

Abstract
We have previously reported that complement activation precedes the development of kidney fibrosis; however, little is known about the cellular mechanisms involved in this transition. We hypothesized that increased expression of C1 complex protease C1r, the initiator of complement activation, contributes to tubulointerstitial fibrosis and tested this idea in mice with global deletion of C1r. Although expression of C1r in untreated wild-type (WT) mice was higher in the liver compared with kidney tissue, administration of folic acid (FA) led to upregulation of C1r mRNA and protein levels only in kidney tissue. Immunohistochemistry and in situ hybridization experiments localized increased expression of C1r and C1s proteases to renal tubular epithelial cells. C1r-null mice had reduced acute tubular injury and inflammation measured 2 days after FA administration compared with WT mice. C1r deletion reduced expression of C1s, C3 fragment formation, and organ fibrosis measured 14 days after FA administration. Differential gene expression performed in kidney tissue demonstrated that C1r-null mice had reduced expression of genes associated with the acute phase response, complement, proliferation of connective tissue cells (e.g., platelet-derived growth factor receptor-β), and reduced expression of genes associated with inflammation compared with FA-treated WT mice. In vitro experiments in renal epithelial cells demonstrated that C1s expression is dependent on increased C1r expression and that interferon-γ induces the expression of these two proteases. We conclude that increased expression of C1 complex proteases is associated with increased tissue inflammation and complement C3 formation and represents an important pathogenic mechanism leading to FA-mediated tubulointerstitial fibrosis.
Keywords: C1r, C1s, C3, renal epithelia
INTRODUCTION
The incidence of chronic kidney disease (CKD), defined as reduced glomerular filtration rate, is increasing, making CKD a public health priority (15). Current interventions to reduce the progression of kidney disease include strict control of blood pressure, correction of dyslipidemia, discontinuation of potentially nephrotoxic drugs, and blockade of the renin-angiotensin system (14). However, despite these interventions, none of these strategies has been shown to reverse progression to end-stage renal disease. Histological changes of tubulointerstitial fibrosis and glomerulosclerosis in kidney tissue represent the final common pathways leading to progression to end-stage renal disease (25).
The complement system, which is composed of >50 different molecules, plays a critical role in host defense and maintenance of normal tissue hemostasis (30). Although complement has a role in host defense and clearance of cell debris, recent studies have reported novel functions of complement activation in processes like embryogenesis, aging (12, 17), and development of CKD (28, 29).
Little is known about how cellular complement is activated in mouse kidney cells during injury; however, three mechanisms have been identified: the classical, lectin, and alternative pathways. While the classical pathway is triggered by antigen-antibody complexes via C1q, the lectin pathway is activated by pattern recognition molecules such as mannose-binding lectins, ficolins, and collectins to oligosaccharides. Activation of the alternative pathway occurs via spontaneous hydrolysis of an ester bond in C3 leading to the formation of C3H2O (8). All three pathways lead to the formation of C3 and C5, anaphylatoxin C3a and C5a, and other C3 fragments that activate various complement receptors. Although these three pathways of complement activation have been documented in the extracellular space, less is known about how intracellular complement activation occurs during kidney injury. Using two different models of kidney fibrosis, we previously reported that isolated platelet-derived growth factor receptor (PDGFR)-β-positive pericytes and macrophages synthesize intracellular complement components, including C1q, C3, C5, and anaphylatoxin receptors C3aR1 and C5aR1 and that the use of C3-null mice and macrophage depletion reduced fibrosis (35).
Given our previous observations that C1q was generated in kidney cells during fibrosis and that the use of C1q-null mice did not prevent the formation of complement C3 or fibrosis (35), in the present study, we examined the role of C1 complement protease C1r on kidney fibrosis.
MATERIALS AND METHODS
Mouse models of renal fibrosis.
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all procedures were approved by the University of Virginia Animal Care and Use Committee. Unilateral ureteral obstruction (UUO) was done in 8- to 10-wk-old male wild-type (WT) or C1r−/− mice. The left kidney was exposed through a midline incision under sterile conditions; the ureter was dissected and securely tied at two places. As control, sham surgery was performed the same as UUO without the ureter being tied. Mice were euthanized at day 7 after UUO. The folic acid (FA) model of fibrosis was performed by intraperitoneal injections of 250 mg/kg FA as previously described (35). To evaluate FA-mediated acute kidney injury (AKI), mice were euthanized at day 2 after FA injections, and plasma and tissues were collected for measurement of creatinine and RNA analysis. For fibrosis experiments, mice were euthanized at day 14. FA induces severe nephrotoxicity as assessed by histological examination (24).
In situ hybridization, immunohistochemistry, and immunofluorescence microscopy.
In situ hybridization was performed on 5-μm paraffin sections using a C1s antisense RNA probe labeled with digoxygenin. Sections were deparaffinized, rehydrated, and treated with proteinase K (10 μg/ml at 37°C). After hybridization at 65°C overnight with 1 μg/ml probe, sections were washed, and slides were incubated with anti-digoxygenin antibodies overnight at 4°C. Color was developed using nitro blue tetrazolium, and slides were washed and mounted using Cytoseal. Imaging was accomplished using a SPOT RT camera (software version 3.3, Diagnostic Instruments). Immunohistochemistry was performed as previously described using rabbit polyclonal anti-C1r antibody (1:400, Biorbyt). Immunofluorescence microscopy for aquaporin-2 was performed on paraffin sections using rabbit monoclonal aquaporin-2 (1:3,000, Abcam) and Alexa Fluor 488 goat anti-rabbit secondary antibody. Immunofluorescence microscopy for C3d and F4/80 was done on 6-μm frozen sections using goat polyclonal anti-mC3d antibody (1:20, R&D Systems) and phycoerythrin-conjugated anti-F4/80 (1:25, Clone BM8, Biolegend). Sections were mounted with ProLong Gold Antifade reagent with DAPI (Molecular Probes, Life Technologies). Images were acquired using the Carl Zeiss Axiovert 200 microscopy system with ApoTome imaging and Axiovision software (Carl Zeiss Microscopy).
Generation of C1r knockout mice.
C1ratm1.1 (KOMP)Vlcg mice were created from C1RA+/− sperm obtained from the Knockout Mouse Project (KOMP) Repository (www.KOMP.org). After in vitro fertilization, pups were genotyped for the presence of the mutant allele by tail genotype analysis. C1R mutant allele designed by KOMP carries the lacZ reporter under the control of the native promoter for C1R. The primers used for lacZ allele genotyping were forward 5′-ACTTGCTTTAAACCTCCCACA-3′ and reverse 5′-CCTGGGCTGTCCTGAGCTTT-3′; the primers used for the wild-type (WT) allele were forward 5′-CAGCCAGGAGACTAAGAAAATAACGAG-3′ and reverse 5′-TTAAAAGCCGCACTCAACTCAGC-3′. The size of the PCR products was 624 bp for the lacZ allele and 266 bp for the WT allele. Homozygote knockouts for C1RA were generated by mating mice heterozygous for C1RA. C1r deletion was confirmed by absence of C1r mRNA expression in total kidneys by real-time PCR analysis and Western blot analysis.
Plasma creatinine.
Whole blood was collected by cardiac puncture using heparinized syringes. Samples were spun at 13,000 rpm for 5 min at room temperature using a microtainer column to separate plasma from cells, separated into aliquots, and frozen immediately. Creatinine was measured using 8 µl sample with the Diazyme Creatinine assay kit (Diazyme Laboratories) following the manufacturer’s protocol, and creatinine was expressed in milligrams per deciliter.
X-gal staining.
Tissues were fixed with paraformaldehyde at 4°C for 1 h, washed in PBS at 4°C for 4 h, and equilibrated in 30% sucrose at 4°C overnight. Tissues were embedded in optimum cutting temperature compound and sectioned at 10 μm thickness. Frozen sections were placed in rinse buffer for 15 min at room temperature followed by incubation in X-gal staining solution for 3 h at 37°C. Slides were rinsed in PBS poststain, counterstained using nuclear fast red (Sigma-Aldrich), dehydrated, and mounted using Cytoseal. Slides were imaged using a SPOT RT camera (software version 3.3, Diagnostic Instruments).
Picrosirious red staining.
Kidneys embedded in paraffin were sectioned (5 µm) and stained as previously described (35).
Periodic acid-Schiff staining.
Kidneys embedded in paraffin were sectioned (5 µm) and stained with periodic acid-Schiff reagent.
RNA analysis.
Total RNA from kidney tissue was isolated, and real-time PCR was performed as previously described (35). Primer sequences are provided in Supplemental Table S1 (Supplemental data can be found at https://doi.org/10.6084/m9.figshare.9170981.v1.).
Western blot analysis.
Western blot analysis for C1r was performed under reducing conditions using mouse liver lysates (60 µg), mouse kidney lysates (75 µg), or serum (0.5 µl) from WT and C1r−/− mice using mouse C1r antibody (1:500, Santa Cruz Biotechnology). Human activated C1r (10 ng) purified from serum (Calbiochem) was used as a positive control. Western blot analysis for collagen type I was performed under reducing conditions using mouse kidney lysates (60 µg) from WT and C1r−/− mice using rabbit polyclonal anti-mouse collagen type I antibody (1:500, Millipore). GAPDH was used as a loading control. C3 fragments were detected as previously described (27, 35).
Microarray-based gene expression experiments.
Total RNA was isolated from snap-frozen whole kidney tissue from WT and C1r−/− mice (vehicle and FA treated) using the TRIzol method. RNA was cleaned using the RNA clean-up kit (Zymo Research). After evaluation of RNA quality and concentration, 3′ in vitro transcription and labeling was done using the GeneChip 3′ IVT plus reagent kit (ThermoFisher) and hybridized to a GeneChip Mouse genome 430A 2.0 array (ThermoFisher) following the manufacturer’s instructions as previously described (2). mRNA gene chips were scanned on a Gene Chip Scanner 3000 G7, and data were obtained. Before analyses, the array quality was assessed by examining 3′-to-5′ ratios for GAPDH and β-actin as previously described (2). Probeset expression summaries were then obtained from arrays that passed quality check using the robust multiarray average method. The Expression and Transcriptome Console was used to analyze the data.
Cell culture.
IMCD3 cells were obtained from the American Type Culture Collection and cultured in DMEM with high glucose supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were plated in six-well dishes and grown to confluence, serum starved overnight, and treated with interferon (IFN)-γ (4 ng/ml, Roche) for 6 h.
Overexpression of human C1r in renal epithelial cells in culture.
The HK-2 (CRL-2190) proximal tubule cell line obtained from the American Type Culture Collection was maintained in keratinocyte serum-free medium supplemented with bovine pituitary extract (0.5 mg/ml) and human recombinant EGF (5 ng/ml). Expression plasmids coding for human C1r or control plasmid were transfected using Lipofectamine 2000. Stably transfected cells were selected in the presence of G418 (600 µg/ml, Life Technologies). Overexpression for C1r was confirmed by real-time PCR and Western blot analysis of cell supernatants (100 µg).
Statistics.
All experiments were performed at least three times. All values are expressed as means + SD. Statistical analysis was performed using an unpaired Student’s t-test. P values of <0.05 were considered to be statistically significant.
RESULTS
Figure 1 shows the study design and measurements performed in whole kidney tissue using both FA and UUO models of fibrosis.
Fig. 1.
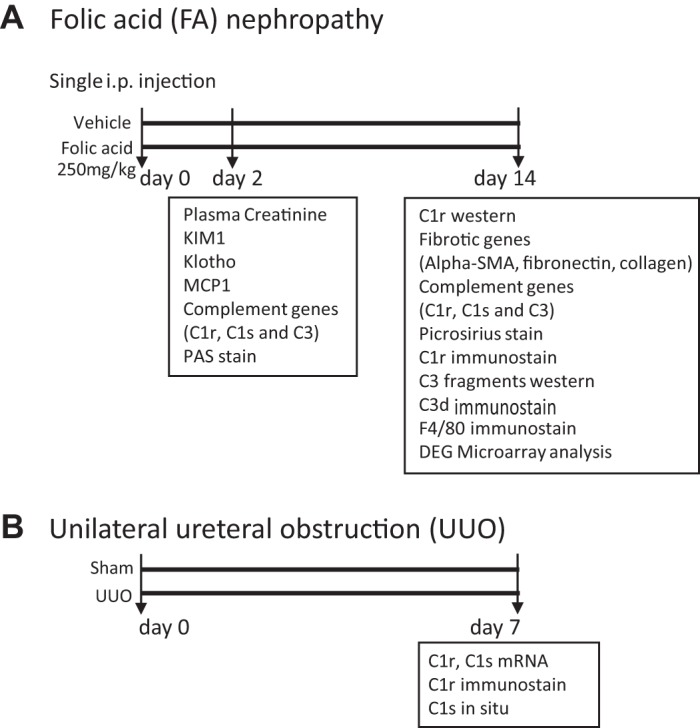
Study design and measurements performed in whole kidney tissue using both folic acid and unilateral ureteral obstruction models of fibrosis. KIM-1, kidney injury molecule-1; MCP-1, monocyte chemoattractant protein 1; PAS, periodic acid-Schiff; α-SMA, α-smooth muscle actin; DEG, differentially expressed gene.
C1r and C1s expression is increased in the distal tubular epithelium during fibrosis.
Kidney tissue C1r and C1s mRNA levels were significantly increased in FA-treated mice at days 2 and 14 compared with vehicle-treated mice (Fig. 1A). Expression of C1r and C1s mRNA was also induced in day 7 UUO mouse kidneys (Fig. 2B). Immunostaining for C1r protein in FA-treated mice at day 14 and UUO mice at day 7 was seen predominantly in dilated collecting duct (CD) cells and in a few interstitial cells, whereas no significant immunostaining was seen in vehicle-treated mice (Fig. 1C). To determine if increased expression of C1r occurs in CD cells, we performed immunostaining for aquaporin-2 (data not shown), which showed that not all of the dilated distal tubules that were positive for C1r in FA-treated mice were CD cells. As shown in Fig. 2D, in situ hybridization for C1s showed positive staining in cortical thick ascending limbs and dilated CDs in UUO kidneys at day 7 (Fig. 2D). To explore the potential contribution of C1r in renal fibrosis, we created mice with C1r ablation, which were used in the next set of experiments.
Fig. 2.

Induction of C1r/C1s in folic acid (FA)- and unilateral ureteral obstruction (UUO)-mediated fibrosis. A: real-time PCR analysis of whole kidney tissue demonstrating increased mRNA expression for both proteases at days 2 and 14 after FA injury (n = 6, *P < 0.01). B: real-time PCR analysis of whole kidney tissue demonstrating increased mRNA expression for C1r and C1s at UUO day 7 (n = 6, *P < 0.01). C: cellular localization of C1r was done in FA-treated mice by immunohistochemical analysis and demonstrated no detectable staining for C1r protein in kidney tissue of vehicle- or sham-treated mice. Increased expression of C1r protein after FA or UUO was observed in distal tubular epithelial cells, including collecting duct (CD) cells and in few interstitial cells (original magnification: ×400). D: digoxigenin-labeled in situ analysis using C1s antisense RNA probe was performed in sham and UUO kidneys. There was no detectable staining for C1s in kidney tissue of sham-treated mice. Increased expression of C1s after UUO was observed in dilated CDs and thick ascending loop of Henle cells (TALs) (original magnification: ×400).
Characterization of C1r-null mice.
The C1R gene targeting strategy used by KOMP and tail genotype analysis for WT heterozygous mice (C1r+/−) and C1r-null mice (C1r−/−) is shown in Fig. 3A. C1r−/− mice were viable, fertile, and had normal renal function compared with WT littermates. Because these mice carry the lacZ gene under the C1R promoter, we performed X-gal staining to identify C1r-expressing cells in the kidney. Figure 3B shows that X-gal-positive staining was seen primarily in the distal tubules and in very few proximal tubules, thus confirming our observations by immunohistochemistry staining using anti-C1r antibody. The absence of C1r expression in C1r−/− mice was validated by RT-PCR analysis in kidney homogenates (Fig. 3C). Surprisingly, mRNA expression of C1s was reduced significantly in kidney tissue of C1r−/− mice. Analysis of expression of C1r demonstrated the absence of C1r mRNA in all organ tissues of C1r−/− mice, whereas WT mice showed high levels of expression in the liver, spleen, heart, and kidney, as shown in Fig. 3D. C1r−/− mice had reduced expression of C1s in the liver and spleen, and C1s was completely absent in the kidney and heart tissue (Fig. 3D). Western blot analysis for C1r in mouse liver lysates and serum from WT and C1r−/− mice detected the 95-kDa protein corresponding to C1r only in WT mice but was absent both in liver tissue and serum of C1r−/− mice (Fig. 3E).
Fig. 3.

Generation of C1r knockout mice. A, top: scheme showing the gene targeting strategy by the Knockout Mouse Project (KOMP) to obtain disruption of exons 2−11 of the C1R gene. A, bottom: gel image of the tail genotype analysis demonstrating the presence of the wild-type (WT) allele using C1R gene-specific primers 1 and 2 as a 266-bp band. Using lacZ gene-specific primers 3 and 4, the C1R mutant allele was detected as a 624-bp fragment in either heterozygous C1r+/− or homozygous C1r−/− mice. B: X-gal staining performed in whole kidney tissue obtained from untreated WT mice did not show any stain; however, X-gal stain was present in distal tubules of C1r−/− mice (original magnification: ×5). C: kidney tissue expression of C1r was quantified by real-time PCR analysis. C1r was reduced in kidney tissue of heterozygous C1r+/− mice and completely absent in kidney tissue of C1r−/− compared with WT mice. C1s expression was also reduced in kidney tissue of heterozygous C1r+/− mice and significantly reduced in C1r−/− mice compared with WT mice (n = 5, *P < 0.001). D: comparison of C1r and C1s expression in various tissues obtained from WT and C1r−/− mice. E: Western blot analysis under reducing conditions for C1r protein in mouse liver tissue (60 µg) and serum (0.5 µl) from WT and C1r−/− mice showed the presence of a 95-kDa band only in WT mice. GAPDH was used as a loading control. Human purified C1r was used as positive control.
FA-induced AKI, inflammation, and fibrosis are reduced in C1r-null mice.
After 2 days, FA administration to WT mice caused a transient elevation in plasma creatinine that was accompanied by increased expression of the proximal tubule marker kidney injury molecule 1 (Kim-1), reduced Klotho expression, and increased expression of monocyte chemoattractant protein 1 (MCP-1). In contrast, C1r−/− mice treated with FA had significantly reduced plasma creatinine at day 2, with reduced expression of the tubular injury marker Kim-1 and no loss of expression of Klotho (Fig. 4, A and B). MCP-1 mRNA levels were also significantly reduced in C1r1−/− mice compared with WT mice. In addition, FA-mediated increased expression of complement genes C1r, C1s, and C3 in WT mice was also significantly reduced in C1r−/− mice (Fig. 4C). Tubular injury in WT mice treated with FA was evident, since dilated tubules containing proteinous casts were detected at day 2 by periodic acid-Schiff staining (Fig. 4D). In contrast, C1r−/− mice treated with FA were protected against tubular injury, since the tubular morphology was well preserved similar to vehicle-treated WT or C1r−/− mice.
Fig. 4.

Deletion of endogenous C1r reduces folic acid (FA)-induced acute kidney injury (AKI), tubular injury, and inflammation. A: plasma creatinine levels at day 2 after FA injection were reduced in C1r−/− mice (red circles) compared with wild-type (WT) mice (black circles). n = 8 WT mice and 4 C1r−/− mice for vehicle treatment; n = 8 WT mice and 6 C1r−/− mice for FA treatment. *P < 0.05. B: real-time PCR analysis of whole kidney tissue showing increased expression of the tubular injury marker kidney injury molecule-1 (Kim-1) and loss of expression of Klotho in WT mice (black circles), whereas C1r−/− mice (red circles) showed decreased expression of Kim-1 and no significant loss of Klotho 2 days after FA administration. Monocyte chemoattractant protein-1 (MCP-1) expression was also decreased in C1r−/− mice (*P < 0.01). C: real-time PCR analysis of whole kidney tissue for complement genes C1r, C1s, and C3 showed the absence of C1r in vehicle- and FA-treated C1r−/− mice (red circles) compared with WT mice (black circles). C1s and C3 expression was also significantly reduced both in vehicle- and FA-treated C1r−/− mice. n = 3 WT mice and 3 C1r−/− mice for vehicle treatment; n = 5–7 WT mice and 6 C1r−/− mice for FA treatment (**P < 0.005). D: images for periodic acid-Schiff (PAS) stain showing reduced tubular injury in FA-treated C1r−/− mice (original magnification: ×200).
FA-mediated fibrosis is reduced in C1r-null mice.
FA administration led to increased C1r protein levels in whole kidney lysates compared with kidney lysates of vehicle-treated WT mice (Fig. 5A). FA-induced increased expression of fibrotic genes α-smooth muscle actin and fibronectin in WT mice was significantly reduced in FA-treated C1r−/− mice (Fig. 5B). Collagen type I mRNA expression and protein were reduced in C1r−/− mice (Fig. 5C). In addition, FA-mediated increased expression of complement genes C1r, C1s, and C3 in WT mice was also significantly reduced in C1r−/− mice (Fig. 5D). These changes in gene expression in whole kidney tissue were associated with reduced collagen production detected by reduced picrosirius red staining and absence of C1r immunostaining in kidney tissue of C1r−/− mice (Fig. 5E), further indicating that C1r−/− mice are protected against FA-mediated kidney fibrosis. To address the effects of reduced expression of C1r on complement activation and formation of C3 fragments, we evaluated effects of FA-mediated C3 fragment formation in both WT and C1r-null mice. Western blot analysis for cleaved C3 fragments (C3b/iC3b/C3c; see Ref. 27) showed increased C3 protein fragments in FA-treated WT mice, which was significantly reduced in FA-treated C1r−/− mice (Fig. 5F). To further examine the effects of C1r deletion on complement activation, we performed immunohistochemical staining using C3d antibodies. There was a peritubular stain in vehicle-treated mice, whereas FA treatment showed increased tubulointerstitial stain, suggesting increased complement activation in interstitial cells. C1r-null mice had significantly reduced expression of C3d, as shown by reduced immunostaining in the tubulointerstitial compartment (Fig. 5G). Recruitment of macrophages after injury was also significantly reduced in C1r−/− mice, indicating reduced inflammation in C1r−/− mice after FA injury (Fig. 5H).
Fig. 5.

C1r-deficient mice show reduced fibrosis following folic acid (FA) injury at day 14. A: Western blot analysis for C1r showing the presence of a 95-kDa band only in wild-type (WT) FA-treated mice and absent in vehicle-treated WT mice and C1r−/− mice (n = 3, *P < 0.05, WT vehicle vs. FA). B: real-time PCR analysis of whole kidney tissue showing reduced expression of fibrotic marker genes α-smooth muscle actin (α-SMA), fibronectin, and collagen in C1r−/− mice (red circles) compared with WT mice (black circles) after FA injury. n = 6–8 WT mice and 5–7 C1r−/− mice for vehicle treatment; n = 7–8 WT mice 8–10 C1r−/− mice for FA treatment. *P < 0.05. C: real-time PCR and Western blot analysis for collagen type IA showing reduced expression of collagen type I in C1r−/− mice after FA injury (n = 3, *P < 0.01). D: images for picrosirious red staining showing reduced fibrosis in C1r−/− mice. Quantitation of fibrosis is shown (n = 7, **P < 0.001), and immunohistochemistry for C1r in WT and C1r−/− FA-treated mouse kidneys showed an absence of C1r stain in C1r−/− mice (original magnification: ×400). E: real-time PCR analysis of whole kidney tissue for complement genes C1r, C1s, and C3 showed an absence of C1r in vehicle- and FA-treated C1r−/− mice (red circles) compared with WT mice (black circles). C1s and C3 expression was also significantly reduced both in vehicle- and FA-treated C1r−/− mice. n = 6–8 WT mice and C1r−/− mice for vehicle treatment; n = 4–6 WT mice and 5–7 C1r−/− mice for FA treatment. **P < 0.005. F: native gel Western blot analysis for C3 active fragments (C3b, iC3b, and C3c) showed reduced C3 fragments in FA-treated C1r−/− mice (n = 3, *P < 0.05 for WT vehicle-treated vs. FA-treated mice). G: images for C3d staining showing reduced immunostain for C3d in FA-treated C1r−/− mice (original magnification: ×200). H: images for F4/80 staining showing reduced infiltration of macrophages in FA-treated C1r−/− mice (original magnification: ×200).
Comparison of differentially expressed genes between C1r-null and WT mice validates the cytoprotective role of reducing C1r expression during fibrosis.
We first compared differentially expressed genes (DEGs) between untreated C1r−/− and WT mice. A total of 341 genes (377 probe sets) were differentially expressed between the two groups (fold change ≥ 1.5, P < 0.05). Of these genes, 235 and 107 genes were up- and downregulated, respectively, in C1r−/− mouse kidneys compared with WT mouse kidneys. DEGs were analyzed using Ingenuity Pathway Analysis (IPA) software, and one of the top canonical pathways that was predicted to be activated in C1r−/− mouse kidneys (z score: ≥2) was the peroxisome proliferator-activated receptor (PPAR) signaling pathway (z score: 2.36, P = 0.014; Fig. 6A). Other pathways that were significantly inhibited in C1r−/− mice included the acute phase response signaling (z score: −1.342, P = 1.0965 × 10−5) and complement (z score: −0.447, P = 2.2387 × 10−4) system (Fig. 5B). Analysis by disease function showed that the top downregulated genes in C1r−/− mice also included those associated with inhibition of proliferation of connective tissue cells (WNT5A, PDGFR-α, PDGFR-β, IGF-1, KL3, cysteine-rich angiogenic inducer 61, and colony-stimulating factor 1 receptor, z score: −2.412, P = 4.66 × 10−4), as shown in Fig. 6B and Supplemental Table S2.
Fig. 6.

Differential gene expression pattern between wild-type (WT) and C1r−/− mouse kidney tissue. A: differentially expressed genes (fold change ≥ 1.5, P ≤ 0.05) analyzed using Ingenuity Pathway Analysis software showng canonical pathways with positive and negative z scores. The peroxisome proliferator-activated receptor (PPAR) signaling pathway was significantly upregulated and complement system significantly downregulated in C1r−/− mice compared with WT mice. The canonical pathways tab displays the most significant canonical pathways across the entire data set. The significance values for the canonical pathways were calculated by Fisher's exact test (right-tailed). The significance indicates the probability of association of molecules from the current data set with the canonical pathway by random chance alone. The orange- and blue-colored bars indicate predicted pathway activation or predicted inhibition, respectively (z score). The intensity of the colors (from light to dark orange and blue) indicates values of the z score for each bar associated with a unique pathway (from lower to higher). The significant canonical pathways for the data set that are involved in the analysis are displayed along the x-axis. The y-axis displays the −log of the P value that was calculated by a Fisher's exact test (right-tailed) such that taller bars equate to increased significance. The orange points connected by a thin orange line represent the ratio. The ratio was calculated as follows: number of genes in a given pathway that meet your cutoff criteria ÷ the total number of genes that make up that pathway and that are in the reference gene set. B: analysis of disease functions showing inhibition of proliferation of connective tissue cells and the genes downregulated or upregulated in C1r−/− mice compared with WT mice. NFTA, nuclear factor of activated T cells; HIV, human immunodeficiency virus; IGF-I, insulin-like growth factor-I; LXR/RXR, liver X receptor/retinoid X receptor; GP6, glycoprotein VI; PAK, p21-activated kinases; PPRAα/RXRα, peroxisome proliferator-activated receptor/retinoid X receptor; NFR2, nuclear factor erythroid 2-related factor 2; IL-8, interleukin-8; PTEN, phosphatase and tensin homolog; CXCr4, chemokine (C-X-C motif) receptor 4; AMPK, AMP-activated protein kinase; eNOS, endothelial nitric oxide synthase.
We also compared DEGs between FA-treated C1r−/− and WT mice. Upon comparison, 518 probe sets corresponding to 416 genes were differentially expressed with a fold change of ≥1.5 and P < 0.05. Among these genes, 364 genes were downregulated, whereas the remaining genes were upregulated, in FA-treated C1r−/− mice compared with FA-treated WT mice. DEGs analyzed by IPA software showed that the most significant canonical pathways were those related to the immune response like dendritic cell maturation (z score: −4.025, P = 3.2359 × 10−10), triggering receptor expressed on myeloid cells 1 signaling (z score: −3.162, P = 8.51138 × 10−7), acute phase response signaling (z score: −2.673, P = 1.09648 × 10−8), IL-6 signaling pathway (z score: −2.496, P = 7.07946 × 10−8), and leukocyte extravasation signaling (z score: −2.138, P = 1.31826 × 10−6). These pathways were predicted to be inhibited in FA-treated C1r−/− mice compared with FA-treated WT mice (Fig. 7A). Further analysis of disease functions showed inhibition of functions related to immune cell trafficking, cell-cell signaling interactions, and the inflammatory response. Inhibition of injury to the kidney (z score: −3.342, P = 1.91 × 10−12) was seen in FA-treated C1r−/− mice compared with FA-treated WT mice, including downregulated genes like C3, lysozyme, Toll-like receptor4, chemokine (C-C motif) ligand 2, Fc fragment of IgG receptor IIa, chemokine (C-C motif) receptor 2, and CD44 (Fig. 7B and Supplemental Table S3). RT-PCR analysis demonstrated significantly reduced expression of IL-1β, IL-6, MCP-1, and macrophage inflammatory protein-1α in FA-treated C1r−/− mice (Fig. 7C), supporting the data provided by microarray analysis.
Fig. 7.

Differential gene expression pattern between folic acid (FA)-treated wild-type (WT) and C1r−/− mouse kidney tissue. A: differentially expressed genes (with fold change cutoff of 1.5 and P value cutoff of 0.05) analyzed using Ingenuity Pathway Analysis software showing canonical pathways with positive and negative z scores. Inflammatory pathways were significantly downregulated with upregulation of peroxisome proliferator-activated receptor (PPAR) signaling in FA-treated C1r−/− mice compared with WT mice. B: analysis of disease functions showed inhibition of injury of kidney and the genes downregulated in FA-treated C1r−/− mice compared with WT mice. C: real-time PCR analysis of whole kidney tissue showing reduced expression of inflammatory genes IL-1β, IL-6, monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein (MIP)-1α in FA-treated C1r−/− mice (red circles) compared with WT mice (black circles) (n = 5–7 for WT and C1r−/− mice, *P < 0.05). TREM1, triggering receptor expressed on myeloid cells 1; HMGB1, high-mobility group box 1 protein; VDR, vitamin D receptor; RXR, retinoid acid receptor; ILK, integrin-linked kinase; MIF, macrophage migration inhibitory factor; TEC, Tec protein tyrosine kinase; P38 MAPK, p38 mitogen-activated protein kinase; NFAT, nuclear factor of activated T cells; PI3K, phosphoinositide 3-kinase; GP6, glycoprotein VI; iNOS, inducible nitric oxide synthase; ERK5, extracellular signal-regulated kinase 5; Rho GTPase, Ras homologous GTPase; RhoGDI, Ras homologous GDP dissociation inhibitors.
IFN-γ increases C1r expression: dependency of C1s expression on C1r.
We next sought to examine the mechanisms by which C1r and C1s expression could be increased during injury using an in vitro system of renal epithelial cells in culture. Because we demonstrated that C1r expression in mouse kidney tissue occurs primarily in distal tubular cells, including the CD, we performed experiments using the mouse inner medullary CD cell line (IMCD3 cells). We first tested whether the presence of an IFN-γ regulatory factor-1 element in the promoter region of mouse C1R could account for its increased expression during kidney fibrosis (4). IMCD3 cells in culture treated with IFN-γ had increased mRNA expression of both C1r and C1s (Fig. 8A), suggesting that increased IFN-γ could account for its increased expression during fibrosis.
Fig. 8.

Regulation of expression of C1r/C1s in renal epithelial cells by interferon (IFN)-γ and dependency of C1s expression on C1r. A: real-time PCR analysis of mIMCD3 cells treated with IFN-γ (4 ng/ml) showing increased expression of both C1r and C1s mRNA (n = 3, *P < 0.05). B: real-time PCR analysis for C1r and C1s in HK-2 cells stably transfected with human C1r showing increased expression of C1r that was accompanied by increased expression of C1s (n = 3, *P < 0.05). C: Western blot analysis under reducing conditions of HK-2 cell supernatants (100 µg) stably transfected with human C1r showing the presence of autoactivated 60-kDa C1r fragments only in C1r-transfected cells, which was not present in vector-transfected cells. Human purified C1r was used as a positive control.
We also generated human C1r stable transfectants using human renal epithelial cells of proximal tubule origin (HK-2 cells). Increased expression of C1r was associated with increased expression of C1s in C1r transfected cells (Fig. 8B). These in vitro data using human C1r confirms the observations in vivo in mouse kidney tissue where we showed that C1s expression was dependent on C1r. Increased expression of C1r protein in stably transfected HK-2 cells was confirmed by Western blot analysis of cell supernatants, demonstrating the presence of the 60-kDa autoactivation fragment (Fig. 8C).
DISCUSSION
The present study expands previous observations from our laboratory with regard to the contribution of complement activation to kidney fibrosis. More specifically, we show that 1) increased synthesis of complement serine protease C1r by renal epithelial cells associates with increased fibrosis, 2) C1r-null mice had reduced kidney fibrosis, and 3) C1r-null mice showed reduced AKI, reduced C3 fragment formation, and reduced cytokine production in the model of FA-induced nephropathy, suggesting that increased inflammation and complement activation represent important mechanisms contributing to tubulointerstitial fibrosis.
C3 formation during tubulointerstitial fibrosis requires increased expression of C1 complex proteases C1r and C1s.
We have previously reported increased synthesis of C1q in PDGFR-β-positive pericytes and CD45+ F4/80+ immune cells; however, the use of global C1q-null mice did not prevent the increase of C1r, C1s, and C3 expression in whole kidney tissue and did not affect fibrosis (35). Our present findings showing that reduced expression of C1r and C1s is cytoprotective in the FA model of tubulointerstitial fibrosis, suggesting that increased expression of C1 complex proteases in renal epithelial cells during injury could have a noncanonical role in the development of inflammation and fibrosis, independent of their well-appreciated role in the C1qC1rC1s complex, as previously reported for other complement components (3, 17, 33). In recent years, several noncomplement targets and activators of C1 complex have been described. C1s protease has been shown to cleave a list of noncomplement proteins that include low-density lipoprotein receptor-related protein 6, major histocompatibility complex class 1, IGF-I-binding protein 5, nucleophosmin 1, high mobility group box 1, and nucleolin (26).
A previous study by Lee et al. (22) supports our observation regarding the cell origin of these two serine proteases. These investigators used RNA sequencing of microdissected nephron segments of healthy rats and mice and demonstrated the presence of C1r and C1s transcripts predominantly in the distal tubule, including the CD and thick ascending loop of Henle (13, 22). To explore the pathophysiological role of increased C1r/C1s expression in kidney fibrosis, we developed mice with genetic deficiency of C1r (Fig. 2). Adult C1r−/− mice had normal growth, normal kidney histology, and normal renal function. Our results show that lack of C1r expression in kidney tissue ameliorates FA-induced renal tubular epithelial cell injury measured by reduced Kim-1 expression and increased Klotho expression, resulting in improved kidney function measured at 2 days after FA administration, as shown in Fig. 4.
Our study supports the notion that increased expression of C1 proteases associates with increased formation of C3 in kidney tissue. C3 is the major complement component expressed in kidney tissue. Previous studies have suggested that epigenetic changes, including increased histone methylation of the C3 gene during ischemia-reperfusion injury to the kidney, could account for increased expression of C3 mRNA levels (31). Our study suggests a direct relationship between C1 complex protease C1r, C1s, and C3 formation, since mice with C1r deletion had reduced C3 formation during FA-mediated tubulointerstitial fibrosis. In contrast, we did not consistently observe reduced C3 formation or reduced inflammation and fibrosis in C1r-null mice subjected to UUO (data not shown). A potential explanation for this difference could be that, during mechanical ligation of the ureter, activation of alternative pathway of complement could represent the major mechanism by which C3 formation occurs rather than just activation of the classical pathway. Differences in gene expression pathways associated with inflammation have been previously reported in these two models of tubulointerstitial fibrosis (7).
Evidence for the protective role of inhibition of the classical pathway during ischemia-reperfusion injury was also recently demonstrated (5). These investigators show that the use of C1 inhibitor (C1-INH), which targets not only classical but also the lectin pathway, was able to reduce pericyte to myofibroblast transformation (5). More recently, the use of a C1s inhibitor in patients with bullous pemphigo showed reduced complement activation, reduced C3 fragment formation, and reduced skin inflammation (9).
DEGs in kidney tissue confirm that reduced expression of complement components and inflammation represent important mechanisms by which C1r-null mice have reduced fibrosis.
Our gene array data further show that untreated C1r−/− mice had reduced complement components, including C3, a reduced acute inflammatory response, and reduced proliferation of connective tissue cells. This is of significance in view of the recent identification of human C1r and C1s mutations in individuals with periodontal Ehlers-Danlos syndrome (16). On the other hand, when we analyzed FA-treated WT mice versus C1r−/− mice, several canonical pathways of inflammation, oxidative injury, and tubular toxicity were significantly reduced in C1r−/− mice. Of interest, the PPAR pathway, known to be cytoprotective, was also upregulated in C1r−/− mice. Our previous work in AKI and renal fibrosis models demonstrated the protective role of increasing mitochondrial fatty acid oxidation via increased PPAR-α expression (6, 23). These findings suggest that progressive inflammation and potential changes in the epithelial and macrophage phenotype could be effectively reduced by C1r deficiency.
Increased formation of IFN-γ could represent a mechanism by which C1r and C1s expression is increased during tubulointerstitial fibrosis.
Our findings support the role of inflammation as a potential mechanism by which expression of kidney proteases C1r and C1s are increased during kidney injury. C1r and C1s are modular proteins each consisting of two CUB domains separated by an EGF-like domain followed by two CCP modules and a serine protease domain (1, 10). C1r dimerizes through interactions between the serine protease domain of one polypeptide and the CCP domains of its partner (19, 20). In the presence of calcium, each chain binds to a C1s polypeptide via the CUB1-EGF domains to form a C1rs tetramer (C1s-C1r-C1r-C1s). When C1 binds to a cellular target via its globular heads, C1r activates through autocleavage. C1r then cleaves and activates C1s (11). A previous study (21) has suggested that increased production of IFN-γ by immune cells such as natural killer T cells contributes to renal fibrosis. Our cell culture experiments using mouse IMCD3 cells support the role of IFN-γ as a potential stimulus to increase C1r and C1s expression in vivo; however, additional studies are needed to prove this. Recent work suggests that increased expression of complement in human T cells is associated with increased T helper 1 responses, including increased production of IFN-γ, which, in turn, perpetuates the inflammatory response by increasing production of C1 complex proteases during kidney injury (18, 34). In addition, our gain-of-function experiments using stable transfection of human C1r cDNA in renal epithelial cells in culture demonstrate that C1r is needed to increase expression of C1s and that, upon increased expression, human C1r is autocleaved intracellularly before it is secreted (32). Taken together, our results suggest that there is transcriptional dependence of C1s mediated by changes in the expression of C1r. Further analysis of the C1s gene promoter could possibly reveal which transcriptional elements/factors are involved in this unique regulation.
Conclusions
To the best of our knowledge, this is the first study to suggest that, in addition to C1q, C1 complex proteases C1r and Cs are involved in the development of both inflammation and renal scarring. Further studies are needed to further define the mechanisms and cell types by which complement activation leads to fibrosis, which would enhance our understanding of the mechanisms of progressive kidney disease. In conclusion, our study supports the role of C1r in kidney fibrosis and identifies C1r as a novel potential therapeutic target for the treatment of progressive kidney disease.
GRANTS
This work was supported by National Institutes of Health (NIH) Grant RO1-DK-75976 and a Veterans Affairs Merit Award (to D. Portilla). NIH Grants to Velocigene at Regeneron (U01-HG-004085) and the Complementary Sex Determiner Consortium (U01-HG-004080) funded the generation of gene-targeted embryonic stem cells for 8,500 genes in the Knockout Mouse Project (KOMP) Program, which were archived and distributed by the KOMP Repository at the University of California-Davis and Children’s Hospital Oakland Research Institute (U42-RR-024244).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.X., R.K.S., S.V.B., and D.P. performed experiments; S.X., R.K.S., S.V.B., V.M., and D.P. analyzed data; S.X., R.K.S., S.V.B., and D.P. interpreted results of experiments; S.X., R.K.S., S.V.B., and D.P. prepared figures; S.X., R.K.S., and D.P. drafted manuscript; S.X., R.K.S., R.P.T., and D.P. edited and revised manuscript; S.X., R.K.S., S.V.B., V.M., R.P.T., J.M., N.M.T., and D.P. approved final version of manuscript; D.P. conceived and designed research.
ACKNOWLEDGMENTS
We thank Susan Landes and Rebekka Groebner for technical assistance. We thank Dr. Albert Amberger (Medical University of Innsbruck) for providing the C1r expression plasmids. We acknowledge Wenhao Xu (University of Virginia) and the Genetically Engineered Murine Model core for technical assistance in generating C1r-null mice. C1RA+/− sperm used for this research project was generated by the trans-NIH Knockout Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org).
REFERENCES
- 1.Arlaud GJ, Thielens NM, Illy C. Arrangement of the C1 complex of complement. Biochem Soc Trans 18: 1148–1151, 1990. doi: 10.1042/bst0181148. [DOI] [PubMed] [Google Scholar]
- 2.Bontha SV, Maluf DG, Archer KJ, Dumur CI, Dozmorov MG, King AL, Akalin E, Mueller TF, Gallon L, Mas VR. Effects of DNA methylation on progression to interstitial fibrosis and tubular atrophy in renal allograft biopsies: a multi-omics approach. Am J Transplant 17: 3060–3075, 2017. doi: 10.1111/ajt.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bröker K, Figge J, Magnusen AF, Manz RA, Köhl J, Karsten CM. A novel role for C5a in B-1 cell homeostasis. Front Immunol 9: 258, 2018. doi: 10.3389/fimmu.2018.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byun SJ, Jeon IS, Lee H, Kim TY. IFN-gamma upregulates expression of the mouse complement C1rA gene in keratinocytes via IFN-regulatory factor-1. J Invest Dermatol 127: 1187–1196, 2007. doi: 10.1038/sj.jid.5700660. [DOI] [PubMed] [Google Scholar]
- 5.Castellano G, Franzin R, Stasi A, Divella C, Sallustio F, Pontrelli P, Lucarelli G, Battaglia M, Staffieri F, Crovace A, Stallone G, Seelen M, Daha MR, Grandaliano G, Gesualdo L. Complement activation during ischemia/reperfusion injury induces pericyte-to-myofibroblast transdifferentiation regulating peritubular capillary lumen reduction through pERK signaling. Front Immunol 9: 1002, 2018. doi: 10.3389/fimmu.2018.01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, Li J, Tran PT, Kaimal V, Huang X, Chang AN, Li S, Kalra A, Grafals M, Portilla D, MacKenna DA, Orkin SH, Duffield JS. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 4: 121ra18, 2012. doi: 10.1126/scitranslmed.3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craciun FL, Bijol V, Ajay AK, Rao P, Kumar RK, Hutchinson J, Hofmann O, Joshi N, Luyendyk JP, Kusebauch U, Moss CL, Srivastava A, Himmelfarb J, Waikar SS, Moritz RL, Vaidya VS. RNA sequencing identifies novel translational biomarkers of kidney fibrosis. J Am Soc Nephrol 27: 1702–1713, 2016. doi: 10.1681/ASN.2015020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest 127: 970–981, 2017. doi: 10.1172/JCI89412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freire PC, Muñoz CH, Derhaschnig U, Schoergenhofer C, Firbas C, Parry GC, Panicker S, Gilbert JC, Stingl G, Jilma B, Heil PM. Specific inhibition of the classical complement pathway prevents C3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol S0022-202X(19)31783-X, 2019. doi: 10.1016/j.jid.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 10.Gaboriaud C, Ling WL, Thielens NM, Bally I, Rossi V. Deciphering the fine details of c1 assembly and activation mechanisms: “mission impossible”? Front Immunol 5: 565, 2014. doi: 10.3389/fimmu.2014.00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garnier G, Circolo A, Xu Y, Volanakis JE. Complement C1r and C1s genes are duplicated in the mouse: differential expression generates alternative isomorphs in the liver and in the male reproductive system. Biochem J 371: 631–640, 2003. doi: 10.1042/bj20021555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol 18: 1288–1298, 2017. doi: 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huling JC, Pisitkun T, Song JH, Yu MJ, Hoffert JD, Knepper MA. Gene expression databases for kidney epithelial cells. Am J Physiol Renal Physiol 302: F401–F407, 2012. doi: 10.1152/ajprenal.00457.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaber BL, Madias NE. Progression of chronic kidney disease: can it be prevented or arrested? Am J Med 118: 1323–1330, 2005. doi: 10.1016/j.amjmed.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 15.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW. Chronic kidney disease: global dimension and perspectives. Lancet 382: 260–272, 2013. doi: 10.1016/S0140-6736(13)60687-X. [DOI] [PubMed] [Google Scholar]
- 16.Kapferer-Seebacher I, Pepin M, Werner R, Aitman TJ, Nordgren A, Stoiber H, Thielens N, Gaboriaud C, Amberger A, Schossig A, Gruber R, Giunta C, Bamshad M, Björck E, Chen C, Chitayat D, Dorschner M, Schmitt-Egenolf M, Hale CJ, Hanna D, Hennies HC, Heiss-Kisielewsky I, Lindstrand A, Lundberg P, Mitchell AL, Nickerson DA, Reinstein E, Rohrbach M, Romani N, Schmuth M, Silver R, Taylan F, Vandersteen A, Vandrovcova J, Weerakkody R, Yang M, Pope FM, Byers PH, Zschocke J, Aleck K, Banki Z, Dudas J, Dumfahrt H, Haririan H, Hartsfield JK, Kagen CN, Lindert U, Meitinger T, Posch W, Pritz C, Ross D, Schroer RJ, Wick G, Wildin R, Wilflingseder D; Molecular Basis of Periodontal EDS Consortium . Periodontal Ehlers-Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Genet 99: 1005–1014, 2016. doi: 10.1016/j.ajhg.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemper C, Köhl J. Back to the future-non-canonical functions of complement. Semin Immunol 37: 1–3, 2018. doi: 10.1016/j.smim.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolev M, West EE, Kunz N, Moseman A, Balmer ML, Dimeloe S, Rosser E, Wedderburn LR, Lavender P, Cope A, Moutsopoulos N, Kaplan MJ, Holland SM, McGavern D, Hess C, Kemper C. Tissue extravasation/LFA-1 induces complement C3-licensing required for successful Th1 responses. Mol Immunol 102: 173, 2018. doi: 10.1016/j.molimm.2018.06.119. [DOI] [Google Scholar]
- 19.Lacroix M, Ebel C, Kardos J, Dobó J, Gál P, Závodszky P, Arlaud GJ, Thielens NM. Assembly and enzymatic properties of the catalytic domain of human complement protease C1r. J Biol Chem 276: 36233–36240, 2001. doi: 10.1074/jbc.M105688200. [DOI] [PubMed] [Google Scholar]
- 20.Lacroix M, Rossi V, Gaboriaud C, Chevallier S, Jaquinod M, Thielens NM, Gagnon J, Arlaud GJ. Structure and assembly of the catalytic region of human complement protease C1r: a three-dimensional model based on chemical cross-linking and homology modeling. Biochemistry 36: 6270–6282, 1997. doi: 10.1021/bi962719i. [DOI] [PubMed] [Google Scholar]
- 21.Law BMP, Wilkinson R, Wang X, Kildey K, Lindner M, Rist MJ, Beagley K, Healy H, Kassianos AJ. Interferon-γ production by tubulointerstitial human CD56bright natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int 92: 79–88, 2017. doi: 10.1016/j.kint.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Mariappan N, Megyesi J, Shank B, Kannan K, Theus S, Price PM, Duffield JS, Portilla D. Proximal tubule PPARα attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol 305: F618–F627, 2013. doi: 10.1152/ajprenal.00309.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long DA, Woolf AS, Suda T, Yuan HT. Increased renal angiopoietin-1 expression in folic acid-induced nephrotoxicity in mice. J Am Soc Nephrol 12: 2721–2731, 2001. [DOI] [PubMed] [Google Scholar]
- 25.López-Novoa JM, Rodríguez-Peña AB, Ortiz A, Martínez-Salgado C, López Hernández FJ. Etiopathology of chronic tubular, glomerular and renovascular nephropathies: clinical implications. J Transl Med 9: 13, 2011. doi: 10.1186/1479-5876-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Kishore U. C1 complex: an adaptable proteolytic module for complement and non-complement functions. Front Immunol 8: 592, 2017. doi: 10.3389/fimmu.2017.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mastellos D, Prechl J, László G, Papp K, Oláh E, Argyropoulos E, Franchini S, Tudoran R, Markiewski M, Lambris JD, Erdei A. Novel monoclonal antibodies against mouse C3 interfering with complement activation: description of fine specificity and applications to various immunoassays. Mol Immunol 40: 1213–1221, 2004. doi: 10.1016/j.molimm.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Mathern DR, Heeger PS. Molecules great and small: the complement system. Clin J Am Soc Nephrol 10: 1636–1650, 2015. doi: 10.2215/CJN.06230614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng Q, Li K, Smyth LA, Xing G, Wang N, Meader L, Lu B, Sacks SH, Zhou W. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 1474–1485, 2012. doi: 10.1681/ASN.2011111072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol 190: 3839–3847, 2013. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodríguez-Romo R, Berman N, Gómez A, Bobadilla NA. Epigenetic regulation in the acute kidney injury to chronic kidney disease transition. Nephrology (Carlton) 20: 736–743, 2015. doi: 10.1111/nep.12521. [DOI] [PubMed] [Google Scholar]
- 32.Rossi V, Bally I, Lacroix M, Arlaud GJ, Thielens NM. Classical complement pathway components C1r and C1s: purification from human serum and in recombinant form and functional characterization. Methods Mol Biol 1100: 43–60, 2014. doi: 10.1007/978-1-62703-724-2_4. [DOI] [PubMed] [Google Scholar]
- 33.Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: a fresh look upon an old molecule. Mol Immunol 89: 73–83, 2017. doi: 10.1016/j.molimm.2017.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West EE, Afzali B, Kemper C. Unexpected roles for intracellular complement in the regulation of Th1 responses. Adv Immunol 138: 35–70, 2018. doi: 10.1016/bs.ai.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Xavier S, Sahu RK, Landes SG, Yu J, Taylor RP, Ayyadevara S, Megyesi J, Stallcup WB, Duffield JS, Reis ES, Lambris JD, Portilla D. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am J Physiol Renal Physiol 312: F516–F532, 2017. doi: 10.1152/ajprenal.00604.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]