

Abstract
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune disorder that is characterized by prevalent hypertension, renal injury, and cardiovascular disease. Numerous studies have reported a low prevalence and/or impaired function of regulatory T (TREG) cells in both patients with SLE and murine models of the disease. Evidence suggests that TREG cell dysfunction in SLE results from a deficiency in IL-2. Recent studies have reported that low-dose IL-2 therapy expands TREG cells in mouse models of SLE, but whether expanding TREG cells protects against hypertension and renal injury during SLE is unclear. To examine this question, female SLE (NZBWF1) and control (NZW) mice were injected with vehicle or recombinant mouse IL-2 three times in 24 h followed by single maintenance doses every 5 days for 4 wk. Treatment with IL-2 effectively expanded TREG cell populations in the peripheral blood, spleen, and kidneys. Circulating levels of anti-dsDNA IgG autoantibodies, a marker of SLE disease activity, were higher in SLE mice compared with control mice but were unaffected by IL-2 treatment. As previously reported by our laboratory, mean arterial pressure, measured in conscious mice by a carotid catheter, was higher in SLE mice than in control mice. Mean arterial pressure was significantly lower in IL-2-treated SLE mice compared with vehicle-treated SLE mice, suggesting that expanding TREG cells using low-dose IL-2 attenuates the development of hypertension. While the mechanism for the protection against hypertension is unclear, it does not appear to be related to the delay of SLE disease progression.
Keywords: autoimmunity, hypertension, interleukin-2, regulatory T cells, systemic lupus erythematosus
INTRODUCTION
T lymphocytes were specifically implicated in the pathogenesis of hypertension in the 1970s by Svendsen, who discovered that the development of DOCA-salt-induced hypertension was blunted in athymic nude mice that lack T cells (52), and these findings were later echoed in studies using other hypertensive models, including the Lyon hypertensive rat (6), the New Zealand Black mouse (53), and the spontaneously hypertensive rat (7). A definitive role of T cells in hypertension was demonstrated by Guzik et al. (20), who showed that the development of angiotensin II-dependent hypertension was blunted in rag−/− mice, which lack B and T lymphocytes. The full hypertensive response was restored with adoptive transfer of T lymphocytes but not B lymphocytes (20). Recently, researchers have sought to discern the relative importance of various T cell subsets, and studies have implicated both CD4+ and CD8+ T lymphocytes in the pathogenesis of hypertension, depending on the animal model and hypertensive stimulus (10, 68). Specifically, several CD4+ T helper (Th) subsets, including Th1, Th17, and regulatory T (TREG) cells have been studied with regard to the pathophysiology of hypertension (68).
TREG cells, which are defined by surface expression of CD4 and CD25 and expression of the transcription factor Foxp3, maintain peripheral tolerance and prevent autoimmunity. TREG cells either develop in the thymus or are induced in the periphery from CD4+CD25− precursors. They use multiple mechanisms to suppress immune responses, including the secretion of cytokines IL-10, transforming growth factor (TGF)-β, and IL-35, consumption of the T cell growth factor IL-2, cytolysis of target cells, and presentation of immunosuppressive ligands (60). Mice that have a mutation in the gene for the foxp3 transcription factor lack functional TREG cells and develop an autoimmune phenotype characterized by lymphoproliferation and multiorgan inflammation, especially in the skin, lung, and liver. This phenotype is reversed by the adoptive transfer of TREG cells (8). A similar phenotype is seen in humans suffering from immunodysregulation polyendocrinopathy enteropathy X-linked, who also lack functional TREG cells due to mutations in Foxp3 (63). Despite discrepancies in the literature, multiple studies have reported impaired TREG cell function and/or numbers in humans and animal models of the autoimmune disease systemic lupus erythematosus (SLE) (23, 41, 48). SLE is a systemic autoimmune disorder that predominantly affects women of childbearing age and is characterized by B and T lymphocyte hyperreactivity and the production of pathogenic autoantibodies to a variety of nuclear components. The prevalent immune system dysfunction in SLE leads to a wide range of disease manifestations, including hypertension, renal injury, and cardiovascular disease (5, 57). Multiple TREG cell-based therapies have been tested to expand TREG cells in patients with SLE and in animal models, including adoptive transfer (48), stem cell transplantation (62, 69), statins (1), retinoids (45, 66), tolerogenic peptide administration (12, 28), and low-dose IL-2 (22, 61). Many of these studies have reported improvements in disease activity (23, 48); however, the ability of these TREG cell-based therapies to ameliorate SLE-associated hypertension is unknown. Various studies have linked abnormal TREG cell numbers and/or function to hypertension, myocardial infarction, and atherosclerosis (40), and the TREG cell abnormalities that are present in SLE may contribute to the development of cardiovascular disease in this patient population. In the present study, we demonstrated that treatment of a hypertensive mouse model of SLE, the female NZBWF1 mouse, with low-dose recombinant mouse IL-2 leads to expansion of TREG cells and the attenuation of hypertension.
MATERIALS AND METHODS
Animals.
Adult (30 wk old) female NZBWF1 (SLE; n = 30) and NZW/LacJ (control; n = 30) mice (Jackson Laboratories, Bar Harbor, ME) were used in this study. Mice were maintained on a 12:12-h light-dark cycle in temperature-controlled rooms with access to chow and water ad libitum. All experiments were performed with the approval of the University of Mississippi Medical Center Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Recombinant mouse IL-2 administration.
Recombinant mouse IL-2 (R&D Systems, Minneapolis, MN) was dissolved in sterile PBS (pH 7.4). Mice received 2 µg recombinant IL-2 or vehicle (PBS) intraperitoneally every 12 h for 24 h (3 times total). Additional single 2-µg doses were given every 5 days for 4 wk. This treatment strategy was previously reported by Humrich et al. (23) to selectively expand TREG cells in NZBWF1 mice.
Blood pressure.
Mean arterial pressure (MAP; in mmHg) was recorded via indwelling carotid artery catheters in freely moving conscious mice as previously described by our laboratory (36–39, 59). Briefly, mice were implanted with catheters, and animals were allowed to recover for 24 h. Blood pressure was recorded in conscious, unrestrained mice via pressure transducers on 2 consecutive days after catheter implantation (1.5 h/day). Blood pressure was measured in all animals between 8:00 and 10:00 AM each day. The final hour of recorded data on each day was used to determine MAP.
Preparation of cells for flow cytometry.
Blood was collected from the retroorbital plexus from IL-2- or vehicle-treated animals 48 h after the first three doses of IL-2 and at the conclusion of the study. The blood was centrifuged at 350 g for 5 min to isolate plasma. Erythrocytes were lysed by adding 10× volume of 1× PharmLyse (BD Biosciences, San Jose, CA). After incubation for 5 min at room temperature, the blood was centrifuged at 200 g for 5 min. Pelleted peripheral blood leukocytes (PBLs) were washed with 1× PBS and 2% FCS and centrifuged at 350 g for 5 min. Cells were immediately used for flow cytometry. Spleens were homogenized using the Spleen Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and GentleMACS Octo Dissociator (Miltenyi Biotec) according to the manufacturer’s instructions. Splenocytes were subsequently used for flow cytometric analyses. For the isolation of renal immune cells, one kidney was homogenized in 5 ml RPMI media containing 200 U/ml DNase and 10 mg/ml collagenase type IV using the GentleMACS and a user-defined protocol for the mouse kidney. The resulting homogenate was filtered through a 70-μm cell strainer and washed with 1× PBS containing 2% FCS and 2 mM EDTA. The single cell suspension was centrifuged at 300 g for 10 min. The resulting cell pellet was then resuspended in 1× PBS and 2% FCS and subjected to downstream analyses.
Flow cytometric analyses.
For all flow cytometric analyses, cells were first washed and resuspended in 1× PBS, 2% FCS, and 0.9% sodium azide at a concentration of 2 × 107 cells/ml. Cells (1 × 106 cells, 50 μl) were aliquoted into a flow cytometry tube and incubated with 0.25 μg anti-mouse CD32/CD16 (FcR block, BD Biosciences) for 5 min on ice. For staining of PBLs, spleen leukoocytes, and kidney leukoocytes, cells were stained with either isotype control antibodies or anti-CD3 phycoerythrin (PE)-Cy7 (clone 145-2C11) and anti-CD4-FITC (clone GK1.5, BD Biosciences). Cells were incubated on ice for 30 min protected from light. For staining of TREG cells, cells were stained with anti-CD3 and anti-CD4 for 30 min on ice. Cells were then fixed and permeabilized using the eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were subsequently incubated with anti-mouse/rat Foxp3 PE (clone FJK-16s, Invitrogen). A subset of spleen and PBL samples was also analyzed for activated CD4 T cells using anti-CD3, anti-CD4, and anti-CD69-PE (clone H1.2F3, BD Biosciences). All samples were analyzed on a Gallios (Becton Dickinson, Franklin Lakes, NJ) flow cytometer at the University of Mississippi Medical Center Flow Cytometry core facility. A total of 100,000 events were acquired for each sample. Data were analyzed using FCS express 6 software.
Autoantibodies and total IgG.
Anti-dsDNA IgG was detected in plasma at 34 wk of age (SLE mice) or 35 wk of age (control mice) using the anti-dsDNA IgG ELISA (Alpha Diagnostic, San Antonio, TX) per the manufacturer’s instructions and as previously described by our laboratory (36, 37, 39, 59). Total plasma IgG concentrations were determined at the same ages using the mouse IgG ELISA kit (Invitrogen) according to the manufacturer’s instructions and as previously described by our laboratory (55).
Renal injury.
Urinary albumin was monitored weekly by dipstick analysis (Albustix, Siemens). Animals were considered positive for albuminuria at ≥100 mg/dl (36–39). The urinary albumin excretion rate (in mg/day) was assessed by ELISA (Alpha Diagnostic) using overnight urine samples collected at the conclusion of the study as previously described (36–39). Glomerulosclerosis and renal interstitial fibrosis were assessed using Masson’s trichrome-stained kidney sections as previously described by our laboratory (39, 55). Slides were analyzed on a Nikon 55I microscope with DS-Fl1 5-Meg Color C digital camera and Nis-Elements Image-analysis software (version 3.03, Nikon Instruments, Melville, NY).
Renal cytokine analysis.
Kidney sections were snap frozen in liquid nitrogen and stored at −80°C until use. Kidneys were combined with 1 ml lysis buffer containing 10 mM Tris·HCl (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 10% glycerol, 5 mM EDTA, and complete mini EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland) and agitated for 2 min in a Bullet Blender tissue homogenizer (Next Advance Laboratory Instruments, Troy, NY). Samples were then centrifuged at 11,000 g for 10 min to remove debris. The supernatant was removed, and total protein was quantified in the supernatant using the BCA method. Cell lysates were then analyzed using the BD CBA Mouse Cytokine Flex Set for IL-10 (BD Biosciences) according to the manufacturer’s instructions. Samples were analyzed on a NovoCyte flow cytometer (ACEA Biosciences, San Diego, CA). A total of 1,500 beads were analyzed per sample, and IL-10 concentrations were determined using FCAP array software (BD Biosciences).
Western blot analyses.
The total abundance of nitric oxide synthase 3 (NOS3) and phosphorylation of NOS3 at Ser1177 were determined by Western blot analysis. The thoracic aorta was homogenized in 250 μl lysis buffer and centrifuged at 10,000 g for 10 min at 4°C, and the homogenate was then mixed with loading buffer and heated to 85°C for 5 min. The rat kidney inner medulla was used as a positive control. Homogenized samples (22.5 μl) were separated by electrophoresis (200 V, 40 min, 4–20% TGX Stain-Free Precast gel, Bio-Rad Laboratories) and transferred onto nitrocellulose membranes (Bio-Rad Trans-Blot Turbo, High MW program). Membranes were stained with Ponceau S for 10 min to determine total protein loading and then blocked for 1 h and cut at the 75-kDa marker. The top half was incubated with rabbit polyclonal phosphorylated (p-)NOS3-Ser1177 antibody (Cell Signaling, 1:250) and mouse NOS3 antibody (BD Transduction, 1:250) overnight at 4°C. Membranes were then incubated with secondary antibodies (BioRad Blue 700 rabbit 1:2,000 for p-NOS3 and BioRad chemiluminescent mouse 1:1,000 for NOS3) for 1 h at room temperature. Bands were then quantified using the BioRad ChemiDoc MP Imaging System and Image Laboratory 6.0 software (Bio-Rad Laboratories). NOS3 phosphorylation was calculated as the integrated optical density (IOD) of the p-NOS3 band to the IOD of the NOS3 band, and NOS3 abundance was calculated as the ratio of the IOD of NOS3 to the IOD of the total lane for Ponceau S staining.
Statistical analysis.
Data are presented as mean ± SE. Statistical analyses were performed using GraphPad Prism 7. Two-way ANOVA was used to analyze treatment (vehicle vs. IL-2) or group (control vs. SLE) interactions. One-way ANOVA was used to analyze individual differences between groups, and Tukey’s post test for multiple comparisons was used to compare groups. An unpaired t-test was used to analyze differences between renal lymphocytes and circulating lymphocytes in vehicle- and IL-2-treated SLE mice. P values of <0.05 were considered statistically significant.
RESULTS
Because administration of low-dose IL-2 has been previously shown to expand CD4+Foxp3+ TREG cells (23), we analyzed TREG cells in the peripheral blood in a subset of animals 48 h after the three doses of IL-2 using flow cytometry. A representative gating strategy is shown in Fig. 1A. Treatment with IL-2 significantly increased percentages of TREG cells in the peripheral blood of both control and SLE mice after 48 h compared with vehicle-treated mice (Fig. 1B). We also analyzed levels of circulating TREG cells at the conclusion of the study in all mice (Fig. 1C), and there were no differences in TREG cell percentages between vehicle- and IL-2-treated control animals. Both vehicle- and IL-2-treated SLE groups had significantly lower TREG cell levels than control groups, and IL-2 administration did not significantly increase percentages of TREG cells. To assess any potential effects of IL-2 treatment on total CD4+ T cells, we also analyzed percentages of CD4+ T cells after 4 wk of treatment, and no differences were found between vehicle- and IL-2-treated groups (Fig. 1D). Overall, SLE mice had lower levels of circulating CD4+ T cells compared with control mice, which has been previously reported by our laboratory (55, 56), but IL-2 treatment had no effect on CD4+ T cell percentages. Additionally, we assessed splenic CD4+ T and TREG cells (Fig. 2). While there were no differences in splenic CD4+ T cells among any of the groups, splenic TREG cells were lower in the vehicle-treated SLE group, and treatment with IL-2 significantly increased percentages of TREG cells in control and SLE mice compared with vehicle-treated SLE mice.
Fig. 1.

Effect of IL-2 treatment on circulating regulator T (TREG) cells in control and systemic lupus erythematosus (SLE) mice. A: representative gating strategy to identify CD4+Foxp3+ TREG cells. B: percentage of CD4+Foxp3+ TREG cells 48 h after three doses of IL-2. *P < 0.05 vs. the vehicle-treated control group; #P < 0.05 vs. all other groups. C: percentage of CD4+Foxp3+ T cells at the conclusion of the study. ^P < 0.05 vs. vehicle- and IL-2-treated control groups. D: percentages of CD3+CD4+ T lymphocytes at the conclusion of the study. ^P < 0.05 vs. vehicle- and IL-2-treated control groups. P values were generated using one-way ANOVA followed by a Tukey’s post test for multiple comparisons.
Fig. 2.

Effect of IL-2 treatment on splenic regulator T (TREG) cells in control and systemic lupus erythematosus (SLE) mice. A: percentages of CD4+Foxp3+ TREG cells at the conclusion of the study. *P < 0.05 vs. IL-2-treated control and IL-2-treated SLE groups. B: percentages of CD3+CD4+ T lymphocytes at the conclusion of the study. P values were generated using one-way ANOVA followed by a Tukey’s post test for multiple comparisons.
To assess the effect of IL-2 administration on SLE disease activity, we analyzed circulating anti-dsDNA IgG levels (Fig. 3A) and circulating IgG concentrations (Fig. 3B). SLE mice had increased levels of anti-dsDNA autoantibodies compared with control mice, as previously reported by our laboratory, but the levels were unaffected by treatment with IL-2. In addition, SLE mice had increased levels of circulating IgG compared with control mice, but low-dose IL-2 administration did not impact circulating IgG. NZBWF1 mice also have increased levels of CD4+ T cell activation, including increases in CD4+ T cells that express the early activation marker CD69 (58). This cell population increases with age in the NZBWF1 mouse model (25). As another measure of SLE disease activity, we analyzed percentages of activated CD69+ T cells using flow cytometry in both PBLs and the spleen. SLE mice treated with IL-2 had significantly lower levels of circulating CD69+CD4+ T cells compared with vehicle-treated SLE mice (Fig. 3C), but there were no differences between CD69+ T cell percentages in the spleens of vehicle- and IL-2-treated SLE mice (P = 0.08; Fig. 3D).
Fig. 3.

Effect of IL-2 treatment on systemic lupus erythematosus (SLE) disease activity. A: anti-dsDNA IgG measured at 34 wk of age in control and SLE mice. Plasma levels of anti-dsDNA IgG were higher in SLE mice than in control mice, but IL-2 treatment had no effect on anti-dsDNA levels. *P < 0.05 vs. vehicle- and IL-2-treated control groups. B: plasma IgG concentrations measured at 34 wk of age in control and SLE mice. IgG concentrations were higher in SLE mice compared with control mice, and IL-2 treatment had no effect on IgG concentration. *P < 0.05 vs. vehicle- and IL-2-treated control groups. C: circulating activated T cells as measured by flow cytometry. Mice treated with IL-2 had lower levels of CD4+CD69+ T cells than vehicle-treated mice. *P < 0.05 vs. the vehicle-treated SLE group. D: CD69+ T cells in the spleen. No differences were detected between the groups. P values were generated using one-way ANOVA followed by a Tukey’s post test for multiple comparisons.
Finally, we tested the effect of IL-2 administration on blood pressure and renal injury. We measured MAP in conscious freely moving mice on 2 consecutive days at the conclusion of the study (Fig. 4). Vehicle-treated SLE mice had elevated blood pressure compared with vehicle-treated control mice. IL-2 treatment had no effect on blood pressure in control mice, but SLE mice that received IL-2 had significantly lower blood pressure compared with vehicle-treated SLE mice. NZBWF1 mice progressively develop albuminuria as they age. In the present study, 29% of the SLE mice administered vehicle developed albuminuria (>100 mg/dl) and 35% of the IL-2 treated mice were positive for urinary albumin (Fig. 5A). The urinary albumin excretion rate was also measured at the conclusion of the study by ELISA (Fig. 5B), and vehicle-treated SLE mice had increased albumin excretion compared with control mice. IL-2 treatment did not affect the albumin excretion rate in either control or SLE animals. Glomerular injury was assessed in Masson’s trichrome-stained kidney sections (Fig. 6, A and B), and SLE mice had increased glomerulosclerosis compared with control mice. IL-2 treatment had no effect on glomerular injury in control mice, but SLE mice treated with IL-2 trended toward decreased glomerular injury. Fibrosis was also assessed in stained kidney sections (Fig. 6, C and D). While there was overall limited evidence of fibrosis in all samples examined, IL-2-treated control animals had increased fibrosis compared with vehicle-treated control animals, but there were no significant differences in tubulointerstitial fibrosis between vehicle- and IL-2-treated SLE animals. To further analyze the effect of IL-2 therapy on renal injury, we also assessed the presence of CD4+ T cells in the kidneys of SLE mice (Fig. 7A). While there were no differences in the percentages of total renal CD4+ T cells, SLE mice administered IL-2 had significantly higher levels of renal TREG cells compared with vehicle-treated mice. Because expansion of TREG cells after IL-2 treatment attenuated the development of hypertension during SLE but did not significantly impact several factors including renal injury and autoantibody production, we examined additional measures that could be affected by expansion of TREG cells, including renal cytokine production and NOS3 expression in the vasculature. We analyzed levels of the cytokines IL-10 and TNF-α in the kidney using a cytometric bead assay (Fig. 7B) and did not detect any differences in renal cytokine levels among the treatment groups. We also analyzed NOS3 and p-NOS3 (Ser1177) expression in the aorta using Western blot analysis (Fig. 8A) and did not find any significant changes in NOS3 expression or the ratio of p-NOS3 to NOS3 (Fig. 8B).
Fig. 4.
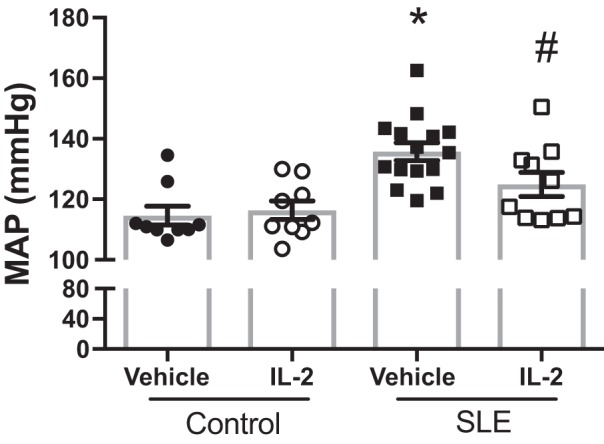
Effect of IL-2 treatment on mean arterial pressure (MAP) in control and systemic lupus erythematosus (SLE) mice. MAP was significantly higher in SLE mice compared with control mice. IL-2 significantly lowered MAP in SLE mice but had no effect in control mice. *P < 0.05 vs. vehicle- and IL-2-treated control groups; #P < 0.05 vs. the vehicle-treated SLE group. P values were generated using one-way ANOVA followed by a Tukey’s post test for multiple comparisons.
Fig. 5.

Effect of IL-2 on albumin excretion in control and systemic lupus erythematosus (SLE) mice. A: albuminuria incidence in SLE mice as measured by a dipstick assay on overnight urine samples. A positive test is considered >100 mg/dl. B: urine albumin in control and SLE mice as measured by albumin ELISA at the conclusion of the study. Urine albumin was similar in control mice treated with vehicle or IL-2. Albumin was higher in SLE mice compared with control mice, and IL-2 treatment had no effect on urinary albumin excretion.
Fig. 6.

Effect of IL-2 on renal injury in control and systemic lupus erythematosus (SLE) mice. A: glomerulosclerosis index assessed in control and SLE mice administered vehicle or IL-2. *P < 0.05 vs. the vehicle-treated control group; #P < 0.01 vs. the IL-2-treated control group. B: representative pictures of glomerulosclerosis (magnification: ×40) from paraffin-embedded kidneys stained with Masson’s trichrome. Vehicle-treated SLE mice had significantly higher glomerular injury. C: tubulointerstitial fibrosis assessed in control and SLE mice. *P < 0.05 vs. the vehicle-treated control group. D: representative images of tubular injury (magnification: ×10) from paraffin embedded kidneys stained with Masson’s trichrome. P values were generated using one-way ANOVA followed by a Tukey’s post test for multiple comparisons.
Fig. 7.

Effect of IL-2 on renal regulatory T cells and cytokines. A: percentages of total CD4+ T cells and regulatory T cells in the kidneys of systemic lupus erythematosus (SLE) mice at the conclusion of the study. *P < 0.05 vs. the vehicle-treated SLE group. P values were determined using an unpaired t-test. B: renal IL-10 and TNF-α levels as measured by a cytokine bead array.
Fig. 8.

Effect of IL-2 on aortic nitric oxide synthase (NOS)3 expression. A: representative Western blot analyses of total NOS3 and Ser1177 phosphorylated NOS3 (p-NOS3) in the aorta of vehicle (Veh)- and IL-2-treated control and systemic lupus erythematosus (SLE) mice. B: densitometric ratio of p-NOS3 to NOS3 and densitometric ratio of NOS3 to total protein loading.
DISCUSSION
Patients with the autoimmune disorder SLE have an increased risk for the development of hypertension and cardiovascular disease, and the prevalent B and T lymphocyte dysfunction present in SLE has been implicated in the development of SLE-associated hypertension. Recent studies have uncovered defects in TREG cell numbers and function in patients with SLE and in murine models of the disease, and one contributing mechanism to the TREG cell dysfunction is a progressive decline in IL-2 production (23). In the present study, we sought to expand the TREG cell population in SLE mice using low-dose IL-2. The major new findings of this study are that 4-week treatment with low-dose IL-2 1) effectively expands TREG cells in the spleen and kidneys of SLE mice independently of changes in CD4+ T cells, 2) does not lower autoantibody production, and 3) attenuates hypertension in SLE mice.
While IL-2 was initially described in the 1970s as a potent growth factor that promoted the clonal expansion of T cells in vitro (43), more recent studies have demonstrated that IL-2 is required for the development of both thymic and peripheral TREG cells (26). Mice deficient in IL-2 or IL-2 receptor subunits develop T cell lymphoproliferation and autoimmunity (47, 51), and Malek and colleagues (34) demonstrated that the autoimmunity was due to an inability to generate thymic TREG cells. Additionally, IL-2/IL-2 receptor signaling is crucial for the function and maintenance of TREG cells in the periphery (13, 15). IL-2 supplementation for TREG cell expansion was first tested in the nonobese diabetic mouse, in which there is a progressive decrease in the TREG cell-to-effector T cell ratio in inflamed pancreatic islets. Tang et al. (54) suggested that the decline in TREG cell numbers was due to increased apoptosis of TREG cells. Low-dose IL-2 treatment promoted TREG cell expansion, likely by increasing the expression of CD25 and Bcl-2, and prevented the development of diabetes (54). Later studies found that only 5 days of low-dose IL-2 starting at the disease onset can reverse disease in nonobese diabetic mice (17). IL-2 therapy has subsequently been tested in other animal models and in humans undergoing hematopoietic stem cell transplantation, those suffering from chronic graft-versus-host disease, hepatitis C-induced vasculitis, alopecia areata, and SLE (67).
NZBWF1 mice have a progressive decline in TREG cell percentages as they age, corresponding with a decrease in IL-2-producing CD4+ T cells (23). In the present study, we treated control and SLE mice with three doses of 2 µg IL-2 in the first 24 h followed by single 2-µg maintenance doses every 5 days. Analysis of circulating TREG cells 48 h after the three doses showed robust expansion of TREG cells in both control (∼2-fold increase) and SLE (∼2.5-fold increase) mice (Fig. 1B). Similar results were previously shown by Humrich et al. (23), who reported a twofold increase in peripheral TREG cells 12 h after three IL-2 injections. We also analyzed circulating and splenic TREG cells at the conclusion of the study. Although not statistically significant, there was a trend toward a sustained increase in circulating TREG cells in SLE animals (vehicle-treated SLE animals: 0.61 ± 0.05% vs. IL-2-treated SLE animals: 0.85 ± 0.1%, P = 0.06), although the increased percentage was still lower than control animals. Splenic TREG cells were significantly elevated in IL-2 treated SLE mice compared with vehicle-treated SLE mice (Fig. 2). IL-2 also effectively expands TREG cells and prolongs survival in the MRL/lpr mouse, another experimental model of SLE (18, 19). Taken together, these data indicate that the treatment strategy used in the present study was sufficient to promote TREG cell expansion in the peripheral blood and spleen.
An increase in TREG cells can have pleiotropic effects on various components of the immune system, including effector T cell activity and antibody production by B cells. TREG cells inhibit expansion of effector T cells by consuming IL-2, due to their higher levels of CD25 expression (33). In addition, TREG cells can prevent effector T cell activation: cytotoxic T lymphocyte-associated protein 4 on the surface of TREG cells binds to the costimulatory molecules CD80/86 on the surface of antigen-presenting cells with a higher affinity than CD28 (46). A breakdown in either of these mechanisms could lead to increases in activated effector T cells in SLE. In the present study, we found a lower percentage of CD4+ T cells that expressed the T cell activation marker CD69 in PBLs in IL-2-treated mice (Fig. 3), suggesting that activated Th cells in the circulation are decreased after IL-2 treatment. SLE is characterized by the presence of autoreactive T cells and abnormal Th1 and Th17 activation (49). While these cell types were not directly measured in the present study, it is likely that an expansion of TREG cells could decrease the activation of these cells. In support of this concept, IL-2 administration in the MRL/lpr mouse model was shown to both expand TREG cells and reduce the number of IL-17-producing T cell receptor αβ+CD4−CD8− T cells in MRL/lpr mice (42). The link between TREG cells and autoantibody production is apparent in Scurfy mice, which have a missense mutation in Foxp3. These mice lack thymic TREG cells and are also incapable of generating TREG cells in the periphery (16). Scurfy mice share many phenotypic characteristics with SLE, including lymphopenia, nephritis, and arthritis, and also show evidence of B cell dysfunction, as they produce many SLE autoantibodies including anti-nuclear, anti-Smith, and anti-dsDNA antibodies (21). In addition, studies have shown that TREG cells can induce apoptosis and cytotoxicity in B cells (24). We did not detect any changes in splenic B cell percentages with IL-2 administration (data not shown), nor did we identify any alterations in plasma IgG concentrations and anti-dsDNA IgG levels (Fig. 3), suggesting that the attenuation of blood pressure by TREG cell expansion may be the result of downstream anti-inflammatory mechanisms unrelated to the progression of SLE disease, including the production of anti-inflammatory cytokines that could impact renal and vascular function.
Previous studies have linked TREG cell alterations to cardiovascular diseases, including hypertension, myocardial infarction, and atherosclerosis (40). Studies in several experimental models have demonstrated the protective role of TREG cells in cardiovascular disease, but the impact on blood pressure is inconsistent. Kvakan and colleagues (30) found that a single adoptive transfer of CD4+CD25+ TREG cells, but not CD4+CD25− T cells, into mice infused with ANG II attenuated cardiac hypertrophy and fibrosis but did not have blood pressure-lowering effects. In a similar study (4), the adoptive transfer of TREG cells both before and during ANG II infusion effectively prevented increases in blood pressure. In an experimental model of atherosclerosis, depletion of peripheral TREG cells using anti-CD25 monoclonal antibodies exacerbated atherosclerotic plaque formation in proatherogenic apolipoprotein E−/− mice (2). The expansion of TREG cells using low-dose IL-2 in the present study attenuated hypertension in SLE mice, although MAP remained elevated compared with control mice (Fig. 4). Similarly, a previous study by our laboratory (35) showed that depletion of T cells using anti-CD3 monoclonal antibodies attenuated hypertension in mice with established renal disease, but not to the levels of control mice. It has been postulated that the use of anti-CD3 antibodies promotes expansion of inducible TREG cells in the periphery (65); thus, it may be that the increased presence of TREG cells in anti-CD3-treated animals also contributed to the attenuation of hypertension in that study.
While the exact mechanisms contributing to the antihypertensive effects of TREG cells during SLE remain unclear, it is likely that the immunosuppressive actions of TREG cells in both the kidney and vasculature plays a role. We demonstrated that TREG cells were expanded in the kidney, and TREG cells have been shown to protect the kidney in a variety of disease states, including forms of autoimmune nephritis, ischemia-reperfusion injury, diabetic nephropathy, and renal transplantation (3). We performed various assessments of renal injury (Figs. 5 and 6) in the present study. We did not detect any changes in albuminuria incidence or urinary albumin excretion rate in SLE animals treated with IL-2. Histological analyses did, however, indicate that vehicle-treated SLE mice had significantly higher glomerular injury scores compared with control mice, and IL-2-treated SLE mice trended toward decreased glomerular injury. Because of the lack of a clear improvement in renal injury, it is possible that the cytokines secreted by the renal TREG cells have disparate effects on kidney function. TREG cells secrete soluble cytokines, including IL-10, IL-35, and TGF-β, that have anti-inflammatory activities. IL-10 and TGF-β each play an important role in renal physiology, with mesangial cells being a major local source of these cytokines under normal conditions (14, 27). IL-10 has been reported to be protective against SLE-induced renal damage (32), but it has also been shown to promote the mesangial deposition of immune complexes (31). In vascular studies using hypertensive mice, microvessels incubated in TREG cell-conditioned media had improved endothelium-dependent relaxation that was mediated by an IL-10-dependent mechanism (29). IL-10 has also been shown to induce the expression of NOS3 in cultured endothelial cells (9); however, we did not detect any changes in NOS3 in the thoracic aorta (Fig. 8) as a result of TREG cell expansion. TGF-β is also a pleiotropic cytokine that can have both pro- and anti-inflammatory effects; experimental models of kidney disease highlight the potential of TGF-β to be either profibrotic or protective (50). IL-35 is a recently discovered member of the IL-12 family of cytokines that potently inhibits Th1 and Th17 cells, promotes TREG cell expansion, and increases IL-10 production (11). In studies of various experimental models of autoimmunity, including collagen-induced arthritis and inflammatory bowel disease, animals treated with IL-35 have reduced disease severity (44, 64). It remains unclear whether the cytokines produced by TREG cells mediate protective effects on the kidney and/or vasculature in SLE.
Perspectives
Altered TREG cell activity and homeostasis have been implicated in many animal models and disease states. SLE is characterized by abnormal TREG cell function and numbers in association with an IL-2 deficiency. Recently, a small clinical study (61) in patients with refractory SLE tested the efficacy of low-dose IL-2 to restore immune tolerance and TREG cell levels. This study (61) showed that a low-dose IL-2 regimen can correct TREG cell defects in vivo. Our present study suggests that low TREG cell levels contribute to the pathogenesis of SLE-associated hypertension in the NZBWF1 mouse; thus, it may be that TREG cell expansion could have the therapeutic benefit of helping to control blood pressure in this vulnerable patient population.
GRANTS
E. B. Taylor was supported by individual National Institutes of Health (NIH) National Research Service Award F32-HL-137393. K. J. Maeda was supported by NIH Grant T32-HL-105324-05. This work was supported by Veteran’s Administration Merit Award BX002604-01A2 (to M. J. Ryan), NIH Grant R01-HL-134711 (to J. M. Sasser), and NIH Grants PO1-HL-051971, P20-GM-104357, and U54-GM-115428 (to the University of Mississippi Medical Center Department of Physiology and Biophysics).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.B.T. and M.J.R. conceived and designed research; E.B.T., J.M.S., and K.J.M. performed experiments; E.B.T., J.M.S., and K.J.M. analyzed data; E.B.T., J.M.S., and M.J.R. interpreted results of experiments; E.B.T. and J.M.S. prepared figures; E.B.T. drafted manuscript; E.B.T., J.M.S., K.J.M., and M.J.R. edited and revised manuscript; E.B.T., J.M.S., K.J.M., and M.J.R. approved final version of manuscript.
REFERENCES
- 1.Abud-Mendoza C, de la Fuente H, Cuevas-Orta E, Baranda L, Cruz-Rizo J, González-Amaro R. Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus 12: 607–611, 2003. doi: 10.1191/0961203303lu429oa. [DOI] [PubMed] [Google Scholar]
- 2.Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med 12: 178–180, 2006. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 3.Alikhan MA, Huynh M, Kitching AR, Ooi JD. Regulatory T cells in renal disease. Clin Transl Immunology 7: e1004, 2018. doi: 10.1002/cti2.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57: 469–476, 2011. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 5.Bartoloni E, Alunno A, Gerli R. Hypertension as a cardiovascular risk factor in autoimmune rheumatic diseases. Nat Rev Cardiol 15: 33–44, 2018. doi: 10.1038/nrcardio.2017.118. [DOI] [PubMed] [Google Scholar]
- 6.Bataillard A, Freiche JC, Vincent M, Sassard J, Touraine JL. Antihypertensive effect of neonatal thymectomy in the genetically hypertensive LH rat. Thymus 8: 321–330, 1986. [PubMed] [Google Scholar]
- 7.Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 99: 600–607, 1981. doi: 10.1016/0006-291X(81)91787-3. [DOI] [PubMed] [Google Scholar]
- 8.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27: 68–73, 2001. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 9.Cattaruzza M, Słodowski W, Stojakovic M, Krzesz R, Hecker M. Interleukin-10 induction of nitric-oxide synthase expression attenuates CD40-mediated interleukin-12 synthesis in human endothelial cells. J Biol Chem 278: 37874–37880, 2003. doi: 10.1074/jbc.M301670200. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Agrawal DK. Dysregulation of T cell subsets in the pathogenesis of hypertension. Curr Hypertens Rep 17: 8, 2015. doi: 10.1007/s11906-014-0521-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J, Leung PS, Bowlus C, Gershwin ME. IL-35 and autoimmunity: a comprehensive perspective. Clin Rev Allergy Immunol 49: 327–332, 2015. doi: 10.1007/s12016-015-8468-9. [DOI] [PubMed] [Google Scholar]
- 12.Eilat E, Dayan M, Zinger H, Mozes E. The mechanism by which a peptide based on complementarity-determining region-1 of a pathogenic anti-DNA auto-Ab ameliorates experimental systemic lupus erythematosus. Proc Natl Acad Sci USA 98: 1148–1153, 2001. doi: 10.1073/pnas.98.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 6: 1142–1151, 2005. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 14.Fouqueray B, Boutard V, Philippe C, Kornreich A, Marchant A, Perez J, Goldman M, Baud L. Mesangial cell-derived interleukin-10 modulates mesangial cell response to lipopolysaccharide. Am J Pathol 147: 176–182, 1995. [PMC free article] [PubMed] [Google Scholar]
- 15.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med 196: 851–857, 2002. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giang S, La Cava A. Regulatory T cells in SLE: biology and use in treatment. Curr Rheumatol Rep 18: 67, 2016. doi: 10.1007/s11926-016-0616-6. [DOI] [PubMed] [Google Scholar]
- 17.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, Piaggio E. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 207: 1871–1878, 2010. doi: 10.1084/jem.20100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Ramos JC, Andreu JL, Marcos MA, Vegazo IR, Martinez C. Treatment with IL2/vaccinia recombinant virus leads to serologic, histologic and phenotypic normalization of autoimmune MRL/lpr-lpr mice. Autoimmunity 10: 15–25, 1991. doi: 10.3109/08916939108997143. [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez-Ramos JC, Andreu JL, Revilla Y, Viñuela E, Martinez C. Recovery from autoimmunity of MRL/lpr mice after infection with an interleukin-2/vaccinia recombinant virus. Nature 346: 271–274, 1990. doi: 10.1038/346271a0. [DOI] [PubMed] [Google Scholar]
- 20.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadaschik EN, Wei X, Leiss H, Heckmann B, Niederreiter B, Steiner G, Ulrich W, Enk AH, Smolen JS, Stummvoll GH. Regulatory T cell-deficient scurfy mice develop systemic autoimmune features resembling lupus-like disease. Arthritis Res Ther 17: 35, 2015. doi: 10.1186/s13075-015-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, Jin Y, Gan Y, Hu X, Jia R, Xu C, Hou Z, Leong YA, Zhu L, Feng J, An Y, Jia Y, Li C, Liu X, Ye H, Ren L, Li R, Yao H, Li Y, Chen S, Zhang X, Su Y, Guo J, Shen N, Morand EF, Yu D, Li Z. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med 22: 991–993, 2016. doi: 10.1038/nm.4148. [DOI] [PubMed] [Google Scholar]
- 23.Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, Kloke L, Heimann J, Gaber T, Brandenburg S, Scheffold A, Huehn J, Radbruch A, Burmester GR, Riemekasten G. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci USA 107: 204–209, 2010. doi: 10.1073/pnas.0903158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iikuni N, Lourenço EV, Hahn BH, La Cava A. Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol 183: 1518–1522, 2009. doi: 10.4049/jimmunol.0901163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishikawa S, Akakura S, Abe M, Terashima K, Chijiiwa K, Nishimura H, Hirose S, Shirai T. A subset of CD4+ T cells expressing early activation antigen CD69 in murine lupus: possible abnormal regulatory role for cytokine imbalance. J Immunol 161: 1267–1273, 1998. [PubMed] [Google Scholar]
- 26.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30: 531–564, 2012. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaname S, Uchida S, Ogata E, Kurokawa K. Autocrine secretion of transforming growth factor-beta in cultured rat mesangial cells. Kidney Int 42: 1319–1327, 1992. doi: 10.1038/ki.1992.423. [DOI] [PubMed] [Google Scholar]
- 28.Kang HK, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol 174: 3247–3255, 2005. doi: 10.4049/jimmunol.174.6.3247. [DOI] [PubMed] [Google Scholar]
- 29.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4+CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol 31: 2534–2542, 2011. doi: 10.1161/ATVBAHA.111.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119: 2904–2912, 2009. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 31.Lakkis FG, Baddoura FK, Cruet EN, Parekh KR, Fukunaga M, Munger KA. Anti-inflammatory lymphokine mRNA expression in antibody-induced glomerulonephritis. Kidney Int 49: 117–126, 1996. doi: 10.1038/ki.1996.16. [DOI] [PubMed] [Google Scholar]
- 32.Ling GS, Cook HT, Botto M, Lau YL, Huang FP. An essential protective role of IL-10 in the immunological mechanism underlying resistance vs. susceptibility to lupus induction by dendritic cells and dying cells. Rheumatology (Oxford) 50: 1773–1784, 2011. doi: 10.1093/rheumatology/ker198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528: 225–230, 2015. doi: 10.1038/nature16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17: 167–178, 2002. doi: 10.1016/S1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 35.Mathis KW, Taylor EB, Ryan MJ. Anti-CD3 antibody therapy attenuates the progression of hypertension in female mice with systemic lupus erythematosus. Pharmacol Res 120: 252–257, 2017. doi: 10.1016/j.phrs.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathis KW, Venegas-Pont M, Flynn ER, Williams JM, Maric-Bilkan C, Dwyer TM, Ryan MJ. Hypertension in an experimental model of systemic lupus erythematosus occurs independently of the renal nerves. Am J Physiol Regul Integr Comp Physiol 305: R711–R719, 2013. doi: 10.1152/ajpregu.00602.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension 59: 673–679, 2012. doi: 10.1161/HYPERTENSIONAHA.111.190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathis KW, Venegas-Pont M, Masterson CW, Wasson KL, Ryan MJ. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am J Physiol Regul Integr Comp Physiol 301: R1281–R1285, 2011. doi: 10.1152/ajpregu.00386.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension 64: 792–800, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol 13: 167–179, 2016. doi: 10.1038/nrcardio.2015.169. [DOI] [PubMed] [Google Scholar]
- 41.Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Nochy D, Debré P, Piette JC, Gorochov G. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol 175: 8392–8400, 2005. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- 42.Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, Tisch R, Tsokos GC. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4-CD8- IL-17-producing T cells. J Immunol 193: 2168–2177, 2014. doi: 10.4049/jimmunol.1400977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 193: 1007–1008, 1976. doi: 10.1126/science.181845. [DOI] [PubMed] [Google Scholar]
- 44.Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, Liew FY. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol 37: 3021–3029, 2007. doi: 10.1002/eji.200737810. [DOI] [PubMed] [Google Scholar]
- 45.Nozaki Y, Yamagata T, Yoo BS, Sugiyama M, Ikoma S, Kinoshita K, Funauchi M, Kanamaru A. The beneficial effects of treatment with all-trans-retinoic acid plus corticosteroid on autoimmune nephritis in NZB/WF mice. Clin Exp Immunol 139: 74–83, 2005. doi: 10.1111/j.1365-2249.2005.02654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, Sansom DM. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332: 600–603, 2011. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sadlack B, Löhler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, Horak I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol 25: 3053–3059, 1995. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- 48.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol 177: 1451–1459, 2006. doi: 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 49.Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol 43: 32–38, 2016. doi: 10.1016/j.coi.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sureshbabu A, Muhsin SA, Choi ME. TGF-β signaling in the kidney: profibrotic and protective effects. Am J Physiol Renal Physiol 310: F596–F606, 2016. doi: 10.1152/ajprenal.00365.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 268: 1472–1476, 1995. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 52.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand [A] 84: 523–528, 1976. doi: 10.1111/j.1699-0463.1976.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 53.Svendsen UG. Spontaneous hypertension and hypertensive vascular disease in the NZB strain of mice. Acta Pathol Microbiol Scand [A] 85: 548–554, 1977. doi: 10.1111/j.1699-0463.1977.tb03887.x. [DOI] [PubMed] [Google Scholar]
- 54.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28: 687–697, 2008. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor EB, Barati MT, Powell DW, Turbeville HR, Ryan MJ. Plasma cell depletion attenuates hypertension in an experimental model of autoimmune disease. Hypertension 71: 719–728, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor EB, Ryan MJ. Immunosuppression with mycophenolate mofetil attenuates hypertension in an experimental model of autoimmune disease. J Am Heart Assoc 6: e005394, 2017. doi: 10.1161/JAHA.116.005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor EB, Ryan MJ. Understanding mechanisms of hypertension in systemic lupus erythematosus. Ther Adv Cardiovasc Dis 2016: 1753944716637807, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Testi R, D’Ambrosio D, De Maria R, Santoni A. The CD69 receptor: a multipurpose cell-surface trigger for hematopoietic cells. Immunol Today 15: 479–483, 1994. doi: 10.1016/0167-5699(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 59.Venegas-Pont M, Mathis KW, Iliescu R, Ray WH, Glover PH, Ryan MJ. Blood pressure and renal hemodynamic responses to acute angiotensin II infusion are enhanced in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 301: R1286–R1292, 2011. doi: 10.1152/ajpregu.00079.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 8: 523–532, 2008. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, Enghard P, Sawitzki B, Hiepe F, Radbruch A, Burmester GR, Riemekasten G, Humrich JY. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 75: 1407–1415, 2016. doi: 10.1136/annrheumdis-2015-207776. [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Qian S, Li J, Che N, Gu L, Wang Q, Liu Y, Mei H. Combined transplantation of autologous hematopoietic stem cells and allogenic mesenchymal stem cells increases T regulatory cells in systemic lupus erythematosus with refractory lupus nephritis and leukopenia. Lupus 24: 1221–1226, 2015. doi: 10.1177/0961203315583541. [DOI] [PubMed] [Google Scholar]
- 63.Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 39: 537–545, 2002. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wirtz S, Billmeier U, Mchedlidze T, Blumberg RS, Neurath MF. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 141: 1875–1886, 2011. doi: 10.1053/j.gastro.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25− LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J Immunol 181: 6038–6050, 2008. doi: 10.4049/jimmunol.181.9.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 181: 2277–2284, 2008. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye C, Brand D, Zheng SG. Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther 3: 2, 2018. doi: 10.1038/s41392-017-0002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang J, Crowley SD. Role of T lymphocytes in hypertension. Curr Opin Pharmacol 21: 14–19, 2015. doi: 10.1016/j.coph.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-beta-producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol 183: 6346–6358, 2009. doi: 10.4049/jimmunol.0901773. [DOI] [PMC free article] [PubMed] [Google Scholar]