

Abstract
Insufficient autophagy has been proposed as a mechanism of cellular aging, as this leads to the accumulation of dysfunctional macromolecules and organelles. Premature vascular aging occurs in hypertension. In fact, many factors that contribute to the deterioration of vascular function as we age are accelerated in clinical and experimental hypertension. Previously, we have reported decreased autophagy in arteries from spontaneously hypertensive rats (SHRs); however, the effects of restoring autophagic activity on blood pressure and vascular function are currently unknown. We hypothesized that reconstitution of arterial autophagy in SHRs would decrease blood pressure and improve endothelium-dependent relaxation. We treated 14- to 18-wk-old Wistar rats (n = 7 vehicle and n = 8 trehalose) and SHRs (n = 7/group) with autophagy activator trehalose (2% in drinking water) for 28 days. Blood pressure was measured by radiotelemetry, and vascular function and structure were measured in isolated mesenteric resistance arteries (MRAs) using wire and pressure myographs, respectively. Treatment with trehalose had no effect on blood pressure in SHRs; however, isolated MRAs presented enhanced relaxation to acetylcholine, in a cyclooxygenase- and reactive oxygen species-dependent manner. Similarly, trehalose treatment shifted the relaxation to the Rho kinase (ROCK) inhibitor Y-27632 to the right, indicating reduced ROCK activity. Finally, trehalose treatment decreased arterial stiffness as indicated by the slope of the stress-strain curve. Overall these data indicate that reconstitution of arterial autophagy in SHRs improves endothelial and vascular smooth muscle function, which could synergize to prevent stiffening. As a result, restoration of autophagic activity could be a novel therapeutic for premature vascular aging in hypertension.
NEW & NOTEWORTHY This work supports the concept that diminished arterial autophagy contributes to premature vascular aging in hypertension and that therapeutic reconstitution of autophagic activity can ameliorate this phenotype. As vascular age is a new clinically used index for cardiovascular risk, understanding this mechanism may assist in the development of new drugs to prevent premature vascular aging in hypertension.
Listen to this article’s corresponding podcast at https://ajpheart.podbean.com/e/behind-the-bench-episode-one-cam-squared/.
Keywords: arterial stiffening, autophagy, hypertension, vascular aging, vascular function
INTRODUCTION
Age is a major risk factor for hypertension (7), and premature aging (relative to actual chronological age) is commonly observed in the vasculature of hypertensive patients and animals (3, 25, 49). In fact, many factors that contribute to the deterioration of vascular function as we age are accelerated and exacerbated in hypertension (12). Nonetheless, the precise mechanisms that underlie the aged phenotype in arteries from hypertensive animals remain elusive.
The aged phenotype is classically viewed as the accumulation of damaged macromolecules and dysfunctional organelles. Although waste products are an inevitable consequence of normal cellular metabolism, multiple systems are devoted to its repair, clearance, or recycling. Autophagy is the evolutionarily conserved catabolic process essential for both maintaining homeostasis via the removal and recycling of damaged macromolecules and dysfunctional organelles and providing micronutrients during times of stress (1). Within the cardiovascular system, most work has focused on how abnormal autophagy affects the heart (53), with only a few studies attempting to understand its contribution to vascular (patho)physiology (2, 38).
Autophagic activity is implicated as a modulator of longevity (57), and it is also known that its induction can extend the life span of mice (40). Therefore, it is plausible that a decline in autophagy contributes to “aged” phenotype of the vasculature associated with hypertension. This notion is supported by studies that showed that an upregulation of autophagy reversed several phenotypes of vascular aging in old mice, including endothelial function (21, 22). Moreover, our own previous data have suggested that autophagic activity is reduced in resistance and conduit vasculature from spontaneously hypertensive rats (SHRs) (29). Collectively, these reports contributed to the hypothesis of the current investigation that reconstitution of autophagic activity in SHRs would decrease blood pressure and improve endothelium-dependent relaxation in mesenteric resistance arteries (MRAs). To test this hypothesis, we treated normotensive Wistar and SHRs with trehalose, a nonreducing disaccharide, and established autophagy activator (43). Trehalose is found in many organisms, including bacteria, yeast, fungi, insects, invertebrates, and plants but not mammals (9). It is able to protect cellular integrity during times of environmental stress, including the stabilization of membranes and protecting proteins from unfolding and aggregation (50). SHRs were our hypertensive model of choice for multiple, compelling reasons, including the following: 1) the development of hypertension in this rat is genetic, and it has many of the features that resemble essential hypertension in humans; 2) the genetic background is homogeneous and environmental factors can be controlled; and 3) we have an established track record in our laboratory using this rat model, including our original observations that SHRs present diminished autophagic indexes (29). In this study, we reveal for the first time that upregulation of arterial autophagy in SHRs reduces several indexes of premature vascular aging associated with hypertension. This article is timely given that vascular age determination has recently been adopted as a clinical index for cardiovascular disease risk (39), and thus autophagy could be a novel therapeutic target.
MATERIALS AND METHODS
Animals
Male Wistar rats and SHRs were used for this investigation (Envigo). Adult rats were 14–18 wk of age (Wistar rats: 419–487 g, and SHRs: 288–367 g). Young rats of both strains were also used in one experiment, and these rats were 5–6 wk of age (Wistar rats: 175–192 g, and SHRs: 94–123 g). The sample size per experiment (see figures and legends) is the number of independent rats used, respective of strain and treatment group. Previous work from our laboratory estimating a large effect size (Cohen’s d > 0.8), as well as power analysis (desired power of 0.80 to 0.85 with a probability of a type I error of 0.05), has provided a basis for the projected number of rats required per experimental group. The predicted number of rats required per experimental group is four to eight, and this indicates the minimum number needed to generate a statistically significant experimental outcome (P < 0.05). In this work, we did not include females because the phenotype of decreased arterial autophagy in hypertension has only been reported in males (29). Therefore, to understand the physiological significance of reconstituting autophagy, we performed this study using male rats. Future studies will include females because it is currently unknown whether female SHRs present a reduction in autophagic activity, relative to normotensive females or SHR males. Therefore, decreased autophagy may be a novel mechanism underlying the well-established sex difference in SHRs (41).
All rats were maintained on a 12-h:12-h light-dark cycle with both chow and water ad libitium. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (36) and were reviewed and approved by the Institutional Animal Care and Use Committee of Augusta University. Euthanasia of rats by thoracotomy and exsanguination via cardiac puncture was performed under isoflurane anesthesia, administered via nose cone (5% in 100% O2), consistent with the American Veterinary Medical Association Guidelines for the Euthanasia of Animals (2013). All euthanasia and tissue harvesting was performed in the laboratory from 9:00 to 11:00 am per experimental day.
Acid Phosphatase Activity
In naïve/untreated adult (n = 5 Wistar rats and n = 7 SHRs) and young (n = 3 Wistar rats and n = 4 SHRs) rats, MRAs and aortas were pooled to isolate a sufficient quantity of crude lysosomal fractions per rat. Arteries were first cleaned of perivascular adipose tissues and washed in ice cold physiological salt solution (PSS) containing the following (in mmol/L): 130.0 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4·7H2O, 14.9 NaHCO3, 5.5 glucose, 1.6 CaCl2·2H2O, and 0.03 EDTA (all MilliporeSigma). Arteries were then homogenized in extraction buffer, plus protease inhibitor cocktail (MilliporeSigma), using a dounce homogenizer. From homogenate, crude lysosomal fractions were isolated according to the manufacturer’s instructions (LYSISO1; MilliporeSigma).
Crude lysosomal fractions were left overnight at 4°C, and subsequently protein quantification was performed. Acid phosphatase activity was then measured in 20 μg of lysosomal protein according to the manufacturer’s instructions and calculations (CS0740; MilliporeSigma).
Treatment
Adult rats were randomly assigned to receive either regular drinking water (vehicle), or 2% trehalose (MilliporeSigma) dissolved in drinking water, for 28 days (Wistar rats: n = 7 vehicle and n = 8 trehalose; SHRs n = 7/group). This concentration of trehalose was determined based on previous literature demonstrating an upregulation of autophagy with this dosage (22, 42, 50).
Western Blot Analysis
Isolated arteries.
After euthanasia, MRAs were removed from rats, cleaned of perivascular adipose tissues in ice-cold PSS, and flash frozen in liquid nitrogen. Frozen arteries were then homogenized using a mortar and pestle and lysed using tissue protein extraction reagent (Thermo Fisher Scientific), with protease inhibitors (sodium orthovanadate, phenylmethylsulfonyl fluoride, and protease inhibitor cocktail) and phosphatase inhibitors (sodium fluoride and sodium pyrophosphate) (all MilliporeSigma). The protein concentration of MRA lysates was subsequently determined, and then, equal quantities of protein (40–50 μg) were loaded into polyacrylamide gels (8–12%).
Plasma.
While the rats were under isoflurane anesthesia, but before thoracotomy and exsanguination via cardiac puncture, arterial blood was collected from the abdominal aorta in tubes containing EDTA. Blood was immediately centrifuged at 1,500 g for 15 min at 4°C; plasma was collected and flash frozen in liquid nitrogen. Samples were diluted 1:20 in deionized water. Equal volumes of diluted plasma (20 μl) were loaded into polyacrylamide gels (12%). After loading, gels were separated by SDS-PAGE and transferred to PVDF membranes (Thermo Fisher Scientific). Antibodies used to determine protein expression were immunoglobulin heavy-chain binding protein (BiP; as known as GRP78), calnexin, cyclooxygenase 1, cyclooxygenase 2, phospho-myosin phosphatase target subunit 1 (MYPT1)Thr696, total MYPT1, NADH dehydrogenase subunit 6 (ND6), protein disulfide isomerase (PDI), Rho kinase (ROCK) II, and sequestosome 1 (SQSTM1/p62). Further details on antibody concentrations, secondary antibody isotype, company from which the antibody was purchased, published studies that have previously validated these antibodies, and the Research Resource Identifiers (RRID) antibody identification number are presented in Supplemental Table S1 (All Supplemental Material is available at https://doi.org/10.6084/m9.figshare.8323301.v4). Phosphorylated proteins were normalized to their total form, and GAPDH was used as the loading control. Densitometric analysis was performed by ImageJ (National Institutes of Health).
Blood Pressure, Heart Rate, and Locomotor Activity
Blood pressure was measured in adult rats (14–18 wk old) by radiotelemetry from the femoral artery using DSI PA-C10 telemeters implanted in the inguinal region under isoflurane anesthesia and using aseptic technique. Approximately 1 wk was allowed for recovery and acclimation before measurements were begun. Pulsatile pressure signals (for mean, systolic, and diastolic pressures) were collected along with locomotor activity signals at 500 Hz for 4 s every 2 min for 19 h per day (1:00 pm through 8:00 am) and averaged to yield daily values for each variable for each rat.
Locomotor activity was calculated based on speed and distance moved by the rats, generating a “count.” More counts/minute equates to more movement, which is measured by the radiotelemetry receiver tracking the signal of the implanted transmitter unit as the animal moves around the cage (it is important to know that the receiver is a platform the same length-width as the cage, and on which the cage rests). Most of the reading is due to movement along the x-y plane, but because signal strength is part of what the system tracks, it may pick up the animal climbing and hanging off the wire cage topper also.
Vascular Function
Third order MRAs were mounted onto Danish Myo Technology (Aarhus, Denmark) wire myographs in PSS. Arteries were normalized to their optimal lumen diameter for active tension development, as described previously (29, 34). All arteries were initially contracted with 120 mmol/L KCl. Endothelium integrity was then tested with a phenylephrine (PE)-induced contraction (3 μmol/L) followed by endothelium-dependent vasodilation with acetylcholine (ACh; 3 μmol/L).
Cumulative concentration-response curves were performed to ACh (0.1 nmol/L to 100 μmol/L), ROCK inhibitor Y-27632 (1 nmol/L to 30 μmol/L), and endothelium-independent vasodilator sodium-nitroprusside (SNP; 0.1 nmol/L to 100 μmol/L) (all Millipore Sigma). All concentration-response curves were performed after an initial contraction with 3 μmol/L PE. Thirty minutes before ACh concentration-response curves in MRAs; some MRAs were incubated with cyclooxygenase 1 and 2 inhibitor indomethacin (10 μmol/L; Millipore Sigma), superoxide dismutase mimetic tempol (100 μmol/L; Tocris), nitric oxide synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME; 100 μmol/L; Millipore Sigma), or potassium channel inhibitor tetraethylammonium (TEA; 10 mmol/L; Millipore Sigma). Results are presented as percent relaxation of the initial precontraction to PE (%PE).
Vascular Structure and Mechanics.
Third- and fourth-order MRAs were mounted in PSS and tied with surgical nylon suture onto glass micropipettes (100- to 125-µm diameter) in an arteriograph (Living Systems Instrumentation), as described previously (54). Briefly, vessel length was carefully adjusted to maintain the artery walls parallel at increasing pressures. Flow in the vessel was generated through the distal pipette with a peristaltic pump (Living Systems Instrumentation). Intraluminal pressure was then raised to 120 mmHg, and again the vessel length was adjusted. The segment was set to a pressure of 60 mmHg (no flow) and allowed to equilibrate for 20 min at 37°C in filtered PSS, gassed with a mixture of 95% O2-5% CO2. Afterward, intraluminal pressure was decreased to 3 mmHg and the PSS was changed for high bicarbonate and calcium free-PSS + EGTA (in mmol/L: 115.0 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4·7H2O, 25.0 NaHCO3, 11.1 glucose, 0.01 EDTA, and 1.0 EGTA) for 20 min and then to bicarbonate and calcium free-PSS without ETGA for the pressure-response curve. Internal (Di0Ca2+) and external (De0Ca2+) diameters were continuously measured in 20-mmHg increments from 3 to 180 mmHg and recorded for 3 min at each intraluminal pressure using a video monitoring system (IonOptix). The final value used was the mean of the three regions of interest, taken during the last 30 s, when the internal and external diameters had reached a steady state.
Calculations of structural properties are the following:
Calculation of mechanical stress-strain is the following:
where D00Ca is the internal diameter at 3 mmHg and Di0Ca2+ is the observed internal diameter for a given intravascular pressure, both measured in 0Ca2+-PSS.
where P is the intraluminal pressure (1 mmHg = 1.334 × 103 dyn/cm2) and WT is wall thickness at each intraluminal pressure in 0Ca2+-PSS.
Arterial stiffness, independent of geometry, is determined by the Young’s elastic modulus (E = stress/strain). Therefore, the stress-strain relationship was analyzed by determining the slope of the stress-strain curve, indicating the tangential or incremental elastic modulus (Einc = δσ/δε). Einc was obtained by fitting the stress-strain data from each animal to an exponential curve using the equation: σ = σorigeβε, where σorig is the stress at the original diameter (3 mmHg). Taking derivatives on the equation, we determine that Einc = βσ. Therefore, for any given σ-value, Einc, is directly proportional to stiffness (β).
Reactive Oxygen Species Measurement
Dihydroethidium (DHE) fluorescence was used to measure reactive oxygen species (ROS), as described previously (29). Briefly, MRAs were embedded in tissue freezing medium (Triangle Biomedical Sciences) and frozen in liquid nitrogen. Transverse cross sections (10 µm) were equilibrated for 10 min in phosphate buffer in a light-protected, humidified chamber at 37°C. Fresh buffer containing hydroethidine (1 µmol/L; Millipore Sigma) was topically applied to each cross section. The slides were then incubated in a light-protected, humidified chamber at 37°C for 30 min. Negative control sections received the same volume of phosphate buffer but in the absence of hydroethidine. Stained cross sections were viewed with a fluorescent microscope using a ×40 objective. Three different cross sections of each artery were examined for the oxidation products of DHE, indicated by red fluorescence. The images were analyzed using ImageJ.
Fibrosis and Extracellular Matrix Measurement
Masson’s trichrome staining was used to measure fibrosis and extracellular matrix deposition. Like DHE fluorescence, MRAs were embedded in tissue freezing medium (Triangle Biomedical Sciences) and frozen in liquid nitrogen. Transverse cross sections (10 µm) were stained by the Augusta University Histology Core following standard Masson’s trichrome staining procedures (48). Stained cross sections were viewed with a light microscope using a ×40 objective. Three different cross sections of each artery were examined for fibrosis and extracellular matrix deposition, indicated by the abundance of blue collagen staining relative to red muscle staining. The images were analyzed using ImageJ.
Statistical Analysis
The statistical procedures used included Student’s t test, one-way and two-way ANOVA, and nonlinear regression analysis (logEC50). Tukey’s post hoc testing was used in all cases using a one-way ANOVA, and the Bonferroni post hoc was used in all cases using a two-way ANOVA. All analyses were performed using data analysis software GraphPad Prism 5.0 (La Jolla, CA). Statistical significance was set at P < 0.05. The concentration-response curves are presented as means ± SE; all other data are presented as means ± SD.
RESULTS
Arteries from Adult SHRs Have Diminished Lysosomal Acid Phosphatase Activity
To measure autophagic activity, we isolated crude lysosomal fractions and measured acid phosphatase activity. Acid phosphatase is the major acid hydrolase in lysosomes. After MRAs and aortas from individual rats were pooled (to obtain sufficient quantity of arterial tissue in which to isolate lysosomes), we observed that acid phosphatase activity increases with age in normotensive Wistar rats. However, this increase does not occur in arteries from SHRs (Fig. 1). Overall, these findings reveal that arteries from adult SHRs have decreased autophagic activity. Therefore, for the subsequent experiments involving the reconstitution of autophagy with trehalose, we used adult rats and SHRs that were in the maintenance phase of high blood pressure.
Fig. 1.
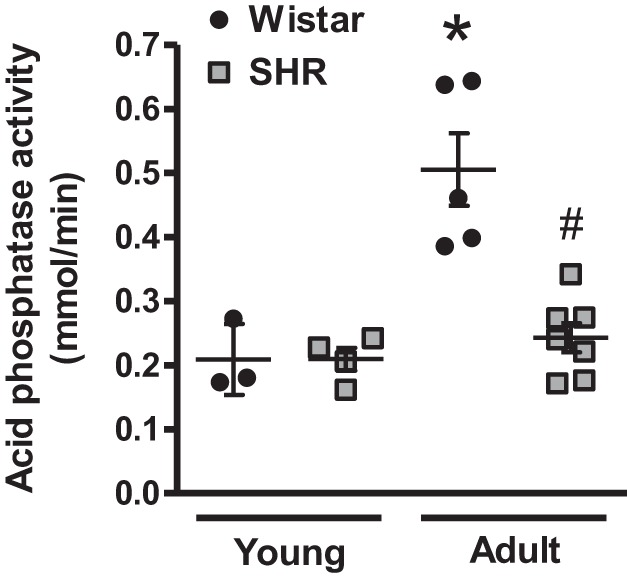
Arteries from adult spontaneously hypertensive rats (SHRs) do not present the age-dependent increase in lysosomal acid phosphatase activity. Acid phosphatase activity in isolated lysosomal fractions from pooled mesenteric resistance arteries and aorta from Wistar and SHRs; n = 3–7 rats. One-way ANOVA: *P < 0.05 vs. young Wistar rats; #P < 0.05 vs. adult Wistar rats.
Trehalose Treatment Increases Arterial Autophagy in SHRs
Next, we wanted to confirm that autophagy was in fact upregulated in MRAs after trehalose treatment. To do this we probed for the expression of SQSTM1/p62, an ubiquitin binding protein that binds the autophagosomal membrane protein LC3/Atg8. Our rationale for focusing on SQSTM1/p62 expression was due to the fact that SQSTM1/p62 is an autophagy substrate that can used as a reporter of autophagy activity (24). As such, active autophagy leads to a decrease in SQSTM1/p62 levels; conversely, inhibition of autophagic activity maintains SQSTM1/p62 expression. Supporting our acid phosphatase assay for decreased autophagic activity in SHR arteries, we observed that SHRs treated with vehicle had significantly increased SQSTM1/p62 expression compared with Wistar treated with vehicle, and SHRs treated with trehalose had a significant reduction in expression (Fig. 2A). However, no differences in SQSTM1/p62 expression were observed in Wistar rats treated with and without trehalose (Fig. 2A). To further support the notion that autophagy is increased after trehalose treatment in SHRs, we probed for the expression for the circulating damage-associated molecular pattern ND6. Our laboratory was the first to identify ND6 as a circulating damage-associated molecular pattern in pathophysiology (55). Under normal, physiological conditions, ND6 is mitochondrial protein housed within cellular membranes. However, upon cell death and mitochondrial breakdown, ND6 can be released into the circulation to unintentionally activate the immune system (28). Therefore, we rationalized that because autophagy is a ubiquitous catabolic process, ND6 (or ND6 degradation) could serve as an indirect biomarker of autophagy. SHRs treated with vehicle had a significant increase in ND6, and this was reduced in SHRs treated with trehalose (Fig. 2B). Finally, we hypothesized that upregulation of autophagy by trehalose would prevent the aggregation of proteins and the unfolded protein response (50). To test this hypothesis we probed for the expression of representative endoplasmic reticulum (ER) chaperones as an indicator of the unfolded protein response and ER stress. We observed that SHRs treated with vehicle had significantly increased BiP, calnexin, and PDI and these proteins were all significantly reduced after trehalose treatment (Fig. 2, C–E). Investigation of these ER chaperones also revealed the unexpected finding that trehalose induced a state of ER stress in MRAs from Wistar rats treated with trehalose (Fig. 2, C–E). Overall, these data demonstrate that arterial autophagy was upregulated after trehalose treatment in SHRs and this can reduce ER stress.
Fig. 2.

Trehalose treatment increases arterial autophagy, decreases circulating damage-associated molecular pattern NADPH dehydrogenase subunit 6 (ND6), and reduces arterial endoplasmic reticulum (ER) stress in mesenteric resistance arteries (MRAs) from spontaneously hypertensive rats (SHRs). A–E: protein expression analysis for sequestosome 1 (SQSTM1/p62; A) in MRAs, ND6 in plasma (B), and ER chaperones immunoglobulin heavy-chain binding protein (BiP; C), calnexin (D), and protein disulfide isomerase (PDI; E) in MRAs; n = 6–8 rats. Left, densitometric analysis; right, representative images of immunoblots. One-way ANOVA: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle.
Trehalose Treatment Does Not Lower Established High Blood Pressure in SHRs
Trehalose treatment did not significantly change any parameter of blood pressure (Fig. 3, A–C), nor heart rate (Fig. 3D) or locomotor activity (Fig. 3E) in either Wistar or SHRs. On the other hand, trehalose treatment tended to increase body mass in Wistar rats (Fig. 4, A and B) and epididymal fat mass, when controlling for strain and treatment differences via tibia length (Fig. 4C). No changes were observed in total heart, left ventricle (LV), or right ventricle (RV) masses (Fig. 4, D–F), and treatment consumption did not differ between strains or treatment groups (Fig. 4G). Overall, these data confirm that both strains were consuming treatments and that trehalose was ineffective at lowering established high blood pressure in SHRs.
Fig. 3.

Trehalose treatment does not lower established high blood pressure in spontaneously hypertensive rats (SHRs). A–E: mean arterial pressure (MAP; A), systolic blood pressure (SBP; B), diastolic blood pressure (DBP; C), heart rate (HR; D), and locomotor activity (E) after 28 days of vehicle or trehalose treatment (2% dissolved in drinking water) in Wistar and SHRs; n = 4–6 rats. A.U., arbitrary units; bpm, beats/min.
Fig. 4.

Trehalose treatment tended to increase body and fat mass in Wistar rats. A–F: body mass (A), the change (∆) in body mass (B), epididymal fat mass normalized to tibia length (C), total heart (D), left ventricle (LV; E), and right ventricle (RV; F) masses normalized to tibia length, and treatment consumption volume (in ml; G) per 24 h; n = 7–8 rats.
Trehalose Treatment Prevents the Generation of Endothelium-Derived Contracting Factors in SHRs
As expected, MRAs from SHRs treated with vehicle presented endothelial dysfunction, specifically via contraction to high concentrations of ACh (Fig. 5A). It was subsequently observed that although trehalose treatment had no effect on the relaxation to ACh in MRAs from Wistar rats (Fig. 5B), it abolished the contractile response to high concentrations of ACh in MRAs from SHRs treated with trehalose (Fig. 5C). The contractile response to high concentrations of ACh is a well-known phenomenon in arteries from SHRs, as a result of cyclooxygenase-derived products (26). To test the hypothesis that trehalose suppresses contraction to ACh via inhibition of the cyclooxygenase enzymes, some arteries were incubated with the cyclooxygenase 1 and 2 inhibitor indomethacin (10 μmol/L) before concentration-response curves with ACh. In vehicle- and trehalose-treated Wistar indomethacin had no effect on the MRA relaxation to ACh (Fig. 5, D and E). In MRAs from vehicle-treated SHRs, the contractile response to ACh was diminished after incubation with indomethacin (Fig. 5F). However, in MRAs from trehalose-treated SHRs, there was no difference in the contractile response to ACh, with or without indomethacin (Fig. 5G). This suggests that trehalose treatment suppressed cyclooxygenase-induced contraction to ACh in SHRs. To support the notion that trehalose suppresses contraction to ACh via inhibition of the cyclooxygenase enzymes, trehalose significantly lowered protein expression of both cyclooxygenase 1 (Fig. 5H) and cyclooxygenase 2 (Fig. 5I) in MRAs from SHRs. Overall these data indicate that trehalose treatment lowers the expression and activity of cyclooxygenase enzymes in SHRs.
Fig. 5.

Trehalose treatment prevents the generation of endothelium-derived contracting factors in mesenteric resistance arteries (MRAs) from spontaneously hypertensive rats (SHRs). A–C: concentration-response curves to ACh in MRAs from vehicle-treated Wistar and SHRs (A), vehicle- and trehalose-treated Wistar rats (B), vehicle- and trehalose-treated SHRs (C). PE, phenylephrine. D–G: concentration-response curves to ACh, with or without indomethacin (10 μmol/L), in MRAs from vehicle-treated Wistar rats (D), trehalose-treated Wistar rats (E), vehicle-treated SHRs (F), and trehalose-treated SHRs (G). H and I: protein expression analysis for cyclooxygenase 1 (H) and cyclooxygenase 2 (I) in MRAs. Left: densitometric analysis; right: representative images of immunoblots. J–M: concentration-response curves to ACh, with or without tempol (100 μmol/L), in MRAs from vehicle-treated Wistar rats (J), trehalose-treated Wistar rats (K), vehicle-treated SHRs (L), and trehalose-treated SHRs (M). N: reactive oxygen species measurement with dihydroethidium (DHE) fluorescent staining in MRAs. A.U., arbitrary units. Left: densitometric analysis; right: representative images. For A–N, n = 4–7. Two-way ANOVA: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle, †P < 0.05 vs. SHR trehalose. One-way ANOVA: Wistar vehicle, #P < 0.05 vs. SHR vehicle.
Another endothelium-derived contracting factor produced from arteries of SHRs is ROS. To test the hypothesis that trehalose suppresses contraction to ACh via inhibition of the ROS, some arteries were incubated with ROS scavenger tempol (100 μmol/L), before concentration-response curves with ACh. Similar to the results observed with indomethacin, tempol had no effect on ACh-induced relaxation in vehicle- and trehalose-treated Wistar rats (Fig. 5, J and K), the contractile response to ACh was diminished after incubation with tempol in SHRs treated with vehicle (Fig. 5L), and there was no difference in the contractile response to ACh, with or without tempol, in SHRs treated with trehalose (Fig. 5M). To support the notion that trehalose suppresses contraction to ACh via inhibition of ROS, DHE fluorescence was significantly lower in MRAs from SHRs treated with trehalose (Fig. 5N). Unexpectedly, the DHE assay revealed that MRAs from Wistar rats treated with trehalose had significantly elevated levels of ROS (Fig. 5N). Overall these data indicate that trehalose treatment in SHRs prevents the production of endothelium-derived contracting factors, such as cyclooxygenase-derived prostaglandins and ROS in arteries from SHRs.
Trehalose Treatment Increases Potassium-Dependent but Not Nitric Oxide-Dependent, Relaxation in SHRs
To evaluate whether trehalose could improve arterial NOS activity in SHRs, some MRAs were incubated with the NOS inhibitor l-NAME (100 μmol/L), before concentration-response curves with ACh. In MRAs from Wistar rats, incubation with l-NAME impaired the relaxation to ACh in both vehicle- and trehalose-treated groups to a similar degree (Fig. 6, A, B, and E). Likewise, in MRAs from SHRs, the effect of l-NAME was similar in both vehicle and trehalose-treated groups (Fig. 6, C, D, and E). These results suggest that trehalose treatment has no effect on NOS activity in MRAs from SHRs.
Fig. 6.

Trehalose treatment increases potassium-dependent (endothelium-derived hyperpolarizing factor), but not nitric oxide-dependent, relaxation in mesenteric resistance arteries (MRAs) from spontaneously hypertensive rats (SHRs). A–D: concentration-response curves to ACh, with or without Nω-nitro-l-arginine methyl ester (l-NAME; 100 μmol/L), in MRAs from vehicle-treated Wistar rats (A), trehalose-treated Wistar rats (B), vehicle-treated SHRs (C), and trehalose-treated SHRs (D). E: the change (∆) in the area under the curve (AUC), with and without l-NAME, for the concentration-response curves to ACh. F–I: concentration-response curves to ACh, with or without TEA (10 mmol/L), in MRAs from vehicle-treated Wistar rats (F), trehalose-treated Wistar rats (G), vehicle-treated SHRs (H), and trehalose-treated SHRs (I). J: the change (∆) in the AUC, with and without TEA, for the concentration-response curves to ACh. For A–J, n = 4–7. Two-way ANOVA: *P < 0.05 vs. Wistar vehicle, +P < 0.05 vs. Wistar trehalose, #P < 0.05 vs. SHR vehicle, †P < 0.05 vs. SHR trehalose. Student’s t test: #P < 0.05 vs. SHR vehicle.
In resistance arteries, the contribution of nonnitric oxide mechanisms to vasodilation increases, particularly potassium efflux [sometimes referred to as endothelium-derived hyperpolarizing factor (EDHF)] (15, 17). To evaluate whether trehalose could improve potassium-dependent hyperpolarization in SHRs, some MRAs were incubated with the potassium channel inhibitor TEA (10 mmol/L) before concentration-response curves with ACh. In MRAs from Wistar rats, incubation with TEA impaired the relaxation to ACh in both vehicle and trehalose-treated groups to a similar degree (Fig. 6, F, G, and J). However, in MRAs from SHRs, TEA impaired the relaxation to ACh to a greater extent in the trehalose-treated group (Fig. 6, H–J). These results reveal that trehalose increases potassium-dependent hyperpolarization in MRAs from SHRs.
Trehalose Treatment Decreases Calcium Sensitization in SHRs but Impairs Endothelium-Independent Relaxation in Wistar Rats
Vascular smooth muscle-dependent function was also evaluated after trehalose treatment. First, we assessed ROCK activity, as an indicator of calcium sensitization, by performing concentration-response curves to Y-27632. As expected, MRAs from SHRs treated with vehicle presented enhanced relaxation to Y-27632, indicating heightened ROCK activity (Fig. 7A). It was subsequently observed that although trehalose treatment had no effect on the relaxation to Y-27632 in MRAs from Wistar rats, relaxation to Y-27632 was reversed in SHRs treated with trehalose (Fig. 7A). To support the observation that trehalose suppresses ROCK activity, trehalose significantly lowered protein expression of both ROCK II (Fig. 7B) and phospho- MYPT1Thr696 (Fig. 7C) in MRAs from SHRs. Overall these data indicate that trehalose treatment decreases ROCK-dependent calcium sensitivity in SHRs.
Fig. 7.

Trehalose treatment decreases calcium sensitization in mesenteric resistance arteries (MRAs) from spontaneously hypertensive rats (SHRs) but impairs endothelium-independent relaxation in MRAs from Wistar rats. A: concentration-response curves to Y-27632 in MRAs from Wistar and SHRs treated with vehicle or trehalose. PE, phenylephrine. B and C: protein expression analysis for Rho kinase (ROCK) II (B) and phospho-myosin phosphatase target subunit 1 (MYPT1; C) in MRAs. Left, densitometric analysis; right, representative images of immunoblots. D: concentration-response curves to SNP in MRAs from Wistar and SHRs treated with vehicle or trehalose, For A–D, n = 5–8. Two-way ANOVA: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle. One-way ANOVA: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle. LogEC50: *P < 0.05 vs. Wistar vehicle.
We also measured endothelium-independent relaxation to SNP. No differences in the relaxation to SNP were observed in MRAs from Wistar rats and SHRs treated with vehicle (logEC50, Wistar vehicle: −7.57 ± 0.08 vs. SHR vehicle: −7.68 ± 0.07; Fig. 7D). Unexpectedly, MRAs from Wistar rats treated with trehalose had decreased sensitivity to SNP (LogEC50, Wistar vehicle: −7.57 ± 0.08 vs. Wistar trehalose: −6.97 ± 0.08), but no effect was observed in SHRs treated with trehalose (logEC50, SHR vehicle: −7.68 ± 0.07 vs. SHR trehalose: −7.77 ± 0.04; Fig. 7D). These data reveal that trehalose treatment impairs vascular smooth muscle-dependent relaxation in MRAs from Wistar rats.
Trehalose Treatment Decreases Vascular Stiffness in SHRs but Increases Vascular Stiffness in Wistar Rats
Finally, we tested whether these changes in vascular reactivity after trehalose treatment would reverse hypertensive vascular remodeling and reduce arterial stiffness. Although SHRs presented decreased lumen diameter (Fig. 8A), increased wall thickness (Fig. 8B), and increased wall-to-lumen ratio (Fig. 8C), trehalose treatment did not alter any of these remodeling indexes in either strain (Fig. 8, A–C). However, trehalose treatment did have an effect on MRA stiffness. As expected, MRAs from SHRs presented an increased vascular stiffness, as shown by the leftward shift in the stress-strain curve and an increase in the elastic modulus β (Fig. 8D). Similarly, MRAs from Wistar rats treated with trehalose also had a leftward shift in the stress-strain curve and an increase in the elastic modulus β (Fig. 8E). However, MRAs from SHRs treated with trehalose had a rightward shift in the stress-strain curve and a decrease in the elastic modulus β (Fig. 8F), indicating decreased vascular stiffness. To support the notion that trehalose alters vascular stiffness in strain-specific manner, Masson’s trichrome staining for fibrosis and extracellular matrix deposition was performed. As expected, SHRs treated with vehicle had significantly increased collagen (blue) staining and this was significantly reduced after trehalose treatment (Fig. 8G). Consistent with our earlier findings, Wistar rats treated with trehalose also had significantly increased fibrosis (Fig. 8G). Overall these data reveal that upregulation of autophagy with trehalose is able to improve mechanical properties of MRAs in SHRs by reducing extracellular matrix deposition but not reverse inward hypertrophic vascular remodeling. Moreover, these data also add to our growing body of evidence that suggests trehalose treatment is deleterious for vascular health in normotensive rats, despite all its beneficial effects in arteries from hypertensive rats (Fig. 9).
Fig. 8.

Trehalose treatment decreases vascular stiffness in mesenteric resistance arteries (MRAs) from spontaneously hypertensive rats (SHRs) but increases vascular stiffness in MRAs from Wistar rats. A–F: structural properties, including lumen diameter (A), wall thickness (B), and wall:lumen ratio (C), as well as mechanical stress-strain (D–F), were plotted via incremental pressure-response curves in calcium free physiological salt solution (0 mmol/L Ca2+). G: fibrosis and extracellular matrix measurement with Masson’s trichrome staining in MRAs. Left, densitometric analysis; right, representative images. For A–G, n = 5–87. Two-way ANOVA: *P < 0.05 vs. Wistar vehicle. One-way ANOVA: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle. Student’s t-test: *P < 0.05 vs. Wistar vehicle, #P < 0.05 vs. SHR vehicle.
Fig. 9.

Upregulation of autophagy with trehalose lowers relative vascular age in hypertension, but induces premature aging in arteries from normotensive rats. For any given chronological age, a hypertensive patient or animal has an increased vascular age, compared with an age-matched normotensive control. The definition of vascular age encompasses a broad range of phenotypes including hypercontractility, stiffening and remodeling, and inflammation and oxidative stress. Reconstitution of autophagy with trehalose reverses premature aging in arteries from spontaneously hypertensive rats (SHRs) by lowering cyclooxygenase activity and expression, preventing oxidative and endoplasmic reticulum (ER) stress, increasing endothelium-derived hyperpolarizing factor, and decreasing stiffness. On the other hand, trehalose accelerates vascular aging in arteries from age-matched Wistar rats by inducing oxidative and ER stress, impairing endothelium-independent relaxation, and increasing stiffness.
DISCUSSION
The major finding of the present investigation was that reconstitution of autophagic activity with trehalose reverses premature aging in resistance arteries from SHRs. Specifically, trehalose treatment lowered cyclooxygenase activity and expression, prevented oxidative and ER stress, increased EDHF, and decreased stiffness. Notably, these changes were independent of blood pressure and therefore can be attributed to the effects of the treatment and autophagy, as opposed to a reduction in vascular load. On the other hand (and unexpectedly), trehalose accelerated vascular aging in resistance arteries from age-matched normotensive Wistar rats by inducing ER and oxidative stress, impairing endothelium-independent relaxation, and increasing stiffness. Whether the deterioration in vascular function and increased stiffness was from exacerbation of autophagy, direct interference with ER homeostasis, and/or adiposity was not determined in the current investigation. Physiologically this study reveals that autophagy could be a cellular process that is targeted to reverse premature vascular aging associated with hypertension. Moreover, it supports the efficacy of trehalose as a therapeutic agent for patients with high blood pressure but not as a preventative measure for individuals without overt cardiovascular disease.
While aging and hypertension are both independent risk factors for arterial stiffness (4), arterial stiffness has been recognized as an important pathophysiological determinant for the age-related rise in blood pressure, demonstrating also an independent predictive value for cardiovascular events (11). Arterial stiffness can arise from remodeling of artery structure, loss of elasticity and compliance, and/or extracellular matrix deposition. In resistance arteries, aging tends to lead to outward hypertrophic remodeling, coupled with collagen deposition and elastin fragmentation (8, 23). On the other hand, resistance artery remodeling in essential hypertension tends to be inward eutrophic remodeling (33) but with similar pathological alterations in elastin and collagen as aging (52). In our investigation, MRAs from SHRs displayed inward hypertrophic remodeling, decreased elasticity as shown by the larger value of β and a leftward shift of the stress-strain relationship, and increased fibrosis compared with Wistar rats. Trehalose treatment reversed this stiffening in SHRs and induced it in Wistar rats by increasing extracellular matrix deposition but not changing wall thickness or wall-to-lumen ratio.
In addition to arterial stiffening, many of the other vascular phenotypes reported in the current study are inextricably linked between aging and hypertension. Specifically, aging and hypertension both promote endothelium-dependent contractions to ACh (19) and in vitro “aging” by prolonged passaging of primary cells (also termed replicative senescence) and increasing endothelium-derived contracting factors, including cyclooxygenase-derived prostaglandins (37, 44) and ROS (10, 13, 51, 56). Furthermore, RhoA/ROCK activity is increased in the vasculature of aged (31) and hypertensive rats (16), participates in the calcification of senescent vascular smooth muscle cells (35), and could participate in the progression of arterial stiffness by mediating stress fiber formation and actin polymerization (14).
Commonly observed in vascular aging studies in vitro are reductions in nitric oxide bioavailability (27, 32, 45). In contrast, our investigation did not reveal any changes in NOS activity in SHRs treated with trehalose. However, we did observe that trehalose treatment increased potassium (EDHF)-dependent relaxation in SHRs. We surmise that this difference is due the relative contributions of nitric oxide-dependent and -independent (e.g., EDHF) relaxation in resistance arteries, with the latter having a greater effect in resistance arteries compared with conduit arteries (15, 17).
The revelation that autophagy can reduce vascular aging has come to prominence recently (2, 38). Specifically, the autophagy activators trehalose and spermadine restored nitric oxide bioavailability by reducing ROS and inflammatory cytokine expression in the vasculature of aged mice and humans (18, 21, 22). Furthermore, with the use of cultured human endothelial cells in vitro, inhibition of autophagy leads to oxidative stress and reduced nitric oxide production, whereas autophagy activation enhanced nitric oxide production via an autophagy-dependent mechanism (21, 22). These seminal reports have been followed up with the observations that low shear stress inhibits endothelial autophagy, albeit, by an unknown mechanism. Impairment of autophagy results in an inhibitory phosphorylation of endothelial NOS (eNOS) at Thr495 and eNOS uncoupling in human umbilical vein endothelial cells. On the other hand, rapamycin restores autophagy to reduce phospho-eNOSThr495 and enhances the activation of eNOS at Ser1177 (59). Nonetheless, further understanding of the mechanisms by which autophagy induction can reduce vascular age is still outstanding, especially nitric oxide-independent factors.
From this work we infer that upregulation of autophagy prevented immune system activation via the clearance of damage-associated molecular patterns and reduced ER stress and that both these phenomena contributed to improvements observed in vascular function and structure in SHRs treated with trehalose. Supporting the use of circulating ND6 expression as biomarker of autophagy, we observed that SHRs treated with trehalose had decreased ND6 expression, which is consistent with our previous publication that ND6 goes up in conditions where cell death is exacerbated (e.g., hypertension or hemorrhagic shock) (55). Furthermore, we did not observe any differences in blood pressure between our SHR groups. Therefore, the differences in ND6 expression cannot be attributed to the release of ND6 into the circulation via pressure-induced cell death.
Although protein misfolding is an inevitable consequence of normal cellular metabolism, multiple proteostasis systems are devoted to the repair or clearance of damaged proteins, including autophagy (20). When these proteostasis systems do not function effectively, misfolded proteins accumulate and are vulnerable to aggregation (5). Misfolded protein aggregation (e.g., amyloid oligomers) has been extensively studied in neurodegenerative diseases (30), and in hypertension our group has previously revealed that alleviation of ER stress lowers blood pressure and improves vascular function and structure in hypertensive rats (47, 48). Interestingly, ER stress can also induce autophagy (58), but in SHRs this does not appear to be the case given the contrasting arterial phenotypes (i.e., increased ER stress but decreased autophagy). Thus our results suggest that the proteotoxicity associated with hypertension (6, 46) can be therapeutically targeted by upregulating autophagy with trehalose.
Limitations to the current investigation were the pooling of MRAs and aortas to then isolate lysosomes and measure acid phosphatase activity. Obviously, the pooling of two functionally different vascular beds does not distinguish whether both or only one of the vascular beds presents diminished acid phosphatase activity. This was a technical limitation due to necessity of obtaining a large quantity of tissue to isolate crude lysosomal fractions and the time-sensitive nature of such activity assays. From our considerable experience, MRAs and aortas are easily accessible vascular beds after euthanasia, and they can be cleaned of perivascular adipose tissue rapidly. Despite this limitation in the current investigation, we have previously reported that both MRAs and aortas both present diminished activity and expression of representative autophagy markers (29). The second limitation we wish to acknowledge is the administration of trehalose systemically via the drinking water. This route of administration does not preclude the possibility that upregulated autophagy in other organs contributed to improved vascular function and reduced stiffness measured in SHRs. Nonetheless, giving the trehalose via drinking water reduces stress on the rats and allows us to treatment them for a much longer period of time, compared with intraperitoneal injections and intravenous infusions, respectively. Furthermore, to the best of our knowledge, there are no means of upregulating autophagy in arteries of SHRs solely.
In conclusion, our results reveal that reconstitution of autophagy can ameliorate several indexes of premature vascular aging in SHRs, independent of reductions in blood pressure. While autophagy has long been implicated as a modulator of longevity and life span (40, 57), only recently was it shown to be inversely associated with chronological vascular aging (21, 22), and we have now extended this notion into the context of premature vascular aging associated with hypertension. Enhancing our understanding of vascular autophagy in health and disease will further refine the prognostic and diagnostic value of vascular age determination as an index of cardiovascular disease risk, as well as offer therapeutic alternatives to hypertensive patients.
GRANTS
This work was supported by American Heart Association Grant 18POST34060003, Fundação de Amparo a Pesquisa do Estado de São Paulo Grant 2016/20592-8, and National Institutes of Health Grants K99-GM-118885, P01-HL-134604, and R01-HL-143082.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.G.M. conceived and designed research; C.G.M., C.F.W., F.B.C., N.S.K., and M.W.B. performed experiments; C.G.M. and F.B.C. analyzed data; C.G.M. interpreted results of experiments; C.G.M. prepared figures; C.G.M. drafted manuscript; C.G.M., C.F.W., F.B.C., N.S.K., B.J., and R.C.W. edited and revised manuscript; C.G.M., C.F.W., F.B.C., N.S.K., M.W.B., B.J., and R.C.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Angela Burton (Augusta University) and Zachary Schreckenberger (University of Toledo College of Medicine and Life Sciences) for assistance with rat treatments.
REFERENCES
- 1.Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep 15: 839–852, 2014. doi: 10.15252/embr.201439076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res 123: 803–824, 2018. doi: 10.1161/CIRCRESAHA.118.312208. [DOI] [PubMed] [Google Scholar]
- 3.Abeywardena MY, Jablonskis LT, Head RJ. Age- and hypertension-induced changes in abnormal contractions in rat aorta. J Cardiovasc Pharmacol 40: 930–937, 2002. doi: 10.1097/00005344-200212000-00015. [DOI] [PubMed] [Google Scholar]
- 4.AlGhatrif M, Strait JB, Morrell CH, Canepa M, Wright J, Elango P, Scuteri A, Najjar SS, Ferrucci L, Lakatta EG. Longitudinal trajectories of arterial stiffness and the role of blood pressure: the Baltimore Longitudinal Study of Aging. Hypertension 62: 934–941, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayyadevara S, Balasubramaniam M, Gao Y, Yu LR, Alla R, Shmookler Reis R. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell 14: 35–48, 2015. doi: 10.1111/acel.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayyadevara S, Mercanti F, Wang X, Mackintosh SG, Tackett AJ, Prayaga SV, Romeo F, Shmookler Reis RJ, Mehta JL. Age- and hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 67: 1006–1013, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UK, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics-2018 update: A Report From the American Heart Association. Circulation 137: e67–e492, 2018. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 8.Briones AM, Salaices M, Vila E. Mechanisms underlying hypertrophic remodeling and increased stiffness of mesenteric resistance arteries from aged rats. J Gerontol A Biol Sci Med Sci 62: 696–706, 2007. doi: 10.1093/gerona/62.7.696. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Haddad GG. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol 207: 3125–3129, 2004. doi: 10.1242/jeb.01133. [DOI] [PubMed] [Google Scholar]
- 10.Deshpande SS, Qi B, Park YC, Irani K. Constitutive activation of rac1 results in mitochondrial oxidative stress and induces premature endothelial cell senescence. Arterioscler Thromb Vasc Biol 23: e1–e6, 2003. doi: 10.1161/01.ATV.0000047869.13737.53. [DOI] [PubMed] [Google Scholar]
- 11.Faconti L, Bruno RM, Ghiadoni L, Taddei S, Virdis A. Ventricular and vascular stiffening in aging and hypertension. Curr Hypertens Rev 11: 100–109, 2015. doi: 10.2174/1573402111666150529131208. [DOI] [PubMed] [Google Scholar]
- 12.Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 70: 660–667, 2017. doi: 10.1161/HYPERTENSIONAHA.117.07802. [DOI] [PubMed] [Google Scholar]
- 13.Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, Dimmeler S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res 94: 768–775, 2004. doi: 10.1161/01.RES.0000121104.05977.F3. [DOI] [PubMed] [Google Scholar]
- 14.Hall A. Rho GTPases and the actin cytoskeleton. Science 279: 509–514, 1998. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 15.Jiang J, Zheng JP, Li Y, Gan Z, Jiang Y, Huang D, Li H, Liu Z, Ke Y. Differential contribution of endothelium-derived relaxing factors to vascular reactivity in conduit and resistance arteries from normotensive and hypertensive rats. Clin Exp Hypertens 38: 393–398, 2016. doi: 10.3109/10641963.2016.1148155. [DOI] [PubMed] [Google Scholar]
- 16.Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, Webb RC. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther 318: 288–295, 2006. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 17.Kang KT. Endothelium-derived relaxing factors of small resistance arteries in hypertension. Toxicol Res 30: 141–148, 2014. doi: 10.5487/TR.2014.30.3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplon RE, Hill SD, Bispham NZ, Santos-Parker JR, Nowlan MJ, Snyder LL, Chonchol M, LaRocca TJ, McQueen MB, Seals DR. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging (Albany NY) 8: 1167–1183, 2016. doi: 10.18632/aging.100962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koga T, Takata Y, Kobayashi K, Takishita S, Yamashita Y, Fujishima M. Age and hypertension promote endothelium-dependent contractions to acetylcholine in the aorta of the rat. Hypertension 14: 542–548, 1989. doi: 10.1161/01.HYP.14.5.542. [DOI] [PubMed] [Google Scholar]
- 20.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10: 524–530, 2000. doi: 10.1016/S0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 21.LaRocca TJ, Gioscia-Ryan RA, Hearon CM Jr, Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev 134: 314–320, 2013. doi: 10.1016/j.mad.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol 590: 3305–3316, 2012. doi: 10.1113/jphysiol.2012.229690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laurant P, Adrian M, Berthelot A. Effect of age on mechanical properties of rat mesenteric small arteries. Can J Physiol Pharmacol 82: 269–275, 2004. doi: 10.1139/y04-026. [DOI] [PubMed] [Google Scholar]
- 24.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 21: 29, 2016. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lüscher TF, Dohi Y, Tschudi M. Endothelium-dependent regulation of resistance arteries: alterations with aging and hypertension. J Cardiovasc Pharmacol 19, Suppl 5: S34–S42, 1992. doi: 10.1097/00005344-199206001-00006. [DOI] [PubMed] [Google Scholar]
- 26.Lüscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 8: 344–348, 1986. doi: 10.1161/01.HYP.8.4.344. [DOI] [PubMed] [Google Scholar]
- 27.Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res 89: 793–798, 2001. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy CG, Goulopoulou S, Webb RC. Paying the Toll for inflammation. Hypertension 73: 514–521, 2019. doi: 10.1161/HYPERTENSIONAHA.118.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107: 119–130, 2015. doi: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyerhof W, Richter D. Neurodegeneration–from multiple sclerosis to Alzheimer’s disease. FEBS J 280: 4337, 2013. doi: 10.1111/febs.12430. [DOI] [PubMed] [Google Scholar]
- 31.Miao L, Calvert JW, Tang J, Parent AD, Zhang JH. Age-related RhoA expression in blood vessels of rats. Mech Ageing Dev 122: 1757–1770, 2001. doi: 10.1016/S0047-6374(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 32.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105: 1541–1544, 2002. doi: 10.1161/01.CIR.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 33.Mulvany MJ. Vascular remodelling of resistance vessels: can we define this? Cardiovasc Res 41: 9–13, 1999. doi: 10.1016/S0008-6363(98)00289-2. [DOI] [PubMed] [Google Scholar]
- 34.Mulvany MJ, Halpern W. Mechanical properties of vascular smooth muscle cells in situ. Nature 260: 617–619, 1976. doi: 10.1038/260617a0. [DOI] [PubMed] [Google Scholar]
- 35.Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol 297: H1673–H1684, 2009. doi: 10.1152/ajpheart.00455.2009. [DOI] [PubMed] [Google Scholar]
- 36.National Research Council Guide for the Care and Use of Laboratory Animals . Washington, DC: The National Academies Press, 2011. doi: 10.17226/12910. [DOI] [Google Scholar]
- 37.Neubert K, Haberland A, Kruse I, Wirth M, Schimke I. The ratio of formation of prostacyclin/thromboxane A2 in HUVEC decreased in each subsequent passage. Prostaglandins 54: 447–462, 1997. doi: 10.1016/S0090-6980(97)00063-4. [DOI] [PubMed] [Google Scholar]
- 38.Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res 116: 480–488, 2015. doi: 10.1161/CIRCRESAHA.116.303805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren WM, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvänne M, Scholte Op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F; Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice; European Association for Cardiovascular Prevention and Rehabilitation . European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Atherosclerosis 223: 1–68, 2012. doi: 10.1016/j.atherosclerosis.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4: 2300, 2013. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reckelhoff JF. Sex differences in regulation of blood pressure. Adv Exp Med Biol 1065: 139–151, 2018. doi: 10.1007/978-3-319-77932-4_9. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, Solano RM, Gómez A, Perucho J, Cuervo AM, García de Yébenes J, Mena MA. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39: 423–438, 2010. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282: 5641–5652, 2007. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 44.Sato I, Kaji K, Morita I, Nagao M, Murota S. Augmentation of endothelin-1, prostacyclin and thromboxane A2 secretion associated with in vitro ageing in cultured human umbilical vein endothelial cells. Mech Ageing Dev 71: 73–84, 1993. doi: 10.1016/0047-6374(93)90036-Q. [DOI] [PubMed] [Google Scholar]
- 45.Sato I, Morita I, Kaji K, Ikeda M, Nagao M, Murota S. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem Biophys Res Commun 195: 1070–1076, 1993. doi: 10.1006/bbrc.1993.2153. [DOI] [PubMed] [Google Scholar]
- 46.Sidorova TN, Mace LC, Wells KS, Yermalitskaya LV, Su PF, Shyr Y, Atkinson JB, Fogo AB, Prinsen JK, Byrne JG, Petracek MR, Greelish JP, Hoff SJ, Ball SK, Glabe CG, Brown NJ, Barnett JV, Murray KT. Hypertension is associated with preamyloid oligomers in human atrium: a missing link in atrial pathophysiology? J Am Heart Assoc 3: e001384, 2014. doi: 10.1161/JAHA.114.001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spitler KM, Matsumoto T, Webb RC. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 305: H344–H353, 2013. doi: 10.1152/ajpheart.00952.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spitler KM, Webb RC. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 63: e40–e45, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taddei S, Virdis A, Mattei P, Ghiadoni L, Fasolo CB, Sudano I, Salvetti A. Hypertension causes premature aging of endothelial function in humans. Hypertension 29: 736–743, 1997. doi: 10.1161/01.HYP.29.3.736. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10: 148–154, 2004. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 51.Unterluggauer H, Hampel B, Zwerschke W, Jansen-Dürr P. Senescence-associated cell death of human endothelial cells: the role of oxidative stress. Exp Gerontol 38: 1149–1160, 2003. doi: 10.1016/j.exger.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 52.Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5: 264–273, 2012. doi: 10.1007/s12265-012-9349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Wang H, Geng QX, Wang HT, Miao W, Cheng B, Zhao D, Song GM, Leanne G, Zhao Z. Augmentation of autophagy by atorvastatin via Akt/mTOR pathway in spontaneously hypertensive rats. Hypertens Res 38: 813–820, 2015. doi: 10.1038/hr.2015.85. [DOI] [PubMed] [Google Scholar]
- 54.Wenceslau CF, McCarthy CG, Szasz T, Calmasini FB, Mamenko M, Webb RC. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol Res 141: 276–290, 2019. doi: 10.1016/j.phrs.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol 308: H768–H777, 2015. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xin MG, Zhang J, Block ER, Patel JM. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech Ageing Dev 124: 911–919, 2003. doi: 10.1016/S0047-6374(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 57.Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 23: 248–262, 2008. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- 58.Yorimitsu T, Klionsky DJ. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3: 160–162, 2007. doi: 10.4161/auto.3653. [DOI] [PubMed] [Google Scholar]
- 59.Zhang JX, Qu XL, Chu P, Xie DJ, Zhu LL, Chao YL, Li L, Zhang JJ, Chen SL. Low shear stress induces vascular eNOS uncoupling via autophagy-mediated eNOS phosphorylation. Biochim Biophys Acta Mol Cell Res 1865: 709–720, 2018. doi: 10.1016/j.bbamcr.2018.02.005. [DOI] [PubMed] [Google Scholar]