

Abstract
Tuberculosis is a bacterial disease caused by Mycobacterium tuberculosis. It is known to be the second-largest cause of death and models a severe risk to public health throughout the world. Though it affects people of almost every age, individuals with weakened immune systems, (e.g., HIV infection) are more likely to get infected. The present study deals with analyzing non-synonymous mutations in anti-tuberculosis drugs, which may have a significant role in causing XDR and MDR tuberculosis drug resistance. Continued use of tuberculosis drugs, discontinuation of medicines and various other factors can promote drug resistance in the host's body. To understand the actual cause of resistance, we have identified some patterns of mutations which might be responsible for a change in the structure of the protein, ultimately causing drug resistance. Here, we aim to present some of the unique mutation patterns in the genes associated with the marketed drugs that might have a deleterious effect. In this study, we have used molecular docking approach for understanding the ligand binding affinity of the mutated drugs. The results are further validated by molecular dynamics studies.
Keywords: Tuberculosis, XDR, MDR, Non-synonymous mutations, Molecular docking, Molecular dynamics
1. Introduction
In today's world, Tuberculosis (TB) is still spreading its notorious activity and resulting in mortality worldwide. It is an infectious disease caused by the bacterium “Mycobacterium tuberculosis” [1], [2], [3], [4]. Studies claim that in the year 2017, 10.0 million people were diagnosed with TB and about 1.3 million died from the disease. The evidence suggests that most of the HIV deaths were due to TB. In recent years, specific tests like GeneXpert, TB Culture and Staining for non-pulmonary type have helped in diagnosing TB at an early stage [5]. However, discontinuing treatment for TB and transmission of drug-resistant TB can result in drug resistance which can further be classified into two categories [6]. The categories include (a) Multidrug Resistance (MDR) TB which occurs when the TB is resistant to both isoniazid and rifampicin, at the least whereas, (b) Extensive drug resistance (XDR) TB involves resistance to any fluoroquinolone, and at least one of three second-line injectable drugs (capreomycin, kanamycin, and amikacin along with multidrug resistance [7]. XDR TB is resistant to most of the effective drugs. Hence, in this case, the patients are left with very minimal treatment options that could help in curing the disease. It is of an utmost threat to patients with HIV infection or other disorder or diseases that can decline the immune system. These patients are prone to developing TB and have a higher risk of fatality [8], [9]. In view of this fact, in the present study, we aim to identify the generative cause of drug resistance patterns in strains of Mycobacterium tuberculosis relating to five marketed drugs. The study involves the analysis of non-synonymous mutations that are responsible for making anti-TB drugs ineffective by obstructing the target's structure. This is followed by examining the corresponding altered interactions between the drug and the mutated structures.
2. Materials and methods
It is widely known that open source softwares’ play an imperative role in the scientific community and provides various advantages to users of molecular modeling applications. Literature also suggests a substantial number of open-source software's are under development and many of them are used significantly by the scientific community. Therefore, to be a part of this scientific community, we have used various open-source software's for molecular docking and molecular dynamics studies [10].
2.1. Collection of data
A list of marketed drugs was collected from the web server known as “Web MD”. From the selected marketed drugs, five were shortlisted for further studies based on the availability of 3-Dimensional structure [11]. In the present work, we emphasize on analysing non-synonymous mutations which were collected from the Genome-wide Mycobacterium tuberculosis variation (GMTV) database [19]. This database includes 1084 genomes and over 69,000 Single nucleotide polymorphisms (SNP) or Indel variants, which can be queried about the causative's genome variation across 1623 Mycobacterium tuberculosis clinical isolates, including 1084 MDR strains. This database also gives us a holistic view of the mutations occurring according to the space access on or the gene name.
2.2. Retrieving PDB structures
The crystal structures of the targets in the present study were collected from the Protein Data Bank (PDB) [12] and the structures of the shortlisted marketed drugs were also downloaded from Drug Bank [13]. The details of the target (gene involved), their functions and the shortlisted marketed drugs are presented in Table 1.
Table 1.
List of known antibiotics their corresponding targets, functions, and PDB ID.
| S. No | Generic name of the marketed drugs | Gene involved | Functions involved | PDB ID |
|---|---|---|---|---|
| 1 | Cycloserine | alr | Catalyzes the interconversion of l-alanine and d-alanine. d-alanine is critical in peptidoglycan cross-linking [14] | 1XFC |
| 2 | Capreomycin | gid | Involved in transferring sugar moieties to the substrates involved [15] | 3CKJ |
| 3 | Amikacin | sirR | Sulfite reductase catalyzes the six-electron reduction of sulfite to sulfide [16] | 5IEB |
| 4 | Streptomycin | rpsL | A homodimer enzyme with two catalytic centers at the dimer interface. The binding after the mutation of substrate-specifying residues is altered by this enzyme [17] | 4G3N |
| 5 | Moxifloxacin | gyrA | Controls and helps the enzyme in DNA recognition [18] | 4HTR |
2.3. Generation of mutation patterns
The mutation data was obtained from the GMTV database [19]. The data was organized based on the high degree of mutations in the bacterial genome to identify the “patterns” of mutations. The “patterns” were obtained corresponding to each strain (a single mutation or a combination of two or more mutations) which were organized in the descending order of their frequency of occurrence in the population. A web server known as “Protein Variation Effect Analyser”, (PROVEAN) was used to identify the nature of the mutations and to predict the effect of a mutation on the amino acid sequence [20].
2.4. Homology modeling of mutants’ structures
The PDB structures of all the mutated patterns were generated using Swiss Model, a web server which is widely used for generating protein models [21]. Swiss Model is a fully automated protein structure homology-modeling server, accessible via ExPASy web server. The input was given as the sequence that is to be modelled and based on sequence identity “Swiss Model” gives the best possible outputs. All the mutant structures (of the obtained patterns) pertaining to each target were generated by Swiss Model and downloaded in .pdb format.
2.5. Molecular docking studies
Molecular docking studies for each target gene were carried out using “PatchDock” which functions by setting patches that match to certain patterns in the molecule. The resulting docking scores (binding score) were checked and compared with the wild type target and its mutants. The results revealed the best ten docked models. Of these ten models, one model was selected for further studies [22]. In order to validate the obtained models, we used “FireDock” which is used for refinement and re-scoring of models [22], [23]. The resulting docked complexes were analysed to check the interacting amino acids in for each target gene using BIOVIA Discovery Studio Visualizer [24]. The interactions were analysed to assess the effect of the non-synonymous mutation on the structure.
2.6. Molecular dynamics simulation studies
2.6.1. Protein modeling and structure prediction
The Phyre2 web portal was used for the generation of the 3-Dimensional structure of the protein and prediction of the biological functions of protein molecules from the specific amino acid sequence. The Protein sequences were submitted to the web server and the protein structures were obtained. The mutated model for the protein corresponding to the respective amino acid substitution was generated by Swiss-PDB Viewer (v4.10). The mutant amino acid sequence was used as an input in the Phyre2 web portal for 3D structure modeling and the PDB files obtained were saved for further analysis [25].
2.6.2. MD simulations in water
The Desmond package from Schrodinger 2018–2 was used to run the molecular dynamic simulation. Predefined TIP4P water model was used for the simulation of water molecules. Orthorhombic periodic boundary conditions buffered at 10 Å distances were set up to specify the shape and size of the repeating unit. To neutralize the system prepared, appropriate counter ions (Na+/Cl-) were added and were placed randomly in the solvated system. The minimization and relaxation of the protein/protein-ligand complex were performed after setting up the solvated system by NPT ensemble using default protocol of Desmond as followed elsewhere; which includes a total of 8 stages which includes series of minimization and short simulation steps (18–21) [26], [27]. For a summary of the Desmond's MD simulation stages, please refer to the supplementary file 1.
2.6.3. Analysis of molecular dynamics trajectory
The molecular dynamics simulation trajectories were analysed using simulation event analysis and simulation interaction diagram programs available with the Desmond Module. The Root mean squared deviation (RMSD) and Root mean squared Fluctuation (RMSF) data for Wild-type and mutated proteins were calculated from the simulation interaction diagram. The total Energy was obtained from the simulation files whereas the number of intra-molecular Hydrogen bonds and Radius of gyration was obtained from the Simulation event analysis. The data obtained was then graphed using R project for Statistical Computing [30]. Simulation Event Analysis (SEA) was used to analyze each frame of simulated trajectory output while Simulation Interaction Diagram (SID) was utilized to analyze parameters during the simulation time [27], [28].
3. Results and discussion
3.1. Molecular docking studies
To explore the nature of the mutations under study, a web server known as “PROVEAN” was used to screen the generated mutant patterns, and as a result, several deleterious and neutral mutations were retrieved [29]. In the case of “alr and gid gene” most of the mutations were found to be deleterious. Since “gid gene” is a bulky protein; the effects were seen at multiple sites. The results of the “sirR” gene were in the ratio of 8:10 (neutral: deleterious) suggesting it to be the least harmful set of mutations observed thereby, making it the least drug-resistant mutations. All the mutations seen in the case of “rpsL” gene were found to be deleterious, whereas the results of “gyrA” gene reveal that most of the mutations were deleterious and about 38% were neutral [30].
This analysis was followed by molecular docking studies which were carried out by using “PatchDock”, [22], [23] and “FireDock” was used later for the refinement of the docked models [23]. This method targets issues of docked models like flexibility and scoring solutions. It generates the best pose of the drug and ligand interactions. The input for FireDock was generated by rigid body docking methods and the analysis and interpretation of the docked models were carried out by using BIOVIA Discovery Studio Visualizer [24].
The analysis of docking results is important to predict how the ligand binds to the receptor's binding site [26]. The analysis of the docked model for the cycloserine drug revealed that the diphosphate ring attached to the benzenoid ring forms hydrogen bonds with Tyr 46, Ile 231, Gly 230 and Ser 213. Serine usually participates in hydrogen bond formations whereas, glycine stays inside the protein core. Arg 228 is a positively charges residue that is usually involved in detox processes and arginine, Trp 88 forms a hydrogen bond with the benzene ring. Lys 42 is the only attractive charge present. Hoh 557, Met 170, Lys 133, Asn 212, Pro 229 showed van der Waals interactions with the complex. This model exhibited global energy of 1.50 kcal/mol.
The interaction analysis of Capreomycin revealed that the hydroxyl, oxygen, and nitro groups form hydrogen bonds with Gly 132, Glu 80, Ile 134 and Val 136. The van der Waals interactions were constituted by Arg 131, Arg 222, Val 53, Val 78, Val 135, Ser 130, Ile 50. Arginine is a charged amino acid often forms salt bridges and the model exhibited global energy of −30.41 kcal/mol.
The interaction analysis of Amikacin revealed that Gln 396 is the only hydrogen bonding interaction. However, many hydrophobic interactions with Cys434, Gly 484, Asn 364, Glu 448 were also seen. Cys 434 and Met 432 were involved in both hydrophobic and covalent interactions and the model exhibited global energy of −11.14 kcal/mol.
The interaction analysis of Streptomycin revealed that a pentanone ring is predominant and Gly 341 formed the only hydrogen bond in the model. However, Lys 300 and Thr 302 formed covalent bonding very close to the ring. There were many hydrophobic and van der Waals interactions with Phe 310, Thr 302, Val 340, Lys 303, Leu 304, Lys 300, Ala 339, Arg 312 and the global energy of the model was found to be −26.54 kcal/mol.
The interaction analysis of Moxifloxacin revealed that a polycyclic ketone and the oxygen, hydroxy and nitro groups at the edges of the ring showed hydrogen bonds with Arg 691 and Leu 693, Ile 648 and Lys 636, Leu 693. Lys 634 and Lys 636 formed covalent bonds with the ring and the global energy obtained for this model was found to be 3.92. The interactions diagram for all the marketed drugs has been presented in (Table 2) (Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5).
Table 2.
Interaction analysis, total binding energies and the variation effects of the marketed drugs.
| S. No | Drug | Gene ID | Global energy of docked model (kcal/mol) | PROVEAN (deleterious: neutral) | Interaction Diagram for the best-docked molecule |
|---|---|---|---|---|---|
| 1 | Cycloserine | alr | 1.50 | 16: 08 | 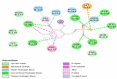 |
| 2 | Capreomycin | giD | −30.41 | 25: 04 | 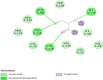 |
| 3 | Amikacin | sirR | −11.14 | 10: 08 |  |
| 4 | Streptomycin | rpsL | −26.54 | 09: 0 | 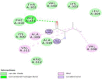 |
| 5 | Moxifloxacin | gyrA | 3.92 | 17: 10 | 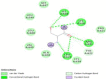 |
Fig. 1.

Plot of the results from PROVEAN and calculation of the Coefficient of determination (R2).
Fig. 2.

The RMSD plot of ALR gene *(Other RMSD plots in Supplementary data 1).
Fig. 3.

The RMSF plot of ALR gene *(Other RMSD plots in Supplementary data 2).
Fig. 4.

The energy plot of ALR gene *(Other energy plots in Supplementary data 3).
Fig. 5.

The Hydrogen bonds plot of ALR gene *(Other energy plots in Supplementary data 4).
3.2. Molecular dynamics (MD simulations)
To make a comparative study the conformational changes in the proteins due to the mutations, molecular dynamics simulation (MDS) was carried out for each protein. Parameters that were analysed include radius of gyration, total energy, total number of intra-molecular hydrogen bonds, RMSD (Root mean squared deviation), as a time-dependent function of MDS. Further, RMSF (Root mean squared fluctuations) of the proteins were also computed. The data obtained were plotted using R statistical tool and a comparative study was made. The time duration of the simulation used is optimal for the protein's native state and to facilitate various conformations. Furthermore, recent studies have shown that the dynamics of a single protein molecule are self-similar and resemble the same, irrespective of duration [28].
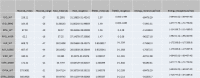
In order to understand the effects of mutations on the structure of the protein, the RMSD values of the wild type/native and other mutant proteins were analysed. The RMSD of the protein alpha carbon was calculated during the simulation with reference to a fixed frame (Table 3).
Table 3.
Statistical analysis for the MD simulations trajectory of wild and mutated proteins.
 |
3.2.1. RMSD
To understand the effect of mutations on the protein structure, their RMSD was calculated throughout the MDS. The RMSD plot of ALR and its mutant reveals that the RMSD of the wild type protein is more stabilized compared to that of the mutant. Further, the wild type RMSD has a lower mean and a narrower range relative to that of the mutant, suggesting that the wild type is more stable than the mutant.
In the case of GID and GYRA, it is clear from the RMSD plot that the wild type has a lower and a more stabilized RMSD compared to the mutant. This clearly reveals that the wild type is protein is more stable than the mutant.
The RMSD plot of RPSL shows that there is a higher variation in the RMSD of wild type compared to that of the mutant. Also, the mean RMSD of the wild type is higher than that of the mutant. This does not provide conclusive evidence comparing the stability of the proteins.
The RMSD plot of SIRR reveals that the RMSD of the mutant is more stabilized and has a higher mean than that of the wild type. This reveals the mutant to be stable relative to the wild type.
3.2.2. RMSF
The RMSF of each residue was also monitored to determine the effects of mutation on the protein residues’ dynamic behavior. The results suggest that residue level fluctuations for wild type are like that of the mutants for ALR and SIRR, indicating similar flexibility. RMSF plot of GYRA indicates that although the Wild type and mutant have similar RMSF for some residues, RMSF of residues 500 – 700 (approx.) reveal wild type to have higher flexibility. The RMSF plot of RPSL reveals that the wild type protein is slightly more flexible and the RMSF of GID shows that the mutant is slightly more flexible than the wild type. In conclusion, the mutant has a higher degree of flexibility in case of GID while the wild type has a higher degree of flexibility in case of GID. Further, no conclusion could be attained from the RMSF plot of ALR and SIRR.
3.2.3. Energy parameters
Analysing the energy parameters for the MDS trajectories revealed that the mean total energy of wild type/native protein to be lower than that of the mutant in the case of ALR and RPSL indicating that the mutant is destabilizing.
The mean total energy of GYRA, SIRR and GID mutants were revealed to be significantly lower than that of their wild type counterparts. This lower total energy of mutants indicates that the mutations have a stabilizing effect on the proteins.
3.2.4. Intra-molecular Hydrogen bonds
Study of the total numbers of intra-molecular Hydrogen bonds of the wild type and mutant proteins (Table 1) revealed that the mutant had the highest mean number of Hydrogen bonds occurred in the case of GID. In the case of all other proteins, the wild type had the highest mean number of hydrogen bonds. In the case of GYRA, the mutant has the largest range for the number of hydrogen bonds. This indicates that mutant has greater flexibility compared to wild type in the case of GYRA.
3.2.5. Radius of gyration
The radiuses of gyration of the wild type/native and mutant proteins were analysed to measure their compactness. From Table 1, it is revealed that wild type protein has the highest compactness in case of all the proteins except RPSL. In RPSL, the mutation K43R has the highest compactness with a radius of 62.2A (Fig. 6).
Fig. 6.

The radius of gyration plot of ALR gene *(Other energy plots in Supplementary data 5) *(Visualization of the protein superimposed of pre (Green) and post (Red) MD structures can be viewed in Supplementary data 6).
4. Conclusions
In the present study, we observed that the obtained mutation patterns led to change in the structure of the protein and eventually the interactions between the drug and the mutated structures were altered. The stability of the target-drug complex was evaluated using docking studies which revealed that the wild type complex was the most stable among all the patterns under investigation. On analysing the stability of the mutated complexes as well as the interactions between the drug and the target(s), it was concluded that the mutations are responsible for making the drug ineffective at the genetic level. The graph plotted from the results obtained from the PROVEAN server as shown in Fig. 1 indicates the coefficient of determination where R² = 0.129 (deleterious mutations) and R² = 0.006 (neutral mutations). This clearly suggests that the deleterious mutations are more significant than neutral ones. Many other factors such as the passage of drug via metabolic networks, pumping out through ion channels etc. might also be responsible for a drug's inefficacy in cases where the docking results were not reasonable. Hence, in the observed data, simple mutations in the genetic basis of life (genome sequence) have been found to initiate the complex changes in the structural and functional aspects of the resulting proteins. Furthermore, the molecular dynamics simulations of these mutant and wild type proteins have demonstrated similar properties and molecular interactions. Subsequently, it can be concluded that the results of molecular docking studies are in line with simulation studies and the mutant protein is structurally and functionally validated. Overall, the current investigation aids to find the mutation patterns which are responsible for the inefficacy of the marketed drugs for Tuberculosis. These results can be taken up for further evaluation and we believe that this approach of identifying the mutation patterns will greatly reduce the chances of drug resistance. We believe that prescribing medicines for Tuberculosis according to the genomic analysis of the individuals will strongly improve efficacy, specificity, fewer side effects with fewer chances of drug resistance.
Ethical statement
Not applicable
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of Competing Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We would like to thank Prof. Samir K Brahmachari for his valuable suggestions. We are also thankful to Ms. Naina Goel for assisting in the project.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jctube.2019.100124.
Appendix. Supplementary materials
References
- 1.Global Tuberculosis Report World health organisation, factsheets. Tuberculosis. 2018 [Google Scholar]
- 2.What is Tuberculosis, Tuberculosis (TB) - What happens, lung disease and respiratory health, WebMD.
- 3.What is the difference between latent tb infection and active tb disease? tuberculosis, Minnesota Department of Health [www.health.state.mn/about/index.html].
- 4.Tuberculosis – Diagnosis and Treatment, Disease and conditions, patient care and health information, Mayo Clinic website.
- 5.Diagnosis Tuberculosis – Clinical Trials, NHS Choices, Patient care and health information, Mayo Clinic website.
- 6.Global Tuberculosis Control 2017, WHO, Geneva, 2017 [http://apps.who.int/medicinedocs/en/m/abstract/Js23360en/].
- 7.TB drug resistance types, Tuberculosis, world health organisation. [https://www.who.int/tb/areas-of-work/drug-resistant-tb/types/en/].
- 8.Drug-Resistant TB, Tuberculosis, Centres for disease control and prevention, U.S. Department of Health and Human Services.
- 9.Joshi J.M. Tuberculosis chemotherapy in the 21st century: back to the basics. Lung India, NCBI. 2011;28(3(July – September)):193–200. doi: 10.4103/0970-2113.83977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pirhadib S., Sunseria J., Koes D.R. Open source molecular modelling. J Molecul Graph Model. 2016 (September);69:127–143. doi: 10.1016/j.jmgm.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Multiple-Drug Therapy for Tuberculosis (TB), Tuberculosis, WebMD.
- 12.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law, V., Knox, C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V., Tang A., Gabriel G., Ly C., Adamjee S., Dame Z.T., Han B., ZhouY Wishart DS. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014 Jan 1;42(1):D1091–D1097. doi: 10.1093/nar/gkt1068. 24203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.PDB ID: 1XFC, LeMagueres P., Im H., Ebalunode J., Strych U., Benedik M.J., Briggs J.M., Kohn H., Krause K.L., The 1.9 a crystal structure of alanine racemase from mycobacterium tuberculosis contains a conserved entryway into the active site, PubMed: 15683232. [DOI] [PubMed]
- 15.PDB ID- 3CKJ. Fulton Z., McAlister A., Wilce M.C., Brammananth R., Zaker-Tabrizi L., Perugini M.A., Bottomley S.P., Coppel R.L., Crellin P.K., Rossjohn J., Beddoe T. Crystal structure of a UDP-glucose-specific glycosyltransferase from a mycobacterium species, (2008) J Biol Chem, pubmed: 18667419. [DOI] [PubMed]
- 16.PDB ID- 5IEB. Campagne S., Dintner S., Gottschlich L., Thibault M., Bortfeld-Miller M., Kaczmarczyk A., Francez-Charlot A., Allain F.H., Vorholt J.A. Role of the PFXFATG[G/Y] Motif in the activation of Sdrg, a response regulator involved in the alphaproteobacterial general stress response, (2016) Structure24: 1237–47, PubMed: 27396826. [DOI] [PubMed]
- 17.PDB ID- 4G3N. Bouige A., Darmon A., Piton J., Roue M., Petrella S., Capton E., Forterre P., Aubry A., Mayer C. Mycobacterium tuberculosis DNA gyrase possesses two functional GyrA-boxes. Biochem J. 2013;455:285–294. doi: 10.1042/BJ20130430. PubMed: 23869946. [DOI] [PubMed] [Google Scholar]
- 18.PDB ID- 4HTR. Smith K.W., Stroupe M.E. Mutational analysis of sulfite reductase hemoproteinreveals the mechanism for coordinated electron and proton transfer. Biochemistry. 2012;51:9857–9868. doi: 10.1021/bi300947a. PubMed: 23153334. [DOI] [PubMed] [Google Scholar]
- 19.Chernyaeva E.N., Shulgina M.V., Rotkevich M.S., Dobrynin P., Simonov S.A., Shitikov E.A., Ischenko D.S., Karpova I.Y., Kostryukova E.S., Ilina E.N., Govorun V.M., Zhuravlev V.Y., Manicheva O.A., Yablonsky P.K., Isaeva Y.D., Nosova E.Y., Mokrousov I., Vyazovaya A.A., Narvskaya O.V., Lapidus A.L., O'Brien S.J. Genome-wide mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics. 2013;15(1):308. doi: 10.1186/1471-2164-15-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi Y., Chan A.P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics (Oxford, England) 2015;31(16):2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., Lepore R., Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucl Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucl Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mashiach E., Schneidman-Duhovny D., Andrusier N., Nussinov R., Wolfson H.J. FireDock: a web server for fast interaction refinement in molecular docking. Nucl Acids Res. 2008;36(Web Server issue):W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dassault Systèmes BIOVIA . v.16.1.0.15350. DassaultSystèmes; San Diego: 2016. (Discovery studio 2016). [Google Scholar]
- 25.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 26.Bowers K.J. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing - SC ’06. 2006. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters. [Google Scholar]
- 27.R Development Core Team R. R: a language and environment for statistical computing. R Found Stat Comput. 2011 [Google Scholar]
- 28.Hu X., Hong L., Smith M.D., Neusius T., Cheng X., Smith J.C. The dynamics of single protein molecules is non-equilibrium and self-similar over thirteen decades in time. Nat. Phys. 2016 [Google Scholar]
- 29.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGrath1 M., Gey van Pittius N.C., van Helden P.D., Warren R.M., Warner D.F. Mutation rate and the emergence of drug resistance in mycobacterium tuberculosis. J Antimicrob Chemother. 2014;69:292–302. doi: 10.1093/jac/dkt364. Advance Access publication 26 September 2013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.