

Abstract
Human monocytes have been classified into three distinct groups, classical (anti-inflammatory; CD14+/CD16−), nonclassical (patrolling; CD14+/CD16++), and intermediate (proinflammatory; CD14++/CD16+). Adhesion of nonclassical/intermediate monocytes with the endothelium is important for innate immunity, and also vascular inflammatory disease. However, there is an incomplete understanding of the mechanisms that regulate CD16+ versus CD16− monocyte adhesion to the inflamed endothelium. Here, we tested the hypothesis that a high-mannose (HM) N-glycoform of intercellular adhesion molecule-1 (ICAM-1) on the endothelium mediates the selective recruitment of CD16+ monocytes. Using TNF-α treatment of human umbilical vein endothelial cells (HUVECs), and using proximity ligation assay for detecting proximity of specific N-glycans and ICAM-1, we show that TNF-α induces HM-ICAM-1 formation on the endothelial surface in a time-dependent manner. We next measured CD16− or CD16+ monocyte rolling and adhesion to TNF-α-treated HUVECs in which HM- or hybrid ICAM-1 N-glycoforms were generated using the α-mannosidase class I and II inhibitors, kifunensine and swainsonine, respectively. Expression of HM-ICAM-1 selectively enhanced CD16+ monocyte adhesion under flow with no effect on CD16− monocytes noted. CD16+ monocyte adhesion was abrogated by blocking either HM epitopes or ICAM-1. A critical role for HM-ICAM-1 in mediating CD16+ monocyte rolling and adhesion was confirmed using COS-1 cells engineered to express HM or complex ICAM-1 N-glycoforms. These data suggest that HM-ICAM-1 selectively recruits nonclassical/intermediate CD16+ monocytes to the activated endothelium.
NEW & NOTEWORTHY Monocyte subsets have been associated with cardiovascular disease, yet it is unknown how different subsets are recruited to the endothelium. This study demonstrates the formation of distinct ICAM-1 N-glycoforms in the activated endothelium and reveals a key role for high mannose ICAM-1 in mediating proinflammatory CD16+ monocyte adhesion. Presented data identify roles for endothelial N-glycans in recruiting specific monocyte subsets during inflammation.
Keywords: inflammation, mannosidase, N-glycan
INTRODUCTION
Monocyte adhesion and migration through the endothelial cell (EC) layer is a key process during inflammation (9). Monocyte trafficking is mediated by EC surface adhesion molecules, the surface expression of which is induced by inflammatory stimuli (8, 15). Once inflammation is resolved, adhesion molecule expression returns to basal levels. However, in chronic inflammation as seen in diseases such as atherosclerosis, diabetes, and autoimmune disorders, adhesion molecule expression remains elevated, leading to excessive adhesion and accumulation of monocytes in the vessel wall (8, 16, 17, 42).
Relative to the understanding that monocytes are important drivers of inflammation, the appreciation that human monocytes are heterogeneous is a more recent advance in knowledge. Human monocytes are grouped into three categories based on the surface markers CD14 and CD16: classical (anti-inflammatory; CD14++/CD16−), nonclassical (patrolling; CD14+/CD16++), and intermediate (proinflammatory; CD14++/CD16+) (7, 44). At rest, classical monocytes are most abundant, accounting for 90% of total monocytes. During chronic inflammation, nonclassical and intermediate subsets can expand from ~10% to 20–30% (39, 40), depending on the disease. Furthermore, higher levels of nonclassical and intermediate monocytes are associated with poorer prognoses in atherosclerosis, diabetes, asthma, stroke, and other diseases (18, 21, 28, 29, 39). Recent studies have extensively characterized the biochemical, molecular, and genetic profiles of the different monocytes (7, 19, 38, 41) and associated these with the distinct functions each monocyte subset mediates (30, 42, 43). Surprisingly, however, little is known about mechanisms that govern how distinct monocyte subsets roll and adhere to an inflamed endothelial surface.
Endothelial intercellular adhesion molecule-1 (ICAM-1) is an important adhesion molecule that recognizes CD11a/CD18 [lymphocyte function-associated antigen 1 (LFA-1)] and CD11b/CD18 [macrophage-1 antigen (Mac-1)] on monocytes to regulate the latter’s rolling, firm adhesion, and transmigration (3, 6, 11, 23, 27). ICAM-1 is heavily N-glycosylated; ~50% of its observed mass is the result of N-glycans (5, 20). Protein N-glycosylation is an enzyme-driven cotranslational modification that attaches a core branched tetradecasaccharide [glucose3-mannose9-N-acetylglucosamine (GlcNAc)2] on the amide residues of Asn in the consensus sequence N-X-S/T (where X cannot be proline). Sugars from this core are sequentially cleaved by a series of glucosidases and mannosidases leading to high-mannose (HM) and hybrid N-glycan structures in the endoplasmic reticulum. Thereafter, fucose, galacatose, GlcNAc, and sialic acid are added, resulting in complex N-glycoproteins (35). Although most glycoproteins are assumed to be processed to complex forms for cell-surface expression, we have previously demonstrated that the endothelial cell surface expresses increased HM and/or hybrid N-glycans during inflammation (10, 33). In particular, using a model cell system where expression of ICAM-1 N-glycoforms could be controlled, we showed that ICAM-1 expressing HM/hybrid N-glycans resulted in increased adhesion of monocytes compared with ICAM-1 bearing more complex type N-glycans (31). The current study tests 1) whether HM/hybrid ICAM-1 is formed and expressed on the surface of activated endothelial cells and, if so, how this compares to complex N-glycoforms and 2) whether HM/hybrid or complex ICAM-1 N-glycoforms selectively mediate adhesion of classical versus nonclassical/intermediate monocytes.
MATERIALS AND METHODS
Materials.
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical veins via collagenase as described (12). COS-1 cells were a generous gift from Dr. Joanne Murphy-Urlich (University of Alabama at Birmingham). MCDB131, RPMI1640, DMEM, heat-inactivated FBS, trypsin, l-glutamine, and penicillin/streptomycin were purchased from Invitrogen (Carlsbad, CA). DuoLink proximity ligation assay kit was purchased from ThermoFisher (Waltham, MA). CD14 and CD16 magnetic beads and magnetic columns were purchased from Miltenyi Biotec (Bergisch Gladbach). Parallel plate flow chambers were purchased from GlycoTech (Gaithersburg, MD). Concanavalin A (ConA), Sambucus nigra (SNA), Hippeastrum hybrid amaryllis (HHL), and maackia amurensis lectin II (MAL-II) lectins were purchased from Vector (Burlingame, CA). Where indicated, ConA or HHL were denatured by heating for 15 min at 95°C. Kifunensine (Kif) and swainsonine (Swain) were purchased form Cayman Chemicals (Ann Arbor, MI). Cell Trackers green [5-chloromethylfluorescein diacetate (CMFDA)] and blue [7-amino-4-chloromethylcourmarin (CMAC)] were purchased from Thermo Fisher (Waltham, MA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Cell culture and treatment.
HUVECs were cultured as previously described (31). Cells were used within 1 day of reaching confluence and were serum-starved in MCDB-131 media containing 1% FBS for 2 h before treatment. In some experiments, cells were pretreated (2 h) with the class I and II α-mannosidase inhibitors Kif or Swain, respectively, before addition of TNF-α (10 ng/ml, 4–18 h). COS-1 cells were transfected with human ICAM-1 as described (31). Once stably transfected, COS-1 cells were cultured in DMEM media containing 10% FBS and 1× penicillin/streptomycin with or without Kif.
Western blot analysis.
Cells were treated as described and lysed in RIPA buffer for 10 min on ice. Protein (20 μg) was loaded on 10% SDS-PAGE and subject to electrophoresis before being transferred to a 0.2-μm nitrocellulose membrane and probed for the described protein overnight at 4°C with rocking. Membranes were incubated with HRP-linked species-appropriate secondary antibodies for 2 h at room temperature (RT) before chemiluminescence measurement. Images were analyzed using ImageQuant software (GE Healthcare Life Sciences).
Proximity-ligation assay.
Following treatment as described, HUVECs or COS-1 cells were washed with ice-cold PBS with 1 mM MgCl2 and CaCl2 before fixation with 4% PFA for 15 min at RT. After fixation, proximity-ligation assay (PLA) staining was carried out as per the manufacturer’s protocols. Briefly, 20 µg of the following biotinylated lectins were added for 30 min at 4°C: HM and hybrid N-glycan-specific lectin ConA, the α2,6-sialyation-specific lectin SNA, the HM-specific lectin HHL, or the α2,3-sialylation-specific lectin MAL-II (see Fig. 1). These lectins were chosen to provide broad coverage of different N-glcyan structures. After 30 min, cells were washed two times with PBS and samples were blocked with 1× Carbo-Free blocking solution (Vector) for 30 min at 20–25°C. Immediately following blocking, samples were incubated with oligo-tagged anti-ConA, avidin, or anti-ICAM-1 (10 μg/mL) for 1 h at 37°C followed by ligation and amplification steps as per the protocol. Human ICAM-1 (clone RR1/1) antibody recognizes the N-glycan-devoid extracellular domain 1 epitopes on ICAM-1. Slides were left to dry and mounted using the DuoLink mounting medium containing DAPI. To quantify, three random fields per group per experiment were selected, and average fluorescence intensity was calculated and plotted per counted nuclei.
Fig. 1.

Lectins used in this study and their specificity for N-glycan structures. Man, mannose; Gal, galactose; GlcNac, N-acetylglucosamine; Sa, sialic acid; R, varying N-glycan structures (adapted from Ref. 34).
α-Mannosidase activity assay.
Total (class I and II) α-mannosidase activity was determined as described (33) with minor modifications. Cells were washed with PBS before lysis in PBS containing 1% Triton X-100 for 0 min on ice before clarification at 10,000 g for 10 min. Cell lysate (50 μL, corresponding to 30–40 μg protein) was prepared in a microtiter plate in 100 mM acetate buffer (pH 6.5), and 200 μM of the α-mannosidase substrate resorufin-α-d-mannopyranoside were added to start the reaction. Plates were read continuously at 550 excitation and 595 emission in a Biotek Synergy plate reader for 18 h at 37°C.
Surface N-glycan immunoprecipitation.
To label surface N-glycans only, after treatment, cells were washed with ice-cold PBS containing 1 mM CaCl2 and MgCl2 before incubation with 10 μg biotinylated ConA for 15 min at 4°C. Cells were lysed with 1% Triton X-100 in PBS and incubated with avidin resin for 2 h at RT. Samples were washed three times and boiled at 95°C for 10 min in 5× SDS loading buffer to release biotin complexes. Samples were then treated as Western blot as described above.
Monocyte and neutrophil isolation.
Primary human monocytes and neutrophils were isolated from freshly drawn whole blood collected by venipuncture from healthy volunteers using magnetic beads as described (32). Briefly, after removal of the plasma layer, blood was layered on top of a Histopaque 1119 to 1077 gradient and centrifuged at 700 g for 20 min at RT. Monocytes appear at the interface of media and the 1077 layer, whereas neutrophils appear between the two Histopaque layers. Monocytes were incubated with a CD16 antibody and isolated using positive selection via a magnetic column. The flow-through (CD16− cells) was then incubated with a CD14 antibody to isolate distinct CD16+ and CD16− monocyte populations. Neutrophils were incubated with a CD15 antibody and isolated in the same manner. Purity of monocyte preparations was confirmed via flow cytometry analysis (data not shown). All protocols were approved by the University of Alabama at Birmingham Institutional Review Board.
Monocyte and neutrophil rolling and adhesion assay.
Isolated monocyte populations were incubated with 1 μM fluorescent CellTracker dyes; CD16− cells were labeled with CellTracker green (CMFDA), and CD16+ cells were labeled with CellTracker blue (CMAC) for 30 min at 37°C. Cells were pelleted, and supernatant was aspirated to remove any unincorporated dye. CD16− and CD16+ monocytes were then combined in equal amounts (final cell count 250,000 cells/mL; 125,000 cells/mL of each subtype), unless otherwise stated. Treated HUVECs or COS-1 cells were exposed to fluorescent monocytes or neutrophils at a flow rate of 100 μL/min, corresponding to 1 dyn/cm2, in a GlycoTech parallel plate flow chamber. Neutrophils were treated with 100 ng/mL PMA (15 min) before adhesion assay. Images were captured on a Biotek Lionheart live cell imager over 2 min at 30 frames/s. Any cell that was stationary for ≥5 s was considered firmly adhered as described (23, 32). Where indicated, 10 min before adhesion assay, HUVECs were treated with ConA or HHL lectins (10 µg/mL) to block surface hybrid and HM-N-glycans.
Statistics.
All statistical analyses were performed using GraphPad Prism software. Paired t-test or one-way ANOVA followed by Tukey’s post hoc test were performed as indicated in the legends for Figs. 1–6. A minimum of three independent experiments were performed for each replicate.
RESULTS
HM-ICAM-1 formation is transient in TNF-α-treated endothelial cells.
Our previous studies have shown that the luminal surface of atherosclerotic lesions is characterized by increased HM/hybrid N-glycan structures that parallel with severity of disease (10, 31, 32). Moreover, we identified ICAM-1 as a candidate adhesion molecule that may harbor HM structures on the endothelial surface. To determine if endothelial inflammation leads to formation of ICAM-1 harboring HM/hybrid N-glycan structures, we used Western blot analysis and PLA. Treatment of HUVECs with TNF-α led to a time-dependent increase in ICAM-1 indicated by a 100-kDa band (Fig. 2A). Also seen is an ~75-kDa band that transiently appears at 4 h, comprising ∼25% of the total ICAM-1 at this time (Fig. 2B), after which it returns to basal levels by 18 h. We have shown previously that the ∼100 and ~75-kDa bands correspond to complex and HM/hybrid ICAM-1 N-glycoforms, respectively (31).
Fig. 2.

TNF-α forms endothelial high-mannose (HM)-intercellular adhesion molecule-1 (ICAM-1) in a time-dependent manner. Human umbilical vein endothelial cells (HUVECs) were treated with 10 ng/mL TNF-α for 0, 4, or 18 h, and either lysates were collected for Western blot analyses or cells were processed for proximity-ligation assay (PLA). A: representative Western blot for ICAM-1 expression. The 100-kDa band represents the fully glycosylated complex ICAM-1, and the 75-kDa band represents the hypoglycosylated high-mannose ICAM-1. Kif, Kifunensine; Swain, swainsonine. B: quantification of HM-ICAM-1 as a percentage of total ICAM-1. Data are means ± SE, n = 3. C: HUVECs were treated as above and subject to a PLA for HM/hybrid, HM, α2,6-sialylated, and α2,3-sialylated ICAM-1. Shown are representative images from each time point. Red puncta represents positive PLA signal, blue staining is DAPI nuclear stain. ConA, concanavalin A; HHL, Hippeastrum hybrid amaryllis; SNA, Sambucus nigra; MAL-II, maackia amurensis lectin II. D: PLA staining controls where one PLA reagent was left out (left, no anti-ICAM-1; right, no avidin). E: quantification of PLA puncta. For each replicate, puncta were counted in three fields and averaged. Each symbol represents an independent replicate. Data are means ± SE, n = 4. *P ≤ 0.05 compared with respective time control by 1-way ANOVA with Tukey’s posttest.
To determine if different ICAM-1 N-glycoforms [HM, hybrid, and complex (via α2,3-sialylated and α2,6-sialylated identification)] were expressed on the cell-surface, we used PLA. HUVECs were treated with TNF-α and cell surface N-glycans labeled with biotinylated lectins (see Fig. 1 for structures recognized by each lectin) followed by treatment with avidin and an anti-ICAM-1 antibody, both tagged with complementary oligonucleotides. Red puncta indicate positive staining where the two epitopes (ICAM-1 and the lectin-identified N-glycan structures) are within 40 nm of each other. Figure 2, C and D, shows representative images, and Fig. 2E shows quantitation. Surface HM/hybrid ICAM-1 transiently increased at 4 h, whereas α2,6-sialylated (SNA) ICAM-1 continued to increase over 18 h. No α2,3-sialyated ICAM-1 (MAL-II) was observed. HUVECs that were not exposed to TNF-α had no detectable staining (data not shown), similar to the staining controls in which one of the PLA reagents was absent (Fig. 2D).
To test whether HM structures selectively bind CD16+ versus CD16− monocytes, we first established conditions to selectively express HM and hybrid structures on the endothelial surface using the α-mannosidase class I and II inhibitors Kif and Swain, respectively (Fig. 3A). Kif and Swain each inhibited α-mannosidase activity in HUVECs to similar extents, indicating approximately equal class I and class II α-mannosidase activities (Fig. 3B). Furthermore, Kif and Swain produced HM and hybrid N-glycoforms of ICAM-1, indicated by the bands at 75 and 100 kDa, respectively (Fig. 3, C and D). Importantly, Kif and Swain did not change total ICAM-1 expression (Fig. 3E) or surface ICAM-1 expression (Fig. 3F).
Fig. 3.

Kifunensine (Kif) and swainsonine (Swain) selectively form high-mannose (HM) and hybrid N-glycans on the activated endothelium. A: scheme showing early and sequential processing of N-glycans from HM to hybrid and complex N-glycans. Also shown are sites where Kif and Swain inhibit class I and II α-mannosidases, respectively. B: α-mannosidase activity in human umbilical vein endothelial cells (HUVECs) exposed to 1 μM Kif or 1 μM Swain. Data show activity relative to control. *P ≤ 0.05 compared with control by 1-way ANOVA with Tukey’s posttest. C and D: Western blot (C) and analysis (D) of intercellular adhesion molecule-1 (ICAM-1) glycoforms in HUVECs when exposed to TNF-α and/or 1 μM Kif or Swain. The 100-kDa band represents the fully glycosylated complex ICAM-1, and the 75-kDs band represents the hypoglycosylated high-mannose ICAM-1. Lane 1, control; lane 2, TNF-α only; lane 3, TNF-α and Swain; lane 4, TNF-α and Kif. E: total band density of ICAM-1 from C. F: ICAM-1 surface stain (left) and quantitation (right) on HUVECs treated with TNF-α and/or 1 μM Kif or Swain.
Endothelial HM epitopes and ICAM-1 enhance CD16+ monocyte rolling and adhesion.
To test if HM or hybrid N-glycans had an effect on monocyte subset adhesion, HUVECs were treated as described and exposed to isolated monocytes under flow. As shown in Fig. 4A, TNF-α increased adhesion for both CD16− and CD16+ monocytes. Pretreatment of HUVECs with Kif or Swain had no effect on TNF-α-dependent CD16− monocyte adhesion. However, Kif pretreatment, but not Swain, significantly further increased CD16+ monocyte adhesion by ∼1.7-fold (Fig. 4A), suggesting that endothelial HM epitopes have higher affinity for CD16+ monocytes. Because neutrophils also adhere to endothelial ICAM-1, we tested the effects of Kif on neutrophil adhesion. Kif did not affect neutrophil adhesion to TNF-α-treated HUVECs (Fig. 4B).
Fig. 4.
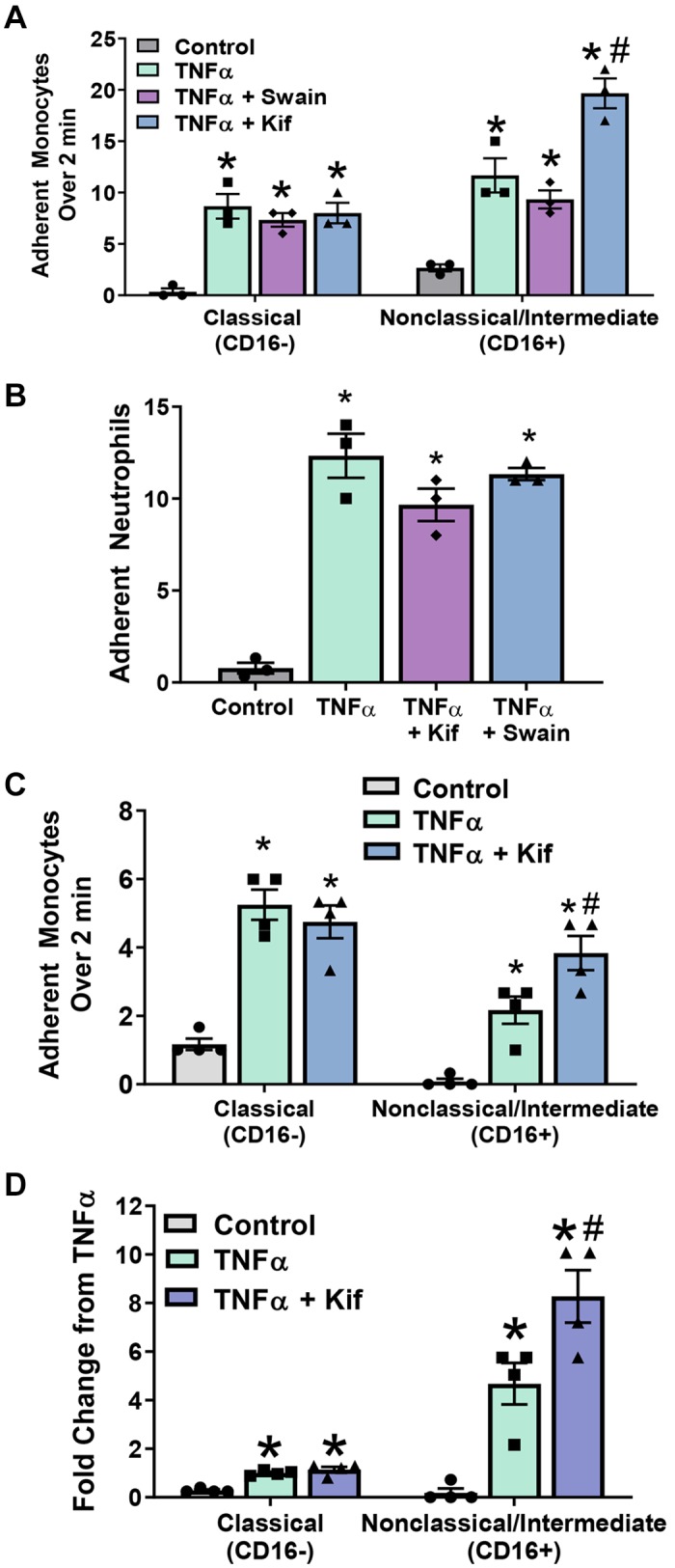
CD16+ monocyte adhesion is enhanced by endothelial high-mannose epitopes. A: human umbilical vein endothelial cells (HUVECs) were treated as above and exposed under flow to fluorescently labeled CD16+ and CD16− monocytes. Each symbol represents the average of an independent experiment. Kif, kifunensine; Swain, swainsonine. Data are means ± SE, n = 3. P ≤ 0.05 compared with control (*) or TNF-α (#) by 1-way ANOVA with Tukey’s posttest within monocyte groups. B: HUVECs were treated, and neutrophil adhesion under flow was determined as described. Data are means ± SE, n = 3. *P ≤ 0.05 compared with control alone by 1-way ANOVA. C: monocyte adhesion under flow to HUVECs at physiological ratios (225,000 cells/mL CD16− and 25,000 cells/mL CD16+). Each bar represents the average of 4 independent experiments. Data are means ± SE, n = 4. P ≤ 0.05 compared with control (*) or TNF-α (#) by 1-way ANOVA with Tukey’s posttest within monocyte groups. D: monocyte adhesion from C as a fold change compared with CD16− adhesion to TNF-α-treated HUVECs. Each symbol represents the average of 4 independent experiments. Data are means ± SE, n = 4. P ≤ 0.05 compared with control (*) or TNF-α (#) by 1-way ANOVA with Tukey’s posttest within monocyte groups.
Physiologically, the relative abundance of circulating CD16+ and CD16− monocytes is ∼10 and 90%, respectively. To evaluate if HM epitopes can selectively recruit CD16+ cells at these physiologic cell number ratios, we performed the same experiment as above using a 10:1 ratio of CD16− to CD16+ monocytes. Figure 4C shows that TNF-α increased both CD16− and CD16+ monocyte adhesion relative to control. Swain had no effect, whereas Kif treatment significantly further increased CD16+ monocyte adhesion relative to TNF-α alone. This effect of Kif was not observed with CD16− monocyte adhesion. Figure 4D shows fold change in monocyte adhesion relative to CD16− cell adhesion to TNF-α-treated ECs. These data suggest that, whereas there are less CD16+ cells relative to CD16− cells, the former are more likely to adhere to HM glycan epitopes.
CD16+ adhesion to activated ECs is dependent on HM epitopes and ICAM-1.
We next tested the effects of ConA- or HHL-dependent blocking of endothelial HM/hybrid type N-glycans on monocyte adhesion. Figure 5A shows ConA decreased adhesion of CD16− cells by 49% and CD16+ cells by 75%. However, blocking with HHL only decreased adhesion of CD16+ monocytes (by ∼80%). Similar inhibitory effects of both lectins were observed in Kif-pretreated cells, although only trends were noted with CD16− cell adhesion. Prior denaturation of Con A or HHL resulted in a loss in their ability to inhibit monocyte adhesion, suggesting that this effect is not because of nonspecific binding of lectins to the endothelium.
Fig. 5.

CD16+ monocyte adhesion is dependent on endothelial high-mannose (HM) epitopes and intercellular adhesion molecule-1 (ICAM-1). A: human umbilical vein endothelial cells (HUVECs) were treated with 10 ng/mL TNF-α for 4 h before monocyte adhesion assay. Some cells were pretreated with Kifunensine (Kif) to form HM epitopes on the cell surface, and then monocyte adhesion under flow was measured. Before adhesion assay, some cells were treated with the lectins Hippeastrum hybrid amaryllis (HHL) or concanavalin A (ConA) to block HM/hybrid N-glycans on the cell surface, or with denatured lectins to assess nonspecific binding effects. Each symbol represents the average of an independent experiment. Data are means ± SE, n = 3 or 4. P ≤ 0.05 compared with control (*), TNF-α alone ($), and TNF-α + Kif (#) by 1-way ANOVA with Tukey’s posttest within monocyte groups. B: HUVECs were treated as described, and some cells were treated with anti-ICAM-1 blocking antibodies that recognize domains 1 (D1) or 3 (D3) of ICAM-1 before monocyte adhesion assay. Each symbol represents the average of an independent experiment. Data are means ± SE, n = 3 or 4. P ≤ 0.05 compared with control (*), TNF-α ($), TNF-α + IgG control (&), TNF-α + Kif (#), and TNF-α + Kif + IgG control (%) by 1-way ANOVA with Tukey’s posttest within monocyte and cell treatment groups.
We hypothesized that ICAM-1 is the primary adhesion molecule harboring HM epitopes that mediate CD16+ adhesion. To test this, we compared the effects of two anti-ICAM-1 blocking antibodies that recognize the LFA-1 [domain 1 (D1)]- or Mac-1 [domain 3 (D3)]-binding domains on ICAM-1 with an IgG control antibody. Figure 5B shows that D1 and D3 blockade inhibits CD16− monocyte adhesion by ∼75 and 35%, respectively. In Kif-treated cells, D1 blockade still inhibited CD16− monocyte adhesion; however, D3 blockade had no effect. With CD16+ monocyte adhesion, in the absence or presence of Kif, D1 blockade had no effect, whereas D3 blockade significantly (75–90%) decreased adhesion.
HM-ICAM-1 enhances CD16+ monocyte adhesion under flow.
To evaluate the potential for ICAM-1 N-glycoforms to regulate CD16+ versus CD16− monocyte rolling and adhesion in the absence of other adhesion molecules, we used COS-1 cells, which inherently do not express ICAM-1. Cells were transfected with [wild type (WT)] or without (empty vector) human ICAM-1 and grown in the presence or absence of Kif. Figure 6A shows that total ICAM-1 expression was similar, but, with Kif, all ICAM-1 migrated at ∼75 kDa, consistent with a HM-ICAM-1 N-glycoform. Immunoprecipitation of surface proteins with ConA confirmed that HM-ICAM-1 was present on the cell surface, with the levels being greater in Kif-treated cells (Fig. 6B). Furthermore, PLA analysis demonstrated the presence of HM-ICAM-1 in Kif-treated WT COS-1 cells, with complex (α2,6-sialyated) ICAM-1 predominating in WT COS-1 cells not treated with Kif (Fig. 6, C and D).
Fig. 6.

High-mannose (HM)-intercellular adhesion molecule-1 (ICAM-1) enhances CD16+ monocyte adhesion under flow. A: Western blot of ICAM-1 from COS-1 cells transfected with or without human ICAM-1 and grown in the presence or absence of Kifunensine (Kif). B: surface concanavalin A (ConA) reactive epitopes were immunoprecipitated from COS-1 cells grown in the presence or absence of Kif, and ICAM-1 levels were determined by Western blotting. WCL, whole cell lysate. C: COS-1 cells transfected with or without human ICAM-1 and grown in the presence or absence of Kif were subjected to a proximity-ligation assay (PLA) for HM- and α2,6-sialylated ICAM-1. Shown are representative images from each time point. Red puncta represents positive PLA signal, and blue staining is DAPI nuclear stain. SNA, Sambucus nigra. D: quantification of PLA signal from COS-1 cells. P ≤ 0.05 compared with wild-type (WT)-Hippeastrum hybrid amaryllis (HHL) signal (*) and WT + Kif SNA signal (#). Data are means ± SE, n = 3 with an average of 2 fields/n. E: COS-1 cells treated as above were exposed to isolated C16+ and CD16− monocytes under flow, and adhesion was measured over 2 min. Data are means ± SE, n = 3. Each symbol represents a single experiment with two replicates per experiment. P ≤ 0.05 compared with empty vector (EV) (*) and compared with WT alone by 1-way ANOVA with Tukey’s posttest within monocyte groups ($). F: monocyte rolling velocities from E were calculated as described above. Data are means ± SE, n = 3 or 4. P ≤ 0.05 compared with EV (*) and compared with WT alone by 1-way ANOVA with Tukey’s posttest within monocyte groups ($). G: COS-1 cells were subjected to monocyte adhesion under flow as described above. Before adhesion assay, some cells were treated with the lectins HHL or ConA to block HM/hybrid N-glycans on the cell surface. Data are means ± SE, n = 3 or 4 with two replicates per experiment. P ≤ 0.05 compared with WT alone (*) and WT + Kif (#) by 1-way ANOVA with Tukey’s posttest within monocyte groups. Broken line represents average of nontransfected cells. H: PLA images from WT and EV COS-1 cells subject to PLA for ICAM-1 and HHL from C collected with longer exposure times. I: velocities calculated from G. Data are means ± SE, n = 4. P ≤ 0.05 compared with WT (*) and WT + Kif (#) by 1-way ANOVA and Tukey’s posttest within monocyte groups. Broken lines represent average of nontransfected cells. J: following native monocyte adhesion to COS-1 cells under flow, cells were fixed and stained for CD16+ and CD16− markers. Data are means ± SE, n = 3. P ≤ 0.05 compared with baseline (*) and WT ($) by 1-way ANOVA with Tukey’s posttest within monocyte groups.
CD16+ monocyte adhesion is higher, and rolling velocity is significantly slower to HM-ICAM-1 compared with complex ICAM-1-expressing COS-1 cells (Fig. 6, E and F). CD16− monocyte adhesion was higher in ICAM-1-expressing COS-1 cells compared with empty vector control. Modulating N-glycoforms using Kif had no effect on CD16− monocyte rolling or adhesion. Treatment of WT or Kif-exposed COS-1 cells with ConA or HHL lectins, before assessing monocyte adhesion, had no effect on CD16− monocyte adhesion but significantly abrogated CD16+ monocyte adhesion (Fig. 6G). Notably, both ConA and HHL abrogated CD16+ cell adhesion to COS-1 cells expressing predominantly complex ICAM-1 also, i.e., without prior Kif treatment (Fig. 6G). This indicates that a basal level of HM epitopes is present on the cell surface for HHL to block, which leads to inhibition of CD16+ monocyte adhesion. Consistent with this conclusion, longer exposure times of PLA staining with HHL and ICAM-1 in WT ICAM-1-transfected COS-1 cells, without Kif treatment, demonstrate clear HM-ICAM-1 staining (Fig. 6H). Furthermore, ConA and HHL had no effect on monocyte rolling velocities in CD16− cells. However, HHL rescued rolling velocity of CD16+ cells in the presence of Kif (Fig. 6I).
Finally, because CD16+ monocytes were purified using positive selection by CD16− magnetic beads, these cells may have been activated, leading to increased adhesivity. Therefore, monocyte adhesion to COS-1 cells was also assessed using monocytes without CD16− based selection. In these experiments, at the end of the adhesion assay, cells were fixed, and levels of CD62L and CD16 were determined to assess relative adhesion of CD16− or CD16+ cells, respectively. Figure 6J shows that both CD62L and CD16 levels increased in ICAM-1-expressing COS-1 cells, with only CD16 levels increasing further in Kif-treated cells.
DISCUSSION
The process of monocyte adhesion to and migration across the EC layer is a key component of inflammatory responses, as is the more recent concept that there are distinct monocyte populations that have proinflammatory or anti-inflammatory (reparative) functions. However, there remains limited insight into whether the mechanisms that govern how CD16− versus CD16+ monocytes adhere to inflamed endothelial cells are the same or are regulated differently. Both CD16− and CD16+ cells express VLA4, LFA-1 (CD11a/18), and Mac-1 (CD11b/18), which comprise the main monocytic ligands for adhesion to endothelial vascular cell adhesion molecule-1 and ICAM-1. Because both monocyte subsets express similar levels of these ligands, it is unlikely that their abundance accounts for differential recruitment of CD16− versus CD16+ monocytes to the endothelium during inflammation (36, 39). Some studies have demonstrated a role for CX3CR1/CX3CL1 (fractalkine) in the recruitment of CD16+ monocytes (1, 2, 4); here we provide evidence that HM N-glycans, specifically residing on endothelial ICAM-1, may selectively recruit CD16+ monocytes.
A role for HM epitopes is indicated by elevated CD16+ monocyte adhesion in EC or COS-1 cells treated with the α-mannosidase class I inhibitor Kif. This was confirmed by data showing that HHL or ConA, lectins that recognize terminal mannose residues, abrogated CD16+ monocyte adhesion. A role for surface HM epitopes was confirmed using COS-1 cells transfected with ICAM-1. CD16+ cell adhesion was higher in COS-1 cells expressing higher levels of HM-ICAM1, and blocking surface HM using HHL completely inhibited CD16+ monocyte interactions. Interestingly, this inhibition was observed with control ICAM-1-transfected COS-1 cells also where HM-ICAM-1 was still detectable, albeit at lower levels compared with Kif-treated cells (see Fig. 6H). These data suggest that surface HM sugars are primary mediators of CD16+ cell adhesion. ConA also, albeit to lesser extent, inhibited CD16− cell adhesion (Fig. 5A), which is likely because of the fact that ConA also recognizes terminal glucose and galactose residues in addition to mannose residues (35). Indeed, ConA inhibited CD16− adhesion with or without Kif treatment, suggesting for this monocyte subset that the anti-adhesive effect of this lectin is HM independent.
Our previous studies identified ICAM-1 as the primary endothelial adhesion molecule whose N-glycosylation is modulated during inflammation (31). We therefore focused on determining the role of HM and complex ICAM-1 N-glycoforms in mediating CD16− and CD16+ monocyte adhesion in this study. A dependence on ICAM-1 was demonstrated using ICAM-1-blocking antibodies. We show that CD16+ monocyte adhesion occurs at the D3 ICAM-1 domain (where Mac-1 binds). These data are consistent with previous reports showing that the Mac-1-binding domain is N-glycosylated, whereas the LFA-1-binding domain is not (13, 26). Notably, a HM dependence was not observed for polymorphonuclear neutrophil adhesion, consistent with studies showing a dominant role for LFA-1-dependent interactions in mediating polymorphonuclear neutrophil-ICAM-1 adhesion (14, 24, 25). The mechanisms by which polymorphonuclear neutrophils preferentially use LFA-1-dependent mechanisms, whereas CD16+ monocytes use Mac-1, remain to be determined and likely are important in understanding how different leukocytes traffic to sites of injury.
A role for HM-ICAM-1 in mediating CD16+ monocyte adhesion was demonstrated with both a mixture of naïve monocytes (without prior CD16 ligation) and with CD16+ monocytes purified by positive selection using anti-CD16-coated magnetic beads. Moreover, in all experiments, total levels of ICAM-1 were similar, but HM composition of ICAM-1 was altered, and the latter regulated CD16+ cell adhesion. This supports the conclusion that, whereas ICAM-1 is necessary, HM glycoforms on ICAM-1 confer selectivity for CD16+ monocyte binding. This conclusion is further supported by experiments in which CD16+ monocyte adhesion was determined at physiological ratios with CD16− cells (Fig. 3, C and D). If binding of CD16− and CD16+ cells was equivalent, based on relative abundance, a 10-fold less adhesion of CD16+ cells is expected. However, only a twofold decrease in CD16+ cells, compared with CD16− cells, was observed in TNF-α-treated ECs, with no difference in Kif-treated cells. Collectively, these data suggest that HM-ICAM-1 selectively recruits low abundant CD16+ monocytes to the endothelium. Because CD16+ monocyte numbers expand during chronic inflammation, we speculate that HM-ICAM-1 plays a significant role in disease settings also (18, 39).
CD16+ monocyte rolling velocity was selectively slowed when COS-1 expressed HM-ICAM-1, consistent with earlier reports of a role for ICAM-1 in both the rolling and adhesion processes (16, 22, 37). However, the effects of HM glycoforms were greater in the firm adhesion step. Further studies are required to identify the precise interactions between HM-ICAM-1 and CD16+ monocyte receptors. Current studies are focusing primarily on the ICAM-1 ligands Mac-1 and LFA-1, their activation states, and the role of CD16 itself.
Notably, surface expression of HM-ICAM-1 on cultured endothelial cells was transient, providing an intriguing insight into the timing of inflammation. Based on our data, we speculate that an initial wave of proinflammatory monocyte infiltration is mediated by the formation of HM-ICAM-1. Thereafter, HM-ICAM1 levels decline, preventing further CD16+ monocyte recruitment and allowing resolution. Further studies testing how HM-ICAM-1 may change in chronic inflammation are warranted and may shed insights into how recruitment of proinflammatory monocytes continues in disease settings. Although we have not investigated the mechanisms by which HM-N-glycan are transiently incorporated into induced ICAM-1, our prior data show that TNF-α inhibits α-mannosidase activity (33). Given that N-glycan biosynthesis is a linear process, and data with Kif treatment, inhibition of α-mannosidase activity likely mediates formation of HM-N-glycans. Ongoing studies are determining the role of TNF-α on the transient inhibition of α-mannosidase activity. Future studies are also required to map the specific N-glycan sites on ICAM-1 where HM structures reside. The term “high-mannose” does not imply that ICAM-1 exclusively expresses HM structures. ICAM-1 contains eight N-glycosylation sites, and any single ICAM-1 is likely to harbor an array of N-glycans, from HM to complex structures. Our data simply demonstrate that a form of ICAM-1 harboring HM structures enhances CD16+ monocyte adhesion.
In conclusion, the presented data suggest that endothelial HM-ICAM-1 is a key mediator for recruiting CD16+ monocytes to the activated endothelium and, in turn, may present a novel therapeutic target to modulate proinflammatory monocyte trafficking during innate immunity and inflammatory disease.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grant HL-007918 (K. Regal-McDonald) and K99/R00 Pathway to Independence Award Grant R00-HL-131866 (to J. W. Barnes).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.R.-M. and R.P.P. conceived and designed research; K.R.-M. performed experiments; K.R.-M. and B.X. analyzed data; K.R.-M., J.W.B., and R.P.P. interpreted results of experiments; K.R.-M. prepared figures; K.R.-M. and R.P.P. drafted manuscript; K.R.-M., J.W.B., and R.P.P. edited and revised manuscript; K.R.-M., B.X., J.W.B., and R.P.P. approved final version of manuscript.
REFERENCES
- 1.Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 197: 1701–1707, 2003. doi: 10.1084/jem.20022156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ancuta P, Wang J, Gabuzda D. CD16+ monocytes produce IL-6, CCL2, and matrix metalloproteinase-9 upon interaction with CX3CL1-expressing endothelial cells. J Leukoc Biol 80: 1156–1164, 2006. doi: 10.1189/jlb.0206125. [DOI] [PubMed] [Google Scholar]
- 3.Argenbright LW, Letts LG, Rothlein R. Monoclonal antibodies to the leukocyte membrane CD18 glycoprotein complex and to intercellular adhesion molecule-1 inhibit leukocyte-endothelial adhesion in rabbits. J Leukoc Biol 49: 253–257, 1991. doi: 10.1002/jlb.49.3.253. [DOI] [PubMed] [Google Scholar]
- 4.Aspinall AI, Curbishley SM, Lalor PF, Weston CJ, Blahova M, Liaskou E, Adams RM, Holt AP, Adams DH. CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of CD16(+) monocytes across human liver sinusoidal endothelium. Hepatology 51: 2030–2039, 2010. doi: 10.1002/hep.23591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloom JW, Madanat MS, Ray MK. Cell line and site specific comparative analysis of the N-linked oligosaccharides on human ICAM-1des454-532 by electrospray ionization mass spectrometry. Biochemistry 35: 1856–1864, 1996. doi: 10.1021/bi952354m. [DOI] [PubMed] [Google Scholar]
- 6.Bourdillon MC, Poston RN, Covacho C, Chignier E, Bricca G, McGregor JL. ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(-/-)/ICAM-1(-/-)) fed a fat or a chow diet. Arterioscler Thromb Vasc Biol 20: 2630–2635, 2000. doi: 10.1161/01.ATV.20.12.2630. [DOI] [PubMed] [Google Scholar]
- 7.Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, Chalasani G, Taboas JM, Lakkis FG, Metes DM. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One 12: e0176460, 2017. doi: 10.1371/journal.pone.0176460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood 84: 2068–2101, 1994. [PubMed] [Google Scholar]
- 9.Čejková S, Kralova-Lesna I, Poledne R. Monocyte adhesion to the endothelium is an initial stage of atherosclerosis development. Cor Vasa 58: e419–e425, 2016. doi: 10.1016/j.crvasa.2015.08.002. [DOI] [Google Scholar]
- 10.Chacko BK, Scott DW, Chandler RT, Patel RP. Endothelial surface N-glycans mediate monocyte adhesion and are targets for anti-inflammatory effects of peroxisome proliferator-activated receptor γ ligands. J Biol Chem 286: 38738–38747, 2011. doi: 10.1074/jbc.M111.247981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med 191: 189–194, 2000. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crampton SP, Davis J, Hughes CC. Isolation of human umbilical vein endothelial cells (HUVEC). J Vis Exp 3: 183, 2007. doi: 10.3791/183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 65: 961–971, 1991. doi: 10.1016/0092-8674(91)90548-D. [DOI] [PubMed] [Google Scholar]
- 14.Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 163: 5029–5038, 1999. [PubMed] [Google Scholar]
- 15.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 27: 2292–2301, 2007. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 16.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res 107: 321–330, 2015. doi: 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilgendorf I, Swirski FK, Robbins CS. Monocyte fate in atherosclerosis. Arterioscler Thromb Vasc Biol 35: 272–279, 2015. doi: 10.1161/ATVBAHA.114.303565. [DOI] [PubMed] [Google Scholar]
- 18.Idzkowska E, Eljaszewicz A, Miklasz P, Musial WJ, Tycinska AM, Moniuszko M. The role of different monocyte subsets in the pathogenesis of atherosclerosis and acute coronary syndromes. Scand J Immunol 82: 163–173, 2015. doi: 10.1111/sji.12314. [DOI] [PubMed] [Google Scholar]
- 19.Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115: e10–e19, 2010. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiménez D, Roda-Navarro P, Springer TA, Casasnovas JM. Contribution of N-linked glycans to the conformation and function of intercellular adhesion molecules (ICAMs). J Biol Chem 280: 5854–5861, 2005. doi: 10.1074/jbc.M412104200. [DOI] [PubMed] [Google Scholar]
- 21.Kapinsky M, Torzewski M, Büchler C, Duong CQ, Rothe G, Schmitz G. Enzymatically degraded LDL preferentially binds to CD14(high) CD16(+) monocytes and induces foam cell formation mediated only in part by the class B scavenger-receptor CD36. Arterioscler Thromb Vasc Biol 21: 1004–1010, 2001. doi: 10.1161/01.ATV.21.6.1004. [DOI] [PubMed] [Google Scholar]
- 22.Kevil CG, Chidlow JH, Bullard DC, Kucik DF. High-temporal-resolution analysis demonstrates that ICAM-1 stabilizes WEHI 274.1 monocytic cell rolling on endothelium. Am J Physiol Cell Physiol 285: C112–C118, 2003. doi: 10.1152/ajpcell.00334.2002. [DOI] [PubMed] [Google Scholar]
- 23.Kevil CG, Patel RP, Bullard DC. Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. Am J Physiol Cell Physiol 281: C1442–C1447, 2001. doi: 10.1152/ajpcell.2001.281.5.C1442. [DOI] [PubMed] [Google Scholar]
- 24.Lefort CT, Ley K. Neutrophil arrest by LFA-1 activation. Front Immunol 3: 157, 2012. doi: 10.3389/fimmu.2012.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li N, Yang H, Wang M, Lü S, Zhang Y, Long M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol Biol Cell 29: 408–418, 2018. doi: 10.1091/mbc.E16-12-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto VI, Damoc E, Cueni LN, Schürpf T, Frei R, Ali S, Callewaert N, Moise A, Leary JA, Folkers G, Przybylski M. N-glycan structures and N-glycosylation sites of mouse soluble intercellular adhesion molecule-1 revealed by MALDI-TOF and FTICR mass spectrometry. Glycobiology 16: 1033–1044, 2006. doi: 10.1093/glycob/cwl032. [DOI] [PubMed] [Google Scholar]
- 27.Patel SS, Thiagarajan R, Willerson JT, Yeh ET. Inhibition of alpha4 integrin and ICAM-1 markedly attenuate macrophage homing to atherosclerotic plaques in ApoE-deficient mice. Circulation 97: 75–81, 1998. doi: 10.1161/01.CIR.97.1.75. [DOI] [PubMed] [Google Scholar]
- 28.Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Große-Dunker G, Heisel I, Hornof F, Jeken J, Rebling NM, Ulrich C, Scheller B, Böhm M, Fliser D, Heine GH. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol 60: 1512–1520, 2012. doi: 10.1016/j.jacc.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Rogacev KS, Ulrich C, Blömer L, Hornof F, Oster K, Ziegelin M, Cremers B, Grenner Y, Geisel J, Schlitt A, Köhler H, Fliser D, Girndt M, Heine GH. Monocyte heterogeneity in obesity and subclinical atherosclerosis. Eur Heart J 31: 369–376, 2010. doi: 10.1093/eurheartj/ehp308. [DOI] [PubMed] [Google Scholar]
- 30.Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. monocyte subsets: phenotypes and function in tuberculosis infection. Front Immunol 9: 1726, 2018. doi: 10.3389/fimmu.2018.01726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott DW, Dunn TS, Ballestas ME, Litovsky SH, Patel RP. Identification of a high-mannose ICAM-1 glycoform: effects of ICAM-1 hypoglycosylation on monocyte adhesion and outside in signaling. Am J Physiol Cell Physiol 305: C228–C237, 2013. doi: 10.1152/ajpcell.00116.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott DW, Chen J, Chacko BK, Traylor JG Jr, Orr AW, Patel RP. Role of endothelial N-glycan mannose residues in monocyte recruitment during atherogenesis. Arterioscler Thromb Vasc Biol 32: e51–e59, 2012. doi: 10.1161/ATVBAHA.112.253203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott DW, Vallejo MO, Patel RP. Heterogenic endothelial responses to inflammation: role for differential N-glycosylation and vascular bed of origin. J Am Heart Assoc 2: e000263, 2013. doi: 10.1161/JAHA.113.000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanley P, Cummings RD. Structures common to different glycans. In: Essentials of Glycobiology (3rd ed.), edited by Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2017, chapt. 14. [Google Scholar]
- 35.Stanley P, Schachter H, Taniguchi N. N. Glycans. In: Essentials of Glycobiology, edited by Varki A, Cummings R, Esko J, Freeze H, Stanley P, Bertozzi C, Hart G, Etzler M. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2009. [PubMed] [Google Scholar]
- 36.Stec M, Weglarczyk K, Baran J, Zuba E, Mytar B, Pryjma J, Zembala M. Expansion and differentiation of CD14+CD16(-) and CD14+ +CD16+ human monocyte subsets from cord blood CD34+ hematopoietic progenitors. J Leukoc Biol 82: 594–602, 2007. doi: 10.1189/jlb.0207117. [DOI] [PubMed] [Google Scholar]
- 37.Steeber DA, Campbell MA, Basit A, Ley K, Tedder TF. Optimal selectin-mediated rolling of leukocytes during inflammation in vivo requires intercellular adhesion molecule-1 expression. Proc Natl Acad Sci USA 95: 7562–7567, 1998. doi: 10.1073/pnas.95.13.7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weber C, Belge KU, von Hundelshausen P, Draude G, Steppich B, Mack M, Frankenberger M, Weber KS, Ziegler-Heitbrock HW. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol 67: 699–704, 2000. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 39.Wildgruber M, Aschenbrenner T, Wendorff H, Czubba M, Glinzer A, Haller B, Schiemann M, Zimmermann A, Berger H, Eckstein HH, Meier R, Wohlgemuth WA, Libby P, Zernecke A. The “Intermediate” CD14++CD16+ monocyte subset increases in severe peripheral artery disease in humans. Sci Rep 6: 39483, 2016. doi: 10.1038/srep39483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wildgruber M, Czubba M, Aschenbrenner T, Wendorff H, Hapfelmeier A, Glinzer A, Schiemann M, Zimmermann A, Eckstein HH, Berger H, Wohlgemuth WA, Meier R, Libby P, Zernecke A. Increased intermediate CD14++CD16++ monocyte subset levels associate with restenosis after peripheral percutaneous transluminal angioplasty. Atherosclerosis 253: 128–134, 2016. doi: 10.1016/j.atherosclerosis.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, Kourilsky P, Wong SC. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118: e16–e31, 2011. doi: 10.1182/blood-2010-12-326355. [DOI] [PubMed] [Google Scholar]
- 42.Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res 53: 41–57, 2012. doi: 10.1007/s12026-012-8297-3. [DOI] [PubMed] [Google Scholar]
- 43.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 7: 77–86, 2010. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziegler-Heitbrock L. Blood monocytes and their subsets: established features and open questions. Front Immunol 6: 423, 2015. doi: 10.3389/fimmu.2015.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]