (GPX4), and lipid peroxidation (Fig. 5) (29). The discovery of ferroptosis in cardiac tissue is even more recent, with proportionately few studies describing ferroptosis specifically in the context of the heart. Despite their limited number, those studies were able to repeat and demonstrate the same characteristic features of ferroptosis in cardiac tissue and successfully use methods of inducing and inhibiting ferroptosis in the heart (11, 12, 63, 74, 148, 153).
Fig. 5.

Signaling pathway of ferroptosis in cardiomyocytes. The defining characteristics of ferroptosis are 1) iron dependency 2), disturbances in glutathione (GSH) and glutathione peroxidase 4 (GPX4)-mediated redox balance, and 3) lipid peroxidation. Pointed arrowheads indicate transport, products, or activation. Blunt arrowheads indicate inhibition. Red arrows indicate ferroptotic activity or features that promote ferroptosis. Blue arrows indicate processes that inhibit ferroptosis. GSSG, glutathione disulfate; ROS, reactive oxygen species; RSL3, Ras-selective lethal 3.
Iron bestows ferroptosis with its namesake and is required for ferroptosis to occur. The importance of iron in ferroptosis was established by experiments that showed that increasing iron within the redox-active labile iron pool (LIP) made cells more prone to ferroptosis (74, 95). The necessity of iron in ferroptosis was established by experiments that showed that chelators and mechanisms that reduce iron in the LIP also inhibit ferroptosis (57, 132). Iron is an essential cofactor in several molecules and enzymes and is especially important in mitochondrial function (141, 238). The constant activity and contractions of cardiac tissue require a constant supply of energy from mitochondria, and thus it follows that the mitochondria compose a significant proportion of the cardiomyocyte volume, which further underscores the importance of iron in cardiac function (15). Iron deficiency impairs the normal functioning of the heart and is associated with cardiovascular disease (262). Conversely, excess iron is also harmful to normal cardiac function, and iron-overload diseases and ischemia-reperfusion injury can also cause accumulation of iron and impaired function in cardiac tissue (46, 71).
Cardiac tissue and cardiomyocytes can import and accumulate excess iron from iron-overload disease or cardiac pathologies (46, 71, 198). Soluble, redox-active free iron in the LIP is thought to be the catalyst that drives increased reactive oxygen species (ROS) production in ferroptosis (29, 250), thus making cardiomyocytes prone to oxidative stress in excess iron conditions (171). In addition to the LIP, excess iron is also absorbed into the mitochondria (63, 198). The mitochondria are already a hotbed of ROS production due to oxidative phosphorylation and H2O2 production within their matrices (141, 187), and therefore, excess iron will also cause mitochondrial ROS overproduction (75). Ferroptosis also results in altered mitochondrial morphology and fragmentation through an unknown mechanism (57). Increased ROS production is already associated with several cardiac pathologies, including ischemia-reperfusion injury, left ventricular remodeling, and cardiomyopathy (121, 185). Adding excess iron to the ROS generated by those pathologies increases ROS production beyond already elevated levels, thus pushing cardiomyocytes further toward oxidative stress and the ravaging of intracellular molecules by ROS. Membrane lipids are one of the targets of ROS, which leads to the production of lipid peroxides, a signature hallmark of ferroptosis (Fig. 5). Specifically, polyunsaturated fatty acids (PUFA) are targeted by lipoxygenases for oxidation into detectable byproducts (6, 135). Lipid peroxidation is also observed in various cardiac pathologies (170) and therefore represents another common intersection between ferroptosis and cardiovascular disease.
Redox homeostasis is crucial for the normal functioning of cardiac tissue and even more so for the protection of cardiomyocytes from the constant redox activity and ROS production in the mitochondria (187). In ferroptosis, GPX4 is an important antioxidant enzyme that uses GSH as a substrate to reduce and prevent the accumulation of ROS, thus protecting cells against oxidative stress and lipid peroxidation (40). Chemicals that cause ferroptosis, such as erastin and Ras-selective lethal 3 (RSL3), target either the production of GSH or GPX4 itself (29). Erastin is one of the first known ferroptosis-inducing agents and does so by targeting and inhibiting system Xc− (cystine/glutamate antiporter) (57). System Xc− imports cystine into the cell, where it is reduced into cysteine, one of the three amino acids needed to synthesize GSH. Erastin inhibits system Xc−, which deprives cells of cysteine and, consequently, GSH, thereby inhibiting the ability of GPX4 to reduce ROS in the cell (Fig. 5) (40). While system Xc− is not expressed in the heart (142), another study showed that erastin is still able to induce ferroptosis in the heart (11). This suggests that despite the absence of system Xc− in cardiac tissue, erastin may target another mechanism of glutathione synthesis or GPX4 activity in the heart. The mechanism of RSL3 is comparatively straightforward: RSL3 directly inhibits GPX4 activity, thus tipping the redox balance of the cell toward ROS accumulation, oxidative stress, and ferroptosis (Fig. 5) (29, 232).
A recent study investigated the mechanisms underlying doxorubicin cardiotoxicity and provided further evidence that ferroptosis is distinct from apoptosis and necrosis. The study used mice that had defective apoptotic or necroptotic signaling and showed that doxorubicin induced ferroptotic cell death in both types of knockout mice (63). This elegantly designed study demonstrated not only that doxorubicin induces ferroptosis but also that ferroptosis can occur in the absence of canonical apoptotic and necroptotic signaling proteins (63). However, although ferroptosis has distinct hallmarks that distinguish it from other forms of cell death in the heart, some cross-talk can occur. Ferroptosis-inducing agents also cause endoplasmic reticulum (ER) stress and the unfolded protein response, which in turn activate the PERK [protein kinase RNA (PKR)-like ER kinase]/eIF2α (eukaryotic initiation factor 2a)/ATF4 (activating transcription factor 4)/CHOP [C/EBP (CCAAT-enhancer-binding protein) homologous protein] pathway (138). The PERK/eIF2α/ATF4/CHOP pathway can also activate apoptotic and autophagic mechanisms and is observed during ischemia-reperfusion injury in the heart (283). Despite ferroptosis and apoptosis having a common intersection at the ER with the PERK/eIF2α/ATF4/CHOP pathway, ferroptosis does not necessarily cause apoptosis, as ferroptosis can occur without caspase activation, DNA laddering, or other classic apoptotic hallmarks (11, 57, 63).
Identification of Ferroptosis
The requirements, redox mechanisms, and byproducts of ferroptosis also serve as the basis for detecting ferroptosis (Table 5). In terms of mechanisms, disturbances in GSH and GPX4 activity are a unique feature of ferroptosis and are not a central pathway for any of the other types of cell death known to occur in the heart. Decreases in GSH and total glutathione can be measured using chromogenic assays that use 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) as a detection reagent and oxidized glutathione (glutathione disulfate) as a standard (201). Total GPX enzymatic activity can be quantified by measuring the reduction rate of tert-butyl hydroperoxide catalyzed by GSH and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) (200). Measurements of GPX activity should also be supported with Western blot data to determine whether ferroptotic decreases in GPX4 activity are due to direct inhibition of enzymatic activity or downregulated expression of the GPX4 protein itself (Table 5) (277).
Table 5.
Evaluation of myocardial ferroptosis
| Characteristics of Ferroptosis | Differences Between Ferroptosis and other Forms of Cell Death | Methods to Detect Characteristic Features | Limitations | References |
|---|---|---|---|---|
| GPX4 and GSH | GPX4 activity should be lowered only in cases of ferroptosis and should not be significantly altered in other forms of cell death like apoptosis; ferroptosis should also result in lowered GSH/total glutathione ratios, lower total glutathione, or both. | 1) GPX4 activity can be assessed via colorimetric assays that measure GPX4-catalyzed consumption of GSH; 2) GPX4 itself can be detected by an antibody against GPX4. | 1) GPX4 activity is often quick in nature, and thus experiments must be time sensitive; 2) antibody detection does not accurately depict the activity level of GPX4. High levels of GPX4 may still have reduced activity when inhibited. | 1) 201; 2) 153 |
| Lipid ROS | General ROS production is present in nonpathological conditions and can be found in other forms of cell death such as apoptosis and necrosis; lipid ROS is more specific to ferroptosis and accumulates in much higher amounts than other forms of cell death due to specific decreases in GPX4 activity. | 1) Detection: Measurement of membrane-associated fluorescent probes (e.g., liperfluo) that fluoresce upon oxidation; lipid ROS downstream byproducts (e.g., 4-HNE protein adducts, MDA) can be quantified using ELISAs and TBARS assays; 2) inhibitors: Specific antioxidant ferroptosis inhibitors such as ferrostatin-1. | 1) Downstream products such as 4-HNE often quickly dilute as time passes and thus create a time sensitive element, and thus fixation and assaying the cells should be done in a timely manner. | 1) 104, 267, 281; 2) 11, 57, 63 |
| Iron dependency | Ferroptosis requires iron but does not need abnormally high levels of iron; elevated levels of iron may be a sign that the cells are undergoing ferroptosis; if ferroptosis is occurring, chelators or other ways of reducing intracellular iron should significantly decrease cell death and lipid ROS and restore GPX4 and GSH; other non-iron-dependent forms of cell death should not be significantly affected if iron is removed via chelators or other techniques. | 1) Iron can be detected via colorimetric assays that use compounds such as ferrozine, which produces a color when bound to iron; 2) chelators or genetic manipulation of proteins that affect iron concentrations (e.g., TfR, ferritin, FtMt) will also affect ferroptosis. | Iron levels may not indicate levels of ferroptosis and need to be paired with another assay. | 1) 100, 218; 2) 63, 74 |
| Supplementary features: mitochondrial morphology change | While mitochondrial fragmentation may not be distinct to ferroptosis; its pairing with high amounts of lipid ROS on the mitochondrial membrane sets it apart. | 1) Smaller mitochondria smaller, with increased membrane densities and reduced cristal; 2) signs of cell fragmentation after a few hours of reducing GPX4 function (the morphology of the mitochondria can be assessed using a mitochondria marker like MitoTracker). | Morphology can vary, depending on the cells used, and must be analyzed using an imaging software for quantitative analysis. | 57, 101 |
GPX4, glutathione peroxidase 4; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; ROS, reactive oxygen species; TBARS, Thiobarbituric acid-reactive substance.
ROS production plays a significant role in ferroptosis, and ROS can be easily detected by using fluorescent probes such as 2′,7′-dichlorodihydrofluorescein diacetate (11) or more organelle-specific probes such as MitoSOX (184). However, simple ROS detection cannot be used alone to implicate ferroptosis in cardiac cell death, as ROS are also produced in necrosis and necroptosis. Therefore, more specific assays are required to distinguish ferroptotic ROS production from ROS produced by other forms of cell death in the heart.
Fortunately, there are assays that can specifically detect lipid peroxides, which are another distinct characteristic of ferroptosis and a downstream consequence of oxidative stress. Liperfluo (104) and C11-BODIPY (60) are both lipophilic fluorescent probes that associate with lipid bilayers and fluoresce when oxidized by lipid peroxides. Additionally, some specific lipid peroxides, such as 4-hydroxynonenal (HNE) and malondialdehyde (MDA), can be detected via various assays. 4-HNE is formed by the oxidation of polyunsaturated ω-6 fatty acids and can form adducts with membrane proteins (267, 281). 4-HNE production can be measured and visualized using various immunodetection methods, including ELISA (267) and immunohistochemistry (281). MDA is byproduct of further degradation of oxidized PUFAs and can be measured with a colorimetric assay using thiobarbituric acid as a detection reagent (Table 5).
Although the presence of redox-active iron is needed for ferroptosis to occur, three important caveats must be stated. 1) Ferroptosis can still occur at normal intracellular iron concentrations; 2) excess iron can induce ferroptosis, but is not necessary for ferroptosis; and 3) depriving cells of soluble, redox-active iron inhibits ferroptosis (11, 57, 63). Nevertheless, total iron can be detected in cell or tissue lysate samples to determine whether ferroptosis occurred due to an accumulation of excess iron. Ferrozine is an iron-detecting compound that forms a magenta-colored complex when bound to ferrous ions and can be used to detect and quantify total iron up to the nanomolar scale (100). Bathophenanthroline and Ferene-S can also detect total iron in samples using colorimetric assays, although Ferene-S is the more sensitive of the two and requires a smaller sample volume (209). For iron detection on an organismal level, T2 and T2* magnetic resonance imaging can be used to visualize iron deposition in the myocardium and other organs and tissues (26).
Several ferroptosis studies used chelators as a straightforward way of removing redox-active iron from cells without needing to alter the expression of genes and proteins that regulate iron homeostasis. In animal models, treatment with a chelator such as deferoxamine, dexrazoxane, or 2,2′-bipyridyl protects the heart against ferroptosis in ischemia-reperfusion injury and other models of cardiovascular disease (32, 63, 74). If ferroptosis is occurring as a result of experimental or pathological conditions, the addition of chelators to those conditions should inhibit cell death, as ferroptosis requires the presence of redox-active free iron (Table 5).
Recommendations and Strategies For Detecting Ferroptosis and Anti-Ferroptotic Effects
In addition to the ferroptosis detection methods and assays described above, anti-ferroptotic agents such as ferrostatin-1, liproxstatin-1, and vitamin E are also useful in determining whether ferroptosis is occurring (11, 63, 148, 293). All of those anti-ferroptotic agents are lipid-soluble antioxidants that scavenge free radicals, thereby reducing and decreasing ROS and lipid peroxides. If ferroptosis is suspected to be occurring in an experiment, the addition of ferrostatin-1 or a similar ferroptosis inhibitor should decrease the amount of lipid peroxidation and cell death.
Conversely, if a drug or intervention is thought to have anti-ferroptotic activity, it can be tested against treatments that induce ferroptosis, such as excess iron, erastin, or RSL3 (11, 12, 63). Excess iron causes ferroptosis due to an increase in overall ROS production, but treatment with erastin and RSL3 is a more targeted way of causing ferroptosis due to affecting glutathione synthesis and GPX4 activity, respectively. If a drug or intervention truly has anti-ferroptotic activity, it should decrease the amount of lipid peroxidation and cell death caused by ferroptotic agents such as erastin and RSL3. The detailed methods to evaluate ferroptosis are elaborated on in Table 5.
AUTOPHAGIC CELL DEATH
Autophagy is a cellular mechanism of bulk degradation of long-lived proteins and damaged organelles and recycling of the molecular components. It commences with nucleation and proceeds through sequestration of these materials in double-membraned structures termed autophagosomes, fusion of autophagosomes with lysosomes where enzymes catalyze the destruction of the cargo and release of the breakdown products into the cytosol (178). In contrast with other forms of cell death reviewed in this paper, autophagy as a process does not universally command the destruction of the cell. Earliest observations of autophagic structures were made in cells undergoing remodeling during differentiation or in response to stress (45). Whereas autophagic activity was reported in association with cellular demise as early as 1977 (17), autophagy often plays a role in a survival effort by supplying building blocks to facilitate cellular adaptation and nutrients for energy production. If these efforts are not successful, cell death may be observed to be concurrent with but not necessarily consequent to autophagy. Therefore, a distinction must be made between autophagy-associated and autophagy-mediated cell death, and experimental strategies should be designed accordingly.
Autophagy-Associated Cell Death
Autophagy-associated cell death is established by the detection of cell death, using standard approaches described herein, in cells in which autophagy is measured. Transmission electron microscopy (TEM) emerged as the earliest reliable method for visualizing autophagic compartments (241) and remains the tool with the highest resolution for characterizing their morphology. The presence of double membranes enclosing electron-dense cytoplasmic material or recognizable organelles is typical of autophagosomes and early autolysosomes (280). At later stages of degradation, definitive identification of these structures requires immuno-EM (electron microscopy) detection of lysosomal structural proteins or hydrolases. Furthermore, imaging of only a single section of the cardiomyocyte makes this identification more difficult due to significant limitations related to serial sections and representative sampling. For these reasons, collecting quantitative data such as autophagosome number and volume is challenging. Therefore, EM is better reserved for morphological characterization of autophagic vacuoles rather than monitoring changes in autophagic flux.
Alternative methods for microscopic visualization of autophagic machinery commonly involve tracking of autophagy-related proteins, such as microtubule-associated proteins 1A/1B light chain 3 (MAP1LC3; hereafter termed LC3). LC3, one of the mammalian homologs of Atg8, is a ubiquitin-like protein that is cleaved by Atg4 to produce LC3-I, which is then converted to LC3-II by Atg3- and Atg7-facilitated lipidation (103). LC3-II is incorporated into both sides of autophagosome membranes and can also be found on the outside of autolysosomes (274), making it a useful marker of these autophagic compartments (Fig. 6A). Given that an antibody capable of binding exclusively to the lipidated isoform does not exist, we rely on punctate localization of LC3-II versus diffuse distribution of LC3-I in the cytosol to differentiate between the two. For accurate interpretation of LC3 distribution, we recommend fixing samples with neutral-buffered formalin and preparing paraffin-embedded instead of frozen sections. Cryosections are not only inferior in morphology but also preserve lipid droplets, which have been shown to harbor LC3 on their surface (231). Because endogenous levels of LC3 are not robust in cardiomyocytes, permeabilization and antigen retrieval are also necessary. A gentle detergent such as digitonin or saponin can be used to avoid damaging membranes during permeabilization, as LC3-II is membrane-bound.
Fig. 6.

Autophagic cell death pathway and autosis. A: steps of adaptive autophagy/autophagic cell death and proteins involved in different steps. B: factors inducing autosis mediated by autophagy machinery and changes in the cell morphology during early and late phases of autosis. LC3, microtubule-associated proteins 1A/1B light chain 3; RER, rough endoplasmic reticulum.
Levels of LC3 expression may not always reach the threshold of detection even with careful tissue processing. To address this issue, transgenic animals expressing fluorescently tagged LC3 have been developed, starting with the ubiquitously expressed GFP-fused LC3 mouse engineered by Mizushima et al. (179) and Dr. Joseph Hill’s cardiomyocyte-specific, α-myosin heavy chain (α-MHC) promoter-driven GFP-LC3 mouse (292). These transgenic animals harbor higher levels of LC3 in the heart, which aids significantly in immunological detection but does not eliminate the need for it despite the fluorescent tag. While autophagosomes can be directly observed as green puncta in these mice, they can be difficult to distinguish from protein aggregates generated by GFP-LC3 overexpression or autofluorescent structures, such as lipofuscin. Additionally, autolysosomes cannot be visualized by the GFP signal alone, as GFP is quenched within their acidic environment. Use of fluorophore with lower pKa, such as RFP, or mCherry, which is also more resistant to degradation, circumvents this issue (98).
Tandem reporter mice expressing two fluorophores with differing spectral properties have proven valuable in differentiating nucleation membranes and autophagosomes from autolysosomes (107, 147). Although the data remain only a snapshot of the number of autophagic components at a given time, quantification of components at earlier versus later stages of autophagy using these mice affords an estimate of flux through the system in what is otherwise a static method.
Further insight into the dynamic process of autophagy can be obtained by time-lapse imaging, which is possible by isolation of cardiomyocytes from these reporter animals or by labeling LC3 in in vitro cardiomyocyte models. Lipid or viral delivery of fluorescent-tagged LC3 has been successfully utilized for microscopy in HL-1 and H9c2 cell lines as well as isolated adult mouse cardiac myocytes and neonatal rat ventricular myocytes (NRVMs) (98, 124, 278, 285). Given that gene delivery to isolated cardiomyocytes in culture cannot be accomplished with high efficiency without the use of viral vectors (158), proper controls to account for cytotoxicity due to infection must be included. This involves optimization of multiplicity of infection to a level that maximizes the proportion of cells infected while maintaining cell viability. Because transfection alone can activate stress pathways and induce autophagy, deferring imaging for 24–48 h after infection can help decrease background. To fully account for this stress, we recommend the use of a control virus sharing the same sequence as the experimental vector, with the exclusion of LC3, for baseline comparison. Mutated versions of LC3 that are defective in lipidation or in interacting with ubiquitinated proteins can be substituted based on experimental goals (234). β-Gal substitution, with preservation of the chosen fluorescent protein(s) between the two viral vectors, is also feasible and commonly used (130). Because Atg4 cleaves the COOH-terminal region of LC3, care should be taken to attach the fluorescence tag to the NH2 terminus. Once the in vitro model is selected, the optimal time frame for expression, adequate for detection with low basal levels of tagged LC3, will need to be determined empirically. Furthermore, to establish that the fluorescence signal truly reflects intrinsic changes in autophagy, it is necessary to evaluate the ratio of endogenous to exogenous LC3 and demonstrate a proportional response to known inhibitors and inducers of autophagic flux using an orthogonal method, such as Western blotting.
Once the in vitro model is established, fluorescence microscopy allows for quantification of autophagic structures at the cell and tissue levels at multiple time points (Table 6). These data can be expressed in many different ways, including but not limited to 1) number of puncta per cell, 2) mean signal intensity per cell, 3) mean puncta size/volume, and 4) total number of cells with puncta (123). We caution against the use of all but the number of puncta per cell divided by the cell area for accurate interpretation of results. Samples from transgenic animals and transfected cardiomyocytes often display fluorescently tagged LC3 puncta even under basal conditions due to high expression levels, meaning a punctum-free state may not be achievable. Furthermore, fluorescence intensity may not directly correlate with levels of autophagy due to the formation of protein aggregates, nonspecific binding of antibodies, and autofluorescence of other cellular structures. Therefore, it is preferable to quantify the number of autophagic puncta with fluorescence intensity over a threshold set to account for the sources of noise listed above. If a tandem reporter system utilizing fluorophores with different pKa is used, colocalization analysis can be carried out to divide autophagic puncta into double-labeled autophagosome and single-labeled autolysosome pools. Although the results are less specific, colocalization analysis can also be performed on endogenous puncta by simultaneous labeling of autophagic and lysosomal structures via immunochemistry and/or commercially available dyes such as LysoTracker, acridine orange, or monodansylcadaverine (98, 174). Complementary data can be collected on the size, or volume in the case of confocal microscopy, of these puncta after verifying that lysosome fusion is intact, as failure in this process can also lead to enlarged puncta.
Table 6.
Evaluation of myocardial autophagy and features of autosis
| Method for Autophagy | In vitro | In vivo | Live Imaging? | Quantitation | Specificity | Flux | Technical difficulty |
|---|---|---|---|---|---|---|---|
| EM | Yes | Yes | No | No | High | No | High |
| Fluorescence microscopy | Yes | Yes | Yes | Yes | Medium-High | Possible with additional tags/treatment | Medium |
| LC3 immunoblotting | Yes | Yes | No | Semi | Low | Possible with additional treatment | Low |
| Labeled substrate degradation | Yes | Yes | No | Yes | Low, requires proteasome inhibition | Yes | Medium |
EM, electron microscropy; LC3, microtubule-associated proteins 1A/1B light chain 3. Features of autosis as follows. Early-stage autosis: 1) increased cell-substrate adherence, 2) nuclear membrane convolution and shrinkage, 3) dysmorphic endoplasmic reticulum. Late-stage autosis: 1) presence of a perinuclear space indicating separation of nuclear membranes focally; 2) ER disappearance; 3) swollen mitochondria.
Out of the four steps of autophagy, 1) nucleation, 2) autophagosome formation, 3) autophagosome to lysosome fusion, and 4) cargo degradation within the autolysosome (Fig. 6A), microscopy-based methods outlined above primarily evaluate steps 2 and 3. Appropriate interpretation of the differences in the number of autophagic compartments requires evaluation of passage of materials through the entire system, as an increase in their steady-state abundance can arise through increased formation (increased flux) and/or decreased clearance (decreased flux). One of the most widely used approaches to distinguish between these possibilities is the deliberate inhibition of flux at the distal end of the flux pathway. A large number of chemicals that inhibit autophagy at each step have been developed (205), but chloroquine, bafilomycin A1, and 3-methyladenine (3-MA) remain the most commonly used in cardiac research (174, 220, 285).
Unlike IHC/ICC, SDS-PAGE-based methods can differentiate between these two isoforms, because the PE moiety of LC3-II increases its hydrophobicity and, despite its larger size, allows it to migrate faster than LC3-I. Given that LC3-II is incorporated into autophagosomes, changes in LC3-II levels as quantified via densitometric analysis of blots are indicative of changes in the number of autophagosomes (Table 6). Alternatively, Western blotting can be used with inhibitors of autophagy to monitor the turnover of p62/SQSTM1, a receptor of polyubiquitinated proteins that interacts with LC3 and is incorporated in autophagosomes to be degraded alongside the protein cargo (202). It should be noted that, because autophagy is an established but not exclusive means of p62 degradation in cardiomyocytes, interpretation of results may be difficult. Furthermore, because expression of p62 can change in response to cellular stress, p62 mRNA levels should be measured in the same samples or synthesis blocked with cycloheximide.
Another approach to assessing autophagic flux involves measuring the breakdown of labeled substances (Table 6). Although diverse methods have been developed for various mammalian cell lines and yeast, the turnover of radioactively labeled long-lived proteins is the most commonly used method in cardiomyocytes (123). Radiolabeled amino acids such as [14C] valine or [3H]leucine are added to the cell culture medium during a pulse period to be incorporated into newly synthesized proteins, followed by a chase phase without radioactivity to allow for the degradation of short-lived proteins by the proteasome. Afterward, the release of radiolabeled amino acids from long-lived proteins being broken down by autophagy can be quantified as acid-soluble radioactivity at multiple time points (143). Further specificity can be obtained by addition of autophagy and proteasome inhibitors to delineate their relative contributions, but caution should be exercised, as suppression of one degradative pathway may activate the other. Radiolabeling of proteins can also be coupled with quantitative mass spectrometry to measure the degradation of thousands of proteins at once (30). To avoid technical difficulties associated with radioactivity, methods utilizing alternative means of labeling newly synthesized proteins are being developed; stable isotope labeling with amino acids in cell culture (SILAC) has been successfully utilized in cardiomyocytes in vitro and in vivo (224, 275). Work adapting SILAC-based labeling to measure autophagic protein degradation has been accomplished in other cell types (128) but remains to be adapted to cardiomyocytes. For further information on the application of proteomics to study autophagy, readers are directed to a comprehensive review by Wong et al. (272).
Autophagy-Mediated Cell Death
Autophagy-mediated cell death, also known as autophagy-dependent cell death, is the only form of autophagic death recognized by the Nomenclature Committee on Cell Death, emphasizing its distinction from cell death with adaptive autophagy (73). As part of the cellular response to stress, autophagy is commonly upregulated in cardiomyocytes that may later undergo death by necrosis, necroptosis, or apoptosis. Therefore, implicating autophagy as the proximal cause of death, as opposed to a compensatory response occurring in the setting of other death-triggering events, requires exclusion of cell death by alternate pathways. Furthermore, causality between autophagic machinery involvement and regulated cell death must be established, preferably by multiple complementary methods.
To determine conclusively that autophagy is the cause of cell death in a given cardiomyocyte, downregulation of autophagy should promote survival. Chemical interventions described above to block flux can be used for this purpose and cell survival evaluated both in vivo and in vitro (7, 98, 143). However, all well-characterized inhibitors of autophagy lack specificity, a fact that must be taken into consideration when designing and interpreting these studies. For instance, lysosomotropic alkaline chloroquine can inhibit apoptosis as well as autophagy, and 3-MA has autophagy-independent effects as a class III PI 3-kinase inhibitor (205). On the other hand, genetic approaches can be tailored to target specific steps in the autophagic process individually or simultaneously. Induction of autophagy and phagophore formation has been successfully downregulated in cardiomyocytes in vitro by adenovirus-delivered short-hairpin RNA targeting beclin1 (124). siRNA has been used to target autophagosome formation by knocking down atg7 (285) or atg5 (220), and lysosome function has been downregulated using siRNA against tfeb (174, 199) and adenovirus against lamp2 (160). Whenever possible, knockout/knockdown studies should be performed targeting more than one gene, as compensation is possible, and some atgs are involved in cellular processes other than autophagy, including apoptosis (161). Knockdown should be quantitatively confirmed both at the transcript level by qRT-PCR and at the protein level by Western blotting.
Because of the essential role of autophagy in cellular homeostasis, full-body knockout animals may either not survive or exhibit multi-organ dysfunction that contributes secondarily to a cardiac phenotype, as seen with atg5−/− and atg7−/− mice (126, 131, 212). Cardiomyocyte-specific phenotypes can be assessed by floxing the gene in question and crossing these mice with a Cre line driven by a cardiomyocyte-specific promoter, e.g., α-MyHC-Cre. Use of various Cre lines, such as MLC2v-Cre and α-MyHC-MerCreMer, also allows for evaluation of the role of a given gene at various developmental stages (189). For all knockout animal studies involving a lox-Cre system, controls should include Flox+/+ Cre−/− and Flox−/− Cre+/− littermates, as either of these genetic manipulations could elicit a baseline phenotype, depending on the lox insertion site. In addition, if an inducible system that is activated by drug delivery is used, experimental groups should include each genotype receiving treatment or the vehicle control, as α-MyHC-MerCreMer mice exposed to high doses of tamoxifen develop dilated cardiomyopathy (21). If knockout studies prove technically challenging, other strategies include knocking down the gene in question with lentiviral injection (285) or utilizing a haploinsufficiency model (292).
Conversely, if autophagy is truly responsible for cardiomyocyte death, its upregulation should hasten cell death. This can be achieved both in vivo and in vitro by starvation or chemical induction, most notably with rapamycin, the canonical inhibitor of mammalian target of rapamycin. Other agents known to activate autophagy in cardiomyocytes include AMPK activators, Sirt1 activators, and trehalose (225), all of which also elicit nonautophagic events. Similarly, hypoxia and exercise can upregulate autophagy, along with many other cellular processes, and therefore, they should be used with caution in studies testing for a causal role of autophagy in cell death. More targeted approaches with genetic overexpression of autophagic machinery elements include adenoviral overexpression of beclin1 (124) in NRVM and atg7 or beclin1 plasmid transfection in HL-1 cells (24, 285). In vivo techniques include overexpression by adeno-associated virus-9 (AAV9) delivery into the jugular vein of mice (220) and Beclin transgenic mice (292). Also, it should be noted that fluoro-tagged LC3 mouse models described above, such as CAG-RFP-GFP-LC3 transgenic mice, although initially designed for evaluating flux, also manifest inherently higher expression of these components and should be assumed to have increased autophagy at baseline unless it is shown otherwise experimentally. Finally, mice receiving an intraperitoneal injection of the Tat-Beclin-1 peptide, comprising HIV-1 Tat protein transduction domain linked to an 18-amino acid fragment of Beclin1, manifest elevated levels of autophagy in heart among other organs (233).
Autosis
Recently, studies employing Tat-Beclin have uncovered a previously unrecognized form of autophagy-dependent cell death termed autosis (155). In addition to the presence of numerous autophagosomes and autolysosomes indicative of autophagy, additional, distinct, morphological features mark autosis; cells in early stages of autosis are characterized by increased cell substrate adherence, nuclear membrane convolution and shrinkage, and dysmorphic endoplasmic reticulum, and at later stages a perinuclear space is observed, indicative of separation of the nuclear membranes focally (Table 6). These changes in nuclear membrane morphology may arise due to the involvement of the Na+,K+-ATPase (Fig. 6B), whose inhibition by cardiac glycosides or α1-subunit knockdown prevent autosis (154). In addition to Tat-Beclin, starvation and hypoxia-ischemia have been shown to trigger autosis in the brain in vivo and in vitro. The role of autosis in cardiomyocyte death remains to be elucidated. Several methods to determine autophagy and autosis are described in Table 6.
Recommendation for Evaluating Autophagic Cell Death
We recommend that the autophagy process should be blocked to conclusively determine autophagic cell death or autosis. Different inhibitors are described above to inhibit autophagy. If blocking autophagy, both in vivo and in vitro, increases survival of myocardial cells, then it can be concluded that autophagic cell death/autosis is involved in the myocardial cell death. The characteristic features of autosis are described in Table 6.
OVERALL DISCUSSION AND CONCLUSIONS
Myocardial cell death is a critical step in the pathophysiology of cardiovascular disease. However, there is a gap in knowledge on how and why a particular form of cell death in preferred in a cardiac disease. Understanding the specific cell death mechanism in cardiomyopathies is important for increasing our fundamental knowledge on how myocardial cells die due to different insults and for developing therapeutic strategy by targeting specific cell death mechanism. One critical step in evaluating cell death in cardiomyopathies is correctly measuring different forms of cell death. It is important to notice limitations of methods during myocardial cell death assessment. Equally important is to pay attention to the common and the distinct/characteristic features of cell death when evaluating myocardial cell death because several forms of cell death can share common features (Fig. 8).
Fig. 8.
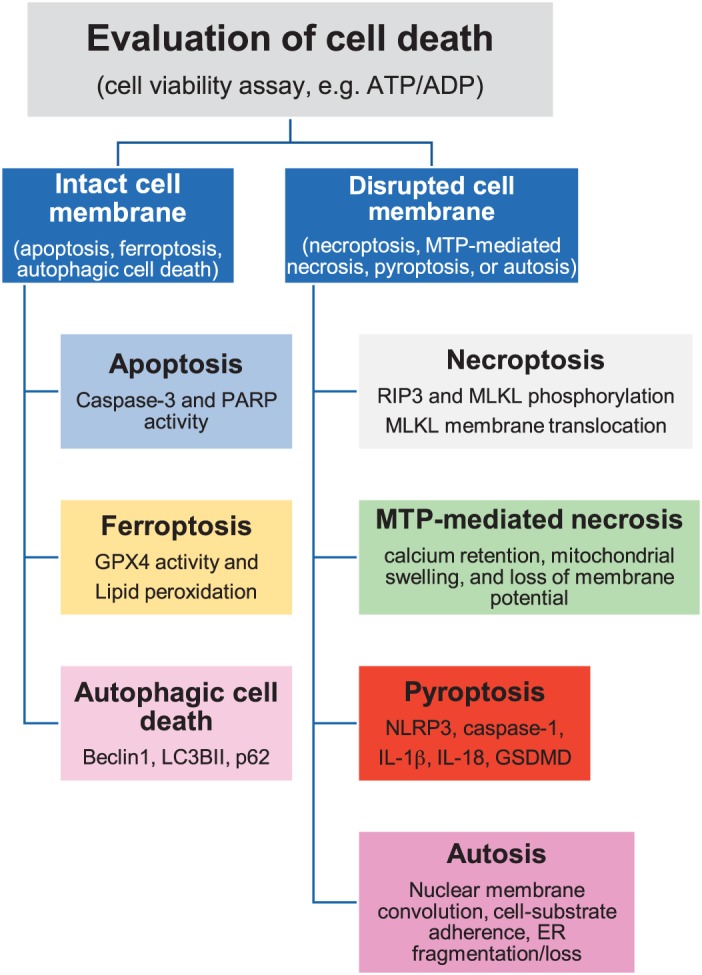
Evaluation of myocardial cell death. Different steps to distinguish and identify cell death mechanisms in the heart. GPX4, glutathione peroxidase 4; GSDMD, gasdermine D; LC3BII microtubule-associated proteins 1A/1B light chain 3BII; MLKL, mixed-lineage kinase domain-like; MTP, mitochondrial permeability transition pore; NLPR3, NACHT, LRR, and PYD domains-containing protein 3; PARP, poly-ADP ribose polymerase; RIP3, receptor-interacting protein kinase 3.
It is plausible that more than one form of cell death can occur in the heart during cardiomyopathies, and there can be cross-talk among these forms such as necroptosis can, which can induce pyroptosis via DAMPs (Fig. 7). Availability of ATP could be a deciding factor in initiating specific cell death signaling such as MPT necrosis in absence of ATP and apoptosis in presence of ATP (Fig. 7). One precursor can initiate more than one form of cell death, such as complex I can signal for both apoptosis and necrosis, and oxidative stress can trigger multiple forms of cell death, including apoptosis, MPT necrosis, pyroptosis, and ferroptosis (Fig. 7). Therefore, it is necessary to determine the specific forms of cell death by using its characteristic features and to understand its shared features with other cell death mechanisms. We have elaborated different approaches and techniques to measure characteristic and overlapping features of myocardial cell death mechanisms in Table 7.
Fig. 7.

Interactions between 6 forms of myocardial cell death. The nature of the signals in the myocardium determines the cell death pathway a cell undergoes. However, oxidative damage to mitochondria can trigger multiple cell death pathways. Different colors denote different forms of cell death. DAMPs, damage-associated molecular patterns; LC3, microtubule-associated proteins 1A/1B light chain 3; MPT, Mitochondrial permeability transition; ROS, reactive oxygen species.
Table 7.
Methods to evaluate myocardial cell death
| Approach (Technique/Method) | Assessment | Cell Death Mechanism |
|---|---|---|
| Imaging | ||
| Observation of cell morphology under a microscope | Cell shrinkage, chromatin condensation | Apoptosis |
| TUNEL assay | DNA fragmentation | Apoptosis, necroptosis/necrosis, normal DNA repair |
| Annexin V staining with/without vital dye, duramycin with/without vital dye | Exteriorization of phosphatidylserine; exteriorization of phosphatidylethanolamine | No vital dye uptake apoptosis, vital dye uptake, necroptosis/necrosis, pyroptosis |
| Annexin V and PI staining | Diffused PI staining and intense Annexin V fluorescence; spots of PI with Annexin V-positive blebs | Necroptosis, late apoptosis/secondary necrosis |
| Immunofluorescence for necrosome formation | Co-localization of RIP3 and MLKL or RIP1-RIP3 | Necroptosis |
| Immunohistochemical/immunocytochemical staining of MLKL | Membrane translocation of MLKL | Necroptosis |
| Decreased ΔΨm | Intensity of TMRE, JC-1, Rhodamine 123 | Necroptosis, MPT-mediated necrosis |
| Decreased calcium retention capacity | Change in the intensity of calcium fluorescence | MPT-mediated necrosis |
| Observation of isolated cardiac mitochondria under a microscope | Mitochondrial swelling | MPT-mediated necrosis, autosis |
| Immunofluorescence of ASC, cleaved caspase-1 | Measurement of fluorescence intensity | Pyroptosis |
| Immunofluorescence of mitoSOX | Measurement of fluorescence intensity | Ferroptosis, necroptosis/necrosis |
| Observing cell under microscope | Increased substrate adherence, ER fragmentation/loss, nuclear convolution and shrinkage, increased space between nuclear membranes, focal expansion of nuclear membrane | Autosis |
| Immunofluorescence of LC3-GFP | LC3B-GFP puncta intensity/cell area | Autophagy cell death, autosis |
| TTC staining | Viable myocardium red, unstained tissue non-viable | Necroptosis/necrosis, pyroptosis, other cell death types |
| Molecular/biochemical | ||
| LDH assay | Colorimetric analyses of releases of cytoplasmic protein LDH | Pyroptosis, necroptosis/necrosis |
| DNA laddering by agarose gel electrophoresis | DNA fragmentation | Apoptosis, necroptosis/necrosis, normal DNA repair |
| Western blotting for caspase-3 activity by | Cleaved to total caspase-3 protein | Apoptosis |
| Western blotting for PARP activity | Cleaved to total PARP protein | Apoptosis |
| Western blotting for BID | Expression of BID protein | Receptor-mediated apoptosis |
| Western blotting for RIP3 and MLKL | Phosphorylated to total protein | Necroptosis |
| Western blotting for MLKL oligomerization | Expression of 160- to 300-kDa proteins of MLKL tetramers and octamers | Necroptosis |
| Coimmunoprecipitation of RIP1-RIP3 and RIP3-MLKL | Protein-protein interactions | Necroptosis |
| Cellular fractionation and Western blotting of MLKL | MLKL translocation to membrane by measuring the levels of MLKL in the membrane and cytosolic fractions | Necroptosis |
| QPCR and Western blotting for NLRP3 | mRNA and protein expression of NLRP3 | Pyroptosis |
| Western blotting for caspase-1 activity | Cleaved to total caspase-1 | Pyroptosis |
| Western blotting for IL-1β, IL-18, and GSDMD activity | Cleaved to total IL-1β, IL-18, and GSDMD | Pyroptosis |
| Western blotting for GSH, GPX4 | Protein levels of GSH, GPX4 | Ferroptosis |
| Western blotting for LC3-I and LC3-II | Protein levels of LC3-I and LC3-II | Autophagy |
Because only 80–250 myocytes per 105 nuclei undergo apoptosis in the failing human heart (270), it is likely that apoptosis may not be the predominant form of cell death in the failing human heart. There is a possibility of contribution of other forms of cell death, and thus understanding their roles in cardiomyopathies is valuable. The predominant cell death mechanism can vary in different types of cardiomyopathies, based on their microenvironment and the intrinsic factors. Thus, targeting the predominant form of cell death is crucial for developing an intervention approach for myocardial cell death in a specific cardiomyopathy.
To evaluate cell death, it is important to first assess whether the cells are dying by using a viability assay such as ATP/ADP ratio. Depending on cell membrane integrity, cell death mechanisms can be broadly categorized into two groups: 1) with intact cell membranes such as apoptosis, ferroptosis and autophagic cell death; and 2) necroptosis, mitochondrial permeability transition pore (MTP)-mediated necrosis, necroptosis, pyroptosis, and autosis with ruptured cell membranes. Thus, determining the cell membrane integrity could help only as an initial screen for cell death mechanisms. In each category, further assessment of the characteristic features will delineate the specific form of cell death (Fig. 8).
We have provided a brief characterization of six forms of myocardial cell death (Figs. 1–8) and elaborated different methods to evaluate key features of cell death (Tables 1–7). As indicated in several sections of this paper, cell death is a pivotal step in cardiac pathology, and developing guidelines to evaluate myocardial cell death is crucial for determining the cause of cardiomyopathies. Here, we have elaborated various approaches to measure apoptosis, necroptosis, MPT-mediated necrosis, pyroptosis, ferroptosis, and autophagic cell death/autosis. We have explained the concept of a particular method of myocardial cell death, different methods to assess myocardial cell death, limitations of these methods, and the best practices to evaluate a myocardial cell death mechanism. This article is intended to provide guidelines to evaluate myocardial cell death in cardiomyopathies using the best approaches, assessments of the data, and interpretations of the results, which will reduce the variability in results obtained from different laboratories in the world.
GRANTS
This work is supported by National Institutes of Health Grants HL-113281 and HL-116205 to P. K. Mishra; APVV-15-607, VEGA-1/0271/16, and 1/0016/20 to A. Adameova; HL-120732, HL-128215, and HL-126012 to J. A. Hill; HL-136816 to A. Abbate; and HL-094404 to CPB and P20GM113134 to T. Matsui.
DISCLOSURES
A. Abbate has served as a consultant to Astra-Zeneca, Janssen, Merck, Novartis, Olatec and Serpin pharma.
AUTHOR CONTRIBUTIONS
P.K.M. conceived and designed research; P.K.M. and S.K. prepared figures; P.K.M., A. Adameova, J.A.H., C.P.B., P.M.K., J.D., J.N., M.T., A. Abbate, H.C.P., S.S., J.K.H., N.K.K., and T.M. drafted manuscript; P.K.M., A. Adameova, J.A.H., C.P.B., P.M.K., J.D., J.N., M.T., A. Abbate, H.C.P., S.K., S.S., J.K.H., N.K.K., and T.M. edited and revised manuscript; P.K.M., A. Adameova, J.A.H., C.P.B., P.M.K., J.D., J.N., M.T., A. Abbate, H.C.P., S.K., S.S., J.K.H., N.K.K., and T.M. approved final version of manuscript.
REFERENCES
- 1.Abbate A, De Falco M, Morales C, Gelpi RJ, Prisco M, De Luca A, Palleiro J, Fedele V, Feroce F, Baldi F, Vetrovec GW, Baldi A. Electron microscopy characterization of cardiomyocyte apoptosis in ischemic heart disease. Int J Cardiol 114: 118–120, 2007. doi: 10.1016/j.ijcard.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 2.Abramov AY, Duchen MR. Measurements of threshold of mitochondrial permeability transition pore opening in intact and permeabilized cells by flash photolysis of caged calcium. Methods Mol Biol 793: 299–309, 2011. doi: 10.1007/978-1-61779-328-8_19. [DOI] [PubMed] [Google Scholar]
- 3.Adameova A, Goncalvesova E, Szobi A, Dhalla NS. Necroptotic cell death in failing heart: relevance and proposed mechanisms. Heart Fail Rev 21: 213–221, 2016. doi: 10.1007/s10741-016-9537-8. [DOI] [PubMed] [Google Scholar]
- 4.Adameova A, Hrdlicka J, Szobi A, Farkasova V, Kopaskova K, Murarikova M, Neckar J, Kolar F, Ravingerova T, Dhalla NS. Evidence of necroptosis in hearts subjected to various forms of ischemic insults. Can J Physiol Pharmacol 95: 1163–1169, 2017. doi: 10.1139/cjpp-2016-0609. [DOI] [PubMed] [Google Scholar]
- 5.Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, Agarwal A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314: F702–F714, 2018. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep 8: 5155, 2018. doi: 10.1038/s41598-018-23408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aki T, Yamaguchi K, Fujimiya T, Mizukami Y. Phosphoinositide 3-kinase accelerates autophagic cell death during glucose deprivation in the rat cardiomyocyte-derived cell line H9c2. Oncogene 22: 8529–8535, 2003. doi: 10.1038/sj.onc.1207197. [DOI] [PubMed] [Google Scholar]
- 8.Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Res Cardiol 93, Suppl 3: 8–12, 1998. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- 9.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol 38: 367–374, 2005. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Audia JP, Yang XM, Crockett ES, Housley N, Haq EU, O’Donnell K, Cohen MV, Downey JM, Alvarez DF. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol 113: 32, 2018. doi: 10.1007/s00395-018-0692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H, Matsui T. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol 314: H659–H668, 2018. doi: 10.1152/ajpheart.00452.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai YT, Chang R, Wang H, Xiao FJ, Ge RL, Wang LS. ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem Biophys Res Commun 499: 44–51, 2018. doi: 10.1016/j.bbrc.2018.03.113. [DOI] [PubMed] [Google Scholar]
- 13.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 14.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24: 669–681, 1992. doi: 10.1016/0022-2828(92)93381-S. [DOI] [PubMed] [Google Scholar]
- 16.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 280: 18558–18561, 2005. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 17.Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J Morphol 154: 39–57, 1977. doi: 10.1002/jmor.1051540104. [DOI] [PubMed] [Google Scholar]
- 18.Beilharz M, De Nardo D, Latz E, Franklin BS. Measuring NLR oligomerization II: detection of ASC speck formation by confocal microscopy and immunofluorescence. Methods Mol Biol 1417: 145–158, 2016. [Erratum in: Methods Mol Biol 1417: E1, 2016.] doi: 10.1007/978-1-4939-3566-6_9. [DOI] [PubMed] [Google Scholar]
- 19.Belizário J, Vieira-Cordeiro L, Enns S. Necroptotic cell death signaling and execution pathway: lessons from knockout mice. Mediators Inflamm 2015: 1–15, 2015. doi: 10.1155/2015/128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kühn B. Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech 6: 1459–1469, 2013. doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boucher D, Chan A, Ross C, Schroder K. Quantifying caspase-1 activity in murine macrophages. Methods Mol Biol 1725: 163–176, 2018. doi: 10.1007/978-1-4939-7568-6_14. [DOI] [PubMed] [Google Scholar]
- 23.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, Schroder K. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 215: 827–840, 2018. doi: 10.1084/jem.20172222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J 274: 3184–3197, 2007. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 25.Briston T, Selwood DL, Szabadkai G, Duchen MR. Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends Pharmacol Sci 40: 50–70, 2019. doi: 10.1016/j.tips.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Bulluck H, Rosmini S, Abdel-Gadir A, White SK, Bhuva AN, Treibel TA, Fontana M, Ramlall M, Hamarneh A, Sirker A, Herrey AS, Manisty C, Yellon DM, Kellman P, Moon JC, Hausenloy DJ. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging 9: e004940, 2016. doi: 10.1161/CIRCIMAGING.116.004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16: 55–65, 2014. [Erratum in: Nat Cell Biol 16: 200, 2014.] doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai Z, Zhang A, Choksi S, Li W, Li T, Zhang XM, Liu ZG. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res 26: 886–900, 2016. doi: 10.1038/cr.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 73: 2195–2209, 2016. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cargile BJ, Bundy JL, Grunden AM, Stephenson JL Jr. Synthesis/degradation ratio mass spectrometry for measuring relative dynamic protein turnover. Anal Chem 76: 86–97, 2004. doi: 10.1021/ac034841a. [DOI] [PubMed] [Google Scholar]
- 31.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 20: 7342–7351, 2001. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 32.Chang HC, Wu R, Shang M, Sato T, Chen C, Shapiro JS, Liu T, Thakur A, Sawicki KT, Prasad SV, Ardehali H. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol Med 8: 247–267, 2016. doi: 10.15252/emmm.201505748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121, 2014. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem 277: 34793–34799, 2002. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 36.Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW, Petrova AV, Warren MD, Pantazides BG, Gandhi K, Kowalski J, Kooby DA, El-Rayes BF, Staley CA III, Adsay NV, Curran WJ Jr, Landry JC, Maithel SK, Yu DS. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 119: 3148–3155, 2013. doi: 10.1002/cncr.28144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins RJ, Harmon BV, Gobé GC, Kerr JF. Internucleosomal DNA cleavage should not be the sole criterion for identifying apoptosis. Int J Radiat Biol 61: 451–453, 1992. doi: 10.1080/09553009214551201. [DOI] [PubMed] [Google Scholar]
- 38.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, Masters SL, Murphy JM, Schroder K, Vaux DL, Lawlor KE, Lindqvist LM, Vince JE. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA 114: E961–E969, 2017. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366, 2016. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol 2: e995047, 2015. doi: 10.4161/23723556.2014.995047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res 95: 957–970, 2004. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 42.Cruz SA, Qin Z, Stewart AFR, Chen HH. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen Res 13: 252–256, 2018. doi: 10.4103/1673-5374.226394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cummings BS, Schnellmann RG. Measurement of cell death in mammalian cells. Curr Protoc Pharmacol Chapter 12: Unit 12.8, 2004. doi: 10.1002/0471141755.ph1208s25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med 48: 306–317, 2010. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 28: 435–492, 1966. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 46.de Montalembert M, Ribeil JA, Brousse V, Guerci-Bresler A, Stamatoullas A, Vannier JP, Dumesnil C, Lahary A, Touati M, Bouabdallah K, Cavazzana M, Chauzit E, Baptiste A, Lefebvre T, Puy H, Elie C, Karim Z, Ernst O, Rose C. Cardiac iron overload in chronically transfused patients with thalassemia, sickle cell anemia, or myelodysplastic syndrome. PLoS One 12: e0172147, 2017. doi: 10.1371/journal.pone.0172147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell 138: 229–232, 2009. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 48.Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol 52: 48–61, 2012. doi: 10.1016/j.yjmcc.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313–321, 2008. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119, 2005. [Erratum in: Nat Chem Biol 1: 234, 2005.] doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 51.Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20: 366, 2013. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis 3: e338, 2012. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 297: F749–F759, 2009. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 54.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571–2575, 2001. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 55.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Reports 1: 401–407, 2012. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dive C, Gregory CD, Phipps DJ, Evans DL, Milner AE, Wyllie AH. Analysis and discrimination of necrosis and apoptosis (programmed cell death) by multiparameter flow cytometry. Biochim Biophys Acta 1133: 275–285, 1992. doi: 10.1016/0167-4889(92)90048-G. [DOI] [PubMed] [Google Scholar]
- 57.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072, 2012. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Reports 7: 971–981, 2014. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 59.Dong W, Li Z, Chen Y, Zhang L, Ye Z, Liang H, Li R, Xu L, Zhang B, Liu S, Wang W, Li C, Luo J, Shi W, Liang X. NADPH oxidase inhibitor, diphenyleneiodonium prevents necroptosis in HK-2 cells. Biomed Rep 7: 226–230, 2017. doi: 10.3892/br.2017.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drummen GP, van Liebergen LC, Op den Kamp JA, Post JA. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic Biol Med 33: 473–490, 2002. doi: 10.1016/S0891-5849(02)00848-1. [DOI] [PubMed] [Google Scholar]
- 61.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22: 245–257, 2006. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 62.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 77: 1111–1122, 2013. doi: 10.1253/circj.CJ-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA 116: 2672–2680, 2019. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M, Gridling M, Parapatics K, Colinge J, Bennett KL, Kubicek S, Krautwald S, Linkermann A, Superti-Furga G. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6: e1767, 2015. doi: 10.1038/cddis.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol 430: 3068–3080, 2018. doi: 10.1016/j.jmb.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19: 2056–2067, 2007. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 67.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43: 449–463, 2011. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907–1916, 2005. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8: 1812–1825, 2006. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 70.Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26: 99–114, 2019. doi: 10.1038/s41418-018-0212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujikura K, Golive AD, Ando T, Corado FM, Shitole SG, Kizer JR, Shah AM, Prince MR, Spevack DM, Garcia MJ. Increased iron deposition is directly associated with myocardial dysfunction in patients with sickle cell disease. JACC Cardiovasc Imaging 11: 279–280, 2018. doi: 10.1016/j.jcmg.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 35: 24–32, 2014. doi: 10.1016/j.semcdb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541, 2018. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 59: 298–308, 2015. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao X, Qian M, Campian JL, Marshall J, Zhou Z, Roberts AM, Kang YJ, Prabhu SD, Sun XF, Eaton JW. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic Biol Med 49: 401–407, 2010. doi: 10.1016/j.freeradbiomed.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gerencser AA, Doczi J, Töröcsik B, Bossy-Wetzel E, Adam-Vizi V. Mitochondrial swelling measurement in situ by optimized spatial filtering: astrocyte-neuron differences. Biophys J 95: 2583–2598, 2008. doi: 10.1529/biophysj.107.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghardashi Afousi A, Gaeini A, Rakhshan K, Naderi N, Darbandi Azar A, Aboutaleb N. Targeting necroptotic cell death pathway by high-intensity interval training (HIIT) decreases development of post-ischemic adverse remodelling after myocardial ischemia / reperfusion injury. J Cell Commun Signal 13: 255–267, 2019. doi: 10.1007/s12079-018-0481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giricz Z, Koncsos G, Rajtík T, Varga ZV, Baranyai T, Csonka C, Szobi A, Adameová A, Gottlieb RA, Ferdinandy P. Hypercholesterolemia downregulates autophagy in the rat heart. Lipids Health Dis 16: 60, 2017. [Erratum in: Lipids Health Dis 16: 133, 2017.] doi: 10.1186/s12944-017-0455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res 108: 1122–1132, 2011. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 80.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther 16: 233–238, 2011. doi: 10.1177/1074248411409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307: 93–98, 1995. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol 25: 1461–1469, 1993. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 83.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol 292: C45–C51, 2007. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 84.Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol 72: 316–325, 2014. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78: 129–141, 2015. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 86.Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong X, Eidam PM, Finger JN, Hoffman SJ, Kang J, Kasparcova V, King BW, Lehr R, Lan Y, Leister LK, Lich JD, MacDonald TT, Miller NA, Ouellette MT, Pao CS, Rahman A, Reilly MA, Rendina AR, Rivera EJ, Schaeffer MC, Sehon CA, Singhaus RR, Sun HH, Swift BA, Totoritis RD, Vossenkämper A, Ward P, Wisnoski DD, Zhang D, Marquis RW, Gough PJ, Bertin J. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem 60: 1247–1261, 2017. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 87.He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci USA 114: 9086–9091, 2017. doi: 10.1073/pnas.1711201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci USA 114: 3409–3414, 2017. doi: 10.1073/pnas.1702357114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 108: 20054–20059, 2011. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41: 1012–1021, 2016. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hengartner MO. The biochemistry of apoptosis. Nature 407: 770–776, 2000. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 92.Herrmann M, Lorenz HM, Voll R, Grünke M, Woith W, Kalden JR. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res 22: 5506–5507, 1994. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hikoso S, Yamaguchi O, Nakano Y, Takeda T, Omiya S, Mizote I, Taneike M, Oka T, Tamai T, Oyabu J, Uno Y, Matsumura Y, Nishida K, Suzuki K, Kogo M, Hori M, Otsu K. The IkappaB kinase beta/nuclear factor kappaB signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circ Res 105: 70–79, 2009. doi: 10.1161/CIRCRESAHA.108.193318. [DOI] [PubMed] [Google Scholar]
- 94.Hou H, Wang Y, Li Q, Li Z, Teng Y, Li J, Wang X, Chen J, Huang N. The role of RIP3 in cardiomyocyte necrosis induced by mitochondrial damage of myocardial ischemia-reperfusion. Acta Biochim Biophys Sin (Shanghai) 50: 1131–1140, 2018. doi: 10.1093/abbs/gmy108. [DOI] [PubMed] [Google Scholar]
- 95.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12: 1425–1428, 2016. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang D, Zheng X, Wang ZA, Chen X, He WT, Zhang Y, Xu JG, Zhao H, Shi W, Wang X, Zhu Y, Han J. The MLKL channel in necroptosis is an octamer formed by tetramers in a dyadic process. Mol Cell Biol 37: e00497–e00417, 2017. doi: 10.1128/MCB.00497-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ishii T, Warabi E, Mann GE. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic Biol Med 133: 169–178, 2019. doi: 10.1016/j.freeradbiomed.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 98.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy 4: 322–329, 2008. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ, van Delft MF, Liu Z, Conos SA, Zhang JG, Huang DC, Silke J, Lessene G, Murphy JM. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis 7: e2051, 2016. doi: 10.1038/cddis.2015.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jeitner TM. Optimized ferrozine-based assay for dissolved iron. Anal Biochem 454: 36–37, 2014. doi: 10.1016/j.ab.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 101.Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, Diederich WE, Culmsee C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med 117: 45–57, 2018. doi: 10.1016/j.freeradbiomed.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 102.Jing L, Song F, Liu Z, Li J, Wu B, Fu Z, Jiang J, Chen Z. MLKL-PITPα signaling-mediated necroptosis contributes to cisplatin-triggered cell death in lung cancer A549 cells. Cancer Lett 414: 136–146, 2018. doi: 10.1016/j.canlet.2017.10.047. [DOI] [PubMed] [Google Scholar]
- 103.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728, 2000. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90, 2017. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288: 31268–31279, 2013. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471: 368–372, 2011. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T, Mizushima N. An autophagic flux probe that releases an internal control. Mol Cell 64: 835–849, 2016. doi: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 108.Kang PM, Izumo S. Apoptosis in heart failure: is there light at the end of the tunnel (TUNEL)? J Card Fail 6: 43–46, 2000. doi: 10.1016/S1071-9164(00)80005-6. [DOI] [PubMed] [Google Scholar]
- 109.Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol Med 9: 177–182, 2003. doi: 10.1016/S1471-4914(03)00025-X. [DOI] [PubMed] [Google Scholar]
- 110.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38: 27–40, 2013. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 111.Kavuri SM, Geserick P, Berg D, Dimitrova DP, Feoktistova M, Siegmund D, Gollnick H, Neumann M, Wajant H, Leverkus M. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J Biol Chem 286: 16631–16646, 2011. doi: 10.1074/jbc.M110.148585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594–604, 2011. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 113.Kawai H, Chaudhry F, Shekhar A, Petrov A, Nakahara T, Tanimoto T, Kim D, Chen J, Lebeche D, Blankenberg FG, Pak KY, Kolodgie FD, Virmani R, Sengupta P, Narula N, Hajjar RJ, Strauss HW, Narula J. Molecular imaging of apoptosis in ischemia reperfusion injury with radiolabeled duramycin targeting phosphatidylethanolamine: effective target uptake and reduced nontarget organ radiation burden. JACC Cardiovasc Imaging 11: 1823–1833, 2018. doi: 10.1016/j.jcmg.2017.11.037. [DOI] [PubMed] [Google Scholar]
- 114.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8: 297–303, 1998. doi: 10.1016/S1074-7613(00)80535-X. [DOI] [PubMed] [Google Scholar]
- 115.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257, 1972. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim CR, Kim JH, Park HL, Park CK. Ischemia Reperfusion Injury Triggers TNFα Induced-Necroptosis in Rat Retina. Curr Eye Res 42: 771–779, 2017. doi: 10.1080/02713683.2016.1227449. [DOI] [PubMed] [Google Scholar]
- 117.Kim H, Zamel R, Bai XH, Lu C, Keshavjee S, Keshavjee S, Liu M. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol 315: L595–L608, 2018. doi: 10.1152/ajplung.00069.2018. [DOI] [PubMed] [Google Scholar]
- 118.Kim S, Dayani L, Rosenberg PA, Li J. RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int J Physiol Pathophysiol Pharmacol 2: 137–147, 2010. [PMC free article] [PubMed] [Google Scholar]
- 119.Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, Monette S, Pauliah M, Gonen M, Zanzonico P, Quinn T, Wiesner U, Bradbury MS, Overholtzer M. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11: 977–985, 2016. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 26: 675–687, 2007. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 121.Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res 87: 392–398, 2000. doi: 10.1161/01.RES.87.5.392. [DOI] [PubMed] [Google Scholar]
- 122.Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog 11: e1004820, 2015. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algul H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, , et al. . Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem 285: 793–804, 2010. doi: 10.1074/jbc.M109.070037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465, 2004. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425–434, 2005. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27: 673–684, 2017. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kristensen AR, Schandorff S, Høyer-Hansen M, Nielsen MO, Jäättelä M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics 7: 2419–2428, 2008. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- 129.Krysko DV, Vanden Berghe T, D’Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods 44: 205–221, 2008. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 130.Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, Gustafsson AB. pink1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS One 10: e0130707, 2015. doi: 10.1371/journal.pone.0130707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 132.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6: 24393–24403, 2015. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 21: 206–214, 2015. doi: 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Laker RC, Taddeo EP, Akhtar YN, Zhang M, Hoehn KL, Yan Z. The Mitochondrial Permeability Transition Pore Regulator Cyclophilin D Exhibits Tissue-Specific Control of Metabolic Homeostasis. PLoS One 11: e0167910, 2016. doi: 10.1371/journal.pone.0167910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta, Gen Subj 1861: 1893–1900, 2017. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 136.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW, Silke J, Vaux DL, Vince JE. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6: 6282, 2015. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lecoeur H, Prévost MC, Gougeon ML. Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: a reconsideration of the specificity of the annexin V/propidium iodide assay. Cytometry 44: 65–72, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 138.Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer Res 16: 1073–1076, 2018. doi: 10.1158/1541-7786.MCR-18-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee YS, Park KM, Yu L, Kwak HH, Na HJ, Kang KS, Woo HM. Necroptosis is a mechanism of death in mouse induced hepatocyte-like cells reprogrammed from mouse embryonic fibroblasts. Mol Cells 41: 639–645, 2018. doi: 10.14348/molcells.2018.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Leers MP, Kölgen W, Björklund V, Bergman T, Tribbick G, Persson B, Björklund P, Ramaekers FC, Björklund B, Nap M, Jörnvall H, Schutte B. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J Pathol 187: 567–572, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 141.Levi S, Rovida E. The role of iron in mitochondrial function. Biochim Biophys Acta 1790: 629–636, 2009. doi: 10.1016/j.bbagen.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 142.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The cystine/glutamate antiporter system xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18: 522–555, 2013. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, Hill JA. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133: 1668–1687, 2016. doi: 10.1161/CIRCULATIONAHA.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem 281: 13636–13643, 2006. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 145.Li J, Yin HL, Yuan J. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J Cell Biol 181: 321–333, 2008. doi: 10.1083/jcb.200711082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5: e1278, 2014. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li L, Wang ZV, Hill JA, Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol 25: 305–315, 2014. doi: 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, Liu X, Hassan A, Tanaka S, Cicka M, Hsiao HM, Ruiz-Perez D, Bredemeyer A, Gross RW, Mann DL, Tyurina YY, Gelman AE, Kagan VE, Linkermann A, Lavine KJ, Kreisel D. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest 129: 2293–2304, 2019. doi: 10.1172/JCI126428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lichý M, Szobi A, Hrdlička J, Horváth C, Kormanová V, Rajtík T, Neckář J, Kolář F, Adameová A. Different signalling in infarcted and non-infarcted areas of rat failing hearts: A role of necroptosis and inflammation. J Cell Mol Med 23: 6429–6441, 2019. doi: 10.1111/jcmm.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther 21: 467–469, 2007. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81: 751–761, 2012. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 152.Linkermann A, Heller JO, Prókai A, Weinberg JM, De Zen F, Himmerkus N, Szabó AJ, Bräsen JH, Kunzendorf U, Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 24: 1545–1557, 2013. doi: 10.1681/ASN.2012121169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun 497: 233–240, 2018. doi: 10.1016/j.bbrc.2018.02.061. [DOI] [PubMed] [Google Scholar]
- 154.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22: 367–376, 2015. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA 110: 20364–20371, 2013. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu Z, Zhao M, Zhu X, Furenlid LR, Chen YC, Barrett HH. In vivo dynamic imaging of myocardial cell death using 99mTc-labeled C2A domain of synaptotagmin I in a rat model of ischemia and reperfusion. Nucl Med Biol 34: 907–915, 2007. doi: 10.1016/j.nucmedbio.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27, Suppl 1: S2–S19, 2008. doi: 10.1038/onc.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol 51: 288–298, 2011. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe HJ, Linkermann A, Wolf MJ, Rose-John S, Lüllmann-Rauch R, Adam D, Flögel U, Heikenwalder M, Luedde T, Frey N. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 103: 206–216, 2014. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 160.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 125: 3170–3181, 2012. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741–752, 2007. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 162.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56: 481–495, 2014. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol 63: 316–322, 2014. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, Nordio A, Carbone S, Mauro AG, Das A, Zalavadia AA, Halquist MS, Federici M, Van Tassell BW, Zhang S, Abbate A. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J Cardiovasc Pharmacol 66: 1–8, 2015. doi: 10.1097/FJC.0000000000000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Marechal X, Montaigne D, Marciniak C, Marchetti P, Hassoun SM, Beauvillain JC, Lancel S, Neviere R. Doxorubicin-induced cardiac dysfunction is attenuated by ciclosporin treatment in mice through improvements in mitochondrial bioenergetics. Clin Sci (Lond) 121: 405–413, 2011. doi: 10.1042/CS20110069. [DOI] [PubMed] [Google Scholar]
- 166.Marton J, Albert D, Wiltshire SA, Park R, Bergen A, Qureshi S, Malo D, Burelle Y, Vidal SM. Cyclosporine A Treatment Inhibits Abcc6-Dependent Cardiac Necrosis and Calcification following Coxsackievirus B3 Infection in Mice. PLoS One 10: e0138222, 2015. doi: 10.1371/journal.pone.0138222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mastrocola R, Collino M, Penna C, Nigro D, Chiazza F, Fracasso V, Tullio F, Alloatti G, Pagliaro P, Aragno M. Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid Med Cell Longev 2016: 1–12, 2016. doi: 10.1155/2016/3480637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matsumoto-Ida M, Akao M, Takeda T, Kato M, Kita T. Real-time 2-photon imaging of mitochondrial function in perfused rat hearts subjected to ischemia/reperfusion. Circulation 114: 1497–1503, 2006. doi: 10.1161/CIRCULATIONAHA.106.628834. [DOI] [PubMed] [Google Scholar]
- 169.Medhora M, Haworth S, Liu Y, Narayanan J, Gao F, Zhao M, Audi S, Jacobs ER, Fish BL, Clough AV. Biomarkers for radiation pneumonitis using noninvasive molecular imaging. J Nucl Med 57: 1296–1301, 2016. doi: 10.2967/jnumed.115.160291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Meerson FZ, Kagan VE, Kozlov YP, Belkina LM, Arkhipenko YV. The role of lipid peroxidation in pathogenesis of ischemic damage and the antioxidant protection of the heart. Basic Res Cardiol 77: 465–485, 1982. doi: 10.1007/BF01907940. [DOI] [PubMed] [Google Scholar]
- 171.Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, Pluhacek T, Spatenka J, Kovalcikova J, Drahota Z, Kautzner J, Pirk J, Houstek J. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail 19: 522–530, 2017. doi: 10.1002/ejhf.640. [DOI] [PubMed] [Google Scholar]
- 172.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol 57: 891–903, 2011. doi: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA 108: 19725–19730, 2011. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miceli C, Santin Y, Manzella N, Coppini R, Berti A, Stefani M, Parini A, Mialet-Perez J, Nediani C. Oleuropein aglycone protects against MAO-A-induced autophagy impairment and cardiomyocyte death through activation of TFEB. Oxid Med Cell Longev 2018: 1–13, 2018. doi: 10.1155/2018/8067592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190, 2003. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 176.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 14: 442–447, 2008. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mizushima N. Autophagy: process and function. Genes Dev 21: 2861–2873, 2007. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 179.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111, 2004. doi: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Montaigne D, Marechal X, Baccouch R, Modine T, Preau S, Zannis K, Marchetti P, Lancel S, Neviere R. Stabilization of mitochondrial membrane potential prevents doxorubicin-induced cardiotoxicity in isolated rat heart. Toxicol Appl Pharmacol 244: 300–307, 2010. doi: 10.1016/j.taap.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 181.Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci 35: 434–441, 2010. doi: 10.1016/j.tibs.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol 196: 407–415, 2016. doi: 10.4049/jimmunol.1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mott JL, Zhang D, Freeman JC, Mikolajczak P, Chang SW, Zassenhaus HP. Cardiac disease due to random mitochondrial DNA mutations is prevented by cyclosporin A. Biochem Biophys Res Commun 319: 1210–1215, 2004. doi: 10.1016/j.bbrc.2004.05.104. [DOI] [PubMed] [Google Scholar]
- 184.Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2: 2295–2301, 2007. doi: 10.1038/nprot.2007.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88: 581–609, 2008. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453, 2013. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 187.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 189.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624, 2007. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 190.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest 117: 2431–2444, 2007. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343: 1357–1360, 2014. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 192.O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol 17: 418–424, 2007. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367, 2011. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol 107: 270, 2012. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 195.Ohno M, Takemura G, Ohno A, Misao J, Hayakawa Y, Minatoguchi S, Fujiwara T, Fujiwara H. “Apoptotic” myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with In situ nick end-labeling. Circulation 98: 1422–1430, 1998. doi: 10.1161/01.CIR.98.14.1422. [DOI] [PubMed] [Google Scholar]
- 196.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med 336: 1131–1141, 1997. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 197.Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290: 11635–11648, 2015. doi: 10.1074/jbc.M115.646042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9: 1187–1194, 2003. doi: 10.1038/nm920. [DOI] [PubMed] [Google Scholar]
- 199.Pan B, Zhang H, Cui T, Wang X. TFEB activation protects against cardiac proteotoxicity via increasing autophagic flux. J Mol Cell Cardiol 113: 51–62, 2017. doi: 10.1016/j.yjmcc.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Panee J, Stoytcheva ZR, Liu W, Berry MJ. Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. J Biol Chem 282: 23759–23765, 2007. doi: 10.1074/jbc.M702267200. [DOI] [PubMed] [Google Scholar]
- 201.Pang X, Panee J, Liu X, Berry MJ, Chang SL, Chang L. Regional variations of antioxidant capacity and oxidative stress responses in HIV-1 transgenic rats with and without methamphetamine administration. J Neuroimmune Pharmacol 8: 691–704, 2013. doi: 10.1007/s11481-013-9454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145, 2007. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 203.Park HH, Park SY, Mah S, Park JH, Hong SS, Hong S, Kim YS. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp Mol Med 50: 125, 2018. doi: 10.1038/s12276-018-0152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Parra V, Rothermel BA. Calcineurin signaling in the heart: The importance of time and place. J Mol Cell Cardiol 103: 121–136, 2017. doi: 10.1016/j.yjmcc.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pasquier B. Autophagy inhibitors. Cell Mol Life Sci 73: 985–1001, 2016. doi: 10.1007/s00018-015-2104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Petit PX, Zamzami N, Vayssière JL, Mignotte B, Kroemer G, Castedo M. Implication of mitochondria in apoptosis. Mol Cell Biochem 174: 185–188, 1997. doi: 10.1023/A:1006848205880. [DOI] [PubMed] [Google Scholar]
- 207.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76: 725–734, 1999. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, Gough PJ, Aloi M, Cucchiara S. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol 109: 279–287, 2014. doi: 10.1038/ajg.2013.403. [DOI] [PubMed] [Google Scholar]
- 209.Pieroni L, Khalil L, Charlotte F, Poynard T, Piton A, Hainque B, Imbert-Bismut F. Comparison of bathophenanthroline sulfonate and ferene as chromogens in colorimetric measurement of low hepatic iron concentration. Clin Chem 47: 2059–2061, 2001. [PubMed] [Google Scholar]
- 210.Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 8: 603–619, 2016. doi: 10.18632/aging.100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Porter GA Jr, Beutner G. Cyclophilin D, somehow a master regulator of mitochondrial function. Biomolecules 8: 176, 2018. doi: 10.3390/biom8040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128: 931–946, 2007. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 213.Qu Y, Tang J, Wang H, Li S, Zhao F, Zhang L, Richard Lu Q, Mu D. RIPK3 interactions with MLKL and CaMKII mediate oligodendrocytes death in the developing brain. Cell Death Dis 8: e2629, 2017. doi: 10.1038/cddis.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 214.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell 61: 589–601, 2016. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165: 792–800, 2016. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 5: e1004, 2014. doi: 10.1038/cddis.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ribble D, Goldstein NB, Norris DA, Shellman YG. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol 5: 12, 2005. doi: 10.1186/1472-6750-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem 331: 370–375, 2004. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 219.Sallmann FR, Bourassa S, Saint-Cyr J, Poirier GG. Characterization of antibodies specific for the caspase cleavage site on poly(ADP-ribose) polymerase: specific detection of apoptotic fragments and mapping of the necrotic fragments of poly(ADP-ribose) polymerase. Biochem Cell Biol 75: 451–456, 1997. doi: 10.1139/o97-075. [DOI] [PubMed] [Google Scholar]
- 220.Santin Y, Sicard P, Vigneron F, Guilbeau-Frugier C, Dutaur M, Lairez O, Couderc B, Manni D, Korolchuk VI, Lezoualc’h F, Parini A, Mialet-Perez J. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal 25: 10–27, 2016. doi: 10.1089/ars.2015.6522. [DOI] [PubMed] [Google Scholar]
- 221.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 274: 1541–1548, 1999. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 222.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010, 2005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schütze S. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21: 415–428, 2004. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 224.Scholten A, Mohammed S, Low TY, Zanivan S, van Veen TA, Delanghe B, Heck AJ. In-depth quantitative cardiac proteomics combining electron transfer dissociation and the metalloendopeptidase Lys-N with the SILAC mouse. Mol Cell Proteomics 10: O111.008474, 2011. doi: 10.1074/mcp.O111.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sciarretta S, Zhai P, Volpe M, Sadoshima J. Pharmacological modulation of autophagy during cardiac stress. J Cardiovasc Pharmacol 60: 235–241, 2012. doi: 10.1097/FJC.0b013e3182575f61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 282: 36321–36329, 2007. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 227.Sharov VG, Todor AV, Imai M, Sabbah HN. Inhibition of mitochondrial permeability transition pores by cyclosporine A improves cytochrome C oxidase function and increases rate of ATP synthesis in failing cardiomyocytes. Heart Fail Rev 10: 305–310, 2005. doi: 10.1007/s10741-005-7545-1. [DOI] [PubMed] [Google Scholar]
- 228.She L, Tu H, Zhang YZ, Tang LJ, Li NS, Ma QL, Liu B, Li Q, Luo XJ, Peng J. Inhibition of phosphoglycerate mutase 5 reduces necroptosis in rat hearts following ischemia/reperfusion through suppression of dynamin-related protein 1. Cardiovasc Drugs Ther 33: 13–23, 2019. doi: 10.1007/s10557-018-06848-8. [DOI] [PubMed] [Google Scholar]
- 229.Shekhar A, Heeger P, Reutelingsperger C, Arbustini E, Narula N, Hofstra L, Bax JJ, Narula J. Targeted Imaging for Cell Death in Cardiovascular Disorders. JACC Cardiovasc Imaging 11: 476–493, 2018. doi: 10.1016/j.jcmg.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 230.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42: 245–254, 2017. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 231.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, Uchiyama Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 382: 419–423, 2009. doi: 10.1016/j.bbrc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 232.Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, Torii S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 108: 2187–2194, 2017. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494: 201–206, 2013. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 121: 2685–2695, 2008. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- 235.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther 21: 227–233, 2007. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 236.Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY, Yang P. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci 8: 564–572, 2011. doi: 10.7150/ijms.8.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sosnovik DE, Schellenberger EA, Nahrendorf M, Novikov MS, Matsui T, Dai G, Reynolds F, Grazette L, Rosenzweig A, Weissleder R, Josephson L. Magnetic resonance imaging of cardiomyocyte apoptosis with a novel magneto-optical nanoparticle. Magn Reson Med 54: 718–724, 2005. doi: 10.1002/mrm.20617. [DOI] [PubMed] [Google Scholar]
- 238.Stehling O, Lill R. The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol 5: a011312, 2013. doi: 10.1101/cshperspect.a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Stennicke HR, Jürgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 273: 27084–27090, 1998. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 240.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227, 2012. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 241.Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab 12: 453–463, 1976. [PubMed] [Google Scholar]
- 242.Szobi A, Farkašová-Ledvényiová V, Lichý M, Muráriková M, Čarnická S, Ravingerová T, Adameová A. Cardioprotection of ischaemic preconditioning is associated with inhibition of translocation of MLKL within the plasma membrane. J Cell Mol Med 22: 4183–4196, 2018. doi: 10.1111/jcmm.13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Szobi A, Gonçalvesová E, Varga ZV, Leszek P, Kuśmierczyk M, Hulman M, Kyselovič J, Ferdinandy P, Adameová A. Analysis of necroptotic proteins in failing human hearts. J Transl Med 15: 86, 2017. [Erratum in: J Transl Med 15: 103, 2017.] doi: 10.1186/s12967-017-1189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Szobi A, Rajtik T, Adameova A. Effects of necrostatin-1, an inhibitor of necroptosis, and its inactive analogue Nec-1i on basal cardiovascular function. Physiol Res 65: 861–865, 2016. [DOI] [PubMed] [Google Scholar]
- 245.Szobi A, Rajtik T, Carnicka S, Ravingerova T, Adameova A. Mitigation of postischemic cardiac contractile dysfunction by CaMKII inhibition: effects on programmed necrotic and apoptotic cell death. Mol Cell Biochem 388: 269–276, 2014. doi: 10.1007/s11010-013-1918-x. [DOI] [PubMed] [Google Scholar]
- 246.Taatjes DJ, Sobel BE, Budd RC. Morphological and cytochemical determination of cell death by apoptosis. Histochem Cell Biol 129: 33–43, 2008. doi: 10.1007/s00418-007-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Reports 5: 878–885, 2013. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J 55: 101–105, 2014. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 249.Takahashi M. Role of the inflammasome in myocardial infarction. Trends Cardiovasc Med 21: 37–41, 2011. doi: 10.1016/j.tcm.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 250.Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep 14: 102–108, 2009. doi: 10.1179/135100009X392566. [DOI] [PubMed] [Google Scholar]
- 251.Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol 15: 203–214, 2018. doi: 10.1038/nrcardio.2017.161. [DOI] [PubMed] [Google Scholar]
- 252.Toldo S, Mauro AG, Cutter Z, Abbate A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 315: H1553–H1568, 2018. doi: 10.1152/ajpheart.00158.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Toldo S, Mezzaroma E, McGeough MD, Peña CA, Marchetti C, Sonnino C, Van Tassell BW, Salloum FN, Voelkel NF, Hoffman HM, Abbate A. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc Res 105: 203–212, 2015. doi: 10.1093/cvr/cvu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, Abbate A. Interleukin-1β blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol 98: 734–745, 2013. doi: 10.1113/expphysiol.2012.069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11: 290–297, 2012. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31: 1–9, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 257.van Heerde WL, Robert-Offerman S, Dumont E, Hofstra L, Doevendans PA, Smits JF, Daemen MJ, Reutelingsperger CP. Markers of apoptosis in cardiovascular tissues: focus on Annexin V. Cardiovasc Res 45: 549–559, 2000. doi: 10.1016/S0008-6363(99)00396-X. [DOI] [PubMed] [Google Scholar]
- 258.Vanden Berghe T, Denecker G, Brouckaert G, Vadimovisch Krysko D, D’Herde K, Vandenabeele P. More than one way to die: methods to determine TNF-induced apoptosis and necrosis. Methods Mol Med 98: 101–126, 2004. doi: 10.1385/1-59259-771-8:101. [DOI] [PubMed] [Google Scholar]
- 259.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 3: re4, 2010. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 260.Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 18: 656–665, 2011. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188: 919–930, 1998. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.von Haehling S, Jankowska EA, van Veldhuisen DJ, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Nat Rev Cardiol 12: 659–669, 2015. doi: 10.1038/nrcardio.2015.109. [DOI] [PubMed] [Google Scholar]
- 263.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146, 2014. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 264.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111: 15438–15443, 2014. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103, 2017. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 266.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148: 228–243, 2012. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 267.Weber D, Milkovic L, Bennett SJ, Griffiths HR, Zarkovic N, Grune T. Measurement of HNE-protein adducts in human plasma and serum by ELISA-Comparison of two primary antibodies. Redox Biol 1: 226–233, 2013. doi: 10.1016/j.redox.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol 1: 6, 2018. doi: 10.1038/s42003-017-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Webster KA, Graham RM, Thompson JW, Spiga MG, Frazier DP, Wilson A, Bishopric NH. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal 8: 1667–1676, 2006. doi: 10.1089/ars.2006.8.1667. [DOI] [PubMed] [Google Scholar]
- 270.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504, 2003. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Whiteside G, Munglani R. TUNEL, Hoechst and immunohistochemistry triple-labelling: an improved method for detection of apoptosis in tissue sections--an update. Brain Res Brain Res Protoc 3: 52–53, 1998. doi: 10.1016/S1385-299X(98)00020-8. [DOI] [PubMed] [Google Scholar]
- 272.Wong YK, Zhang J, Hua ZC, Lin Q, Shen HM, Wang J. Recent advances in quantitative and chemical proteomics for autophagy studies. Autophagy 13: 1472–1486, 2017. doi: 10.1080/15548627.2017.1313944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23: 994–1006, 2013. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19: 3290–3298, 2008. doi: 10.1091/mbc.e07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Xu ZW, Chen X, Jin XH, Meng XY, Zhou X, Fan FX, Mao SY, Wang Y, Zhang WC, Shan NN, Li YM, Xu RC. SILAC-based proteomic analysis reveals that salidroside antagonizes cobalt chloride-induced hypoxic effects by restoring the tricarboxylic acid cycle in cardiomyocytes. J Proteomics 130: 211–220, 2016. doi: 10.1016/j.jprot.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 276.Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinters HV, Frautschy SA, Cole GM. Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer’s disease. Am J Pathol 152: 379–389, 1998. [PMC free article] [PubMed] [Google Scholar]
- 277.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331, 2014. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yang YF, Wu CC, Chen WP, Chen YL, Su MJ. Prazosin induces p53-mediated autophagic cell death in H9C2 cells. Naunyn Schmiedebergs Arch Pharmacol 384: 209–216, 2011. doi: 10.1007/s00210-011-0657-3. [DOI] [PubMed] [Google Scholar]
- 279.Yin B, Xu Y, Wei RL, He F, Luo BY, Wang JY. Inhibition of receptor-interacting protein 3 upregulation and nuclear translocation involved in Necrostatin-1 protection against hippocampal neuronal programmed necrosis induced by ischemia/reperfusion injury. Brain Res 1609: 63–71, 2015. doi: 10.1016/j.brainres.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 280.Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol 452: 143–164, 2009. doi: 10.1016/S0076-6879(08)03610-0. [DOI] [PubMed] [Google Scholar]
- 281.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93: 2696–2701, 1996. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ytrehus K, Liu Y, Tsuchida A, Miura T, Liu GS, Yang XM, Herbert D, Cohen MV, Downey JM. Rat and rabbit heart infarction: effects of anesthesia, perfusate, risk zone, and method of infarct sizing. Am J Physiol 267: H2383–H2390, 1994. doi: 10.1152/ajpheart.1994.267.6.H2383. [DOI] [PubMed] [Google Scholar]
- 283.Yu L, Li B, Zhang M, Jin Z, Duan W, Zhao G, Yang Y, Liu Z, Chen W, Wang S, Yang J, Yi D, Liu J, Yu S. Melatonin reduces PERK-eIF2α-ATF4-mediated endoplasmic reticulum stress during myocardial ischemia-reperfusion injury: role of RISK and SAFE pathways interaction. Apoptosis 21: 809–824, 2016. doi: 10.1007/s10495-016-1246-1. [DOI] [PubMed] [Google Scholar]
- 284.Yu PW, Huang BC, Shen M, Quast J, Chan E, Xu X, Nolan GP, Payan DG, Luo Y. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFκB. Curr Biol 9: PS1–PS3, 1999. doi: 10.1016/S0960-9822(99)80239-5. [DOI] [PubMed] [Google Scholar]
- 285.Yuan Y, Zhao J, Gong Y, Wang D, Wang X, Yun F, Liu Z, Zhang S, Li W, Zhao X, Sun L, Sheng L, Pan Z, Li Y. Autophagy exacerbates electrical remodeling in atrial fibrillation by ubiquitin-dependent degradation of L-type calcium channel. Cell Death Dis 9: 873, 2018. doi: 10.1038/s41419-018-0860-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Zhang L, Feng Q, Wang T. Necrostatin-1 Protects Against Paraquat-Induced Cardiac Contractile Dysfunction via RIP1-RIP3-MLKL-Dependent Necroptosis Pathway. Cardiovasc Toxicol 18: 346–355, 2018. doi: 10.1007/s12012-017-9441-z. [DOI] [PubMed] [Google Scholar]
- 287.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med 22: 175–182, 2016. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 288.Zhang Y, Rong H, Zhang FX, Wu K, Mu L, Meng J, Xiao B, Zamponi GW, Shi Y. A membrane potential- and calpain-dependent reversal of caspase-1 inhibition regulates canonical NLRP3 inflammasome. Cell Reports 24: 2356–2369.e5, 2018. doi: 10.1016/j.celrep.2018.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, Zhang F, Zheng S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14: 2083–2103, 2018. doi: 10.1080/15548627.2018.1503146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 109: 5322–5327, 2012. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zhao M, Li Z, Bugenhagen S. 99mTc-labeled duramycin as a novel phosphatidylethanolamine-binding molecular probe. J Nucl Med 49: 1345–1352, 2008. doi: 10.2967/jnumed.107.048603. [DOI] [PubMed] [Google Scholar]
- 292.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117: 1782–1793, 2007. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 3: 232–243, 2017. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zoratti M, De Marchi U, Biasutto L, Szabò I. Electrophysiology clarifies the megariddles of the mitochondrial permeability transition pore. FEBS Lett 584: 1997–2004, 2010. doi: 10.1016/j.febslet.2010.01.012. [DOI] [PubMed] [Google Scholar]