

Abstract
Voltage-dependent L-type Ca2+ channels (L-VDCCs) and the RhoA/Rho kinase pathway are two predominant intracellular signaling pathways that regulate renal microvascular reactivity. Traditionally, these two pathways have been thought to act independently; however, recent evidence suggests that these pathways could be convergent. We hypothesized that Rho kinase inhibitors can influence L-VDCC signaling. The effects of Rho kinase inhibitors Y-27632 or RKI-1447 on KCl-induced depolarization or the L-VDCC agonist Bay K8644 were assessed in afferent arterioles using an in vitro blood-perfused rat juxtamedullary nephron preparation. Superfusion of KCl (30–90 mM) led to concentration-dependent vasoconstriction of afferent arterioles. Administration of Y-27632 (1, 5, and 10 µM) or RKI-1447 (0.1, 1, and 10 µM) significantly increased the starting diameter by 16–65%. KCl-induced vasoconstriction was markedly attenuated with 5 and 10 µM Y-27632 and with 10 µM RKI-1447 (P < 0.05 vs. KCl alone). Y-27632 (5 µM) also significantly attenuated Bay K8644-induced vasoconstriction (P < 0.05). Changes in intracellular Ca2+ concentration ([Ca2+]i) were estimated by fura-2 fluorescence during KCl-induced depolarization in cultured A7r5 cells and in freshly isolated preglomerular microvascular smooth muscle cells. Administration of 90 mM KCl significantly increased fura-2 fluorescence in both cell types. KCl-mediated elevation of [Ca2+]i in A7r5 cells was suppressed by 1–10 µM Y-27632 (P < 0.05), but 10 µM Y-27632 was required to suppress Ca2+ responses in preglomerular microvascular smooth muscle cells. RKI-1447, however, significantly attenuated KCl-mediated elevation of [Ca2+]i. Y-27632 markedly inhibited Bay K8644-induced elevation of [Ca2+]i in both cell types. The results of the present study indicate that the Rho kinase inhibitors Y-27632 and RKI-1447 can partially inhibit L-VDCC function and participate in L-VDCC signaling.
Keywords: afferent arteriole, Bay K8644, calcium signaling, RKI-1447, voltage-dependent L-type Ca2+ channels, Y-27632
INTRODUCTION
For a healthy individual, maintaining appropriate vascular tone and vasoreactivity is crucial for the control of arterial pressure and organ perfusion (2, 29). There is a consensus that vascular smooth muscle contraction occurs via Ca2+-dependent and Ca2+ sensitization mechanisms (7, 17). Ca2+-dependent signaling occurs through Ca2+ influx from the extracellular fluid space via voltage-dependent Ca2+ channels (VDCCs), stimulation of nonselective cation channels, or receptor-operated Ca2+ channels or through release of Ca2+ from intracellular stores (2, 29, 33). Alternatively, vascular smooth muscle contraction can also involve Ca2+ sensitization perhaps through the RhoA/Rho kinase pathway by inhibition of myosin light chain phosphatase (17, 43). In the kidneys, voltage-dependent L-type Ca2+ channels (L-VDCCs) are the predominant VDCCs that regulate preglomerular microvascular tone and reactivity to various vasoconstrictor stimuli (2, 3, 8, 21, 23, 27, 29). In addition to VDCCs, afferent arterioles also use RhoA/Rho kinase signaling to control renal vascular resistance and vascular responses to vasoconstrictor or pressure stimuli (6, 19, 27). Accordingly, a better understanding of the respective roles of VDCC and RhoA/Rho kinase pathways in regulating renal microvascular function is important for understanding renal hemodynamic control under physiological conditions and their impact on renal microvascular dysfunction under pathophysiological conditions. Traditionally, these two pathways were thought to act independently; however, growing evidence suggests that these signaling pathways might converge to regulate vascular reactivity (1, 11–13, 24, 32, 37–39, 41). For example, studies in small nonrenal arteries have shown that the Rho kinase inhibitor Y-27632 (3 × 10−6 to 1 × 10−5 M) attenuated the vasoconstriction stimulated by KCl-induced membrane depolarization or by the L-VDCC agonist FPL64176 (11, 12, 38). Exposure to KCl (70 mM) or FPL64176 increased RhoA activity in basilar arteries or aortas of rabbits, mice, or rats, but the increased RhoA activity could be blocked by the L-VDCC blocker nifedipine (11, 12, 32). These studies suggested that there may be an interaction between L-VDCCs and the RhoA/Rho kinase pathway in regulating vascular contraction; however, it is unknown whether such changes occur in renal microvessels.
Recently, we applied nifedipine or Y-27632 to determine the contribution of L-VDCCs and Rho kinase signaling in sphingosine-1-phosphate (S1P)-mediated vasoconstriction of the renal microvasculature (15). We observed that although nifedipine or Y-27632 alone significantly suppressed S1P-mediated vasoconstriction of afferent arterioles, combined treatment with both drugs produced no additional suppressive effects on S1P-induced vasoconstriction. These observations suggest that activation of L-VDCCs or Rho kinase activity by exogenous S1P in the renal microvasculature may involve similar intracellular signaling mechanisms rather than two independent pathways. This idea is supported by a prior study (19) in which sustained afferent arteriole vasoconstriction induced by P2X1 receptor activation with α,β-methylene ATP was completely abolished by 10 µM Y-27632. P2X1 receptor activation is known to stimulate nonselective cation influx, leading to membrane depolarization and L-VDCC activation (20, 40). However, P2Y2-mediated afferent arteriole vasoconstriction by UTP, which increases intracellular Ca2+ concentration ([Ca2+]i) primarily by mobilization of intracellular Ca2+, was not affected by Y-27632 (19, 20). These observations raise the question of whether L-VDCCs and the Rho kinase pathway share downstream intracellular signaling mechanisms in mediating afferent arteriole vasoconstrictor responses to S1P, α,β-methylene ATP, or other vasoconstrictors. It is therefore essential to ascertain whether there are important interactions between these two intracellular signaling mechanisms impacting renal microvascular reactivity.
Y-27632 is a pyridine derivative and the most widely used inhibitor of Rho kinases (22, 28, 36). Y-27632 selectively inhibits smooth muscle cell contraction by inhibiting Ca2+ sensitization with IC50 values of 140–220 nM for Rho kinase 1 and 2 (22). Y-27632 inhibits vascular smooth muscle contraction induced by agonists including phenylephrine, endothelin-1, angiotensin II, ATP, α,β-methylene ATP, and S1P (6, 15, 19, 27, 31, 39). RKI-1447 is a newly developed Rho kinase inhibitor with IC50 values of 14.5 and 6.2 nM for Rho kinase 1 and 2, respectively (30), which is more potent than Y-27632 in inhibiting Rho kinase activity. Protein expression of Rho kinase isoform-α (Rho kinase 2) and isoform-β (Rho kinase 1) was detected in preglomerular microvessels (19), suggesting a role for Rho kinase in regulating renal microvascular function.
Given that Ca2+ and Rho kinase signaling play important roles in regulating vasoreactivity, we decided to determine whether VDCCs and Rho kinase signaling pathways might overlap or interact. Accordingly, we postulated that Rho kinase inhibitors can influence L-VDCC signaling. We used the in vitro blood-perfused rat juxtamedullary nephron technique to assess afferent arteriole reactivity to membrane depolarization induced by application of KCl with or without Y-27632 or RKI-1447. We also measured [Ca2+]i estimated by fura-2 fluorescence [ratio of fluorescence at 340 nm to that at 380 nm (F340-to-F380 ratio)] during KCl-induced depolarization in cultured rat aortic smooth muscle cells (A7r5 cells) and in freshly isolated preglomerular microvascular smooth muscle cells (PMVSMCs) in the presence or absence of Rho kinase inhibitors. The impact of Y-27632 was further assessed on afferent arteriole vasoconstriction and [Ca2+]i signaling in cells stimulated by the L-VDCC agonist Bay K8644. Our results indicate that both Y-27632 and RKI-1447 partially inhibit L-VDCC function and participate in L-VDCC signaling.
METHODS
Animals
A total of 132 male Sprague-Dawley rats (300–400 g body wt, Charles River Laboratories, Raleigh, NC) were used to assess afferent arteriole reactivity and renal microvascular smooth muscle cell isolation. All rats had free access to standard chow (LabDiet, PMI Nutrition, Brentwood, MO) and water, and they were maintained according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
General Methods
In vitro blood-perfused juxtamedullary nephron preparation.
Afferent arteriole reactivity was assessed in vitro using the blood-perfused juxtamedullary nephron technique as previously described (14, 18). Briefly, two rats were anesthetized with pentobarbital sodium (50 mg/kg body wt ip, Diamondback, Scottsdale, AZ) for each experiment. The left renal vascular bundle of the kidney donor rat was tied off, and the right kidney was then cannulated and continuously perfused with Tyrode buffer (Sigma-Aldrich, St. Louis, MO) containing 5.2% BSA (Calbiochem, Billerica, MA). Perfusate blood was collected via a carotid artery cannula and processed with blood collected from a blood donor as previously described (14, 18). Briefly, the combined blood was centrifuged at 2,700 g for 13 min. The plasma was collected and filtered through a 0.2-µm filter (Corning). The buffy coat was removed from the packed cells, and the packed erythrocytes were washed with 0.9% saline and centrifuged at 320 g for 14 min and 2,700 g for 13 min, respectively. The washed erythrocytes were mixed with the plasma to yield a hematocrit of ~33%. The reconstituted blood was filtered through 5-µm nylon mesh for kidney perfusion. The right kidney was harvested and sectioned along the longitudinal axis on the dorsal two-thirds of the kidney and then was positioned with pins on the silicone platform of the perfusion chamber. The main renal arterial branches were exposed after the pelvic mucosa was removed. The ends of the intrarenal arteries and renal vascular branches that were cut during the dissection were tied with 10-0 nylon suture to restore renal perfusion pressure.
After completion of the dissection, the kidney was moved to the stage of a Nikon Eclipse E600FN microscope (Nikon, Tokyo, Japan) fitted with a Nikon water-immersion objective, and perfusion was switched to the reconstituted blood from a sealed reservoir pressurized with 95% O2-5% CO2. The inner cortical surface was superfused with 37°C Tyrode buffer containing 1% BSA. The image of the kidney was displayed on a video monitor and recorded on DVD for later analysis. Perfusion pressure was held constant at 100 mmHg during equilibration. Afferent arteriole inner diameters were measured at a single site at 12-s intervals using a calibrated image-shearing monitor, and the mean diameter effect was averaged from all diameter measurements obtained during the final 2 min of each period.
Measurement of [Ca2+]i in cultured rat aortic smooth muscle A7r5 cells.
To determine whether Rho kinase inhibitors blocked L-VDCC-dependent Ca2+ influx, we measured [Ca2+]i in cultured rat aortic smooth muscle A7r5 cells (CRL-1444, American Type Culture Collection, Manassas, VA) to determine the impact of Y-27632 or RKI-1447 on L-VDCCs induced by 90 mM KCl-mediated depolarization. Briefly, A7r5 cells were cultured with DMEM (Life Technologies, Grand Island, NY) containing 10% FBS (Sigma-Aldrich) at 37°C in a 5% CO2 chamber. A7r5 cells were subcultured using 0.25% trypsin-EDTA solution (Life Technologies). Cells (passages 4–10) were seeded on glass coverslips in 60-mm culture dishes 24 h before experiments. Before Ca2+ measurements, cultured A7r5 cells were incubated with the Ca2+-sensitive fluorescent dye fura-2 AM (5 µM loading concentration, Molecular Probes, Eugene, OR) for 40–45 min at room temperature in darkness. To measure cytosolic free Ca2+, coverslips were transferred to a laminar flow perfusion chamber (Warner Instrument, Hamden, CT) and mounted to the stage of an Olympus IX73 inverted microscope fitted with ×40 fluorescence objectives. The chamber was continuously perfused with physiological salt solution (PSS) containing 1.8 mM Ca2+ at a flow rate of 1.5 ml/min, and cells were randomly selected for study. Fluorescence was measured using EasyRatioPro (Photon Technology, Edison, NJ) with excitation wavelengths of 340 and 380 nm and the emission wavelength set at 510 nm. Changes in [Ca2+]i were estimated by the F340-to-F380 ratio.
Isolation of PMVSMCs for [Ca2+]i measurement.
We also measured [Ca2+]i using freshly isolated PMVSMCs collected from preglomerular microvessels as previously described (42). Briefly, rats were anesthetized for retrograde perfusion through the abdominal aorta. The kidneys were perfused with Tyrode buffer containing 5.2% BSA followed by 5% Evans blue, removed, and transferred to ice-cold PSS. The cortical tissue was pressed through a 180-µm sieve and incubated in a digestion solution (0.016% collagenase D, 0.016% trypsin inhibitor, 0.016% dithiothreitol, and 0.016% albumin) at 36.5°C for 35–45 min. The preglomerular microvessels, consisting of interlobular arteries [also defined as cortical radial arteries (26)] and afferent arterioles, were carefully separated from tubular tissue under a stereomicroscope and transferred to a digestion solution containing 0.05% dithiothreitol, 0.14% papain, and 4% albumin dissolved in a low-Ca2+ (50 μM) PSS at 36.5°C. After a 15- to 25-min incubation period, the mixture was centrifuged (900 g for 2–3 min) to pellet the dispersed cells. Cells were resuspended in 1 ml DMEM plus 20% FBS and incubated with 10 µM fura-2 AM in the dark for Ca2+-signaling analysis.
Experimental Design
Experiment 1: effect of Y-27632 on KCl-mediated afferent arteriolar vasoconstriction.
After completion of the in vitro juxtamedullary nephron preparation, the kidney perfusate was switched to reconstituted blood. An equilibration period (at least 15 min) was allowed to establish steady-state arteriolar diameter while renal perfusion pressure was held constant at 100 mmHg. Each protocol began with a 5-min period to establish the afferent arteriole starting diameter, referred to as the “starting diameter.” The inner cortical surface was continuously superfused with Tyrode buffer containing 1% BSA (control group, n = 7 kidneys) or switched to a superfusate containing Y-27632 (1, 5, or 10 µM, n = 6 kidneys) for 15 min until a new steady-state arteriolar diameter was achieved (baseline diameter). Afferent arterioles were then exposed to increasing KCl concentrations (30, 60, and 90 mM) at 5-min intervals with or without Y-27632. The diameter obtained during the last 2 min of each period was compared against the baseline diameter.
Experiment 2: effect of RKI-1447 on KCl-mediated afferent arteriolar vasoconstriction.
Afferent arteriole reactivity was also assessed in the presence of a more potent Rho kinase inhibitor, RKI-1447, with increasing KCl concentrations (30, 60, and 90 mM). A protocol similar to that of experiment 1 was performed. Briefly, after a 5-min period to establish the starting diameter, the kidney was superfused with RKI-1447 (0.1, 1, or 10 µM) for 15 min before the KCl concentration response was determined (n = 6 kidneys/group). The KCl concentration response, without any intervention in experiment 1, served as the control group.
Experiment 3: effect of Y-27632 on afferent arteriolar vasoconstriction induced by the L-VDCC agonist Bay K8644.
We also assessed the effect of Y-27632 on the afferent arteriole vasoconstrictor response to the L-VDCC opener Bay K8644 (4). A protocol similar to that of experiment 1 was performed. Briefly, after a 5-min period to establish the starting diameter, the kidney was continuously superfused with Tyrode buffer containing 1% BSA (control group, n = 6 kidneys) or switched to a superfusate containing Y-27632 (5 µM, n = 6 kidneys) for 15 min until a new steady-state diameter was achieved (baseline diameter). Afferent arterioles were subsequently exposed to increasing log concentrations of Bay K8644 (1 nM−10 μM) at 5-min intervals with or without Y-27632.
Experiment 4: effects of Y-27632 and RKI-1447 on [Ca2+]i during KCl-mediated membrane depolarization in cultured rat aortic smooth muscle A7r5 cells.
To determine whether Y-27632 or RKI-1447 impaired VDCC Ca2+ influx, we measured [Ca2+]i during cell membrane depolarization induced by 90 mM KCl in cultured A7r5 cells. Measurement of [Ca2+]i was performed as previously described (42). Briefly, cells were superfused with PSS containing 1.8 mM Ca2+ to measure baseline [Ca2+]i and then exposed to PSS containing 90 mM KCl for 200 s to assess the influence of depolarization on [Ca2+]i (control group). To determine the effect of Rho kinase inhibition, cells were exposed to PSS containing Y-27632 (0.1, 0.5, 1, 5, or 10 µM) or RKI-1447 (0.1, 1, 5, or 10 µM) for 200 s after a baseline [Ca2+]i measurement. PSS was then switched to KCl solution (90 mM, with Na+ substitution) containing Y-27632 or RKI-1447 for 200 s. The changes in KCl-induced [Ca2+]i elevation (F340-to-F380 ratio) were recorded.
Experiment 5: effects of Y-27632 and RKI-1447 on [Ca2+]i during KCl-mediated membrane depolarization in PMVSMCs.
We determined the impact of the Rho kinase inhibitors Y-27632 and RKI-1447 on L-VDCCs in freshly isolated PMVSMCs according to the procedures outlined in methods, General Methods, Isolation of PMVSMCs for [Ca2+]i measurement. Similar to measurement of [Ca2+]i in cultured A7r5 cells in experiment 4, PMVSMCs were superfused with PSS containing 1.8 mM Ca2+ to measure baseline [Ca2+]i and then exposed to PSS containing 90 mM KCl for 200 s to assess the influence of depolarization on [Ca2+]i (control group). To determine the effect of Rho kinase inhibition, PMVSMCs were exposed to PSS containing Y-27632 (1, 5, or 10 µM) or RKI-1447 (0.1, 1, 5, or 10 µM) for 200 s after a baseline [Ca2+]i measurement. PSS was then switched to KCl (90 mM) solution containing Y-27632 or RKI-1447 for 200 s. The changes in KCl-induced [Ca2+]i elevation (F340-to-F380 ratio) were recorded.
Experiment 6: effect of Y-27632 on [Ca2+]i response induced by the L-VDCC agonist Bay K8644 in A7r5 cells and PMVSMCs.
Similar to the measurement of [Ca2+]i performed in experiments 4 and 5, Bay K8644 (10 μM) was used instead of 90 mM KCl. In our pilot studies, we found that administration of Bay K8644 up to 10 μM failed to evoke significant changes in [Ca2+]i in A7r5 cells and PMVSMCs. The reason for this phenomenon could be due to the cells being in a quiescent state rather than a depolarized state in arterioles under myogenic tone, an observation of Alvarez et al. (1). Therefore, we exposed A7r5 cells or PMVSMCs to PSS containing 10 mM KCl to increase membrane potential slightly but without increasing baseline [Ca2+]i. The following two groups were studied: Bay K8644 and Bay K8644 plus Y-27632. A7r5 cells or PMVSMCs were exposed to PSS containing 10 mM KCl for 200 s after baseline [Ca2+]i was established. PSS was then switched to PSS containing KCl (10 mM) plus Bay K8644 (10 µM) with or without Y-27632 (5 µM) for 200 s. The changes in Bay K8644-induced [Ca2+]i elevation (F340-to-F380 ratio) were recorded.
Drug Preparation
Y-27623 (Chemdea, Ridgewood, NJ) was freshly prepared in water to yield a concentration of 5 mM on the day of experiments. RKI-1447 (Bio-Techne, Minneapolis, MN) was dissolved in water to produce a stock solution of 2.0 mM and then aliquoted in Eppendorf tubes for storage at −20°C. Bay K8644, trypsin inhibitor, dithiothreitol, albumin, and papain were purchased from Sigma-Aldrich. Bay K8644 was dissolved in ethanol to produce a stock solution of 50 mM and aliquoted for storage at −80°C. For experiments, Y-27632, RKI-1447, and Bay K8644 were diluted with Tyrode solution containing 1% BSA or PSS to produce the desired working concentrations. Collagenase D was purchased from Worthington Biochemical (Lakewood, NJ).
Statistical Analysis
All data are expressed as means ± SE. One-way ANOVA for repeated measures was used for within-group analyses followed by post hoc analyses with Dunnett’s multiple-range test. Significant differences between groups, within each series, were determined using one-way ANOVA and Dunnett’s multiple-comparison test. For the Bay K8644 experiments, a two-tailed, unpaired t-test was used for the comparison between two groups. P values of <0.05 were considered to indicate significant differences.
RESULTS
Effects of Y-27632 and RKI-1447 on Starting Afferent Arteriole Diameters
Figure 1 shows the effects of increasing concentrations of the Rho kinase inhibitors Y-27632 and RKI-1447 on the starting diameters of afferent arterioles compared with the control group. Starting diameters were similar across all groups (P > 0.05 vs. control). In the control group, the starting diameter remained stable during the 15-min incubation period with 1% BSA (P > 0.05). In contrast, in the groups treated with Y-27632 (1, 5, and 10 µM), afferent arteriole diameter increased significantly to 116 ± 4%, 159 ± 10%, and 165 ± 7% of the respective starting diameters (P < 0.05; Fig. 1A). Similarly, addition of RKI-1447 also significantly increased arteriole diameter to 117 ± 3%, 164 ± 8%, and 159 ± 12% for the 0.1, 1, and 10 µM concentrations, respectively (P < 0.05; Fig. 1B).
Fig. 1.

Effect of the Rho kinase inhibitors Y-27632 and RKI-1447 (RKI) on starting afferent arteriole diameters. The effect of Y-27632 (1, 5, and 10 µM, A) and RKI-1447 (0.1, 1, and 10 µM, B) on the starting diameter of afferent arterioles compared with control is shown. Data are expressed as a percentage of the starting diameter. Values are expressed as means ± SE; n, number of kidneys. *P < 0.05 vs. baseline in the same group.
Effects of Y-27632 on KCl-Induced Afferent Arteriole Vasoconstriction
Figure 2 shows the effect of Y-27632 on KCl-induced afferent arteriole vasoconstriction. Superfusion of KCl led to concentration-dependent vasoconstriction of afferent arterioles (P < 0.05 vs. baseline diameter; Fig. 2). Although 1 µM Y-27632 significantly dilated afferent arterioles (Fig. 1A), the KCl-induced vasoconstriction (Fig. 2) was similar to that of the control group (P > 0.05 vs. KCl alone). Higher concentrations of Y-27632 (5 and 10 µM) significantly attenuated the response to KCl (P < 0.05 vs. KCl alone; Fig. 2B).
Fig. 2.

Effect of the Rho kinase inhibitor Y-27632 on KCl-induced afferent arteriole vasoconstriction. A: afferent arteriolar responses to increasing concentrations of KCl in the absence and presence of the Rho kinase inhibitor Y-27632 at 1, 5, and 10 µM. Data represent the actual diameters from each group. B: same data normalized as a percentage of the control diameter for each group. Values are expressed as means ± SE; n, number of kidneys. *P < 0.05 vs. baseline diameter in the same group; †P < 0.05 vs. vasoconstriction with KCl alone at the same concentration.
Effects of RKI-1447 on KCl-Induced Afferent Arteriole Vasoconstriction
Figure 3 shows the effect of RKI-1447 on KCl-induced afferent arteriole vasoconstriction. Similar to 1 µM Y-27632, 0.1 µM RKI-1447 did not alter the concentration-dependent vasoconstrictor response to KCl. The profile of the KCl-induced vasoconstriction was identical to that of the control group (Fig. 3, A and B). Afferent arterioles seemed to reach a maximal vasodilation (Fig. 1B) with 1 µM RKI-1447; however, the KCl-induced vasoconstriction was not statistically different from that of the control group (P > 0.05 vs. KCl alone). In contrast, the KCl-induced vasoconstriction was significantly blunted with 10 µM RKI-1447 (P < 0.05 vs. KCl alone).
Fig. 3.

Effect of the Rho kinase inhibitor RKI-1447 (RKI) on KCl-induced afferent arteriole vasoconstriction. A: afferent arteriolar responses to increasing concentrations of KCl in the absence and presence of the Rho kinase inhibitor RKI-1447 at 0.1, 1, and 10 µM. Data represent the actual diameters from each group. B: same data normalized as a percentage of the control diameter for each group. Values are expressed as means ± SE; n, number of kidneys. *P < 0.05 vs. baseline diameter in the same group; †P < 0.05 vs. vasoconstriction with KCl alone at the same concentration.
Effects of Y-27632 on Bay K8644-Induced Afferent Arteriole Vasoconstriction
Figure 4 shows the effect of Y-27632 on Bay K8644-induced afferent arteriole responsiveness. Superfusion of Bay K8644 led to concentration-dependent vasoconstriction of afferent arterioles (P < 0.05 vs. baseline diameter; Fig. 4), similar to that previously reported by Carmines et al. (4). Y-27632 at 5 µM dilated arterioles by 40 ± 7% and significantly attenuated Bay K8644-induced vasoconstriction (P < 0.05 vs. Bay K8644 alone).
Fig. 4.

Effect of the Rho kinase inhibitor Y-27632 on the voltage-dependent L-type Ca2+ channel agonist Bay K8644-induced afferent arteriole vasoconstriction. A: afferent arteriolar responses to increasing concentrations of Bay K8644 in the absence and presence of the Rho kinase inhibitor Y-27632 at 5 µM. Data represent the actual diameters from each group. B: same data normalized as a percentage of the control diameter for each group. Values are expressed as means ± SE; n, number of kidneys. *P < 0.05 vs. baseline diameter in the same group; †P < 0.05 vs. vasoconstriction with Bay K8644 alone at the same concentration.
Effects of Y-27632 and RKI-1447 on [Ca2+]i in Cultured Rat Aortic Smooth Muscle A7r5 Cells and Rat PMVSMCs
Figure 5 shows the effects of 10 µM Y-27632 and 10 µM RKI-1447 on [Ca2+]i in cultured A7r5 and freshly isolated PMVSMCs. Incubation with Y-27632 or RKI-1447 did not significantly impact baseline [Ca2+]i in A7r5 cells or PMVSMCs. Similarly, no effect was observed to lower inhibitor concentrations (data not shown).
Fig. 5.
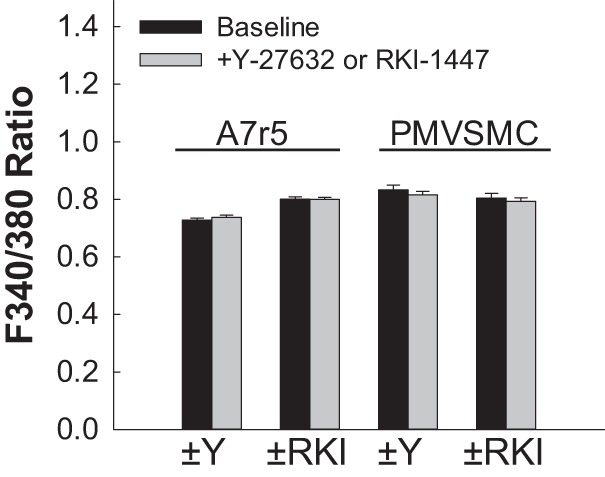
Effect of the Rho kinase inhibitors Y-27632 and RKI-1447 on intracellular Ca2+ concentration ([Ca2+]i) in rat cultured A7r5 aortic smooth muscle cells and rat preglomerular microvascular smooth muscle cells (PMVSMCs). Changes in [Ca2+]i were estimated by fura-2 fluorescence [ratio of fluorescence at 340 nm to that at 380 nm (F340-to-F380 ratio)] before and after incubation with 10 µM Y-27632 (±Y) or 10 µM RKI-1447 (±RKI) in cultured rat aortic A7r5 smooth muscle cells or in PMVSMCs, respectively. Each bar represents the mean ± SE.
Effects of Y-27632 and RKI-1447 on KCl-Induced Elevation of [Ca2+]i in Rat Cultured A7r5 Aortic Smooth Muscle Cells
Figure 6 shows the effects of Y-27632 (Fig. 6A) and RKI-1447 (Fig. 6B) on the [Ca2+]i response to 90 mM KCl in A7r5 cells. In the Y-27632 experiments, KCl alone (control) increased the F340-to-F380 ratio by 0.18 ± 0.01 (n = 118 cells, P < 0.05; Fig. 6A). Low concentrations of Y-27632 did not significantly impact the Ca2+ response to KCl compared with control cells, whereas Y-27632 concentrations of 1.0 µM and higher significantly reduced the F340-to-F380 ratio response by ~50% (P < 0.05 vs. control cells with KCl alone). With RKI-1447, KCl-induced increases in the F340-to-F380 ratio were significantly attenuated (P < 0.05 vs. KCl alone; Fig. 6B). Figure 6C shows traces of the fura-2 fluorescence response to 90 mM KCl in the absence (black line) and in the presence of Y-27632 (10 µM, gray line) in different cultured A7r5 cells.
Fig. 6.
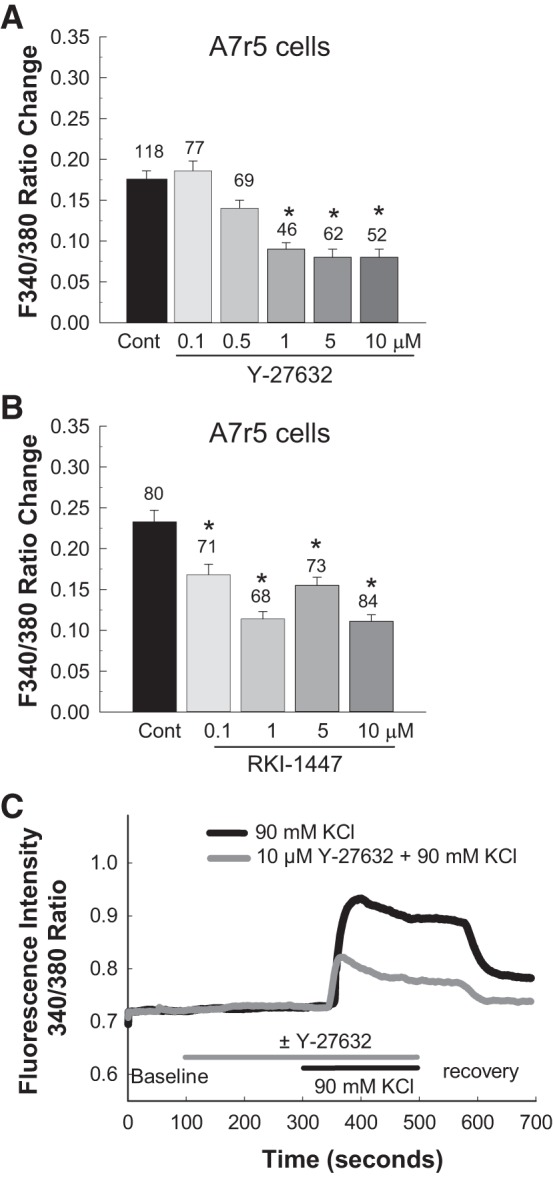
Effect of the Rho kinase inhibitors Y-27632 and RKI-1447 on KCl-induced elevation of intracellular Ca2+ concentration ([Ca2+]i) in cultured rat aortic A7r5 smooth muscle cells. Changes in [Ca2+]i were estimated by fura-2 fluorescence [ratio of fluorescence at 340 nm to that at 380 nm (F340-to-F380 ratio)] during KCl-induced depolarization of cultured rat aortic A7r5 smooth muscle cells. A: effect of Y-27632 on KCl-induced elevation of [Ca2+]i. B: effect of RKI-1447 on KCl-induced elevation of [Ca2+]i. C: representative [Ca2+]i traces of 90 mM KCl alone and with 10 µM Y-27632 in cultured rat aortic A7r5 smooth muscle cells. Each bar (A and B) represents the mean ± SE. The number above each bar indicates the number of cells studied in each group. *P < 0.05 vs. the control (Cont) group.
Effects of Y-27632 or RKI-1447 on KCl-Induced Elevation of [Ca2+]i in Rat PMVSMCs
Figure 7 shows the effects of Y-27632 and RKI-1447 on the [Ca2+]i response to 90 mM KCl in freshly isolated PMVSMCs. KCl alone (control) increased the F340-to-F380 ratio by 0.21 ± 0.02 (n = 118 cells from 10 rats, P < 0.05; Fig. 7A). The KCl-induced elevation of [Ca2+]i was unaffected by Y-27632 until a concentration of 10 µM was reached (P < 0.05 vs. KCl alone; Fig. 7A).
Fig. 7.

Effect of the Rho kinase inhibitors Y-27632 and RKI-1447 on KCl-induced elevation of intracellular Ca2+ concentration ([Ca2+]i) in preglomerular microvascular smooth muscle cells (PMVSMCs). Changes in [Ca2+]i were estimated by fura-2 fluorescence [ratio of fluorescence at 340 nm to that at 380 nm (F340-to-F380 ratio)] during KCl-induced depolarization of freshly isolated PMVSMCs. A: effect of Y-27632 on KCl-induced elevation of [Ca2+]i. B: effect of RKI-1447 on KCl-induced elevation of [Ca2+]i. Each bar represents the mean ± SE. The number above each bar indicates the number of cells studied in each group. *P < 0.05 vs. the control (Cont) group.
Similar to A7r5 cells, KCl-induced increases in the F340-to-F380 ratio were significantly attenuated by application of RKI-1447 in freshly isolated PMVSMCs regardless of the concentration tested (Fig. 7B). The F340-to-F380 ratio was reduced by ~50% compared with the response of control cells to KCl alone (P < 0.05).
Effects of Y-27632 on Bay K8644-Induced Elevation of [Ca2+]i in Rat A7r5 Cells and PMVSMCs
Figure 8 shows the impact of Y-27632 on Bay K8644-induced elevation of [Ca2+]i in rat A7r5 cells (Fig. 8A) and in PMVSMCs (Fig. 8B). Application of 10 µM Bay K8644 significantly increased the F340-to-F380 ratio by 0.034 ± 0.002 in rat A7r5 cells and by 0.014 ± 0.002 in PMVSMCs. In contrast, Y-27632 (5 µM) completely suppressed the Bay K8644-induced elevation of [Ca2+]i in both types of cells (P < 0.05 vs. Bay K8644 alone).
Fig. 8.
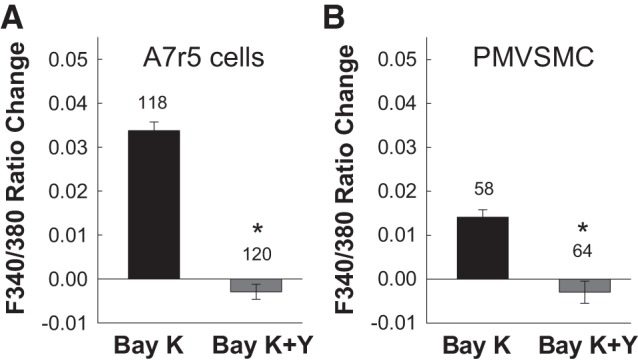
Effect of the Rho kinase inhibitor Y-27632 (Y) on Bay K8644 (Bay K)-induced elevation of intracellular Ca2+ concentration ([Ca2+]i) in rat cultured A7r5 aortic smooth muscle cells and preglomerular microvascular smooth muscle cells (PMVSMCs). Changes in [Ca2+]i were estimated by fura-2 fluorescence [ratio of fluorescence at 340 nm to that at 380 nm (F340-to-F380 ratio)] in response to Bay K8644 (10 µM) with and without Y-27632 (5 µM) in rat cultured A7r5 cells (A) or in freshly isolated PMVSMCs (B). Each bar represents the mean ± SE. The number above each bar indicates the number of cells studied in each group. *P < 0.05 vs. the control group.
DISCUSSION
The present study was designed to determine whether Rho kinase inhibitors impact VDCC signaling in PMVSMCs. Activation of L-VDCCs and stimulation of RhoA/Rho kinase-mediated Ca2+-sensitive pathways are two predominant intracellular signaling mechanisms used for afferent arteriolar vasoconstriction in response to various agonist or pressure stimuli (2, 29). Traditionally, these two pathways were thought to act independently; however, there is growing evidence suggesting that interactions may exist between these pathways in the vasculature (11–13, 24, 32, 39, 41). In the present study, we showed that treatment with two chemically dissimilar Rho kinase inhibitors, Y-27632 and RKI-1447, substantially dilated the starting diameter of afferent arterioles, consistent with previous reports using Y-27632 (6, 19, 27). Moreover, both Y-27632 and RKI-1447 inhibited afferent arteriole vasoconstriction induced by KCl (30–90 mM)-mediated membrane depolarization, implying that VDCC and RhoA/Rho kinase signaling pathways may be overlapping or perhaps convergent. This led to additional studies to evaluate the effect of Rho kinase inhibitors on [Ca2+]i during KCl-mediated membrane depolarization. We found that neither Y-27632 nor RKI-1447 impacted baseline [Ca2+]i but both significantly attenuated the elevation of [Ca2+]i during depolarization with 90 mM KCl in cultured rat aortic smooth muscle A7r5 cells and freshly isolated PMVSMCs. We further showed that Y-27632 significantly blunted afferent arteriole vasoconstriction and blocked the elevation of [Ca2+]i in both types of cells in response to the L-VDCC agonist Bay K8644. Taken together, these results suggest that both Rho kinase inhibitors, Y-27632 and RKI-1447, do blunt VDCC function to some degree, thereby partially blocking VDCC-mediated Ca2+ influx in afferent arterioles.
Increased [Ca2+]i is a major mechanism by which vascular smooth muscle contracts. Elevation of [Ca2+]i can occur by Ca2+ influx and/or by Ca2+ release from the sarcoplasmic reticulum (2, 9, 29, 33). In the preglomerular microvasculature, L-VDCCs represent an important signaling mechanism that can control renal vascular tone and regulate preglomerular microvascular responses to pressure and vasoconstrictor stimuli (2, 3, 8, 21, 23, 27, 29). In the present study, we applied increasing concentrations of KCl (30, 60, and 90 mM) to depolarize vascular smooth muscle and activate VDCCs. KCl caused concentration-dependent vasoconstriction of afferent arterioles, demonstrating the existence of VDCCs in afferent arterioles, consistent with earlier reports (3, 8, 10, 21, 23). This KCl-mediated vasoconstriction was unaltered by 1 µM Y-27632 but significantly attenuated after the Y-27632 concentration increased to 5 and 10 µM, suggesting that Y-27632 may negatively impact VDCC function. Y-27632 is widely used to explore the role of the RhoA/Rho kinase pathway (22, 28, 36). It is unclear whether this inhibitory influence of Y-27632 on KCl-induced vasoconstriction is caused by a nonspecific effect of Y-27632, by inhibition of VDCC signaling pathways, or by desensitization of the contractile apparatus by cytoskeletal derangement. In an effort to exclude the possibility that Y-27632 was exerting a nonspecific effect on KCl-mediated depolarization, we also tested a more potent, chemically dissimilar Rho kinase inhibitor, RKI-1447. RKI-1447 is a recently developed Rho kinase inhibitor with effects on both the α- and β-isoforms of Rho kinase (30). Crystal structure studies revealed that RKI-1447 is a cell-permeable pyridylthiazolyl-urea targeting the ATP-binding site on Rho kinase and shows a greater potency than Y-27632 in inhibiting Rho kinase activity (30). RKI-1447 has been used in cancer research (30), but, to date, there are no reports of its use in vascular studies. Similar to the vasodilatory effect of Y-27632 on baseline afferent arteriole diameter, RKI-1447 also led to dilatation of afferent arterioles with concentrations as low as 0.1 µM and reached saturation of afferent arteriole dilatation at 1 µM. Although 1 µM RKI-1447 showed only limited inhibitory effect on KCl-induced vasoconstriction of afferent arterioles, 10 µM RKI-1447 almost completely blocked the vasoconstriction induced by KCl. These data support the idea that the Rho kinase inhibitors Y-27632 and RKI-1447 can interfere with Ca2+ signaling through VDCCs in renal microvessels in a concentration-dependent manner.
The mechanisms underlying the inhibitory effects of Y-27632 or RKI-1447 on KCl-induced vasoconstriction are uncertain. This could be due to inhibition of VDCC signaling in concert with Rho kinase inhibition. Several studies have reported that administration of a high KCl concentration or the L-VDCC agonist FPL64176 increased RhoA/Rho kinase activity in different vascular beds (11, 12, 32). Alternatively, blocking concentrations of Y-27632 or RKI-1447 on KCl-induced vasoconstriction may inhibit VDCC function by blocking Ca2+ influx, and, therefore, the pathways are not independent of each other. To evaluate the interrelationship between the Rho kinase pathway and VDCCs, we determined the effects of Y-27632 or RKI-1447 on the [Ca2+]i response during KCl-induced depolarization of vascular smooth muscle cells. We found that Y-27632 suppressed KCl-induced elevation of [Ca2+]i in both cultured A7r5 cells and in native PMVSMCs, albeit Y-27632 had a greater inhibitory effect on cultured A7r5 cells than on PMVSMCs. Similarly, RKI-1447 significantly attenuated KCl-mediated elevation of [Ca2+]i in both A7r5 cells and PMVSMCs. Since L-VDCCs are the predominant VDCCs in afferent arterioles, we further examined the influence of Y-27632 on L-VDCC function by direct application of the L-VDCC opener Bay K8644. Bay K8644 caused concentration-dependent vasoconstriction of afferent arterioles, consistent with a previous report (4); however, Bay K8644-induced vasoconstriction was significantly attenuated by 5 µM Y-27632. Furthermore, Y-27632 completely blocked the Bay K8644-induced elevation of [Ca2+]i in both A7r5 cells and PMVSMCs. These results indicate that the Rho kinase inhibitors tested here do have an inhibitory influence on VDCC function, at least some of which are L-VDCCs, thereby blunting influx of extracellular Ca2+. This finding is potentially important because Y-27632 and RKI-1447 are commonly used to probe the role of Rho kinase in cellular signaling processes (22, 30, 35, 36) without consideration of their effect on Ca2+ signaling. Our data provide evidence that the physiological impact of Y-27632 or RKI-1447 could include both inhibition of the Rho kinase pathway as well as inhibitory effects on L-VDCC function.
Inhibition of KCl- and Bay K8644-induced vasoconstriction and elevation of [Ca2+]i by Rho kinase inhibitors Y-27632 or RKI-1447 suggest an association between Rho kinase-dependent vasoconstriction of afferent arterioles and voltage-dependent Ca2+ influx through L-VDCCs. This conclusion is consistent with several reports showing that inhibition of RhoA/Rho kinase activity with Y-27632 or HA1077 blunted KCl-induced vasoconstriction of the aorta or basilar or mesenteric arteries from rabbits, rats, and mice (11–13, 32, 37, 39). Similar to our findings, Alvarez et al. (1) also reported that application of Y-27632 (3 µM) inhibited Bay K8644-induced contraction in rabbit renal arteries. Some of the studies showed that a high concentration of KCl (60–70 mM) or activation of L-VDCCs with FPL64176 stimulated RhoA/Rho kinase activation but that activation was prevented by nifedipine or a calmodulin antagonist (11, 12, 32). However, in contrast to our finding, the KCl-induced increase in [Ca2+]i was not affected by addition of Y-27632 (10 µM) (11, 12, 32), suggesting that Y-27632 does not block VDCCs induced by KCl-induced depolarization. This suggests that functional VDCCs are important for activation of the RhoA/Rho kinase pathway in mediating vasoconstriction during depolarization induced by high K+ concentration. A study (41) in cardiac myocytes, however, suggested that RhoA/Rho kinase activation might stimulate Ca2+ influx via L-VDCCs. For example, Yatani et al. (41) reported that cardiac myocytes from transgenic mice with cardiac-specific inhibition of the Rho family of small G proteins exhibited a significant reduction of basal L-VDCC current density, suggesting that L-VDCCs are downstream targets of the RhoA signaling pathway in cardiac myocytes (41). Overall, these studies suggested that the Rho kinase pathway can interact with VDCCs contributing to the control of smooth muscle cell contraction and relaxation. Therefore, results generated using Y-27632 or RKI-1447 should be interpreted with caution and include consideration of Ca2+-signaling possibilities. For example, one of our previous studies showed that 1 µM Y-27632 completely blocked P2X1 receptor-mediated afferent arteriole vasoconstriction with 1 mM α,β-methylene ATP. Furthermore, Y-27632 also concentration dependently blunted the pressure-mediated vasoconstriction of afferent arterioles (19). Stimulation of P2X1 receptors causes membrane depolarization leading to activation of L-VDCC function (20, 40), and activation of L-VDCCs is a prerequisite for afferent arteriolar autoregulatory responses (2, 16, 29, 34). There is a possibility that the inhibitory effect of Y-27632 on the P2X1 receptor activation-induced vasoconstriction and pressure-mediated vasoconstriction might also reflect some partial impact on L-VDCC activation.
A potentially important consideration relates to the vascular elements being studied. Our prior work in the renal microcirculation revealed that 1–10 μM Y-27632 attenuated, or blocked, pressure-mediated afferent arteriole vasoconstriction in a concentration-dependent manner (19). Rat afferent arterioles range in diameter from 8 to 20 μm and represent a unique microcirculatory bed. With these blood-perfused arterioles, pressurized at 100 mmHg, UTP-mediated vasoconstriction (P2Y2 agonist) is not affected by Y-27632 treatment, but α,β-methylene ATP (P2X1 agonist), which largely requires L-VDCC Ca2+ influx, is almost completely blocked. UTP-dependent arteriole vasoconstriction and Ca2+ signaling were unaffected by Ca2+ channel blockade or by removal of Ca2+ from the extracellular medium (19, 20). In contrast, assessment of the effect of Rho kinase inhibition on cerebral artery (~220 μm) reactivity indicated that UTP-stimulated voltage-dependent vasoconstriction was inhibited by 30 μM Y-27632 (24, 25). UTP-dependent vasoconstriction of cerebral arteries involved suppression of voltage-dependent delayed rectifying K+ current, a sequence that was inhibited by Rho kinase inhibition with Y-27632 (24). In the cerebral arteries, the inhibitory effect of Y-27632 on UTP-dependent vasoconstriction also involved cytoskeletal disorganization (25). The explanation for these disparate results related to Rho kinase inhibition of UTP-mediated vasoconstriction between the renal and cerebral microvessels is unclear but may pertain to important differences between the two beds. Afferent arterioles are much smaller than cerebral arteries, and there is no de facto reason to assume that these vascular elements function in the same way. The experimental conditions were very different, with the arterioles being blood perfused at physiological pressure, 100 mmHg, while lying intact in the surrounding native renal tubular tissue. In contrast, the cerebral arteries are large and were isolated, cannulated, and bathed in cell-free PSS and pressurized to 15 mmHg to minimize inherent myogenic tone. Nevertheless, although there are differences in some of the conditions and mechanisms observed, the broader conclusion is in general agreement. Rho kinase appears to affect vascular (microvascular) function by modulating Ca2+ sensitivity and voltage-dependent Ca2+ signaling and perhaps even altering cytoskeletal organization/assembly.
We noted that the inhibitory impact of Y-27632 or RKI-1447 on KCl-induced afferent arteriole vasoconstriction was different from its impact on [Ca2+]i during KCl exposure. Y-27632 or RKI-1447 acted in a concentration-dependent manner on intact afferent arterioles, whereas this concentration-dependent response was not as clearly apparent in A7r5 cells and PMVSMCs. This could reflect the fact that afferent arterioles and cells were under different experimental conditions involving different states of activation compared with A7r5 cells and PMVSMCs. In the juxtamedullary nephron preparation setting, the kidneys were continuously perfused with reconstituted blood at a renal perfusion pressure of ~100 mmHg mimicking physiological conditions. Under these circumstances, afferent arterioles exhibit significant endogenous tone and intrinsic myogenic reactivity (5, 15, 18). In contrast, in the Ca2+ experiments, A7r5 cells and PMVSMCs were superfused with PSS in an unstimulated condition. This is also evident by the observation that administration of Y-27632 or RKI-1447 evoked clear vasodilation of pressurized afferent arterioles but had no detectable effect on resting [Ca2+]i in PMVSMCs consistent with marked differences in basal channel activity in the two settings. This is further demonstrated in the Bay K8644 experiments, where A7r5 cells and PMVSMCs had to be superfused with PSS containing 10 mM KCl for Bay K8644 to yield detectable Ca2+ responses, whereas blood-perfused afferent arterioles perfused at physiological pressure showed strong vasoconstrictor responses to Bay K8644. So, certainly, the experimental conditions were different, but this still does not negate the observations that Y-27632 or RKI-1447 can suppress Ca2+ signaling responses in a concentration-dependent manner and that concentration dependency may be predicated on basal channel activity and active force generation.
Although this study provides substantial physiological evidence that Rho kinase inhibitors do suppress L-VDCC-dependent Ca2+ influx, it remains uncertain whether the inhibitory impact of Rho kinase blockers on [Ca2+]i is due to alteration of the subconductance state of vascular smooth muscle cells or a reduction of L-VDCC density (24). Furthermore, inhibition of Rho kinase with Y-27632 or RKI-1477 could also reduce actin polymerization or alter cytoskeletal organization/assembly in renal microvascular smooth muscle cells in a manner similar to that reported for cerebral arteries (25). Therefore, further study is needed to determine the mechanisms involved in the interaction between the Rho kinase pathway and VDCCs in renal microvessels.
In conclusion, this study confirms previous observations that both KCl and Bay K8644 evoke concentration-dependent afferent arteriole vasoconstriction. Rho kinase inhibition using two different Rho kinase inhibitors, Y-27632 and RKI-1447, can increase baseline afferent arteriole diameter and attenuate afferent arteriole vasoconstriction induced by KCl-mediated membrane depolarization in a concentration-dependent manner. We also reveal that both Y-27632 and RKI-1447 suppress KCl-induced elevation of [Ca2+]i in cultured rat aortic smooth muscle cells and freshly isolated PMVSMCs. We further show that Y-27632 attenuates afferent arteriole vasoconstriction and diminishes elevation of [Ca2+]i stimulated by the L-VDCC opener Bay K8644. These results support the idea that Rho kinase-mediated vasoconstriction of afferent arterioles is also influenced by voltage-dependent Ca2+ influx mediated significantly by L-VDCC activation. These findings are particularly important because these two intracellular signaling pathways are often considered to function independently in the renal microvasculature. These findings demonstrate that data generated using Rho kinase inhibitors must be interpreted with caution as effects involving L-VDCCs and even cytoskeletal reorganization may also be contributing to the results observed.
GRANTS
This work was supported by American Heart Association Great Southeast Affiliate Grant-In-Aid 15GRNT25240015 and National Institutes of Health Grants DK-106500 (to Z. Guan) and DK-044628 and HL-095499 (to E. W. Inscho).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.G. and E.W.I. conceived and designed research; Z.G., J.J.B., S.Z., and C.E.R. performed experiments; Z.G., J.J.B., S.Z., C.E.R., and E.W.I. analyzed data; Z.G., J.J.B., and E.W.I. interpreted results of experiments; Z.G. and S.Z. prepared figures; Z.G. drafted manuscript; Z.G., J.J.B., S.Z., C.E.R., and E.W.I. edited and revised manuscript; Z.G., J.J.B., S.Z., C.E.R., and E.W.I. approved final version of manuscript.
REFERENCES
- 1.Alvarez SM, Miner AS, Browne BM, Ratz PH. Failure of Bay K 8644 to induce RhoA kinase-dependent calcium sensitization in rabbit blood vessels. Br J Pharmacol 160: 1326–1337, 2010. doi: 10.1111/j.1476-5381.2010.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlström M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev 95: 405–511, 2015. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmines PK, Fowler BC, Bell PD. Segmentally distinct effects of depolarization on intracellular [Ca2+] in renal arterioles. Am J Physiol Renal Physiol 265: F677–F685, 1993. doi: 10.1152/ajprenal.1993.265.5.F677. [DOI] [PubMed] [Google Scholar]
- 4.Carmines PK, Ohishi K, Ikenaga H. Functional impairment of renal afferent arteriolar voltage-gated calcium channels in rats with diabetes mellitus. J Clin Invest 98: 2564–2571, 1996. doi: 10.1172/JCI119075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casellas D, Carmines PK, Navar LG. Microvascular reactivity of in vitro blood perfused juxtamedullary nephrons from rats. Kidney Int 28: 752–759, 1985. doi: 10.1038/ki.1985.194. [DOI] [PubMed] [Google Scholar]
- 6.Cavarape A, Endlich N, Assaloni R, Bartoli E, Steinhausen M, Parekh N, Endlich K. Rho-kinase inhibition blunts renal vasoconstriction induced by distinct signaling pathways in vivo. J Am Soc Nephrol 14: 37–45, 2003. doi: 10.1097/01.ASN.0000039568.93355.85. [DOI] [PubMed] [Google Scholar]
- 7.Cole WC, Welsh DG. Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys 510: 160–173, 2011. doi: 10.1016/j.abb.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 8.Conger JD, Falk SA. KCl and angiotensin responses in isolated rat renal arterioles: effects of diltiazem and low-calcium medium. Am J Physiol Renal Physiol 264: F134–F140, 1993. doi: 10.1152/ajprenal.1993.264.1.F134. [DOI] [PubMed] [Google Scholar]
- 9.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79: 387–423, 1999. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 10.Feng MG, Li M, Navar LG. T-type calcium channels in the regulation of afferent and efferent arterioles in rats. Am J Physiol Renal Physiol 286: F331–F337, 2004. doi: 10.1152/ajprenal.00251.2003. [DOI] [PubMed] [Google Scholar]
- 11.Fernández-Tenorio M, Porras-González C, Castellano A, Del Valle-Rodríguez A, López-Barneo J, Ureña J. Metabotropic regulation of RhoA/Rho-associated kinase by L-type Ca2+ channels: new mechanism for depolarization-evoked mammalian arterial contraction. Circ Res 108: 1348–1357, 2011. doi: 10.1161/CIRCRESAHA.111.240127. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Tenorio M, Porras-González C, Castellano A, López-Barneo J, Ureña J. Tonic arterial contraction mediated by L-type Ca2+ channels requires sustained Ca2+ influx, G protein-associated Ca2+ release, and RhoA/ROCK activation. Eur J Pharmacol 697: 88–96, 2012. doi: 10.1016/j.ejphar.2012.09.047. [DOI] [PubMed] [Google Scholar]
- 13.Ghisdal P, Vandenberg G, Morel N. Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. J Physiol 551: 855–867, 2003. doi: 10.1113/jphysiol.2003.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan Z, Singletary ST, Cook AK, Hobbs JL, Pollock JS, Inscho EW. Sphingosine-1-phosphate evokes unique segment-specific vasoconstriction of the renal microvasculature. J Am Soc Nephrol 25: 1774–1785, 2014. doi: 10.1681/ASN.2013060656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Z, Wang F, Cui X, Inscho EW. Mechanisms of sphingosine-1-phosphate-mediated vasoconstriction of rat afferent arterioles. Acta Physiol (Oxf) 222: e12913, 2018. doi: 10.1111/apha.12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi K, Epstein M, Saruta T. Altered myogenic responsiveness of the renal microvasculature in experimental hypertension. J Hypertens 14: 1387–1401, 1996. doi: 10.1097/00004872-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3: a004549, 2011. doi: 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inscho EW, Carmines PK, Navar LG. Juxtamedullary afferent arteriolar responses to P1 and P2 purinergic stimulation. Hypertension 17: 1033–1037, 1991. doi: 10.1161/01.HYP.17.6.1033. [DOI] [PubMed] [Google Scholar]
- 19.Inscho EW, Cook AK, Webb RC, Jin LM. Rho-kinase inhibition reduces pressure-mediated autoregulatory adjustments in afferent arteriolar diameter. Am J Physiol Renal Physiol 296: F590–F597, 2009. doi: 10.1152/ajprenal.90703.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inscho EW, LeBlanc EA, Pham BT, White SM, Imig JD. Purinoceptor-mediated calcium signaling in preglomerular smooth muscle cells. Hypertension 33: 195–200, 1999. doi: 10.1161/01.HYP.33.1.195. [DOI] [PubMed] [Google Scholar]
- 21.Inscho EW, Mason MJ, Schroeder AC, Deichmann PC, Stiegler KD, Imig JD. Agonist-induced calcium regulation in freshly isolated renal microvascular smooth muscle cells. J Am Soc Nephrol 8: 569–579, 1997. [DOI] [PubMed] [Google Scholar]
- 22.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of Rho-associated kinases. Mol Pharmacol 57: 976–983, 2000. [PubMed] [Google Scholar]
- 23.Loutzenhiser R, Hayashi K, Epstein M. Divergent effects of KCl-induced depolarization on afferent and efferent arterioles. Am J Physiol Renal Physiol 257: F561–F564, 1989. doi: 10.1152/ajprenal.1989.257.4.F561. [DOI] [PubMed] [Google Scholar]
- 24.Luykenaar KD, Brett SE, Wu BN, Wiehler WB, Welsh DG. Pyrimidine nucleotides suppress KDR currents and depolarize rat cerebral arteries by activating Rho kinase. Am J Physiol Heart Circ Physiol 286: H1088–H1100, 2004. doi: 10.1152/ajpheart.00903.2003. [DOI] [PubMed] [Google Scholar]
- 25.Luykenaar KD, El-Rahman RA, Walsh MP, Welsh DG. Rho-kinase-mediated suppression of KDR current in cerebral arteries requires an intact actin cytoskeleton. Am J Physiol Heart Circ Physiol 296: H917–H926, 2009. doi: 10.1152/ajpheart.01206.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsh DJ, Postnov DD, Rowland DJ, Wexler AS, Sosnovtseva OV, Holstein-Rathlou NH. Architecture of the rat nephron-arterial network: analysis with micro-computed tomography. Am J Physiol Renal Physiol 313: F351–F360, 2017. doi: 10.1152/ajprenal.00092.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura A, Hayashi K, Ozawa Y, Fujiwara K, Okubo K, Kanda T, Wakino S, Saruta T. Vessel- and vasoconstrictor-dependent role of Rho/Rho-kinase in renal microvascular tone. J Vasc Res 40: 244–251, 2003. doi: 10.1159/000071888. [DOI] [PubMed] [Google Scholar]
- 28.Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK-specific inhibitor Y-27632. Methods Enzymol 325: 273–284, 2000. doi: 10.1016/S0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- 29.Navar LG, Arendshorst WJ, Pallone TL, Inscho EW, Imig JD, Bell PD. The renal microcirculation. In: Comprehensive Physiology, edited by Tuma RF, Wa LK. San Diego, CA: Elsevier, 2011, p. 550–684. [Google Scholar]
- 30.Patel RA, Forinash KD, Pireddu R, Sun Y, Sun N, Martin MP, Schönbrunn E, Lawrence NJ, Sebti SM. RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res 72: 5025–5034, 2012. doi: 10.1158/0008-5472.CAN-12-0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roos MH, van Rodijnen WF, van Lambalgen AA, ter Wee PM, Tangelder GJ. Renal microvascular constriction to membrane depolarization and other stimuli: pivotal role for rho-kinase. Pflügers Arch 452: 471–477, 2006. doi: 10.1007/s00424-006-0053-x. [DOI] [PubMed] [Google Scholar]
- 32.Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res 93: 548–556, 2003. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 33.Salomonsson M, Sorensen CM, Arendshorst WJ, Steendahl J, Holstein-Rathlou NH. Calcium handling in afferent arterioles. Acta Physiol Scand 181: 421–429, 2004. doi: 10.1111/j.1365-201X.2004.01314.x. [DOI] [PubMed] [Google Scholar]
- 34.Takenaka T, Harrison-Bernard LM, Inscho EW, Carmines PK, Navar LG. Autoregulation of afferent arteriolar blood flow in juxtamedullary nephrons. Am J Physiol Renal Physiol 267: F879–F887, 1994. doi: 10.1152/ajprenal.1994.267.5.F879. [DOI] [PubMed] [Google Scholar]
- 35.Thompson JM, Nguyen QH, Singh M, Pavesic MW, Nesterenko I, Nelson LJ, Liao AC, Razorenova OV. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene 36: 1080–1089, 2017. doi: 10.1038/onc.2016.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389: 990–994, 1997. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 37.Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol 285: C1377–C1385, 2003. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- 38.Ureña J, López-Barneo J. Metabotropic regulation of RhoA/Rho-associated kinase by L-type Ca2+ channels. Trends Cardiovasc Med 22: 155–160, 2012. doi: 10.1016/j.tcm.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Villalba N, Stankevicius E, Simonsen U, Prieto D. Rho kinase is involved in Ca2+ entry of rat penile small arteries. Am J Physiol Heart Circ Physiol 294: H1923–H1932, 2008. doi: 10.1152/ajpheart.01221.2007. [DOI] [PubMed] [Google Scholar]
- 40.White SM, Imig JD, Kim TT, Hauschild BC, Inscho EW. Calcium signaling pathways utilized by P2X receptors in freshly isolated preglomerular MVSMC. Am J Physiol Renal Physiol 280: F1054–F1061, 2001. doi: 10.1152/ajprenal.2001.280.6.F1054. [DOI] [PubMed] [Google Scholar]
- 41.Yatani A, Irie K, Otani T, Abdellatif M, Wei L. RhoA GTPase regulates L-type Ca2+ currents in cardiac myocytes. Am J Physiol Heart Circ Physiol 288: H650–H659, 2005. doi: 10.1152/ajpheart.00268.2004. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Cook AK, Field M, Edwards B, Zhang S, Zhang Z, Pollock JS, Imig JD, Inscho EW. Impaired Ca2+ signaling attenuates P2X receptor-mediated vasoconstriction of afferent arterioles in angiotensin II hypertension. Hypertension 46: 562–568, 2005. doi: 10.1161/01.HYP.0000179584.39937.41. [DOI] [PubMed] [Google Scholar]
- 43.Zicha J, Behuliak M, Pintérová M, Bencze M, Kuneš J, Vaněčková I. The interaction of calcium entry and calcium sensitization in the control of vascular tone and blood pressure of normotensive and hypertensive rats. Physiol Res 63, Suppl 1: S19–S27, 2014. [DOI] [PubMed] [Google Scholar]